

Abstract
Problem statement
Infection with retroviruses such as human immunodeficiency virus type 1 (HIV-1) and human T cell leukemia virus type 1 (HTLV-1) have been shown to lead to neurodegenerative diseases such as HIV-associated dementia (HAD) or neuroAIDS and HTLV-1-Associated Myelopathy/Tropical Spastic Paraparesis (HAM/TSP), respectively.
Approach
HIV-1-induced neurologic disease is associated with an influx of HIV-infected monocytic cells across the blood-brain barrier. Following neuroinvasion, HIV-1 and viral proteins, in addition to cellular mediators released from infected and uninfected cells participate in astrocytic and neuronal dysregulation, leading to mild to severe neurocognitive disorders.
Results
The molecular architecture of viral regulatory components including the Long Terminal Repeat (LTR), genes encoding the viral proteins Tat, Vpr and Nef as well as the envelope gene encoding gp120 and gp41 have been implicated in ‘indirect’ mechanisms of neuronal injury, mechanisms which are likely responsible for the majority of CNS damage induced by HIV-1 infection. The neuropathogenesis of HAM/TSP is linked, in part, with both intra-and extracellular effectors functions of the viral transactivator protein Tax and likely other viral proteins. Tax is traditionally known to localize in the nucleus of infected cells serving as a regulator of both viral and cellular gene expression.
Conclusion/Recommendations
However, recent evidence has suggested that Tax may also accumulate in the cytoplasm and be released from the infected cell through regulated cellular secretion processes. Once in the extracellular environment, Tax may cause functional alterations in cells of the peripheral blood, lymphoid organs and the central nervous system. These extracellular biological activities of Tax are likely very relevant to the neuropathogenesis of HTLV-1 and represent attractive targets for therapeutic intervention.
Keywords: HIV-1, HTLV-1, neuropathogenesis, viral proteins
Introduction
Human Immunodeficiency Virus type 1 (HIV-1) and Human T cell Leukemia Virus type 1 (HTLV-1) both belong to the retroviridae family of viruses. These two retroviruses by definition infect cells as RNA genomes and utilize viral encoded reverse transcriptase to synthesize a DNA copy of the genome to integrate into the host cell, chromosomal material thereby allowing them to utilize the host cell transcriptional machinery to synthesize components necessary for viral replication. In the case of both of these viruses, this integration ultimately results in productive viral replication that can under certain circumstances lead to an immunodeficiency marked by depletion of CD4+ T cells and neurodegenerative diseases such as HIV-Associated Dementia (HAD) or neuroAIDS and HTLV-1-Associated Myelopathy/Tropical Spastic Paraparesis (HAM/TSP), respectively. In the case of HTLV-1, the integration event may also initiate a series of molecular and signaling events that leads to oncogenic processes within the CD4+ T cell compartment leading to malignancy within this critical immune cell population. Interestingly, these two disease processes share some similarity in that the resulting neurodegeneration is caused at least in part by (1) a dysregulation of inflammatory cytokines and chemokines, (2) recruitment of immune cells to the CNS albeit monocyte-macrophages in HIV-1 and T cells in HTLV-1 and (3) functional properties of viral transactivator proteins, Tat in the case of HIV-1 and Tax with HTLV-1-induced neurologic disease. In both of these retroviral-induced neurologic diseases, the shared mechanistic processes lead to devastating and progressive neurodegenerative consequences. In addition, both diseases have unique qualities. HIV-1 also involves other viral proteins such as Viral protein R (Vpr), Nef and the envelope proteins gp120 and gp41, all of which can be secreted and have been shown to have neurotoxic properties. HTLV-1 on the other hand, has been shown to involve gp46-specific antibodies directed against a number of cellular determinants. This review will provide an overview of the mechanisms of both HIV-1 and HTLV-1 neurodegeneration providing the current knowledge on the similarities and differences of these mechanisms.
Overview of HIV-1-associated neurologic disease
HIV-1 infection, in addition to resulting in the eventual destruction of the host immune system, may induce the establishment of a spectrum of neurologic disorders including severe dementia. Opportunistic infections resulting from Acquired Immunodeficiency Syndrome (AIDS) may affect the Central Nervous System (CNS), however, HIV-1 itself is also able to induce neuropathology[1-3]. Conditions directly induced by HIV-1 include peripheral neuropathies, vacuolar myelopathies, as well as a devastating cognitive and motor disorder known as HIV-Associated Dementia (HAD)[4-7]. With the advent of combination AntiRetroviral Therapy (cART), a mild, more subtle form of CNS dysfunction termed Minor Cognitive Motor Disorder (MCMD) has been described[5,7]. MCMD is characterized by a memory loss, decreased computational ability and much less pronounced higher cortical functions[8]. It has been suggested that ∼10% of adults infected with HIV-1 suffer from HAD and that the prevalence of individuals with MCMD may be much higher, possibly approaching 30% of the HIV-1-positive population[7]. The clinical presentation of MCMD is associated with neuropathogical alterations characteristic of HIV encephalitis (HIVE) and this syndrome is associated with a poorer general prognosis for HIV-1-infected patients.
It is widely accepted that HIV-1 neuroinvasion occurs via transmigration of infected cells of the monocyte-macrophage lineage from the peripheral circulation, across the Blood-Brain Barrier (BBB) and into the CNS. Importantly, although macrophages and microglia are the only cells in the brain to be productively infected by HIV-1, neuronal injury and apoptotic death occurs as a result of HIV-1 infection of the CNS[9,10]. The activation of the monocytic cells in the brain due to infection by HIV-1, viral proteins, or inflammatory mediators generated by the host in response to viral infection seemingly results in the release of neurotoxic viral factors which ultimately lead to astrocytic and neuronal dysfunction, thus driving neuropathogenesis and the establishment of HAD[11]. Additionally, viral proteins are also likely directly involved in neuronal damage and death. Neurotoxic viral proteins like Tat and gp120 are known to excessively stimulate neurons, resulting in excitotoxicity and the loss of critical cellular processes in a way corresponding to other neurodegenerative diseases[1,3,9,12-14].
HIV-1 replication and the long terminal repeat
The initiating step of the HIV-1 lifecycle within an infected host involves a high-affinity interaction between the HIV-1 envelope (env) glycoprotein gp120 and the CD4 antigen expressed on target cells including TH cells and cells of the monocyte-macrophage lineage[15,16]. Viral attachment and subsequent entry into the target cell is then facilitated by the aid of an interaction between the HIV-1 env glycoprotein gp41 and either the chemokine receptor CXCR4 or CCR5, which are predominately expressed on the surface of T cells and mononuclear phagocytes, respectively[15,16]. The determination that CXCR4 and CCR5 function as coreceptors for HIV-1 attachment and entry into target cells led to a generalized but not entirely comprehensive understanding as to why some HIV-1 strains preferentially infect T cells, while others seem to prefer macrophages or both cell populations[15,16]. Once HIV-1 has gained entry into a target cell, its double-stranded RNA genome is reverse transcribed by the viral enzyme Reverse Transcriptase (RT), an RNA-dependent DNA polymerase. This process results in the formation of viral cDNA, which is subsequently integrated into the host's chromosomal DNA by the viral enzyme integrase. The integrated viral genome is referred to as a provirus, which is subsequently transcribed by the host enzyme RNA polymerase II (pol II) resulting in a polycistronic RNA message that is multiply spliced prior to translation. The translated viral proteins combined with two copies of the complete HIV-1 RNA genome allow for the formation of new viral particles, or virions able to infect adjacent cells within the host[17-27].
The process of transcription from proviral DNA is driven by the viral promoter region or Long Terminal Repeat (LTR), a duplicated ∼640 bp DNA region located at the 5′ and 3′ ends of the proviral genome resulting from the mechanism of reverse transcription[28]. HIV-1 transcription is modulated in part by interaction of the LTR with various host factors including members of the CCAAT/Enhancer Binding Protein family (C/EBP), the Specificity protein (Sp) family and Nuclear Factor-kappa B (NF-κB) isoforms, all of which bind to specific cis-acting elements within the HIV-1 LTR facilitating viral gene expression[29-38]. The HIV-1 LTR is also known to interact with viral factors including the trans-activating protein Tat and the accessory protein viral protein R (Vpr), factors which enable expression of viral gene products within multiple host cell populations under selected biological conditions[32,39-41].
HIV-1 Vpr has been shown to interact directly with the LTR. Electophorectic Mobility Shift (EMS) analyses have demonstrated an association with LTR sequences including C/EBP site I, the promoter-distal NF-κB site, as well as the upstream ATF-CREB binding site[33,39]. This interaction between the LTR and Vpr was found to be sequence-specific with respect to C/EBP site I[39], with Vpr preferentially binding a 3T (C-to-T change at nucleotide position 3) C/EBP site I variant, which is known to bind C/EBP factors with low affinity[33]. Importantly, affinity of Vpr for C/EBP binding site sequence variants within the HIV-1 LTR have been correlated to HAD, with the observation that C/EBP binding site variants which bind Vpr at high affinity being more prevalent in proviruses derived from brain tissue of autopsied dementia victims[39].
HIV-1 transcription involves an early, Tat-independent and a late, Tat-dependent phase and transactivation of the viral genome is a critical step in the viral replication cycle[42]. The presence of Tat has been shown to increase LTR-mediated transcriptional activity by several hundred-fold and in the absence of Tat, viral replication falls to nearly undetectable levels[43-45]. Tat is a unique transcription factor in that it binds to the “UCU” bulge of the Transactivation Response Element (TAR), a cis-acting RNA enhancer element contained within the 5′ end of all viral transcripts[42,46]. The interaction of HIV-1 Tat with TAR RNA increases viral transcription and elongation[47,48]. Specifically, HIV-1 Tat is known to promote the binding of pTEF-b (cyclin T1 and cdk9) to the TAR region located within the viral promoter, which is immediately downstream of the transcriptional initiation site. The interaction of Tat with pTEF-b and the TAR element results in hyperphosphorylation of the C-terminal domain and subsequent increased processivity of RNA polymerase II (pol II)[47]. Additionally, recent evidence has suggested that HIV-1 Tat may also be involved with the formation of the transcriptional preinitiation complex[47]. In addition to the HIV-1 LTR, Tat is known to upregulate several other viral as well as cellular genes. Within the CNS, Tat has been shown to stimulate HIV-1 LTR-mediated viral gene expression in the absence of the TAR region[49], an activity that may result from its ability to enhance the activity of cytokines like TNF-α[50]. TNF-α also has the ability to activate the HIV-1 LTR via activation of cytoplasmic NF-κB[50-52] and this positive feedback mechanism may lead to constitutive TNF-α expression within HIV-1-infected cells.
The molecular diversity of HIV-1 is a key mediator of viral replication and fitness[39,53] and sequence variation within Tat appears to influence its effects on HIV-1 LTR activity[54]. Studies have demonstrated tat sequence heterogeneity among brain-derived HIV-1 clones from patients with AIDS[55,56] and while phylogenetic analyses of tat sequences have not revealed clustering among individual clinical groups, genetic diversity has been shown to be greatest among HAD patients[55]. Therefore, sequence variation within Tat likely impacts viral replication and possibly host responses to viral infection[57,58]. A recent study involving astrocytic and monocytoid cells co-transfected with tat clones derived from Non-Demented (ND) and demented (HAD) AIDS patients and varying LTR constructs revealed a decrease in Tat-mediated LTR transactivation[59]. Interestingly, both brain-derived HAD and ND tat constructs induced expression of MCP-1 and IL-1β and microarray analysis revealed that upregulation of several host genes, including an enzyme involved in mediating heparan sulphate synthesis, which has been shown to be upregulated in the brains of HAD patients[59]. These studies suggest that mutations within the tat gene may result in neuropathogenic effects leading to the development of HAD that are independent of its ability to transactivate the HIV-1 LTR[59].
The role of HIV-1 proteins in neurodegenerative disease
The mechanism by which HIV-1 infection induces neuronal damage and subsequent motor and neurocognitive impairment is a controversial subject, however it is generally accepted that HIV-1 does directly infect neurons to a limited extent[6]; although HIV-1 infection of neurons is not considered an important component of the etiology of neurodegenerative disease. HIV-1-associated neuropathology appears to be, in large part, due to neurotoxic viral proteins released into the extracellular environment by infected cells[60]. There is evidence suggesting a role for multiple HIV-1 proteins in CNS deregulation and neuronal injury including Tat, Vpr, Nef and the Env proteins 120 and gp41[6]. These observations have lead to the formulation of two primary theories for how HIV-1 infection results in neuropathology, the ‘direct injury’ theory and the ‘indirect’ or ‘bystander effect’ theory[61]. Clearly, the two theories proposed to explain the HIV-1-induced neuronal damage are by no means mutually exclusive and studies have demonstrated a role for both, however the ‘indirect’ mechanism appears to be the predominate means of neurodegenerative disease since the number of HIV-1-infected cells within the CNS does not always correlative with the extent of CNS pathology[3,10,62].
The HIV-1 envelope proteins 120 and gp41
Several studies have provided evidence for both a direct and indirect role for gp 120 in neuronal injury and death[63-65]. HIV-1 gp120 has been shown to induce neuronal apoptosis by interacting with chemokine receptors, CC-chemokine receptor 5 (CCR5) and CXC-chemokine receptor 4 (CXCR4), expressed on the surface of neurons and glial cells[6,66] (Fig. 1). Additionally, several studies have shown gp120-mediated neuronal apoptosis occurs through interaction with the seven-transmembrane chemokine receptor CXCR4[67-71]. Binding of gp120 to CCR5 and CXCR4 activates intracellular signal transduction pathways that mediate neuronal apoptosis[66,72,73]. Several studies have shown that gp120 disrupts calcium homeostasis in neurons and induces disruption of mitochondrial membrane integrity leading to release of cytochrome c and activation of caspases and endonucleases[73-75]. Caspase-3 activation by gp120 has been demonstrated in rat cerebellar granule cells[76], human embryonic kidney cells[77] and human endothelial cells[78]. Conversely, studies have shown that caspase-3 inhibition protects neurons from apoptosis, suggesting that therapeutic candidates aimed at inhibiting caspase-3 activation may prove valuable in preventing gp120-induced neurotoxicity[79].
Fig. 1.
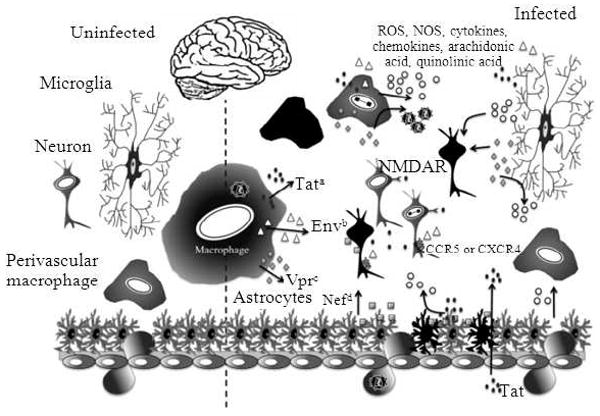
Mechanism of HIV-1-induced neurodegeneration. Neurodegeneration caused by HIV-1 infection is the result of both direct infection of cells, as well as the release of HIV-1 proteins that cause additional neurodegeneration. Infected perivascular macrophages and microglia, in addition to producing more infectious virus, can also release these viral proteins. These proteins include Tat, Env (120 and gp41) and Vpr; (a) Tat (represented as ●) exhibits effects on both infected and uninfected cells, because secreted Tat can be taken up by neighboring cells. Tat has been demonstrated to play a role in oxidative stress-dependent apoptosis of neurons (represented by black cells). It has also been shown that Tat can cause excitotoxicity in neurons by activating the NMDAR; (b) Env (represented as △) also plays both a direct and indirect role in neurodegeneration. The Env protein gp120 has been shown to interact with both CCR5 and CXCR4 expressed on the surface of neurons and glial cells and subsequently induce neuronal apoptosis. Macrophages and microglia activated by gp120 secrete various proinflammatory factors including cytokines, chemokines and arachidonic acid, resulting in neuronal damage; (c) Vpr (represented as grey ◆) has been shown to increase viral transcription and production in cells of the monocyte/macrophage lineage. Extracellular Vpr has also been demonstrated to induce apoptosis in undifferentiated and mature neuronal precursor cells; (d) Nef (represented as grey ■) within the brain has been shown to be predominantly in astrocytes. Stable expression of Nef may alter the growth properties of astrocytes. Nef also demonstrates direct neurotoxicity. When cells of the monocyte-macrophage lineage are exposed to Nef, proinflammatory mediators are released
Calcium homeostasis is disrupted in neurons by gp120-induced perturbation of calcium-regulating systems in the plasma membrane and Endoplasmic Reticulum (ER)[80,81]. In addition, gp120 activation of tissue macrophages and microglia initiates glutamate-related hyperactivation of the N-Methyl-D-Aspartic Acid Receptor (NMDAR), which has been shown to be involved in ER stress and Ca2+ release[82,83]. ER stress may be involved early in the process of cellular apoptosis[84] and it is associated with multiple neurodegenerative diseases[85].
Macrophages and microglia activated by gp120 secrete various factors including TNF-α, arachidonic acids and various β-chemokines, resulting in neuronal damage[6] (Fig. 1). Exposure of astrocytes to gp120 inhibits their ability to take up glutamate, leading to increased glutamate concentrations within the neuronal microenvironment, which results in excitotoxicity[86] (Fig. 1). Additionally, gp120 has been demonstrated to alter gene expression patterns in astrocytes[87,88] and neurons[65], providing evidence that alterations in cellular processes may contribute to neuronal damage.
The HIV-1 transmembrane protein gp41, a protein which links gp120 to the viral envelope has been observed at elevated levels in patients with HAD. Studies have demonstrated gp41 to be lethal to neurons at very low concentrations in the presence of glia and it has been shown that astrocytes exposed to the carboxy-terminus of gp41 exhibit defects in glutamate transport and release[89] (Fig. 1). The mechanisms of neurotoxicity induced by gp41 involves activation of iNOS, NO formation, glutathione depletion and interruption of mitochondrial function[90-92].
The HIV-1 accessory protein Vpr
Viral protein R (Vpr) is one of six auxillary proteins encoded for by HIV-1 and several studies have suggested that this protein may have a role in HIV-1-associated neuropathogenesis[13,93-95]. HIV-1 Vpr is a critical accessory protein of 96 amino acids (14 kDa) synthesized from a singly spliced viral mRNA, which may oligomerize[95,96]. HIV-1 infection of non-dividing cells such as monoctyes and macrophages is critically dependent on Vpr[97,98] and the addition of extracellular Vpr to latently infected T lymphocytes has been shown to dramatically increase viral replication[99]. Vpr has been shown to increase viral transcription and production in cells of the monocyte-macrophage lineage and it may be involved in the translocation of the viral Preintegration Complex (PIC) from the cytoplasm to the nucleus[96,98,100,101]. It has been demonstrated that HIV-1 Vpr arrests T lymphocytes in the G2 phase of the cell cycle[97,102-108] and induces adherent cell differentiation[109]. Interestingly, Vpr has also been shown to suppress the immune response in humans by inhibiting clonal expansion of T cells[110], providing an ideal physiological environment for efficient viral replication[103,111].
Vpr, by binding to the p6 portion of the Gag polyprotein, is incorporated into HIV-1 virons in significant quantities[112] and both purified Vpr, isolated from HIV-1-infected patients and recombinant Vpr has been shown to activate viral replication from latently infected cells. HIV-1 Vpr is primarily localized in the nucleus of the infected cells[113], likely due to the strength of its two nuclear localization signals[114]. This nucleophilic property of Vpr in combination with its presence in the PIC has led to the suggestion that Vpr may facilitate increased viral replication in non-dividing cells like cells of the monocyte-macrophage lineage[98,100,101,115]. Some in vitro studies have provided evidence in support of a direct role for Vpr in PIC import[116,117]. However other studies have placed this observation in question[118] and have suggested that Vpr is not required for HIV-1 infection of non-dividing T lymphocytes[119].
The mechanism by which Vpr is incorporated into budding virions, which are assembled in the cytoplasm near the plasma membrane, is not well understood; however, studies have shown that the nuclear export property of Vpr is required for its efficient incorporation into virions, which is critically important for productive viral replication in tissue macrophages[120]. The absence of Vpr does not prevent HIV-1 infection of tissue macrophages, however, its presence has been demonstrated to greatly enhance infection of these cells[120]. Studies have suggested that Vpr may function like an importin-β homologue by directly binding to nucleoporins within the Nuclear Pore Complex (NPC)[116,121,122]. Matrix (MA) and Integrase (IN) proteins are known to utilize the importin-α/β-dependent pathway for nuclear import, however, Vpr contains a noncanonical NLS and therefore does not use these classical pathways exclusively for nuclear import[101,114,123,124]. It may be that HIV-1 has evolved a novel strategy for avoiding cellular defense mechanisms aimed at preventing viruses from entering the nucleus of infected cells. Considering the large and significant role that HIV-1-infected macrophages play both in peripheral blood and CNS disease as well as HIV disease in other organ systems, it is reasonable to suggest that interrupting Vpr function in vivo may lead to a marked decrease in viral burden within HIV-1-infected patients and ameliorate some of the deleterious effects of HIV-1 infection.
Mutational studies have revealed that the region of the Vpr responsible for cell-cycle arrest is located in the carboxy-terminal basic domain, while the virion incorporation and nuclear translocation functions of Vpr are elicited by the α-helical amino-terminus, suggesting that Vpr likely effects cellular functions in multiple ways[125-129]. Additionally, investigations have shown that Vpr appears to induce apoptosis in T lymphocytes[130,131] and studies have revealed that extracellular Vpr induces apoptosis in undifferentiated as well as mature, differentiated NT2 CNS neuronal precursor cells (derived from a human teratocarcinoma cell line)[95]. Importantly, Vpr has also been shown to both activate and differentiate monocytic cells in the bone marrow[132], a cellular compartment believed to be critically important in the neuropathogenesis of the HIV-1. Due to the multiple effects of HIV-1 Vpr on both T cells and cells of the monocyte-macrophage lineage, in addition to the potential apoptotic effect of extracellular Vpr on neurons, it has been suggested the Vpr very likely plays an important role in CNS damage during the course of HIV-1 infection[13] (Fig. 1).
The HIV-1 auxillary protein Nef
HIV-1 Nef is a nonstructural protein and one of its best known properties is its ability to impact the trafficking of proteins expressed on the surface of cells. Nef has been shown to down-regulate CD4 expression in addition to down-regulating expression of a subset of MHC class molecules[133-135], mature MHC class II molecules, CD8, CD28, CCR5, CXCR4 and the transferring receptor[136-142]. The downregulation of CD4 in HIV-1-infected cells has been shown to facilitate Env incorporation into infectious virions by inhibiting CD4/gp120 complex formation on the surface of the cells[143-145].
Much like Vpr, Nef is also incorporated into HIV-1 virions[146-149]. Nef has been suggested to play a major role in HIV-1 disease progression toward AIDS[150-156] and this increase in viral infectivity associated with Nef may explain the increased pathogenicity of wild-type (wt) HIV-1 compared to ΔNef HIV-1 variants[134,157,158]. Studies have suggested that Nef may play a role in the fusion of HIV-1 to target cells[159] and that Nef may be involved in certain post-fusion events such as facilitating the trafficking of viral core particles through the cortical actin network[160].
Nef affects cellular function in various ways and Nef has been detected in the supernatants of HIV-1-infected cell cultures as well as in sera of AIDS patients[161]. Within the brain of HIV-1-infected patients, Nef is found predominately in astrocytes[162]. Direct neurotoxicity of Nef has been shown by exposure of neuronal cell cultures to recombinant Nef (rNef)[163] (Fig. 1) and studies have revealed that Nef increases total K+ current of chick dorsal root ganglions similar to scorpion neurotoxin[164,165], suggesting that Nef is able to alter the electrophysiological properties of neurons. Exposure of human monocytic cells to extracellular Nef results in the release of inflammatory mediators (Fig. 1) and exposure of neuronal and astrocytic cell lines to rNef leads to the upregulation of complement factor C3[166]. In addition, studies have indicated that stable expression of Nef may alter growth properties of human astrocytes[162].
HIV-1 transactivator protein Tat
HIV-1 Tat is a viral nonstructural protein of 86-101 amino acids in length and it is the product of two exons[3]. Tat, a transactivating nuclear regulatory protein, is critical for viral replication and is secreted by HIV-1-infected cells. Secreted Tat may be taken up by neighboring cells and by this mechanism, Tat is able to elicit affects on both infected and uninfected cells[167,168]. Tat has been found in the brains of HIV-1-infected individuals with known CNS pathology[169] and Tat is known to trigger oxidative stress-dependent apoptosis of neurons both in vitro and in vivo.
Previous investigations have shown that HIV-1 subtype B-derived Tat can cause excitotoxcity in neurons by activating NMDAR[170], however there is some controversy as to whether Tat is able to bind NMDAR directly. Studies have suggested that Tat neurotoxicity is dependent on binding to low-density receptor (LPR) with subsequent activation of NMDAR[171]. A more recent study aimed at comparing the neurotoxic potential of subtype B and C Tat protein has suggested that Tat may be able to directly bind to NMDAR[172]. More specifically, this study demonstrated that both subtype B and C Tat bind directly to NMDAR, however, subtype C Tat was significantly less neurotoxic. Further analysis of sequence differences between the two Tat subtypes revealed that the attenuated neurotoxicity was due to a Cys31Ser mutation found in subtype C Tat. Furthermore, engineering this mutation into a subtype B Tat resulted in decreased neurotoxicity similar to that observed with subtype C Tat. Interestingly, the Cys31Ser mutation had no observable effect on the ability of Tat to bind NMDAR[172], suggesting that other regions of the Tat protein are likely responsible for binding, while the Cys residue is involved in NMDAR activation[172]. These studies underscore potential importance of differences in HIV-1 subtypes from varying geographical regions and are consistent with the observation that geographical regions infected with subtype B report more severe forms of HIV-1-associated neurocognitive impairment as opposed to areas of the world infected with subtype C.
Several studies have shown that NMDAR function may be modulated by dopamine D1-like receptors, composed of D1- and D5-like receptors[173-178] and a recent study has implicated D1-mediated pathways in the mechanism of Tat-induced neurotoxicity[3]. HIV-1 Tat may influence the activity of D1 receptors in postsynaptic neurons, thereby resulting in NMDAR-regulated apoptotic cascades via D1/NMDAR interaction, or alternatively, NMDAR activation in D1-expressing neurons exposed to Tat may up regulate pro-apoptotic D1-mediated signaling[3].
Tat has been detected in the brains of patients with HIV-1 encephalitis (HIVE) by both mRNA and immunoblot analysis, however the source of the Tat detected in these circumstances is unclear[179]. The Tat observed in the brains of these patients may be secreted by infected cells within the CNS, or it may be specifically transported across the BBB from the periphery[180]. Either way, once in the brain, Tat may be taken-up by resident cells of the CNS, often resulting in toxic consequences including neuronal apoptosis[181-183] (Fig. 1). Importantly, studies have shown that Tat can be transported along anatomical pathways within the CNS, indicating that sites of neuronal injury and the site of actual viral infection may be distinct from one another[184].
The uptake of Tat by uninfected cells results in deleterious events in both the cytoplasm and nucleus, including altered gene transcription, cytokine secretion, NMDAR activation in neurons and the initiation of apoptotic cascades[3,46,167,168]. A combination of these cellular events are likely involved in neuronal apoptosis in response to HIV-1 infection and the secretion of Tat, however the exact mechanism is not well understood. It is generally accepted that glutamate and glutamate receptors are involved in the process of neuronal cell death, which suggests that HIV-1-associated neurologic disease pathogenesis may involve mechanisms similar to those of other neurodegenerative disease processes with respect to glutamate dysregulation and excitotoxicity[3].
HIV-1 Tat is known to interact with various receptors expressed on different cell types including integrins, VEGF receptor (KDR/flk) and possibly CXCR4[185,186]. With respect to neurons, Tat uptake and internalization occurs primarily via the Lipoprotein Related Protein (LRP) receptor, which is expressed on the cell surface[187]. There are at least 16 ligands for LRP and in the brain it is expressed on both neurons and activated astrocytes[185]. Tat is the only known ligand of LRP that induces significant levels of apoptosis, the reasons for which are not known. Following ligand binding to LRP, the receptor-ligand complex is internalized. However, in the case of Tat, when it binds to LRP it escapes from endosomes to the cytoplasm and it can be found localized in both the cytoplasm and nucleus where it may affect cellular signaling pathways[188].
Exposure of astrocytes to Tat, either extracellularly through viral infection or intracellularly via transient transfection, results in the induction of cellular inflammatory mediators (Fig. 1). Treatment of human fetal astrocytes with extracellular Tat has been shown in induce production of CCL2 (monoctye chemoattractant protein-1, MCP-1)[189]. CCL2 is involved in the recruitment of monocytes to sites of inflammation in the CNS and it induces monocytic cells to cross the BBB and traffic to the site of secretion. Furthermore, it has been demonstrated that individuals with HIVE have elevated levels of CCL2 in their CSF and even higher levels have been associated with HAD[189,190]. Interestingly, human astrocytes take up Tat and become activated to produce chemokines as well as Nitric Oxide (NO), however, these cells do not undergo apoptosis unless they are co-cultured with neurons[168,181,191]. It has been suggested that Tat-induced apoptosis of astrocytes requires a yet to be identified signal from NMDAR-positive neurons[171].
Monocytes recruited to the CNS by CCL2 secrete cytokines and neurotoxic mediators[6]. A primary function of astrocytes is to regulate extracellular glutamate levels in the brain and it has been demonstrated that monocyte-secreted TNF-α impedes glutamate metabolism by human astrocytes, resulting in altered glutamate homeostasis, accumulation of extracellular glutamate and potentially neuronal injury[192]. In addition to the effect of Tat on glutamate metabolism in astrocytes and the resulting neuronal glutamate excitotoxicity, other mechanisms are known by which astrocytes contribute to neuronal toxicity and apoptosis. Tat-treated astrocytes express inducible Nitric Oxide Synthase (iNOS) via activation of the NF-κB and C/EBPβ pathways and the ERK/MAPK pathway contributes to NO production in astrocytes by activation of C/EBPβ[191]. The NO produced by this mechanism may initiate apoptotic events in neighboring neurons.
Microglia are resident macrophages in the CNS and the most predominate cell type to be productively infected by HIV-1 in the brain[171]. Despite the relatively high level of HIV-1 infection of resident microglia, neurocognitive impairment does not correlate with viral load in the brain, but rather with neuroinflammation[193]. Activated microglial cells secrete pro-inflammatory factors including cytokines and chemokines, reactive oxygen species, reactive nitrogen species and excitatory amino acids, which leads to the recruitment of additional inflammatory cells to the brain. Due to the fact that some of the inflammatory mediators secreted by microglia are known to be neurotoxic, like quinolinic and arachidonic acids[194], it has been suggested that activation of microglia may contribute both directly and indirectly to neuronal damage and apoptosis within the context of the HIV-1 infection of the CNS[171] (Fig. 1).
Overview of HTLV-1 neuropathogenesis
The debilitating neuroinflammatory disease, HAM/TSP, is characterized by over-stimulation of the immunologic compartment, including increased expression of a repertoire of inflammatory cytokines and chemokines, HTLV-1 transactivator protein Tax- and gp46-specific antibodies directed against a number of cellular determinants (including the heteronuclear ribonuclear protein A1, hnRNP A1)[195] and an increase in the number of highly activated circulating CD8+ T cells directed against the Tax11-19 epitope in both Peripheral Blood (PB) and Cerebrospinal Fluid (CSF)[196]. In the acute stage of HAM/TSP, both CD4+ and CD8+ T cells have been shown to accumulate in lesions of the spinal cord; however, during chronic disease, CD8+ T cells are the predominant cellular infiltrate in regions of demyelination[197-199]. In some HLA-A*201 HAM/TSP patients, the frequency of Tax11-19-specific CTLs is as high as 20% of all CD8+ T cells in the PB and even higher in the CSF[195,196,200-203]. During the course of HAM/TSP, various cells of the immune system are infected by HTLV-1 in the PB and tissues. These cells are also likely infected as BM progenitor cells as a result of viral spread from HTLV-1-infected CD4+/CD25+ T cells as these cells traffic back into the BM from the blood. Trafficking of HTLV-1-infected T cells back into the BM over long periods of time has led to the development of a large population of HTLV-1 proviral DNA+/RNA- latently infected cells in the BM of HAM/TSP patients[204]. Additional studies involving HTLV-1-infected CD34+ human BM progenitor cells transplanted into immunocompromised mice led to the development of equivalent numbers of proviral DNA+ cells within defined immune cell lineages, suggesting that maintenance of the proviral genome does not have any detectable impact on lineage development[205]. In addition to the route of exposure (mucosal versus blood-borne) and the inherent differences in the associated primary immune response, the following factors likely play important roles in the genesis and progression of cancer and/or neurologic disease caused by HTLV-1: (i) Viral genomic architecture, (ii) Genetic background of the human host, (iii) HTLV-1 proviral DNA load, (iv) Relative levels of HTLV-1-specific RNA-positive CD4+/CD25+ T cells, (v) Concentrations of secreted extracellular viral proteins such as Tax, (vi) The impact of extracellular viral proteins on cells of immune and nervous system origin, (vii) Relative levels of HTLV-1-specific antibodies that exhibit cross-reactivity to cellular proteins, (viii) Relative levels of HTLV-1-specific CD8+ T lymphocytes and (ix) Relative levels of HTLV-1-susceptible primary and secondary target cells in the immune and nervous system[200,201,206-210]. Figure 2 depicts these scenarios in the context of molecular pathogenesis of HAM/TSP.
Fig. 2.
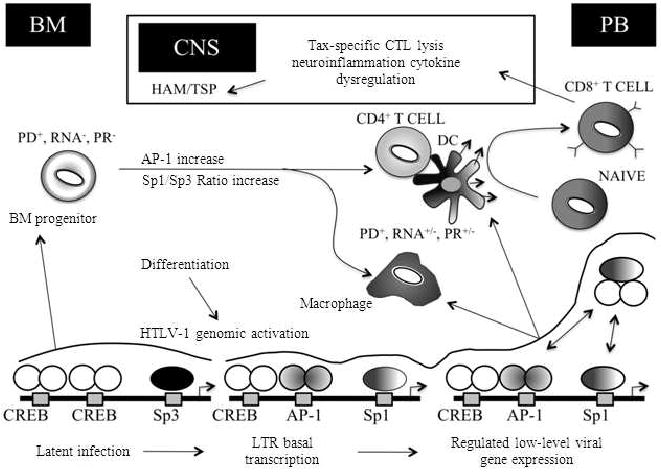
Overview of the HTLV-1 LTR regulation as it relates to HAM/TSP pathogenesis. The progressive stages of HAM/TSP are characterized by the presence of activated CD4+ and CD8+ T cells and macrophages in demyelinating lesions. At these sites an array of proinflammatory cytokines are produced facilitating further recruitment of inflammatory cells into the CNS. CD4+ T cells represent the chief source of viral gene expression and along with the help of antigen presenting cells such as dendritic cells activate CD8+ T cells. HTLV-1-specific CD8+ T cells traffic to and accumulate within the CNS throughout the course of neurologic disease. Therefore, the presence of activated HTLV-1-specific CD8+ CTL and macrophage populations in the CNS may result in the maintenance of a persistent CTL response against infected cells expressing viral antigens and proinflammatory cytokine-mediated bystander damage. During this process, differentiation of infected bone marrow progenitor cells lead to HTLV-1 genomic activation and an increased viral gene expression mediated by Tax-CREB dimer formation and the cell type-specific activity of different transcription factors such as AP-1, C/EBP and Sp1/Sp3. BM, bone marrow; CNS; central nervous system; PB, peripheral blood; PD, proviral DNA; PR, proviral replication; DC, dendritic cell
Nuclear localization and Tax-mediated regulation of cellular genes and viral promoter
The HTLV-1 oncoprotein Tax has been studied extensively with respect to its role in regulating cellular and viral gene expression during the course of HTLV-1 infection of human CD4+ T ells (primary target ell opulation)[211-213]. In particular, these studies have focused on the molecular mechanisms pertaining to the interaction of Tax with nuclear proteins during transcriptional transactivation of cellular and viral gene expression prior to and/or subsequent to Tax localization to the nucleus. One of the major roles of Tax in productive viral replication involves the interaction with host transcriptional machinery to facilitate the binding of cellular transcription factors to the HTLV-1 LTR. Viral gene expression occurs by the binding of Tax to the three 21-bp Tax responsive elements collectively referred to a Tax Responsive Element 1 (TRE-1) that are located within the U3 region of the LTR[214-216]. Each of the three 21-bp repeat elements is comprised of three domains A, B and C. Among these, the central domain B has been shown to closely mimic a cyclic AMP (cAMP) responsive element (CRE) with a conserved 8-nucleotide (nt) core sequence (TGACGTCA) that is flanked by 5′ and 3′ G/C rich sequences[217]. The recruitment of the cellular transcription factors-CRE Binding protein (CREB) and serum response factor (SRF or p67SRF)-to the CRE by Tax, facilitates transcription[218,219]. The interaction of Tax with dimeric CREB as a homodimer, results in the formation of a ternary complex that in turn helps to stabilize the CREB/21-bp repeat complex formation[217,220].
As a consequence of the formation of this stabilized complex, the two cellular coactivators-p300/CREB-Binding Protein (p300/CBP) and p300/CBP-associated factor (P/CAF) are then recruited independently by Tax by binding to the two distinct regions in the amino and carboxy terminus of Tax, respectively. Transcription is then finally initiated by histone acetylation mediated through remodeling of the chromatin structure[221-223]. In addition, Tax is also known to activate various cellular signaling pathways such as the NF-κB and ATF/CREB pathways. The NF-κB is a cytoplasmic transcription factor which when activated by Tax leads to the phosphorylation and degradation of the bound inhibitory proteins (IκBs) thereby allowing the release and eventual translocation of the NF-κB to the nuclei[224,225]. The ATF/CREB family plays an important role in viral transcription as well as Tax-mediated transcription of cellular genes. CREB is activated by phosphorylation in response to the intracellular secondary-messenger cAMP via the cAMP dependent Protein Kinase A (PKA) signaling pathway[226-228]. Numerous studies have focused on the interaction between Tax and CREB-1 with TRE-1 and the importance of this interaction in the activation of viral gene transcription mediated by the TRE-1 21-bp repeats[216,229]. These studies have demonstrated that direct and indirect interaction of Tax with a spectrum of cellular transcription factors may be involved in the differential regulation of both cellular gene expression and viral LTR activation in T cells and other secondary target cell populations.
Mechanism of HTLV-1 LTR activation in secondary target populations
Clearly, the interaction of HTLV-1 with the CD4+/CD25+ T lymphocyte compartment represents a key encounter that leads to either cell death resulting from programmed viral gene expression and production of progeny virus, or oncogenic transformation based on monoclonal expansion of infected cells. In addition to the CD4+/CD25+ T cells, other immune cell populations, such as CD34+ BM progenitor cells[230,231], cells of the monocyte-macrophage lineage[232], antigen presenting cells such as Dendritic Cells (DCs), or cells in the nervous system such as astrocytes and microglial cells[232-236] are also susceptible to productive HTLV-1 infection[237]. Viral-induced alterations in these cell compartments likely play important roles in the genesis of HAM/TSP. However, despite the demonstrated susceptibility of these cells very little information exists concerning the molecular mechanisms regulating HTLV-1 LTR activation in these pathogenically relevant secondary target cell populations as compared to CD4+/CD25+ T cells. Therefore, we have initiated efforts to define the mechanism of HTLV-1 LTR activation during monocytic differentiation in parallel with our studies on HIV-1 LTR regulation in a similar system[33,38,238]. Several members of the CCAAT/Enhancer-Binding Protein (C/EBP) family, including C/EBPβ, are expressed at high levels in cells of the monocyte-macrophage lineage[239] and are intimately involved in the regulation of myelocytic-monocytic gene expression. Results of these studies have shown that low-level basal activation of the HTLV-1 LTR was significantly enhanced by overexpression of C/EBPβ, C/EBPδ, or C/EBPε, whereas transactivation of the HTLV-1 LTR by Tax was inhibited by overexpression of C/EBPα and C/EBPβ and to a lesser extent by C/EBPδ[240]. In addition, the Activator Protein 1 (AP-1) family of transcription factors was also shown to modulate HTLV-1 LTR activation during phorbol ester-induced differentiation of monocytes[241]. The binding sites for the Stimulating protein (Sp) family of transcription factors (Sp1 and Sp3) have been identified within the U3 region of the HTLV-1 LTR[242]. Recent results have suggested that both Sp1 and Sp3 binding to HTLV-1 TRE-1 promoter proximal repeat III within the viral LTR participate in regulation of the LTR by Tax. However, a majority of studies related to HTLV-1 LTR activation[221,243-245] have been performed with transiently transfected LTR plasmids as opposed to chromosomally integrated LTRs that represent an obligatory step in the HTLV-1 life cycle. A report by Okada and Jeang indicated differential requirements for activation of integrated and transiently transfected HTLV-1 LTRs in HeLa and CHOK1 cells by CREB, p300 and P/CAF transcription factors[246]. Our recent studies also revealed critical differences in the regulation of transiently transfected and chromosomally integrated HTLV-1 LTR in T cells (Rahman, Wigdahl and Jain, unpublished observations). Therefore, understanding how the HTLV-1 LTR is regulated when formatted in the context of chromatin is important for elucidating the biology of the provirus within cell populations representative of those encountered during the course of disease.
Tax nuclear export, cytoplasmic trafficking and cellular secretion
Tax is localized in both the nucleus and cytoplasm within cells (reviewed in[247]). The nuclear accumulation of Tax is promoted by an NLS found within the first 58 amino acids of the amino-terminus of the protein, a signal that is unique when compared to classical NLSs[248,249] in that the signal is suspected to involve some form of conformational element. In addition to being an intracellular/nuclear protein, Tax has been shown to be present in the serum and CSF of HAM/TSP patients[250]. However, it is unclear whether the cell-free Tax was the result of apoptosis or necrosis of HTLV-1-infected cells or if it was secreted from the infected cell populations. Consistent with the concept of cellular secretion, we have reported the presence of a leucine rich NES between amino acids 188-200 of Tax. We have also reported that the Tax NES may function as a masked NES and may be hidden by a protein-protein interaction, allowing the Tax NLS to remain the predominant localization signal and directing protein localization to the nucleus. Furthermore, Tax tertiary structure may be altered during the successive steps in which the NES is subsequently exposed to cellular export machinery. Alternatively, Tax nuclear export may be regulated through a secondary protein modification such as phosphorylation or acetylation. In this regard, Tax phosphorylation has been studied in various cell types and alterations in the pattern of Tax accumulation within the cytoplasm have been reported[251,252]. The release of Tax in the extracellular environment has also been reported from HTLV-1-infected cells[253]. We have also demonstrated the secretion of full-length Tax in the cell culture media of Tax-transfected cells where Tax secretion was, at least in part, dependent on a formal cellular secretory pathway[254]. Proteins destined for the cell surface or those released into the extracellular environment proceed through the cellular secretory pathway after being synthesized and inserted into the Endoplasmic Reticulum (ER)[255]. We have previously demonstrated that Tax co-localizes with cytoplasmic organelles relevant to secretion such as ER and golgi complex and the movement of Tax within the cytoplasm was found to be characteristic of secretory vesicles[254]. Evidence was also presented demonstrating that microtubules and the conventional motor protein kinesin are likely involved in shuttling Tax-containing secretory vesicles from the Golgi to the plasma membrane[254]. Subsequent to proper folding in the ER, secretory proteins are then sorted from resident ER proteins and concentrated into ER exit sites that form coat protein complex II (COPII)-coated vesicles. COPII vesicles mediate transport of proteins from the ER to the cis-Golgi[256]. Concentration of proteins into COPII vesicles within the ER occurs through a cargo selection motif found on most secreted proteins. This motif has been shown to consist of a di-acidic signal comprised of an aspartic or glutamic acid bordering a variable residue (D/ExE/D) also known as a DXE signal[257-259]. Table 1 shows the known amino acid signals implicated in targeting proteins to the cellular secretory pathway. While this signal only partially accounts for efficient exit from the ER, mutation of this signal has been demonstrated to reduce accumulation of protein in COPII vesicles[260]. Analysis of the Tax amino acid sequence has revealed the presence of a number of putative secretory signals within the carboxy-terminal domain, of which two putative secretory signals, DHE and a four amino acid di-hydrophobic tyrosine-based motif (YTNI), were found to be essential for Tax secretion[261]. Additionally, Tax was shown to interact with a number of proteins (Fig. 3) involved in the cellular secretory pathway[261,262] suggesting that release of Tax into the extracellular environment is a regulated event that is facilitated by the interaction of Tax with cellular secretory pathway proteins and the presence of critical secretory signals within the carboxy-terminal domain of Tax. Once released, Tax could work as an extracellular effectors molecule, the minute quantity of which may cause major pathogenic changes.
Table 1.
Amino acid signals implicated in targeting proteins to the cellular secretory pathway
| Sorted protein | Single sequence | Role of signal sequence |
|---|---|---|
| Di-acidic: | ||
| VSV-g (virus) | DXE | Cargo concentration and exit from ER |
| Syspl (yeast) | DXE | Binding to 23 sec/24 sec of COPII coat |
| Kir 1.1 (mammalian) | DXE | ER export |
| Kir 2.1 (mammalian) | DXE | ER export |
| Di-hydrophobic tyrosine-based SIV env (viral) | YRPV | ? |
| HIV gp 160 (viral) | YSPL | ? |
| TGN38 (mammalian) | YQRL | Targets protein to the TGN |
| Emp46p (yeast) | YYMF | ER export/localization to Golgi |
| D-hydrophobic tyrosine-based: | ||
| ERGIC-53 (mammalian) | FF | ER export |
| Sysp 1(yeast) | FF | Non-functional |
| P24 family (mammalian) | FF | Binding to 23 sec COPII component in vitro |
| D-hydrophobic Di-leucine: | ||
| Emp24 (yeast) | LV | ER export/incorporation into COPII vesicles |
| Vam3p (yeast) | LL | Sorting to golgi |
| Erv41p-Erv46p (yeast) | IL | Packing into COPII vesicles |
Fig. 3.

Components of the cellular secretory pathway important for nucleocytoplsmic shuttling, endoplasmic reticulum to golgi transport and post-golgi transport to the plasma membrane. While NTF p97 may facilitate nuclear import of Tax, CRM1 and calreticulin may facilitate its export through the nuclear pore complex. Tax is likely targeted to the secretory pathway by being transported into the Endoplasmic Reticulum (ER). Once in the ER, Tax likely moves to the Golgi by inclusion into COPII vesicles. Amino acid signals within Tax including 330DHE332 and 312YTNI315 were shown to be important for targeting Tax to the Golgi. A portion of Tax may also be returned to the ER via retrograde transport by inclusion into COPI vesicles. In the Golgi, Tax is likely included into secretory vesicles through its interaction with SCAMPs and vSNAREs including SNAP23
Role of extracellular tax in HTLV-1 neuropathogenesis
Extracellular Tax has been shown to induce the production of TNF-α from a human neuronal cell line at a concentration that has been shown to be produced by HTLV-1-infected cells[263,264]. Release of TNF-α may result in both an autocrine and paracrine cytokine-mediated destruction of neuronal tissue. Other pathologic processes observed in HAM/TSP patients include demyelination of CNS neurons, which may also be a direct effect of extracellular Tax[263,265]. In addition to neurons, adult human microglial cells were also shown to secrete TNF-α, IL-1β and IL-6 in response to Tax[266]. These observations correlate with additional studies demonstrating that HTLV-1-infected microglial cells secrete both TNF-α and IL-6 but not IL-1β, suggesting that Tax may have a paracrine effect on other Tax-producing cells[266]. The effects of extracellular Tax have not been limited to the CNS, primary human PB macrophages have also been shown to secrete TNF-α, IL-1 and IL-6 in response to extracellular Tax[266]. Recently, cell-free Tax has been demonstrated in the CSF of HAM/TSP patients[250] indicating that Tax is available for immune recognition by APCs. Extracellular Tax released from Tax-producing cells by secretion or apoptosis and necrosis may be internalized by professional APCs. Tax peptides presented in the context of MHC by APCs would result in lysis of Tax-expressing cells by Tax-specific CD8+ T cells. Either production of toxic molecules or specific cell lysis could result in significant CNS damage similar to that observed in HAM/TSP. DCs are the most potent APCs and are of particular significance in the context of HTLV-1 pathogenesis. Development of HAM/TSP is associated with rapid maturation of DCs[267], while ATL is associated with a defect in their maturation[268]. DCs obtained from the PB of HAM/TSP patients were found to be infected with HTLV-1[269]. Similarly, DCs can be infected with HTLV-1 in vitro and when subsequently matured, can stimulate autologous CD4+ and CD8+ T cells[270]. Therefore, we hypothesized that DCs and other APCs might play a critical in the induction of Tax-specific immune response during the progression of HAM/TSP. In initial studies, the effects of intracellular versus extracellular and retrovirally-introduced Tax on murine DCs[271] have been analyzed. We have also studied activation and maturation of primary human monocyte-derived DCs as well as myeloid DCs in response to purified Tax and observed that Tax induces the secretion of proinflammatory cytokines and β chemokines from DCs[272,273] and modulates DC function toward a Th1 type immune response[274]. Moreover, DC-mediated priming of Tax-specific CTL response was demonstrated both in vitro and in vivo (Manuel and Jain, unpublished observations). These studies strongly suggest that DCs represent a major factor in HAM/TSP pathogenesis. The well-defined target of Tax activity, NF-κB coupled with protein kinases and phospholipase C, was also found to be critical for Tax-mediated DC activation and maturation[274]. Collectively, these studies have provided important insight into the molecular and immunologic mechanisms underlying the development of neuroinflammatory syndromes associated with HTLV-1 and other retroviruses. However, several outstanding questions still remain unanswered relative to the restricted epidemiological distribution of HAM/TSP, viral control mechanisms in asymptomatic carriers, progression to HAM/TSP and possible therapeutic interventions.
Conclusion
HIV-1 CNS disease is largely dependent on the trafficking of infected monocytic cells from the periphery across the blood-brain barrier, where virus is subsequently disseminated to susceptible cell populations within the brain including microglia, perivascular macrophages and astrocytes, which produce neurotoxic viral proteins such as Tat, gp120, Nef and Vpr and cytokines and chemokines that induce a positive feedback mechanism for further recruitment during the course of pathogenesis. HTLV-1 during neuroinflammatory disease is also characterized by an over-stimulation of the immunologic compartment, however instead of monocytes and macrophages it is CD8+T cells specific for Tax11-19 along with increased inflammatory cytokines and chemokines. Interestingly, as described above and restated here in both cases, pathogenesis is caused primarily from the inflammatory response to the neurotoxic proteins.
Unfortunately, in the case of both diseases there are very few treatment options for patients with retrovirus-induced neurologic disease. In the case of HIV-1 while antiretroviral therapy has had great impact on the life expectancy of patients and impacted their viral load and peripheral disease, it has increased the prevalence of neurologic disease[4]. With the widespread use of Highly Active Antiretroviral Therapy (HAART), a more subtle form of CNS dysfunction, termed Minor Cognitive Motor Disorder (MCMD), has become more common[7,8]. In the HAART era, it is estimated that ∼10% of HIV-infected adults develop HAD, however, the appearance of MCMD may be several times more common, involving as many as 30% of the HIV-infected population[8,275]. Furthermore, the clinical presentation of MCMD has been associated with neuropathological changes characteristic of HIV encephalitis (HIVE) and MCMD is associated with a worse overall prognostic outlook[7,8,275]. One means of explaining the development of MCMD is that the low-level viral replication associated with successful HARRT regimens, may lead to slowly progressing neurodegeneration. This is consistent with the longer lifespans of patients receiving HAART and possibly with the inability of certain antiretroviral drugs to effectively penetrate into the brain[276]. In addition to this, what is known about HIV-1 CNS disease is mostly from studying subtype B viruses. HIV-1 Subtype B viruses only cause a fraction of the worlds infection with subtype C infection being responsible for approximately half of the world's HIV-1 infections. Unfortunately, little is known about subtype C viruses and there role in neurologic disease and it is here where a majority of the future research will need to be performed.
HTLV-1 neurologic disease is also poorly understood with little or no treatment available. Many questions still remain unanswered as to what drives an infected individual to develop ATL versus HAM/TSP. These have been discussed above, however, the viral protein Tax plays a important role in neuropathogenesis. The levels of this protein being secreted and its presentation by dendritic cells to T cells plays a major role in HAM/TSP. Consequently, this immunologic pathway may provide an opportunity to identify novel therapeutic interventions.
Overall these two human retroviruses share many similarities. Their ability to cause neurologic disease is just one of the many. The fact that they do so in very similar ways is a testament to how these viruses may have evolved within their human hosts. Tat and Tax are just another example of this similarity, in that, both of these viral proteins interact with their viral LTRs and ultimately act as secreted proteins potentially causing dysregulation of a number of cell populations. It is through the study of the similarities and differences between these two viruses that scientists continue to learn more information concerning the pathogenic mechanisms of these two important human pathogens and their role in neurologic disease.
Acknowledgments
These studies were funded in part by the Public Health Service, National Institutes of Health through grants (B. Wigdahl, Principal Investigator) from the National Institute of Neurological Disorders and Stroke (NS32092 and NS46263) and the National Institute of Drug Abuse (DA19807).
References
- 1.Chen W, Tang Z, Fortina P, Patel P, Addya S, Surrey S, Acheampong EA, Mukhtar M, Pomerantz RJ. Ethanol potentiates HIV-1 gp120-induced apoptosis in human neurons via both the death receptor and NMDA receptor pathways. Virology. 2005;334:59–73. doi: 10.1016/j.virol.2005.01.014. http://www.ncbi.nlm.nih.gov/pubmed/15749123. [DOI] [PubMed]
- 2.McArthur JC. Neurologic manifestations of AIDS. Medicine (Baltimore) 1987;66:407–437. doi: 10.1097/00005792-198711000-00001. http://www.be-md.ncbi.nlm.nih.gov/pubmed/3316921. [DOI] [PubMed]
- 3.Silvers JM, Aksenova MV, Aksenov MY, Mactutus CF, Booze RM. Neurotoxicity of HIV-1 Tat protein: Involvement of D1 dopamine receptor. Neurotoxicology. 2007;28:1184–1190. doi: 10.1016/j.neuro.2007.07.005. http://www.ncbi.nlm.nih.gov/pubmed/17764744. [DOI] [PMC free article] [PubMed]
- 4.Childs EA, Lyles RH, Selnes OA, Chen B, Miller EN, et al. Plasma viral load and CD4 lymphocytes predict HIV-associated dementia and sensory neuropathy. Neurology. 1999;52:607–613. doi: 10.1212/wnl.52.3.607. http://www.neurology.org/cgi/content/abstract/52/3/607. [DOI] [PubMed]
- 5.Gendelman HE, Persidsky Y. Infections of the nervous system. Lancet Neurol. 2005;4:12–13. doi: 10.1016/S1474-4422(04)00951-2. http://linkinghub.elsevier.com/retrieve/pii/S1474442204009512. [DOI] [PMC free article] [PubMed]
- 6.Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–994. doi: 10.1038/35073667. http://www.ncbi.nlm.nih.gov/pubmed/11309629. [DOI] [PubMed]
- 7.McArthur JC, Haughey N, Gartner S, Conant K, Pardo C, Nath A, Sacktor N. Human immunodeficiency virus-associated dementia: An evolving disease. J Neurovirol. 2003;9:205–221. doi: 10.1080/13550280390194109. http://www.ncbi.nlm.nih.gov/pubmed/12707851. [DOI] [PubMed]
- 8.Cherner M, Masliah E, Ellis RJ, Marcotte TD, Moore DJ, Grant I, Heaton RK. Neurocognitive dysfunction predicts postmortem findings of HIV encephalitis. Neurology. 2002;59:1563–1567. doi: 10.1212/01.wnl.0000034175.11956.79. http://www.ncbi.nlm.nih.gov/pubmed/12451198. [DOI] [PubMed]
- 9.Alirezaei M, Watry DD, Flynn CF, Kiosses WB, Masliah E, Williams BR, Kaul M, Lipton SA, Fox HS. Human immunodeficiency virus-1/surface glycoprotein 120 induces apoptosis through RNA-activated protein kinase signaling in neurons. J Neurosci. 2007;27:11047–11055. doi: 10.1523/JNEUROSCI.2733-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gartner S. HIV infection and dementia. Science. 2000;287:602–604. doi: 10.1126/science.287.5453.602. [DOI] [PubMed] [Google Scholar]
- 11.Power C, Gill MJ, Johnson RT. Progress in clinical neurosciences: The neuropathogenesis of HIV infection: host-virus interaction and the impact of therapy. Can J Neurol Sci. 2002;29:19–32. doi: 10.1017/s0317167100001682. http://www.ncbi.nlm.nih.gov/pubmed/11858531. [DOI] [PubMed]
- 12.Kaul M, Lipton SA. Signaling pathways to neuronal damage and apoptosis in human immunodeficiency virus type 1-associated dementia: Chemokine receptors, excitotoxicity and beyond. J Neurovirol. 2004;1:97–101. doi: 10.1080/753312759. http://www.cababstractsplus.org/abstracts/Abstract.aspx?AcNo=20043207292. [DOI] [PubMed]
- 13.Patel CA, Mukhtar M, Pomerantz RJ. Human immunodeficiency virus type 1 Vpr induces apoptosis in human neuronal cells. J Virol. 2000;74:9717–9726. doi: 10.1128/jvi.74.20.9717-9726.2000. http://www.ncbi.nlm.nih.gov/pubmed/11000244. [DOI] [PMC free article] [PubMed]
- 14.Turchan J, Pocernich CB, Gairola C, Chauhan A, Nath A, et al. Oxidative stress in HIV demented patients and protection ex vivo with novel antioxidants. Neurology. 2003;60:307–314. doi: 10.1212/01.wnl.0000042048.85204.3d. http://www.ncbi.nlm.nih.gov/pubmed/12552050. [DOI] [PubMed]
- 15.Dalgleish AG. Human retroviruses. Aust N Z J Med. 1985;15:375–385. doi: 10.1111/j.1445-5994.1985.tb04066.x. http://www.ncbi.nlm.nih.gov/pubmed/2998317. [DOI] [PubMed]
- 16.Kozak SL, Platt EJ, Madani N, Ferro FE, Jr, Peden K, Kabat D. CD4, CXCR-4 and CCR-5 dependencies for infections by primary patient and laboratory-adapted isolates of human immunodeficiency virus type 1. J Virol. 1997;71:873–882. doi: 10.1128/jvi.71.2.873-882.1997. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=191134. [DOI] [PMC free article] [PubMed]
- 17.Al-Harthi L, Roebuck KA. Human immunodeficiency virus type-1 transcription: Role of the 5′-untranslated leader region (review) Int J Mol Med. 1998;1:875–881. doi: 10.3892/ijmm.1.5.875. http://www.ncbi.nlm.nih.gov/pubmed/9852310. [DOI] [PubMed]
- 18.Feinberg MB, Baltimore D, Frankel AD. The role of Tat in the human immunodeficiency virus life cycle indicates a primary effect on transcriptional elongation. Proc Natl Acad Sci USA. 1991;88:4045–4049. doi: 10.1073/pnas.88.9.4045. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=51590. [DOI] [PMC free article] [PubMed]
- 19.Gaynor R. Cellular transcription factors involved in the regulation of HIV-1 gene expression. Aids. 1992;6:347–363. doi: 10.1097/00002030-199204000-00001. http://www.ncbi.nlm.nih.gov/pubmed/1616633. [DOI] [PubMed]
- 20.Golub EI, Li GR, Volsky DJ. Induction of dormant HIV-1 by sodium butyrate: Involvement of the TATA box in the activation of the HIV-1 promoter. Aids. 1991;5:663–668. http://www.ncbi.nlm.nih.gov/pubmed/1883541. [PubMed]
- 21.Jordan HL, Pereira AS, Cohen MS, Kashuba AD. Domestic cat model for predicting human nucleoside analogue pharmacokinetics in blood and seminal plasma. Antimicrob Agent Chemother. 2001;45:2173–2176. doi: 10.1128/AAC.45.7.2173-2176.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kao SY, Calman AF, Luciw PA, Peterlin BM. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature. 1987;330:489–493. doi: 10.1038/330489a0. http://www.ncbi.nlm.nih.gov/pubmed/2825027. [DOI] [PubMed]
- 23.Ladias JA. Convergence of multiple nuclear receptor signaling pathways onto the long terminal repeat of human immunodeficiency virus-1. J Biol Chem. 1994;269:5944–5951. http://www.ncbi.nlm.nih.gov/pubmed/8119938. [PubMed]
- 24.Ou SH, Garcia-Martinez LF, Paulssen EJ, Gaynor RB. Role of flanking E box motifs in human immunodeficiency virus type 1 TATA element function. J Virol. 1994;68:7188–7199. doi: 10.1128/jvi.68.11.7188-7199.1994. http://jvi.asm.org/cgi/content/abstract/68/11/7188. [DOI] [PMC free article] [PubMed]
- 25.Roebuck KA, Saifuddin M. Regulation of HIV-1 transcription. Gene Expr. 1999;8:67–84. http://www.ncbi.nlm.nih.gov/pubmed/10551796. [PMC free article] [PubMed]
- 26.Van Lint C, Emiliani S, Ott M, Verdin E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996;15:1112–1120. http://www.ncbi.nlm.nih.gov/pubmed/8605881. [PMC free article] [PubMed]
- 27.Zhang L, Waters C, Nichols J, Crumpacker C. Inhibition of HIV-1 RNA production by the diphtheria toxin-related IL-2 fusion proteins DAB486IL-2 and DAB389IL-2. J. Acquir. Immune Defic Syndr. 1992;5:1181–1187. http://www.ncbi.nlm.nih.gov/pubmed/1453329?dopt=Abstract. [PubMed]
- 28.Smith JS, Roth MJ. Specificity of human immunodeficiency virus-1 reverse transcriptase-associated ribonuclease H in removal of the minus-strand primer, tRNA(Lys3) J Biol Chem. 1992;267:15071–15079. http://www.jbc.org/cgi/content/abstract/267/21/15071. [PubMed]
- 29.Calame A, Fawer CL. Neurodevelopmental follow-up of children after severe neonatal asphyxia. Pediatr Pulmonol Suppl. 1997;16:254–255. doi: 10.1002/ppul.19502308131. http://www.ncbi.nlm.nih.gov/pubmed/9443299. [DOI] [PubMed]
- 30.Gowda SD, Stein BS, Steimer KS, Engleman EG. Expression and processing of human immunodeficiency virus type 1 gag and pol genes by cells infected with a recombinant vaccinia virus. J Virol. 1989;63:1451–1454. doi: 10.1128/jvi.63.3.1451-1454.1989. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=247850. [DOI] [PMC free article] [PubMed]
- 31.Henderson AJ, Zou X, Calame KL. C/EBP proteins activate transcription from the human immunodeficiency virus type 1 long terminal repeat in macrophages/monocytes. J Virol. 1995;69:5337–5344. doi: 10.1128/jvi.69.9.5337-5344.1995. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=189374. [DOI] [PMC free article] [PubMed]
- 32.Hogan TH, Nonnemacher MR, Krebs FC, Henderson A, Wigdahl B. HIV-1 Vpr binding to HIV-1 LTR C/EBP cis-acting elements and adjacent regions is sequence-specific. Biomed Pharmacother. 2003;57:41–48. doi: 10.1016/s0753-3322(02)00333-5. http://www.ncbi.nlm.nih.gov/pubmed/12642036. [DOI] [PubMed]
- 33.Hogan TH, Stauff DL, Krebs FC, Gartner S, Quiterio SJ, Wigdahl B. Structural and functional evolution of human immunodeficiency virus type 1 long terminal repeat CCAAT/enhancer binding protein sites and their use as molecular markers for central nervous system disease progression. J Neurovirol. 2003;9:55–68. doi: 10.1080/13550280390173292. http://www.ncbi.nlm.nih.gov/pubmed/12587069. [DOI] [PubMed]
- 34.Krebs FC, Goodenow MM, Wigdahl B. Neuroglial ATF/CREB factors interact with the human immunodeficiency virus type 1 long terminal repeat. J Neurovirol. 1997;1:S28–32. http://www.ncbi.nlm.nih.gov/pubmed/9179787. [PubMed]
- 35.McAllister JJ, Phillips D, Millhouse S, Conner J, Hogan T, Ross HL, Wigdahl B. Analysis of the HIV-1 LTR NF-kappaB-proximal Sp site III: Evidence for cell type-specific gene regulation and viral replication. Virology. 2000;274:262–277. doi: 10.1006/viro.2000.0476. [DOI] [PubMed] [Google Scholar]
- 36.McDougal JS, Hubbard M, Nicholson JK, Jones BM, Holman RC, et al. Immune complexes in the Acquired Immunodeficiency Syndrome (AIDS): Relationship to disease manifestation, risk group and immunologic defect. J Clin Immunol. 1985;5:130–138. doi: 10.1007/BF00915011. http://cat.inist.fr/?aModele=afficheN&cpsidt=9111311. [DOI] [PubMed]
- 37.Ross HL, Gartner S, McArthur JC, Corboy JR, McAllister JJ, Millhouse S, Wigdahl B. HIV-1 LTR C/EBP binding site sequence configurations preferentially encountered in brain lead to enhanced C/EBP factor binding and increased LTR-specific activity. J Neurovirol. 2001;7:235–249. doi: 10.1080/13550280152403281. http://www.ncbi.nlm.nih.gov/pubmed/11517398. [DOI] [PubMed]
- 38.Ross HL, Nonnemacher MR, Hogan TH, Quiterio SJ, Henderson A, McAllister JJ, Krebs FC, Wigdahl B. Interaction between CCAAT/enhancer binding protein and cyclic AMP response element binding protein 1 regulates human immunodeficiency virus type 1 transcription in cells of the monocyte/macrophage lineage. J Virol. 2001;75:1842–1856. doi: 10.1128/JVI.75.4.1842-1856.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burdo TH, Gartner S, Mauger D, Wigdahl B. Region-specific distribution of human immunodeficiency virus type 1 long terminal repeats containing specific configurations of CCAAT/enhancer-binding protein site II in brains derived from demented and nondemented patients. J Neurovirol. 2004;1:7–14. doi: 10.1080/753312746. http://www.ncbi.nlm.nih.gov/pubmed/14982733. [DOI] [PubMed]
- 40.Gatignol A, Buckler-White A, Berkhout B, Jeang KT. Characterization of a human TAR RNA-binding protein that activates the HIV-1 LTR. Science. 1991;251:1597–1600. doi: 10.1126/science.2011739. http://www.ncbi.nlm.nih.gov/pubmed/2011739. [DOI] [PubMed]
- 41.Sawaya BE, Khalili K, Rappaport J, Serio D, Chen W, Srinivasan A, Amini S. Suppression of HIV-1 transcription and replication by a Vpr mutant. Gene Ther. 1999;6:947–950. doi: 10.1038/sj.gt.3300907. http://www.ncbi.nlm.nih.gov/pubmed/10505122. [DOI] [PubMed]
- 42.Brady J, Kashanchi F. Tat gets the “green” light on transcription initiation. Retrovirology. 2005;2:69–69. doi: 10.1186/1742-4690-2-69. http://www.ncbi.nlm.nih.gov/pubmed/16280076. [DOI] [PMC free article] [PubMed]
- 43.Doppler C, Schalasta G, Amtmann E, Sauer G. Binding of NF-kB to the HIV-1 LTR is not sufficient to induce HIV-1 LTR activity. AIDS Res Hum Retroviruses. 1992;8:245–252. doi: 10.1089/aid.1992.8.245. http://www.ncbi.nlm.nih.gov/pubmed/1540410. [DOI] [PubMed]
- 44.Green M, Ishino M, Loewenstein PM. Mutational analysis of HIV-1 Tat minimal domain peptides: Identification of trans-dominant mutants that suppress HIV-LTR-driven gene expression. Cell. 1989;58:215–223. doi: 10.1016/0092-8674(89)90417-0. http://www.ncbi.nlm.nih.gov/pubmed/2752420. [DOI] [PubMed]
- 45.Rice AP, Mathews MB. Transcriptional but not translational regulation of HIV-1 by the tat gene product. Nature. 1988;332:551–553. doi: 10.1038/332551a0. http://www.ncbi.nlm.nih.gov/pubmed/2833703. [DOI] [PubMed]
- 46.Rappaport J, Joseph J, Croul S, Alexander G, Del Valle L, Amini S, Khalili K. Molecular pathway involved in HIV-1-induced CNS pathology: Role of viral regulatory protein, Tat. J Leukoc Biol. 1999;65:458–465. doi: 10.1002/jlb.65.4.458. http://www.ncbi.nlm.nih.gov/pubmed/10204574. [DOI] [PubMed]
- 47.Raha T, Cheng SW, Green MR. HIV-1 Tat stimulates transcription complex assembly through recruitment of TBP in the absence of TAFs. PLoS Biol. 2005;3:44–44. doi: 10.1371/journal.pbio.0030044. http://www.ncbi.nlm.nih.gov/pubmed/15719058. [DOI] [PMC free article] [PubMed]
- 48.Selby MJ, Bain ES, Luciw PA, Peterlin BM. Structure, sequence and position of the stem-loop in tar determine transcriptional elongation by tat through the HIV-1 long terminal repeat. Genes Dev. 1989;3:547–558. doi: 10.1101/gad.3.4.547. http://genesdev.cshlp.org/content/3/4/547.abstract. [DOI] [PubMed]
- 49.Taylor JP, Khalili K. Activation of HIV-1 transcription by Tat in cells derived from the CNS: Evidence for the participation of NF-kappa B--a review. Adv Neuroimmunol. 1994;4:291–303. doi: 10.1016/s0960-5428(06)80270-6. http://www.biomedexperts.com/Abstract.bme/7874398/Activation_of_HIV-1_transcription_by_Tat_in_cells_derived_from_the_CNS_evidence_for_the_participation_of_NF-kappa_B--a. [DOI] [PubMed]
- 50.Sawaya BE, Khalili K, Amini S. Transcription of the human immunodeficiency virus type 1 (HIV-1) promoter in central nervous system cells: effect of YB-1 on expression of the HIV-1 long terminal repeat. J Gen Virol. 1998;79:239–246. doi: 10.1099/0022-1317-79-2-239. http://vir.sgmjournals.org/cgi/content/abstract/79/2/239. [DOI] [PubMed]
- 51.Nabel GJ. Activation of human immunodeficiency virus. J Lab Clin Med. 1988;111:495–500. http://www.ncbi.nlm.nih.gov/pubmed/3283280. [PubMed]
- 52.Nabel GJ, Rice SA, Knipe DM, Baltimore D. Alternative mechanisms for activation of human immunodeficiency virus enhancer in T cells. Science. 1988;239:1299–1302. doi: 10.1126/science.2830675. http://www.sciencemag.org/cgi/content/abstract/239/4845/1299. [DOI] [PubMed]
- 53.Andreoni M. Viral phenotype and fitness. New Microbiol. 2004;27:71–76. http://www.ncbi.nlm.nih.gov/pubmed/15646067. [PubMed]
- 54.Roof P, Ricci M, Genin P, Montano MA, Essex M, Wainberg MA, Gatignol A, Hiscott J. Differential regulation of HIV-1 clade-specific B, C and E long terminal repeats by NF-kappaB and the Tat transactivator. Virology. 2002;296:77–83. doi: 10.1006/viro.2001.1397. http://www.ncbi.nlm.nih.gov/pubmed/12036319. [DOI] [PubMed]
- 55.Bratanich AC, Liu C, McArthur JC, Fudyk T, Glass JD, Mittoo S, Klassen GA, Power C. Brain-derived HIV-1 tat sequences from AIDS patients with dementia show increased molecular heterogeneity. J Neurovirol. 1998;4:387–393. doi: 10.3109/13550289809114537. http://www.ncbi.nlm.nih.gov/pubmed/9718130. [DOI] [PubMed]
- 56.Mayne M, Bratanich AC, Chen P, Rana F, Nath A, Power C. HIV-1 tat molecular diversity and induction of TNF-alpha: implications for HIV-induced neurological disease. Neuroimmunomodulation. 1998;5:184–192. doi: 10.1159/000026336. http://www.ncbi.nlm.nih.gov/pubmed/9730685. [DOI] [PubMed]
- 57.Johnston JB, Zhang K, Silva C, Shalinsky DR, Conant K, Ni W, Corbett D, Yong VW, Power C. HIV-1 Tat neurotoxicity is prevented by matrix metalloproteinase inhibitors. Ann Neurol. 2001;49:230–241. doi: 10.1002/1531-8249(20010201)49:2<230::aid-ana43>3.0.co;2-o. http://cat.inist.fr/?aModele=afficheN&cpsidt=887916. [DOI] [PubMed]
- 58.Silva C, Zhang K, Tsutsui S, Holden JK, Gill MJ, Power C. Growth hormone prevents human immunodeficiency virus-induced neuronal p53 expression. Ann Neurol. 2003;54:605–614. doi: 10.1002/ana.10729. http://www.ncbi.nlm.nih.gov/pubmed/14595650. [DOI] [PubMed]
- 59.Boven LA, Noorbakhsh F, Bouma G, Van der Zee R, Vargas DL, et al. Brain-derived human immunodeficiency virus-1 Tat exerts differential effects on LTR transactivation and neuroimmune activation. J Neurovirol. 2007;13:173–184. doi: 10.1080/13550280701258399. http://www.ncbi.nlm.nih.gov/pubmed/17505986. [DOI] [PubMed]
- 60.Nath A, Hauser KF, Wojna V, Booze RM, Maragos W, Prendergast M, et al. Molecular basis for interactions of HIV and drugs of abuse. J Acquir Immune Defic Syndr. 2002;2:S62–S69. doi: 10.1097/00126334-200210012-00006. http://www.ncbi.nlm.nih.gov/pubmed/12394784. [DOI] [PubMed]
- 61.Kaul M, Zheng J, Okamoto S, Gendelman HE, Lipton SA. HIV-1 infection and AIDS: Consequences for the central nervous system. Cell Death Differ. 2005;1:878–892. doi: 10.1038/sj.cdd.4401623. http://www.ncbi.nlm.nih.gov/pubmed/15832177. [DOI] [PubMed]
- 62.Zheng J, Thylin MR, Cotter RL, Lopez AL, Ghorpade A, Persidsky Y, et al. HIV-1 infected and immune competent mononuclear phagocytes induce quantitative alterations in neuronal dendritic arbor: Relevance for HIV-1-associated dementia. Neurotox Res. 2001;3:443–459. doi: 10.1007/BF03033203. [DOI] [PubMed] [Google Scholar]
- 63.Hesselgesser J, Taub D, Baskar P, Greenberg M, Hoxie J, Kolson DL, Horuk R. Neuronal apoptosis induced by HIV-1 gp120 and the chemokine SDF-1 alpha is mediated by the chemokine receptor CXCR4. Curr Biol. 1998;8:595–598. doi: 10.1016/s0960-9822(98)70230-1. http://www.ncbi.nlm.nih.gov/pubmed/9601645. [DOI] [PubMed]
- 64.Kaul M, Lipton SA. Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc Natl Acad Sci USA. 1999;96:8212–8216. doi: 10.1073/pnas.96.14.8212. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=22214. [DOI] [PMC free article] [PubMed]
- 65.Xu Y, Kulkosky J, Acheampong E, Nunnari G, Sullivan J, Pomerantz RJ. HIV-1-mediated apoptosis of neuronal cells: Proximal molecular mechanisms of HIV-1-induced encephalopathy. Proc Natl Acad Sci USA. 2004;101:7070–7075. doi: 10.1073/pnas.0304859101. http://www.ncbi.nlm.nih.gov/pubmed/15103018. [DOI] [PMC free article] [PubMed]
- 66.Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc Natl Acad Sci USA. 1998;95:14500–14505. doi: 10.1073/pnas.95.24.14500. http://www.ncbi.nlm.nih.gov/pubmed/9826729. [DOI] [PMC free article] [PubMed]
- 67.Davis CB, Dikic I, Unutmaz D, Hill CM, Arthos J, et al. Signal transduction due to HIV-1 envelope interactions with chemokine receptors CXCR4 or CCR5. J Exp Med. 1997;186:1793–1798. doi: 10.1084/jem.186.10.1793. http://www.ncbi.nlm.nih.gov/pubmed/9362541. [DOI] [PMC free article] [PubMed]
- 68.Herbein G, Mahlknecht U, Batliwalla F, Gregersen P, Pappas T, et al. Apoptosis of CD8+ T cells is mediated by macrophages through interaction of HIV gp120 with chemokine receptor CXCR4. Nature. 1998;395:189–194. doi: 10.1038/26026. [DOI] [PubMed] [Google Scholar]
- 69.Hesselgesser J, Liang M, Hoxie J, Greenberg M, Brass LF, Horuk R, et al. Identification and characterization of the CXCR4 chemokine receptor in human T cell lines: Ligand binding, biological activity and HIV-1 infectivity. J Immunol. 1998;160:877–883. http://www.ncbi.nlm.nih.gov/pubmed/9551924. [PubMed]
- 70.Klein RS, Williams KC, Alvarez-Hernandez X, Westmoreland S, et al. Chemokine receptor expression and signaling in macaque and human fetal neurons and astrocytes: Implications for the neuropathogenesis of AIDS. J Immunol. 1999;163:1636–1646. http://www.jimmunol.org/cgi/content/abstract/163/3/1636. [PubMed]
- 71.Kozak SL, Kuhmann SE, Platt EJ, Kabat D. Roles of CD4 and coreceptors in binding, endocytosis and proteolysis of gp120 envelope glycoproteins derived from human immunodeficiency virus type 1. J Biol Chem. 1999;274:23499–23507. doi: 10.1074/jbc.274.33.23499. http://cat.inist.fr/?aModele=afficheN&cpsidt=10178652. [DOI] [PubMed]
- 72.Kaul M, Ma Q, Medders KE, Desai MK, Lipton SA. HIV-1 coreceptors CCR5 and CXCR4 both mediate neuronal cell death but CCR5 paradoxically can also contribute to protection. Cell Death Differ. 2007;14:296–305. doi: 10.1038/sj.cdd.4402006. http://www.ncbi.nlm.nih.gov/pubmed/16841089. [DOI] [PubMed]
- 73.Lannuzel A, Lledo PM, Lamghitnia HO, Vincent JD, Tardieu M. HIV-1 envelope proteins 120 and gp160 potentiate NMDA-induced [Ca2+]i increase, alter [Ca2+]i homeostasis and induce neurotoxicity in human embryonic neurons. Eur J Neurosci. 1995;7:2285–2293. doi: 10.1111/j.1460-9568.1995.tb00649.x. [DOI] [PubMed] [Google Scholar]
- 74.Dreyer EB, Kaiser PK, Offermann JT, Lipton SA. HIV-1 coat protein neurotoxicity prevented by calcium channel antagonists. Science. 1990;248:364–367. doi: 10.1126/science.2326646. http://www.ncbi.nlm.nih.gov/pubmed/2326646. [DOI] [PubMed]
- 75.Mattson MP, Haughey NJ, Nath A. Cell death in HIV dementia. Cell Death Differ. 2005;12:893–904. doi: 10.1038/sj.cdd.4401577. http://www.nature.com/cdd/journal/vaop/ncurrent/full/4401577a.html. [DOI] [PubMed]
- 76.Campus G, Lumbau A, Bachisio SL. Caries experience and streptococci and lactobacilli salivary levels in 6-8-year-old Sardinians. Int J Paediatr Dent. 2000;10:306–312. doi: 10.1046/j.1365-263x.2000.00206.x. http://www.ncbi.nlm.nih.gov/pubmed/11310244. [DOI] [PubMed]
- 77.Biard-Piechaczyk M, Robert-Hebmann V, Richard V, Roland J, Hipskind RA, Devaux C. Caspase-dependent apoptosis of cells expressing the chemokine receptor CXCR4 is induced by cell membrane-associated human immunodeficiency virus type 1 envelope glycoprotein (gp120) Virology. 2000;268:329–344. doi: 10.1006/viro.1999.0151. http://www.ncbi.nlm.nih.gov/pubmed/10704341. [DOI] [PubMed]
- 78.Ullrich CK, Groopman JE, Ganju RK. HIV-1 gp120- and gp160-induced apoptosis in cultured endothelial cells is mediated by caspases. Blood. 2000;96:1438–1442. http://www.ncbi.nlm.nih.gov/pubmed/10942389. [PubMed]
- 79.Peng F, Dhillon N, Callen S, Yao H, Bokhari S, Zhu X, Baydoun HH, Buch S. Platelet-derived growth factor protects neurons against gp120-mediated toxicity. J Neurovirol. 2008;14:62–72. doi: 10.1080/13550280701809084. http://www.ncbi.nlm.nih.gov/pubmed/18300076. [DOI] [PMC free article] [PubMed]
- 80.Haughey NJ, Mattson MP. Calcium dysregulation and neuronal apoptosis by the HIV-1 proteins Tat and gp120. J Acquir Immune Defic Syndr. 2002;31:S55–61. doi: 10.1097/00126334-200210012-00005. http://cat.inist.fr/?aModele=afficheN&cpsidt=13978804. [DOI] [PubMed]
- 81.Medina I, Ghose S, Ben-Ari Y. Mobilization of intracellular calcium stores participates in the rise of [Ca2+]i and the toxic actions of the HIV coat protein GP120. Eur. J Neurosci. 1999;11:1167–1178. doi: 10.1046/j.1460-9568.1999.00550.x. http://www.ncbi.nlm.nih.gov/pubmed/10103113. [DOI] [PubMed]
- 82.Lei SZ, Pan ZH, Aggarwal SK, Chen HS, Hartman J, Sucher NJ, Lipton SA. Effect of nitric oxide production on the redox modulatory site of the NMDA receptor-channel complex. Neuron. 1992;8:1087–1099. doi: 10.1016/0896-6273(92)90130-6. http://www.ncbi.nlm.nih.gov/pubmed/1376999. [DOI] [PubMed]
- 83.Lipton SA, Sucher NJ, Kaiser PK, Dreyer EB. Synergistic effects of HIV coat protein and NMDA receptor-mediated neurotoxicity. Neuron. 1991;7:111–118. doi: 10.1016/0896-6273(91)90079-f. http://www.ncbi.nlm.nih.gov/pubmed/1676893. [DOI] [PubMed]
- 84.Williams SB, Flanigan TP, Artenstein AW, VanCott TC, Smith D, Mayer K, Koup RA. CCR5 genotype and human immunodeficiency virus (HIV)-specific mucosal antibody in seronegative women at high risk for HIV infection. J Infect Dis. 1999;179:1310–1312. doi: 10.1086/314743. http://www.biomedexperts.com/Abstract.bme/10191415/CCR5_genotype_and_human_immunodeficiency_virus_HIV_-specific_mucosal_antibody_in_seronegative_women_at_high_risk_for_HI. [DOI] [PubMed]
- 85.Bando Y, Katayama T, Taniguchi M, Ishibashi T, Matsuo N, Ogawa S, Tohyama M. RA410/Sly1 suppresses MPP+ and 6-hydroxydopamine-induced cell death in SH-SY5Y cells. Neurobiol Dis. 2005;18:143–151. doi: 10.1016/j.nbd.2004.09.008. http://www.ncbi.nlm.nih.gov/pubmed/15649705. [DOI] [PubMed]
- 86.Wang Z, Pekarskaya O, Bencheikh M, Chao W, Volsky DJ, et al. Reduced expression of glutamate transporter EAAT2 and impaired glutamate transport in human primary astrocytes exposed to HIV-1 or gp120. Virology. 2003;312:60–73. doi: 10.1016/s0042-6822(03)00181-8. [DOI] [PubMed] [Google Scholar]
- 87.Galey D, Becker K, Haughey N, Kalehua A, Nath A, et al. Differential transcriptional regulation by human immunodeficiency virus type 1 and gp120 in human astrocytes. J Neurovirol. 2003;9:358–371. doi: 10.1080/13550280390201119. http://www.ncbi.nlm.nih.gov/pubmed/12775419. [DOI] [PubMed]
- 88.Su ZZ, Kang DC, Chen Y, Pekarskaya O, Chao W, Volsky DJ, Fisher PB. Identification and cloning of human astrocyte genes displaying elevated expression after infection with HIV-1 or exposure to HIV-1 envelope glycoprotein by rapid subtraction hybridization, RaSH. Oncogene. 2002;21:3592–3602. doi: 10.1038/sj.onc.1205445. http://www.nature.com/onc/journal/v21/n22/abs/1205445a.html. [DOI] [PubMed]
- 89.Kort JJ. Impairment of excitatory amino acid transport in astroglial cells infected with the human immunodeficiency virus type 1. AIDS Res Hum Retroviruses. 1998;14:1329–1339. doi: 10.1089/aid.1998.14.1329. http://www.ncbi.nlm.nih.gov/pubmed/9788674. [DOI] [PubMed]
- 90.Adamson DC, McArthur JC, Dawson TM, Dawson VL. Rate and severity of HIV-associated dementia (HAD): Correlations with Gp41 and iNOS. Mol Med. 1999;5:98–109. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2230416. [PMC free article] [PubMed]
- 91.Adamson DC, Wildemann B, Sasaki M, Glass JD, Dawson VL, et al. Immunologic NO synthase: Elevation in severe AIDS dementia and induction by HIV-1 gp41. Science. 1996;274:1917–1921. doi: 10.1126/science.274.5294.1917. http://cat.inist.fr/?aModele=afficheN&cpsidt=2534954. [DOI] [PubMed]
- 92.Sung JH, Shin SA, Park HK, Montelaro RC, Chong YH. Protective effect of glutathione in HIV-1 lytic peptide 1-induced cell death in human neuronal cells. J Neurovirol. 2001;7:454–465. doi: 10.1080/135502801753170318. http://www.ncbi.nlm.nih.gov/pubmed/11582518. [DOI] [PubMed]
- 93.Huang MB, Weeks O, Zhao LJ, Saltarelli M, Bond VC. Effects of extracellular human immunodeficiency virus type 1 vpr protein in primary rat cortical cell cultures. J Neurovirol. 2000;6:202–220. doi: 10.3109/13550280009015823. http://cat.inist.fr/?aModele=afficheN&cpsidt=1392756. [DOI] [PubMed]
- 94.Piller SC, Jans P, Gage PW, Jans DA. Extracellular HIV-1 virus protein R causes a large inward current and cell death in cultured hippocampal neurons: implications for AIDS pathology. Proc Natl Acad Sci USA. 1998;95:4595–4600. doi: 10.1073/pnas.95.8.4595. http://cat.inist.fr/?aModele=afficheN&cpsidt=10407323. [DOI] [PMC free article] [PubMed]
- 95.Pomerantz RJ. Effects of HIV-1 Vpr on neuroinvasion and neuropathogenesis. DNA Cell Biol. 2004;23:227–238. doi: 10.1089/104454904773819815. http://www.ncbi.nlm.nih.gov/pubmed/15142380. [DOI] [PubMed]
- 96.Emerman M. HIV-1, Vpr and the cell cycle. Curr Biol. 1996;6:1096–1103. doi: 10.1016/s0960-9822(02)00676-0. http://direct.bl.uk/bld/PlaceOrder.do?UIN=020191296&ETOC=RN&from=searchengine. [DOI] [PubMed]
- 97.He J, Choe S, Walker R, Di Marzio P, Morgan DO, Landau NR. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J Virol. 1995;69:6705–6711. doi: 10.1128/jvi.69.11.6705-6711.1995. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=189580. [DOI] [PMC free article] [PubMed]
- 98.Heinzinger NK, Bukinsky MI, Haggerty SA, Ragland AM, Emerman M. The Vpr protein of human immunodeficiency virus type 1 influences nuclear localization of viral nucleic acids in nondividing host cells. Proc Natl Acad Sci USA. 1994;91:7311–7315. doi: 10.1073/pnas.91.15.7311. http://www.ncbi.nlm.nih.gov/pubmed/8041786. [DOI] [PMC free article] [PubMed]
- 99.Levy DN, Refaeli Y, MacGregor RR, Weiner DB. Serum Vpr regulates productive infection and latency of human immunodeficiency virus type 1. Proc Natl Acad Sci USA. 1994;91:10873–10877. doi: 10.1073/pnas.91.23.10873. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=45128. [DOI] [PMC free article] [PubMed]
- 100.Connor RI, Chen BK, Choe S, Landau NR. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. 1995;206:935–944. doi: 10.1006/viro.1995.1016. http://www.ncbi.nlm.nih.gov/pubmed/7531918. [DOI] [PubMed]
- 101.Gallay P, Stitt V, Mundy C, Oettinger M, Trono D. Role of the karyopherin pathway in human immunodeficiency virus type 1 nuclear import. J Virol. 1996;70:1027–1032. doi: 10.1128/jvi.70.2.1027-1032.1996. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=189908. [DOI] [PMC free article] [PubMed]
- 102.Bartz SR, Rogel ME, Emerman M. Human immunodeficiency virus type 1 cell cycle control: Vpr is cytostatic and mediates G2 accumulation by a mechanism which differs from DNA damage checkpoint control. J Virol. 1996;70:2324–2331. doi: 10.1128/jvi.70.4.2324-2331.1996. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=190074. [DOI] [PMC free article] [PubMed]
- 103.Huard S, Elder RT, Liang D, Li G, Zhao RY. Human immunodeficiency virus type 1 Vpr induces cell cycle G2 arrest through Srk1/MK2-mediated phosphorylation of Cdc25. J Virol. 2008;82:2904–2917. doi: 10.1128/JVI.01098-07. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2259012. [DOI] [PMC free article] [PubMed]
- 104.Jowett JB, Planelles V, Poon B, Shah NP, Chen ML, Chen IS. The human immunodeficiency virus type 1 vpr gene arrests infected T cells in the G2 + M phase of the cell cycle. J Virol. 1995;69:6304–6313. doi: 10.1128/jvi.69.10.6304-6313.1995. http://jvi.asm.org/cgi/content/abstract/69/10/6304. [DOI] [PMC free article] [PubMed]
- 105.Planelles V, Bachelerie F, Jowett JB, Haislip A, Xie Y, et al. Fate of the human immunodeficiency virus type 1 provirus in infected cells: A role for vpr. J Virol. 1995;69:5883–5889. doi: 10.1128/jvi.69.9.5883-5889.1995. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=189467. [DOI] [PMC free article] [PubMed]
- 106.Poon B, Jowett JB, Stewart SA, Armstrong RW, Rishton GM, Chen IS. Human immunodeficiency virus type 1 vpr gene induces phenotypic effects similar to those of the DNA alkylating agent, nitrogen mustard. J Virol. 1997;71:3961–3971. doi: 10.1128/jvi.71.5.3961-3971.1997. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=191548. [DOI] [PMC free article] [PubMed]
- 107.Re F, Braaten D, Franke EK, Luban J. Human immunodeficiency virus type 1 Vpr arrests the cell cycle in G2 by inhibiting the activation of p34cdc2-cyclin B. J Virol. 1995;69:6859–6864. doi: 10.1128/jvi.69.11.6859-6864.1995. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=189600. [DOI] [PMC free article] [PubMed]
- 108.Rogel ME, Wu LI, Emerman M. The human immunodeficiency virus type 1 vpr gene prevents cell proliferation during chronic infection. J Virol. 1995;69:882–888. doi: 10.1128/jvi.69.2.882-888.1995. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=188655. [DOI] [PMC free article] [PubMed]
- 109.Levy DN, Fernandes LS, Williams WV, Weiner DB. Induction of cell differentiation by human immunodeficiency virus 1 vpr. Cell. 1993;72:541–550. doi: 10.1016/0092-8674(93)90073-y. http://www.ncbi.nlm.nih.gov/pubmed/8440020. [DOI] [PubMed]
- 110.Poon B, Grovit-Ferbas K, Stewart SA, Chen IS. Cell cycle arrest by Vpr in HIV-1 virions and insensitivity to antiretroviral agents. Science. 1998;281:266–269. doi: 10.1126/science.281.5374.266. http://cat.inist.fr/?aModele=afficheN&cpsidt=2376135. [DOI] [PubMed]
- 111.Goh WC, Rogel ME, Kinsey CM, Michael SF, Fultz PN, et al. HIV-1 Vpr increases viral expression by manipulation of the cell cycle: A mechanism for selection of Vpr in vivo. Nat Med. 1998;4:65–71. doi: 10.1038/nm0198-065. http://www.ncbi.nlm.nih.gov/pubmed/9427608. [DOI] [PubMed]
- 112.Paxton W, Connor RI, Landau NR. Incorporation of Vpr into human immunodeficiency virus type 1 virions: Requirement for the p6 region of gag and mutational analysis. J Virol. 1993;67:7229–7237. doi: 10.1128/jvi.67.12.7229-7237.1993. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=238185. [DOI] [PMC free article] [PubMed]
- 113.Lu YL, Spearman P, Ratner L. Human immunodeficiency virus type 1 viral protein R localization in infected cells and virions. J Virol. 1993;67:6542–6550. doi: 10.1128/jvi.67.11.6542-6550.1993. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=238091. [DOI] [PMC free article] [PubMed]
- 114.Sherman MP, De Noronha CM, Heusch MI, Greene S, Greene WC. Nucleocytoplasmic shuttling by human immunodeficiency virus type 1 Vpr. J Virol. 2001;75:1522–1532. doi: 10.1128/JVI.75.3.1522-1532.2001. http://www.ncbi.nlm.nih.gov/pubmed/11152524. [DOI] [PMC free article] [PubMed]
- 115.Bukrinsky M, Zhao Y. Heat-shock proteins reverse the G2 arrest caused by HIV-1 viral protein R. DNA Cell Biol. 2004;23:223–225. doi: 10.1089/104454904773819806. [DOI] [PubMed] [Google Scholar]
- 116.Popov S, Rexach M, Ratner L, Blobel G, Bukrinsky M. Viral protein R regulates docking of the HIV-1 preintegration complex to the nuclear pore complex. J Biol Chem. 1998;273:13347–13352. doi: 10.1074/jbc.273.21.13347. http://www.ncbi.nlm.nih.gov/pubmed/9582382. [DOI] [PubMed]
- 117.Popov S, Rexach M, Zybarth G, Reiling N, Lee MA, Bukrinsky M, et al. Viral protein R regulates nuclear import of the HIV-1 pre-integration complex. EMBO J. 1998;17:909–917. doi: 10.1093/emboj/17.4.909. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1170440. [DOI] [PMC free article] [PubMed]
- 118.Bouyac-Bertoia M, Dvorin JD, Fouchier RA, Jenkins Y, Malim MH, et al. HIV-1 infection requires a functional integrase NLS. Mol Cell. 2001;7:1025–1035. doi: 10.1016/s1097-2765(01)00240-4. http://www.ncbi.nlm.nih.gov/pubmed/11389849. [DOI] [PubMed]
- 119.Eckstein DA, Sherman MP, Penn ML, Chin PS, Goldsmith MA, et al. HIV-1 Vpr enhances viral burden by facilitating infection of tissue macrophages but not nondividing CD4+ T cells. J Exp Med. 2001;194:1407–1419. doi: 10.1084/jem.194.10.1407. http://www.ncbi.nlm.nih.gov/pubmed/11714748. [DOI] [PMC free article] [PubMed]
- 120.Sherman MP, de Noronha CM, Eckstein LA, Hataye J, Greene WC, et al. Nuclear export of Vpr is required for efficient replication of human immunodeficiency virus type 1 in tissue macrophages. J Virol. 2003;77:7582–7589. doi: 10.1128/JVI.77.13.7582-7589.2003. http://cat.inist.fr/?aModele=afficheN&cpsidt=14886735. [DOI] [PMC free article] [PubMed]
- 121.Fouchier RA, Meyer BE, Simon JH, Fischer U, Albright AV, et al. Interaction of the human immunodeficiency virus type 1 Vpr protein with the nuclear pore complex. J Virol. 1998;72:6004–6013. doi: 10.1128/jvi.72.7.6004-6013.1998. http://www.ncbi.nlm.nih.gov/pubmed/9621063. [DOI] [PMC free article] [PubMed]
- 122.Vodicka MA, Koepp DM, Silver PA, Emerman M. HIV-1 Vpr interacts with the nuclear transport pathway to promote macrophage infection. Genes Dev. 1998;12:175–185. doi: 10.1101/gad.12.2.175. http://www.ncbi.nlm.nih.gov/pubmed/9436978. [DOI] [PMC free article] [PubMed]
- 123.Jenkins Y, McEntee M, Weis K, Greene WC. Characterization of HIV-1 vpr nuclear import: Analysis of signals and pathways. J Cell Biol. 1998;143:875–885. doi: 10.1083/jcb.143.4.875. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2132945. [DOI] [PMC free article] [PubMed]
- 124.Karni O, Friedler A, Zakai N, Gilon C, Loyter A. A peptide derived from the N-terminal region of HIV-1 Vpr promotes nuclear import in permeabilized cells: Elucidation of the NLS region of the Vpr. FEBS Lett. 1998;429:421–425. doi: 10.1016/s0014-5793(98)00645-0. http://www.ncbi.nlm.nih.gov/pubmed/9662462. [DOI] [PubMed]
- 125.Di Marzio P, Choe S, Ebright M, Knoblauch R, Landau NR. Mutational analysis of cell cycle arrest, nuclear localization and virion packaging of human immunodeficiency virus type 1 Vpr. J Virol. 1995;69:7909–7916. doi: 10.1128/jvi.69.12.7909-7916.1995. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=189735. [DOI] [PMC free article] [PubMed]
- 126.Macreadie IG, Castelli LA, Hewish DR, Kirkpatrick A, Ward AC, Azad AA. A domain of human immunodeficiency virus type 1 Vpr containing repeated H(S/F)RIG amino acid motifs causes cell growth arrest and structural defects. Proc Nat Acad Sci USA. 1995;92:2770–2774. doi: 10.1073/pnas.92.7.2770. http://www.pnas.org/content/92/7/2770.abstract. [DOI] [PMC free article] [PubMed]
- 127.Mahalingam S, Collman RG, Patel M, Monken CE, Srinivasan A. Role of the conserved dipeptide Gly75 and Cys76 on HIV-1 Vpr function. Virology. 1995;210:495–500. doi: 10.1006/viro.1995.1368. http://www.ncbi.nlm.nih.gov/pubmed/7618286. [DOI] [PubMed]
- 128.Mahalingam S, Khan SA, Jabbar MA, Monken CE, Collman RG, Srinivasan A. Identification of residues in the N-terminal acidic domain of HIV-1 Vpr essential for virion incorporation. Virology. 1995;207:297–302. doi: 10.1006/viro.1995.1081. http://www.ncbi.nlm.nih.gov/pubmed/7871742. [DOI] [PubMed]
- 129.Mahalingam S, Patel M, Collman RG, Srinivasan A. The carboxy-terminal domain is essential for stability and not for virion incorporation of HIV-1 Vpr into virus particles. Virology. 1995;214:647–652. doi: 10.1006/viro.1995.0079. http://linkinghub.elsevier.com/retrieve/pii/S0042682285700797. [DOI] [PubMed]
- 130.Stewart SA, Poon B, Jowett JB, Chen IS. Human immunodeficiency virus type 1 Vpr induces apoptosis following cell cycle arrest. J Virol. 1997;71:5579–5592. doi: 10.1128/jvi.71.7.5579-5592.1997. http://jvi.asm.org/cgi/content/abstract/71/7/5579. [DOI] [PMC free article] [PubMed]
- 131.Stewart SA, Poon B, Song JY, Chen IS. Human immunodeficiency virus type 1 vpr induces apoptosis through caspase activation. J Virol. 2000;74:3105–3111. doi: 10.1128/jvi.74.7.3105-3111.2000. http://www.ncbi.nlm.nih.gov/pubmed/10708425. [DOI] [PMC free article] [PubMed]
- 132.Kulkosky J, Laptev A, Shetty S, Srinivasan A, BouHamdan M, Prockop DJ, Pomerantz RJ. Human immunodeficiency virus type 1 Vpr alters bone marrow cell function. Blood. 1999;93:1906–1915. http://www.ncbi.nlm.nih.gov/pubmed/10068663. [PubMed]
- 133.Cohen GB, Gandhi RT, Davis DM, Mandelboim O, Chen BK, Strominger JL, Baltimore D. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity. 1999;10:661–671. doi: 10.1016/s1074-7613(00)80065-5. http://www.ncbi.nlm.nih.gov/pubmed/10403641. [DOI] [PubMed]
- 134.Garcia JV, Foster JL. Structural and functional correlates between HIV-1 and SIV Nef isolates. Virology. 1996;226:161–166. doi: 10.1006/viro.1996.0642. http://cat.inist.fr/?aModele=afficheN&cpsidt=10710281. [DOI] [PubMed]
- 135.Schwartz O, Marechal V, Le Gall S, Lemonnier F, Heard JM. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat Med. 1996;2:338–342. doi: 10.1038/nm0396-338. http://www.ncbi.nlm.nih.gov/pubmed/8612235. [DOI] [PubMed]
- 136.Hrecka K, Swigut T, Schindler M, Kirchhoff F, Skowronski J. Nef proteins from diverse groups of primate lentiviruses downmodulate CXCR4 to inhibit migration to the chemokine stromal derived factor 1. J Virol. 2005;79:10650–10659. doi: 10.1128/JVI.79.16.10650-10659.2005. http://www.ncbi.nlm.nih.gov/pubmed/16051857. [DOI] [PMC free article] [PubMed]
- 137.Madrid R, Janvier K, Hitchin D, Day J, Coleman S, Benichou S, et al. Nef-induced alteration of the early/recycling endosomal compartment correlates with enhancement of HIV-1 infectivity. J Biol Chem. 2005;280:5032–5044. doi: 10.1074/jbc.M401202200. http://www.jbc.org/cgi/content/abstract/280/6/5032. [DOI] [PubMed]
- 138.Michel N, Allespach I, Venzke S, Fackler OT, Keppler OT. The Nef protein of human immunodeficiency virus establishes superinfection immunity by a dual strategy to downregulate cell-surface CCR5 and CD4. Curr Biol. 2005;15:714–723. doi: 10.1016/j.cub.2005.02.058. http://linkinghub.elsevier.com/retrieve/pii/S0960982205002319. [DOI] [PubMed]
- 139.Michel N, Ganter K, Venzke S, Bitzegeio J, Fackler OT, Keppler OT. The Nef protein of human immunodeficiency virus is a broad-spectrum modulator of chemokine receptor cell surface levels that acts independently of classical motifs for receptor endocytosis and Galphai signaling. Mol Biol Cell. 2006;17:3578–3590. doi: 10.1091/mbc.E06-02-0117. http://www.molbiolcell.org/cgi/content/abstract/17/8/3578. [DOI] [PMC free article] [PubMed]
- 140.Stove V, Van de Walle I, Naessens E, Coene E, Stove C, Plum J, Verhasselt B. Human immunodeficiency virus Nef induces rapid internalization of the T-cell coreceptor CD8alphabeta. J Virol. 2005;79:11422–11433. doi: 10.1128/JVI.79.17.11422-11433.2005. http://www.ncbi.nlm.nih.gov/pubmed/16103193. [DOI] [PMC free article] [PubMed]
- 141.Swigut T, Shohdy N, Skowronski J. Mechanism for down-regulation of CD28 by Nef. EMBO J. 2001;20:1593–1604. doi: 10.1093/emboj/20.7.1593. http://www.ncbi.nlm.nih.gov/pubmed/11285224. [DOI] [PMC free article] [PubMed]
- 142.Venzke S, Michel N, Allespach I, Fackler OT, Keppler OT. Expression of Nef downregulates CXCR4, the major coreceptor of human immunodeficiency virus, from the surfaces of target cells and thereby enhances resistance to superinfection. J Virol. 2006;80:11141–11152. doi: 10.1128/JVI.01556-06. http://jvi.asm.org/cgi/content/abstract/80/22/11141?etoc. [DOI] [PMC free article] [PubMed]
- 143.Arganaraz ER, Schindler M, Kirchhoff F, Cortes MJ, Lama J. Enhanced CD4 down-modulation by late stage HIV-1 nef alleles is associated with increased Env incorporation and viral replication. J Biol Chem. 2003;278:33912–33919. doi: 10.1074/jbc.M303679200. http://gateway.nlm.nih.gov/MeetingAbstracts/102262631.html. [DOI] [PubMed]
- 144.Cortes MJ, Wong-Staal F, Lama J. Cell surface CD4 interferes with the infectivity of HIV-1 particles released from T cells. J Biol Chem. 2002;277:1770–1779. doi: 10.1074/jbc.M109807200. http://www.ncbi.nlm.nih.gov/pubmed/11704677. [DOI] [PubMed]
- 145.Schiavoni I, Trapp S, Santarcangelo AC, Piacentini V, Pugliese K, Baur A, Federico M. HIV-1 Nef enhances both membrane expression and virion incorporation of Env products. A model for the Nef-dependent increase of HIV-1 infectivity. J Biol Chem. 2004;279:22996–23006. doi: 10.1074/jbc.M312453200. http://direct.bl.uk/bld/PlaceOrder.do?UIN=150824276&ETOC=RN&from=searchengine. [DOI] [PubMed]
- 146.Bukovsky AA, Dorfman T, Weimann A, Gottlinger HG. Nef association with human immunodeficiency virus type 1 virions and cleavage by the viral protease. J Virol. 1997;71:1013–1018. doi: 10.1128/jvi.71.2.1013-1018.1997. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=191151. [DOI] [PMC free article] [PubMed]
- 147.Forshey BM, Aiken C. Disassembly of human immunodeficiency virus type 1 cores in vitro reveals association of Nef with the subviral ribonucleoprotein complex. J Virol. 2003;77:4409–4414. doi: 10.1128/JVI.77.7.4409-4414.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kotov A, Zhou J, Flicker P, Aiken C. Association of Nef with the human immunodeficiency virus type 1 core. J Virol. 1999;73:8824–8830. doi: 10.1128/jvi.73.10.8824-8830.1999. http://www.ncbi.nlm.nih.gov/pubmed/10482638. [DOI] [PMC free article] [PubMed]
- 149.Welker R, Hohenberg H, Tessmer U, Huckhagel C, Krausslich HG. Biochemical and structural analysis of isolated mature cores of human immunodeficiency virus type 1. J Virol. 2000;74:1168–1177. doi: 10.1128/jvi.74.3.1168-1177.2000. http://www.ncbi.nlm.nih.gov/pubmed/10627527. [DOI] [PMC free article] [PubMed]
- 150.Chakrabarti LA, Metzner KJ, Ivanovic T, Cheng H, Louis-Virelizier J, Connor RI, Cheng-Mayer C. A truncated form of Nef selected during pathogenic reversion of simian immunodeficiency virus SIVmac239Deltanef increases viral replication. J Virol. 2003;77:1245–1256. doi: 10.1128/JVI.77.2.1245-1256.2003. http://www.ncbi.nlm.nih.gov/pubmed/12502842. [DOI] [PMC free article] [PubMed]
- 151.Deacon NJ, Tsykin A, Solomon A, Smith K, Mills J. Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science. 1995;270:988–991. doi: 10.1126/science.270.5238.988. http://www.ncbi.nlm.nih.gov/pubmed/7481804. [DOI] [PubMed]
- 152.Gorry PR, Churchill M, Learmont J, Cherry C, Dyer WB, Wesselingh SL, Sullivan JS. Replication-dependent pathogenicity of attenuated nef-deleted HIV-1 in vivo. J Acquir Immune Defic Syndr. 2007;46:390–394. doi: 10.1097/QAI.0b013e31815aba08. http://www.ncbi.nlm.nih.gov/pubmed/17993857. [DOI] [PubMed]
- 153.Gorry PR, McPhee DA, Verity E, Dyer WB, Wesselingh SL, Churchill MJ. Pathogenicity and immunogenicity of attenuated, nef-deleted HIV-1 strains in vivo. Retrovirology. 2007;4:66–66. doi: 10.1186/1742-4690-4-66. http://www.ncbi.nlm.nih.gov/pubmed/17888184. [DOI] [PMC free article] [PubMed]
- 154.Kestler HW, 3rd, Ringler DJ, Mori K, Panicali DL, Sehgal PK, Daniel MD, Desrosiers RC. Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell. 1991;65:651–662. doi: 10.1016/0092-8674(91)90097-i. http://www.ncbi.nlm.nih.gov/pubmed/2032289. [DOI] [PubMed]
- 155.Kirchhoff F, Greenough TC, Brettler DB, Sullivan JL, Desrosiers RC. Brief report: Absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N Engl J Med. 1995;332:228–232. doi: 10.1056/NEJM199501263320405. http://www.ncbi.nlm.nih.gov/pubmed/7808489. [DOI] [PubMed]
- 156.Sawai ET, Hamza MS, Ye M, Shaw KE, Luciw PA. Pathogenic conversion of live attenuated simian immunodeficiency virus vaccines is associated with expression of truncated Nef. J Virol. 2000;74:2038–2045. doi: 10.1128/jvi.74.4.2038-2045.2000. http://www.ncbi.nlm.nih.gov/pubmed/10644378. [DOI] [PMC free article] [PubMed]
- 157.Aiken C, Trono D. Nef stimulates human immunodeficiency virus type 1 proviral DNA synthesis. J Virol. 1995;69:5048–5056. doi: 10.1128/jvi.69.8.5048-5056.1995. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=189322. [DOI] [PMC free article] [PubMed]
- 158.Bertsch C, Cluet D, Beyer C, Gloeckler L, Cecile A, Gut JP, Aubertin AM. Properties of a chimeric simian-human immunodeficiency virus expressing an hybrid HIV-1 Nef/SIV mac Nef protein. Arch Virol. 2002;147:1963–1975. doi: 10.1007/s00705-002-0857-8. [DOI] [PubMed] [Google Scholar]
- 159.Schaeffer E, Geleziunas R, Greene WC. Human immunodeficiency virus type 1 Nef functions at the level of virus entry by enhancing cytoplasmic delivery of virions. J Virol. 2001;75:2993–3000. doi: 10.1128/JVI.75.6.2993-3000.2001. http://www.ncbi.nlm.nih.gov/pubmed/11222724. [DOI] [PMC free article] [PubMed]
- 160.Campbell EM, Nunez R, Hope TJ. Disruption of the actin cytoskeleton can complement the ability of Nef to enhance human immunodeficiency virus type 1 infectivity. J Virol. 2004;78:5745–5755. doi: 10.1128/JVI.78.11.5745-5755.2004. http://direct.bl.uk/bld/PlaceOrder.do?UIN=150805604&ETOC=RN&from=searchengine. [DOI] [PMC free article] [PubMed]
- 161.Fujii Y, Otake K, Tashiro M, Adachi A. Human immunodeficiency virus type 1 Nef protein on the cell surface is cytocidal for human CD4+ T cells. FEBS Lett. 1996;393:105–108. doi: 10.1016/0014-5793(96)00862-9. http://linkinghub.elsevier.com/retrieve/pii/0014579396008629. [DOI] [PubMed]
- 162.Kramer-Hammerle S, Hahn A, Brack-Werner R, Werner T. Elucidating effects of long-term expression of HIV-1 Nef on astrocytes by microarray, promoter and literature analyses. Gene. 2005;358:31–38. doi: 10.1016/j.gene.2005.05.011. http://cat.inist.fr/?aModele=afficheN&cpsidt=17117109. [DOI] [PubMed]
- 163.Trillo-Pazos G, McFarlane-Abdulla E, Campbell IC, Pilkington GJ, Everall IP. Recombinant nef HIV-IIIB protein is toxic to human neurons in culture. Brain Res. 2000;864:315–326. doi: 10.1016/s0006-8993(00)02213-7. http://www.ncbi.nlm.nih.gov/pubmed/10802040. [DOI] [PubMed]
- 164.Garry RF, Kort JJ, Koch-Nolte F, Koch G. Similarities of viral proteins to toxins that interact with monovalent cation channels. AIDS. 1991;5:1381–1384. doi: 10.1097/00002030-199111000-00017. http://www.ncbi.nlm.nih.gov/pubmed/1722677. [DOI] [PubMed]
- 165.Werner T, Ferroni S, Saermark T, Brack-Werner R, Banati RB, Erfle V. HIV-1 Nef protein exhibits structural and functional similarity to scorpion peptides interacting with K+ channels. AIDS. 1991;5:1301–1308. doi: 10.1097/00002030-199111000-00003. http://www.ncbi.nlm.nih.gov/pubmed/1768378. [DOI] [PubMed]
- 166.Speth C, Schabetsberger T, Mohsenipour I, Stockl G, et al. Mechanism of human immunodeficiency virus-induced complement expression in astrocytes and neurons. J Virol. 2002;76:3179–3188. doi: 10.1128/JVI.76.7.3179-3188.2002. http://cat.inist.fr/?aModele=afficheN&cpsidt=13548976. [DOI] [PMC free article] [PubMed]
- 167.Kumar A, Dhawan S, Mukhopadhyay A, Aggarwal BB. Human immunodeficiency virus-1-tat induces matrix metalloproteinase-9 in monocytes through protein tyrosine phosphatase-mediated activation of nuclear transcription factor NF-kappaB. FEBS Lett. 1999;462:140–144. doi: 10.1016/s0014-5793(99)01487-8. http://linkinghub.elsevier.com/retrieve/pii/S0014579399014878. [DOI] [PubMed]
- 168.McManus CM, Weidenheim K, Woodman SE, Nunez J, Hesselgesser J, Nath A, Berman JW. Chemokine and chemokine-receptor expression in human glial elements: Induction by the HIV protein, Tat and chemokine autoregulation. Am J Pathol. 2000;156:1441–1453. doi: 10.1016/S0002-9440(10)65013-4. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1876886. [DOI] [PMC free article] [PubMed]
- 169.Del Valle L, Croul S, Morgello S, Amini S, Rappaport J, Khalili K. Detection of HIV-1 Tat and JCV capsid protein, VP1, in AIDS brain with progressive multifocal leukoencephalopathy. J Neurovirol. 2000;6:221–228. doi: 10.3109/13550280009015824. http://www.ncbi.nlm.nih.gov/pubmed/10878711. [DOI] [PubMed]
- 170.Song L, Nath A, Geiger JD, Moore A, Hochman S. Human immunodeficiency virus type 1 Tat protein directly activates neuronal N-methyl-D-aspartate receptors at an allosteric zinc-sensitive site. J Neurovirol. 2003;9:399–403. doi: 10.1080/13550280390201704. http://www.ncbi.nlm.nih.gov/pubmed/12775422. [DOI] [PubMed]
- 171.King JE, Eugenin EA, Buckner CM, Berman JW. HIV tat and neurotoxicity. Microbes Infect. 2006;8:1347–1357. doi: 10.1016/j.micinf.2005.11.014. http://www.ncbi.nlm.nih.gov/pubmed/16697675. [DOI] [PubMed]
- 172.Li W, Huang Y, Reid R, Steiner J, Malpica-Llanos T, Nath A, et al. NMDA receptor activation by HIV-Tat protein is clade dependent. J Neurosci. 2008;28:12190–12198. doi: 10.1523/JNEUROSCI.3019-08.2008. http://www.ncbi.nlm.nih.gov/pubmed/19020013. [DOI] [PMC free article] [PubMed]
- 173.Cepeda C, Levine MS. Dopamine and N-methyl-D-aspartate receptor interactions in the neostriatum. Dev Neurosci. 1998;20:1–18. doi: 10.1159/000017294. [DOI] [PubMed] [Google Scholar]
- 174.Cepeda C, Levine MS. Where do you think you are going? The NMDA-D1 receptor trap. Sci STKE. 2006;333:20–20. doi: 10.1126/stke.3332006pe20. [DOI] [PubMed] [Google Scholar]
- 175.Lee FJ, Liu F. Direct interactions between NMDA and D1 receptors: A tale of tails. Biochem Soc Trans. 2004;32:1032–1036. doi: 10.1042/BST0321032. http://www.ionchannels.org/showabstract.php?pmid=15506956. [DOI] [PubMed]
- 176.Lezcano N, Bergson C. D1/D5 dopamine receptors stimulate intracellular calcium release in primary cultures of neocortical and hippocampal neurons. J Neurophysiol. 2002;87:2167–2175. doi: 10.1152/jn.00541.2001. http://www.ncbi.nlm.nih.gov/pubmed/11929934. [DOI] [PubMed]
- 177.Missale C, Fiorentini C, Busi C, Collo G, Spano PF. The NMDA/D1 receptor complex as a new target in drug development. Curr Top Med Chem. 2006;6:801–808. doi: 10.2174/156802606777057562. http://www.ncbi.nlm.nih.gov/pubmed/16719818. [DOI] [PubMed]
- 178.Pei L, Lee FJ, Moszczynska A, Vukusic B, Liu F. Regulation of dopamine D1 receptor function by physical interaction with the NMDA receptors. J Neurosci. 2004;24:1149–1158. doi: 10.1523/JNEUROSCI.3922-03.2004. http://www.ncbi.nlm.nih.gov/pubmed/14762133. [DOI] [PMC free article] [PubMed]
- 179.Hudson L, Liu J, Nath A, Jones M, Raghavan R, Narayan O, Male D, Everall I. Detection of the human immunodeficiency virus regulatory protein tat in CNS tissues. J Neurovirol. 2000;6:145–155. doi: 10.3109/13550280009013158. http://www.ncbi.nlm.nih.gov/pubmed/10822328. [DOI] [PubMed]
- 180.Banks WA, Robinson SM, Nath A. Permeability of the blood-brain barrier to HIV-1 Tat. Exp Neurol. 2005;193:218–227. doi: 10.1016/j.expneurol.2004.11.019. http://linkinghub.elsevier.com/retrieve/pii/S0014488604004613. [DOI] [PubMed]
- 181.Eugenin EA, D'Aversa TG, Lopez L, Calderon TM, Berman JW. MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. J Neurochem. 2003;85:1299–1311. doi: 10.1046/j.1471-4159.2003.01775.x. http://www.ncbi.nlm.nih.gov/pubmed/12753088. [DOI] [PubMed]
- 182.Haughey NJ, Nath A, Mattson MP, Slevin JT, Geiger JD. HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J Neurochem. 2001;78:457–467. doi: 10.1046/j.1471-4159.2001.00396.x. http://cat.inist.fr/?aModele=afficheN&cpsidt=14094389. [DOI] [PubMed]
- 183.Nath A, Anderson C, Jones M, Maragos W, Booze R, Mactutus C, Bell J, Hauser KF, Mattson M. Neurotoxicity and dysfunction of dopaminergic systems associated with AIDS dementia. J Psychopharmacol. 2000;14:222–227. doi: 10.1177/026988110001400305. http://www.ncbi.nlm.nih.gov/pubmed/11106300. [DOI] [PubMed]
- 184.Bruce-Keller AJ, Chauhan A, Dimayuga FO, Gee J, Keller JN, Nath A. Synaptic transport of human immunodeficiency virus-Tat protein causes neurotoxicity and gliosis in rat brain. J Neurosci. 2003;23:8417–8422. doi: 10.1523/JNEUROSCI.23-23-08417.2003. http://www.ncbi.nlm.nih.gov/pubmed/12968004. [DOI] [PMC free article] [PubMed]
- 185.Albini A, Soldi R, Giunciuglio D, Giraudo E, Benelli R, Primo L, Bussolino F, et al. The angiogenesis induced by HIV-1 tat protein is mediated by the Flk-1/KDR receptor on vascular endothelial cells. Nat Med. 1996;2:1371–1375. doi: 10.1038/nm1296-1371. http://www.ncbi.nlm.nih.gov/pubmed/8946838. [DOI] [PubMed]
- 186.Ghezzi S, Noonan DM, Aluigi MG, Vallanti G, Cota M, Albini A. Inhibition of CXCR4-dependent HIV-1 infection by extracellular HIV-1 Tat. Biochem Biophys Res Commun. 2000;270:992–996. doi: 10.1006/bbrc.2000.2523. http://www.ncbi.nlm.nih.gov/pubmed/10772939. [DOI] [PubMed]
- 187.Liu Y, Jones M, Hingtgen CM, Bu G, Laribee N, He JJ, et al. Uptake of HIV-1 tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat Med. 2000;6:1380–1387. doi: 10.1038/82199. [DOI] [PubMed] [Google Scholar]
- 188.Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med. 2004;10:310–315. doi: 10.1038/nm996. [DOI] [PubMed] [Google Scholar]
- 189.Conant K, Garzino-Demo A, Nath A, McArthur JC, Major EO, et al. Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proc Natl Acad Sci USA. 1998;95:3117–3121. doi: 10.1073/pnas.95.6.3117. http://www.ncbi.nlm.nih.gov/pubmed/9501225. [DOI] [PMC free article] [PubMed]
- 190.Cinque P, Vago L, Mengozzi M, Torri V, Ceresa D, Poli G. Elevated cerebrospinal fluid levels of monocyte chemotactic protein-1 correlate with HIV-1 encephalitis and local viral replication. AIDS. 1998;12:1327–1332. doi: 10.1097/00002030-199811000-00014. http://cat.inist.fr/?aModele=afficheN&cpsidt=2341128. [DOI] [PubMed]
- 191.Liu X, Jana M, Dasgupta S, Koka S, He J, Wood C, Pahan K. Human immunodeficiency virus type 1 (HIV-1) tat induces nitric-oxide synthase in human astroglia. J Biol Chem. 2002;277:39312–39319. doi: 10.1074/jbc.M205107200. http://www.ncbi.nlm.nih.gov/pubmed/12167619. [DOI] [PMC free article] [PubMed]
- 192.Fine SM, Angel RA, Perry SW, Epstein LG, Rothstein JD, Dewhurst S, Gelbard HA. Tumor necrosis factor alpha inhibits glutamate uptake by primary human astrocytes. Implications for pathogenesis of HIV-1 dementia. J Biol Chem. 1996;271:15303–15306. doi: 10.1074/jbc.271.26.15303. http://www.ncbi.nlm.nih.gov/pubmed/8663435. [DOI] [PubMed]
- 193.Sevigny JJ, Albert SM, McDermott MP, McArthur JC, Sacktor N, Marder K. Evaluation of HIV RNA and markers of immune activation as predictors of HIV-associated dementia. Neurology. 2004;63:2084–2090. doi: 10.1212/01.wnl.0000145763.68284.15. http://www.neurology.org/cgi/content/abstract/63/11/2084. [DOI] [PubMed]
- 194.Nath A, Geiger J. Neurobiological aspects of human immunodeficiency virus infection: Neurotoxic mechanisms. Prog Neurobiol. 1998;54:19–33. doi: 10.1016/s0301-0082(97)00053-1. http://www.ncbi.nlm.nih.gov/pubmed/9460791. [DOI] [PubMed]
- 195.Levin MC, Jacobson S. Cellular and humoral immune responses associated with HTLV-I associated myelopathy/tropical spastic paraparesis. Ann NY Acad Sci. 1997;835:142–152. doi: 10.1111/j.1749-6632.1997.tb48625.x. http://www.ncbi.nlm.nih.gov/pubmed/9616769. [DOI] [PubMed]
- 196.Kubota R, Nagai M, Kawanishi T, Osame M, Jacobson S. Increased HTLV type 1 tax specific CD8+ cells in HTLV type 1-asociated myelopathy/tropical spastic paraparesis: Correlation with HTLV type 1 proviral load. AIDS Res Hum Retroviruses. 2000;16:1705–1709. doi: 10.1089/08892220050193182. http://www.ncbi.nlm.nih.gov/pubmed/11080814. [DOI] [PubMed]
- 197.Daenke S, Kermode AG, Hall SE, Taylor G, Weber J, Nightingale S, Bangham CR. High activated and memory cytotoxic T-cell responses to HTLV-1 in healthy carriers and patients with tropical spastic paraparesis. Virology. 1996;217:139–146. doi: 10.1006/viro.1996.0101. http://www.ncbi.nlm.nih.gov/pubmed/8599198. [DOI] [PubMed]
- 198.Jacobson S, McFarlin DE, Robinson S, Voskuhl R, Martin R, Brewah A, Newell AJ, Koenig S. HTLV-I-specific cytotoxic T lymphocytes in the cerebrospinal fluid of patients with HTLV-I-associated neurological disease. Ann Neurol. 1992;32:651–657. doi: 10.1002/ana.410320508. http://www.ncbi.nlm.nih.gov/pubmed/1449245. [DOI] [PubMed]
- 199.Umehara F, Izumo S, Nakagawa M, Ronquillo AT, Takahashi K, Matsumuro K, Sato E, Osame M. Immunocytochemical analysis of the cellular infiltrate in the spinal cord lesions in HTLV-I-associated myelopathy. J Neuropathol Exp Neurol. 1993;52:424–430. doi: 10.1097/00005072-199307000-00010. http://www.ncbi.nlm.nih.gov/pubmed/8355031. [DOI] [PubMed]
- 200.Levin MC, Lee SM, Kalume F, Morcos Y, Dohan FC, Jr, Stuart JM, et al. Autoimmunity due to molecular mimicry as a cause of neurological disease. Nat Med. 2002;8:509–513. doi: 10.1038/nm0502-509. http://www.ncbi.nlm.nih.gov/pubmed/11984596. [DOI] [PMC free article] [PubMed]
- 201.Levin MC, Lee SM, Morcos Y, Brady J, Stuart J. Cross-reactivity between immunodominant human T lymphotropic virus type I tax and neurons: Implications for molecular mimicry. J Infect Dis. 2002;186:1514–1517. doi: 10.1086/344734. http://www.ncbi.nlm.nih.gov/pubmed/12404172. [DOI] [PubMed]
- 202.Elovaara I, Utz U, Smith S, Jacobson S. Limited T cell receptor usage by HTLV-I tax-specific, HLA class I restricted cytotoxic T lymphocytes from patients with HTLV-I associated neurological disease. J Neuroimmunol. 1995;63:47–53. doi: 10.1016/0165-5728(95)00129-8. http://www.ncbi.nlm.nih.gov/pubmed/8557824. [DOI] [PubMed]
- 203.Elovaara I, Koenig S, Brewah AY, Woods RM, Lehky T, Jacobson S. High human T cell lymphotropic virus type 1 (HTLV-1)-specific precursor cytotoxic T lymphocyte frequencies in patients with HTLV-1-associated neurological disease. J Exp Med. 1993;177:1567–1573. doi: 10.1084/jem.177.6.1567. http://cat.inist.fr/?aModele=afficheN&cpsidt=4844986. [DOI] [PMC free article] [PubMed]
- 204.Muraro PA, Wandinger KP, Bielekova B, Gran B, Marques A, Martin R. Molecular tracking of antigen-specific T cell clones in neurological immune-mediated disorders. Brain. 2003;126:20–31. doi: 10.1093/brain/awg021. http://www.ncbi.nlm.nih.gov/pubmed/12477694. [DOI] [PMC free article] [PubMed]
- 205.Liu Y, Dole K, Stanley JR, Richard V, Rosol TJ, Ratner L, Lairmore M, Feuer G. Engraftment and tumorigenesis of HTLV-1 transformed T cell lines in SCID/bg and NOD/SCID mice. Leuk Res. 2002;26:561–567. doi: 10.1016/s0145-2126(01)00169-2. http://www.ncbi.nlm.nih.gov/pubmed/12007504. [DOI] [PubMed]
- 206.Bangham CR. HTLV-1 infections. J Clin Pathol. 2000;53:581–586. doi: 10.1136/jcp.53.8.581. http://jcp.bmj.com/cgi/content/abstract/53/8/581. [DOI] [PMC free article] [PubMed]
- 207.Levin MC, Jacobson S. HTLV-I associated myelopathy/tropical spastic paraparesis (HAM/TSP): A chronic progressive neurologic disease associated with immunologically mediated damage to the central nervous system. J Neurovirol. 1997;3:126–140. doi: 10.3109/13550289709015802. http://www.biomedexperts.com/Abstract.bme/9103896/HTLV-I-associated_myelopathy_tropical_spastic_paraparesis_HAM_TSP. [DOI] [PubMed]
- 208.Jeffery KJ, Usuku K, Hall SE, Matsumoto W, Taylor GP, Bangham CR, et al. HLA alleles determine human T-lymphotropic virus-I (HTLV-I) proviral load and the risk of HTLV-I-associated myelopathy. Proc Natl Acad Sci USA. 1999;96:3848–3853. doi: 10.1073/pnas.96.7.3848. http://www.ncbi.nlm.nih.gov/pubmed/10097126. [DOI] [PMC free article] [PubMed]
- 209.Furukawa Y, Yamashita M, Usuku K, Izumo S, Nakagawa M, Osame M. Phylogenetic subgroups of human T cell lymphotropic virus (HTLV) type I in the tax gene and their association with different risks for HTLV-I-associated myelopathy/tropical spastic paraparesis. J Infect Dis. 2000;182:1343–1349. doi: 10.1086/315897. http://www.ncbi.nlm.nih.gov/pubmed/11010842. [DOI] [PubMed]
- 210.Barmak K, Harhaj EW, Wigdahl B. Mediators of central nervous system damage during the progression of human T-cell leukemia type I-associated myelopathy/tropical spastic paraparesis. J Neuroviro. 2003;9:522–529. doi: 10.1080/13550280390218689. http://cat.inist.fr/?aModele=afficheN&cpsidt=15172925. [DOI] [PubMed]
- 211.Grassmann R, Berchtold S, Radant I, Alt M, Fleckenstein B, Sodroski JG, Haseltine WA, Ramstedt U. Role of human T-cell leukemia virus type 1 X region proteins in immortalization of primary human lymphocytes in culture. J Virol. 1992;66:4570–4575. doi: 10.1128/jvi.66.7.4570-4575.1992. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=241270. [DOI] [PMC free article] [PubMed]
- 212.Sodroski JG, Rosen CA, Haseltine WA. Trans-acting transcriptional activation of the long terminal repeat of human T lymphotropic viruses in infected cells. Science. 1984;225:381–385. doi: 10.1126/science.6330891. http://www.ncbi.nlm.nih.gov/pubmed/6330891. [DOI] [PubMed]
- 213.Sodroski J, Rosen C, Goh WC, Haseltine W. A transcriptional activator protein encoded by the x-lor region of the human T-cell leukemia virus. Science. 1985;228:1430–1434. doi: 10.1126/science.2990028. http://cat.inist.fr/?aModele=afficheN&cpsidt=8572669. [DOI] [PubMed]
- 214.Beimling P, Moelling K. Direct interaction of CREB protein with 21 bp Tax-response elements of HTLV-ILTR. Oncogene. 1992;7:257–262. http://www.ncbi.nlm.nih.gov/pubmed/1532242. [PubMed]
- 215.Paca-Uccaralertkun S, Zhao LJ, Adya N, Cross JV, Cullen BR, Boros IM, Giam CZ. In vitro selection of DNA elements highly responsive to the human T-cell lymphotropic virus type I transcriptional activator, Tax. Mol Cell Biol. 1994;14:456–462. doi: 10.1128/mcb.14.1.456. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=358395. [DOI] [PMC free article] [PubMed]
- 216.Zhao LJ, Giam CZ. Human T-cell lymphotropic virus type I (HTLV-I) transcriptional activator, Tax, enhances CREB binding to HTLV-I 21-base-pair repeats by protein-protein interaction. Proc Natl Acad Sci USA. 1992;89:7070–7074. doi: 10.1073/pnas.89.15.7070. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=49647. [DOI] [PMC free article] [PubMed]
- 217.Lenzmeier BA, Giebler HA, Nyborg JK. Human T-cell leukemia virus type 1 Tax requires direct access to DNA for recruitment of CREB binding protein to the viral promoter. Mol Cell Biol. 1998;18:721–731. doi: 10.1128/mcb.18.2.721. http://www.ncbi.nlm.nih.gov/pubmed/9447968. [DOI] [PMC free article] [PubMed]
- 218.Adya N, Giam CZ. Distinct regions in human T-cell lymphotropic virus type I tax mediate interactions with activator protein CREB and basal transcription factors. J Virol. 1995;69:1834–1841. doi: 10.1128/jvi.69.3.1834-1841.1995. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=188794. [DOI] [PMC free article] [PubMed]
- 219.Suzuki T, Hirai H, Fujisawa J, Fujita T, Yoshida M. A trans-activator Tax of human T-cell leukemia virus type 1 binds to NF-kappa B p50 and Serum Response Factor (SRF) and associates with enhancer DNAs of the NF-kappa B site and CArG box. Oncogene. 1993;8:2391–2397. http://www.ncbi.nlm.nih.gov/pubmed/8361755. [PubMed]
- 220.Azran I, Schavinsky-Khrapunsky Y, Aboud M. Role of Tax protein in human T-cell leukemia virus type-I leukemogenicity. Retrovirology. 2004;1:20–20. doi: 10.1186/1742-4690-1-20. http://www.ncbi.nlm.nih.gov/pubmed/15310405. [DOI] [PMC free article] [PubMed]
- 221.Harrod R, Kuo YL, Tang Y, Yao Y, Vassilev A, Nakatani Y, Giam CZ. p300 and p300/cAMP-responsive element-binding protein associated factor interact with human T-cell lymphotropic virus type-1 Tax in a multi-histone acetyltransferase/activator-enhancer complex. J Biol Chem. 2000;275:20853–20860. doi: 10.1074/jbc.275.16.11852. http://www.ncbi.nlm.nih.gov/pubmed/10779504. [DOI] [PubMed]
- 222.Harrod R, Tang Y, Nicot C, Lu HS, Vassilev A, Nakatani Y, Giam CZ. An exposed KID-like domain in human T-cell lymphotropic virus type 1 Tax is responsible for the recruitment of coactivators CBP/p300. Mol Cell Biol. 1998;18:5052–5061. doi: 10.1128/mcb.18.9.5052. http://cat.inist.fr/?aModele=afficheN&cpsidt=10544243. [DOI] [PMC free article] [PubMed]
- 223.Tie F, Adya N, Greene WC, Giam CZ. Interaction of the human T-lymphotropic virus type 1 Tax dimer with CREB and the viral 21-base-pair repeat. J Virol. 1996;70:8368–8374. doi: 10.1128/jvi.70.12.8368-8374.1996. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=190925. [DOI] [PMC free article] [PubMed]
- 224.Arima N, Molitor JA, Smith MR, Kim JH, Daitoku Y, Greene WC. Human T-cell leukemia virus type I Tax induces expression of the Rel-related family of kappa B enhancer-binding proteins: evidence for a pretranslational component of regulation. J Virol. 1991;65:6892–6899. doi: 10.1128/jvi.65.12.6892-6899.1991. http://jvi.asm.org/cgi/reprint/65/12/6892.pdf. [DOI] [PMC free article] [PubMed]
- 225.Sun SC, Elwood J, Beraud C, Greene WC. Human T-cell leukemia virus type I Tax activation of NF-kappa B/Rel involves phosphorylation and degradation of I kappa B alpha and RelA (p65)-mediated induction of the c-rel gene. Mol Cell Biol. 1994;14:7377–7384. doi: 10.1128/mcb.14.11.7377. http://mcb.asm.org/cgi/content/abstract/14/11/7377. [DOI] [PMC free article] [PubMed]
- 226.Arias J, Alberts AS, Brindle P, Claret FX, Smeal T, Karin M, Feramisco J, Montminy M. Activation of cAMP and mitogen responsive genes relies on a common nuclear factor. Nature. 1994;370:226–229. doi: 10.1038/370226a0. http://www.ncbi.nlm.nih.gov/pubmed/8028671. [DOI] [PubMed]
- 227.Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. http://www.nature.com/nature/journal/v365/n6449/abs/365855a0.html. [DOI] [PubMed]
- 228.Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. http://www.ncbi.nlm.nih.gov/pubmed/2573431. [DOI] [PubMed]
- 229.Zhao LJ, Giam CZ. Interaction of the human T-cell lymphotrophic virus type I (HTLV-I) transcriptional activator Tax with cellular factors that bind specifically to the 21-base-pair repeats in the HTLV-I enhancer. Proc Natl Acad Sci USA. 1991;88:11445–11449. doi: 10.1073/pnas.88.24.11445. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=53152. [DOI] [PMC free article] [PubMed]
- 230.Feuer G, Fraser JK, Zack JA, Lee F, Feuer R, Chen IS. Human T-cell leukemia virus infection of human hematopoietic progenitor cells: Maintenance of virus infection during differentiation in vitro and in vivo. J Virol. 1996;70:4038–4044. doi: 10.1128/jvi.70.6.4038-4044.1996. http://jvi.asm.org/cgi/content/abstract/70/6/4038. [DOI] [PMC free article] [PubMed]
- 231.Mikovits J, Ruscetti F, Zhu W, Bagni R, Dorjsuren D, Shoemaker R. Potential cellular signatures of viral infections in human hematopoietic cells. Dis Marker. 2001;17:173–178. doi: 10.1155/2001/896953. http://cat.inist.fr/?aModele=afficheN&cpsidt=14155476. [DOI] [PMC free article] [PubMed]
- 232.Hoffman PM, Dhib-Jalbut S, Mikovits JA, Robbins DS, et al. Human T-cell leukemia virus type I infection of monocytes and microglial cells in primary human cultures. Proc Natl Acad Sci USA. 1992;89:11784–11788. doi: 10.1073/pnas.89.24.11784. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=50641. [DOI] [PMC free article] [PubMed]
- 233.Szymocha R, Brisson C, Bernard A, Akaoka H, Belin MF, Giraudon P. Long-term effects of HTLV-1 on brain astrocytes: Sustained expression of Tax-1 associated with synthesis of inflammatory mediators. J Neurovirol. 2000;6:350–357. doi: 10.3109/13550280009030761. http://www.aegis.com/aidsline/2000/nov/A00B0250.html. [DOI] [PubMed]
- 234.Hirayama M, Miyadai T, Yokochi T, Sato K, Kubota T, Iidaand M, Fujiki N. Infection of human T-lymphotropic virus type I to astrocytes in vitro with induction of the class II major histocompatibility complex. Neurosci Lett. 1988;92:34–39. doi: 10.1016/0304-3940(88)90738-0. http://www.ncbi.nlm.nih.gov/pubmed/3185977. [DOI] [PubMed]
- 235.Yamada M, Watabe K, Saida T, Kim SU. Increased susceptibility of human fetal astrocytes to human T-lymphotropic virus type I in culture. J Neuropathol Exp Neurol. 1991;50:97–107. doi: 10.1097/00005072-199103000-00001. http://cat.inist.fr/?aModele=afficheN&cpsidt=4505407. [DOI] [PubMed]
- 236.Mendez E, Kawanishi T, Clemens K, Siomi H, Soldan SS, et al. Astrocyte-specific expression of human T-cell lymphotropic virus type 1 (HTLV-1) Tax: Induction of tumor necrosis factor alpha and susceptibility to lysis by CD8+ HTLV-1-specific cytotoxic T cells. J Virol. 1997;71:9143–9149. doi: 10.1128/jvi.71.12.9143-9149.1997. http://cat.inist.fr/?aModele=afficheN&cpsidt=10858480. [DOI] [PMC free article] [PubMed]
- 237.Koyanagi Y, Itoyama Y, Nakamura N, Takamatsu K, Kira J, Iwamasa T, Goto I, Yamamoto N. In vivo infection of human T-cell leukemia virus type I in non-T cells. Virology. 1993;196:25–33. doi: 10.1006/viro.1993.1451. http://www.ncbi.nlm.nih.gov/pubmed/8356797. [DOI] [PubMed]
- 238.Quiterio S, Grant C, Hogan TH, Krebs FC, Wigdahl B. C/EBP-and Tat-mediated activation of the HIV-1 LTR in CD34+ hematopoietic progenitor cells. Biomed Pharmacother. 2003;57:49–56. doi: 10.1016/s0753-3322(02)00332-3. http://www.ncbi.nlm.nih.gov/pubmed/12642037. [DOI] [PubMed]
- 239.Hivin P, Gaudray G, Devaux C, Mesnard JM. Interaction between C/EBPbeta and Tax down-regulates human T-cell leukemia virus type I transcription. Virology. 2004;318:556–565. doi: 10.1016/j.virol.2003.10.027. http://www.ncbi.nlm.nih.gov/pubmed/14972524. [DOI] [PubMed]
- 240.Grant C, Nonnemacher M, Jain P, Pandya D, Irish B, Williams SC, Wigdahl B. CCAAT/enhancer-binding proteins modulate human T cell leukemia virus type 1 long terminal repeat activation. Virology. 2006;348:354–369. doi: 10.1016/j.virol.2005.12.024. http://www.ncbi.nlm.nih.gov/pubmed/16458341. [DOI] [PubMed]
- 241.Grant C, Jain P, Nonnemacher M, Flaig KE, Irish B, Wigdahl B, et al. AP-1-directed human T cell leukemia virus type 1 viral gene expression during monocytic differentiation. J Leukoc Biol. 2006;80:640–650. doi: 10.1189/jlb.1205723. http://www.jleukbio.org/cgi/content/abstract/80/3/640. [DOI] [PubMed]
- 242.Wessner R, Yao J, Wigdahl B. Sp family members preferentially interact with the promoter proximal repeat within the HTLV-I enhancer. Leukemia. 1997;11:10–13. http://www.ncbi.nlm.nih.gov/pubmed/9209281. [PubMed]
- 243.Barnhart MK, Connor LM, Marriott SJ. Function of the human T-cell leukemia virus type 1 21-base-pair repeats in basal transcription. J Virol. 1997;71:337–344. doi: 10.1128/jvi.71.1.337-344.1997. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=191056. [DOI] [PMC free article] [PubMed]
- 244.Jiang H, Lu H, Schiltz RL, Pise-Masison CA, Ogryzko VV, Nakatani Y, Brady JN. PCAF interacts with tax and stimulates tax transactivation in a histone acetyltransferase-independent manner. Mol Cell Biol. 1999;19:8136–8145. doi: 10.1128/mcb.19.12.8136. http://www.ncbi.nlm.nih.gov/pubmed/10567539. [DOI] [PMC free article] [PubMed]
- 245.Kashanchi F, Duvall JF, Kwok RP, Lundblad JR, Goodman RH, Brady JN. The coactivator CBP stimulates human T-cell lymphotrophic virus type I Tax transactivation in vitro. J Biol Chem. 1998;273:34646–34652. doi: 10.1074/jbc.273.51.34646. http://www.ncbi.nlm.nih.gov/pubmed/9852138. [DOI] [PubMed]
- 246.Okada M, Jeang KT. Differential requirements for activation of integrated and transiently transfected human T-cell leukemia virus type 1 long terminal repeat. J Virol. 2002;76:12564–12573. doi: 10.1128/JVI.76.24.12564-12573.2002. http://www.ncbi.nlm.nih.gov/pubmed/12438582. [DOI] [PMC free article] [PubMed]
- 247.Alefantis T, Jain P, Ahuja J, Mostoller K, Wigdahl B. HTLV-1 Tax nucleocytoplasmic shuttling, interaction with the secretory pathway, extracellular signaling and implications for neurologic disease. J Biomed Sci. 2005;12:961–974. doi: 10.1007/s11373-005-9026-x. http://cat.inist.fr/?aModele=afficheN&cpsidt=17762490. [DOI] [PubMed]
- 248.Smith MR, Greene WC. Characterization of a novel nuclear localization signal in the HTLV-I tax transactivator protein. Virology. 1992;187:316–320. doi: 10.1016/0042-6822(92)90320-o. http://www.ncbi.nlm.nih.gov/pubmed/1736534. [DOI] [PubMed]
- 249.Gitlin SD, Lindholm PF, Marriott SJ, Brady JN. Transdominant human T-cell lymphotropic virus type I TAX1 mutant that fails to localize to the nucleus. J Virol. 1991;65:2612–2621. doi: 10.1128/jvi.65.5.2612-2621.1991. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=240619. [DOI] [PMC free article] [PubMed]
- 250.Cartier L, Ramirez E. Presence of HTLV-I Tax protein in cerebrospinal fluid from HAM/TSP patients. Arch Virol. 2005;150:743–753. doi: 10.1007/s00705-004-0443-3. [DOI] [PubMed] [Google Scholar]
- 251.Bex F, Murphy K, Wattiez R, Burny A, Gaynor R. Phosphorylation of the human T-cell leukemia virus type 1 transactivator tax on adjacent serine residues is critical for tax activation. J Virol. 1999;73:738–745. doi: 10.1128/jvi.73.1.738-745.1999. http://jvi.asm.org/cgi/content/abstract/73/1/738. [DOI] [PMC free article] [PubMed]
- 252.Fontes J, Strawhecker J, Bills N, Lewis R, Hinrichs S. Phorbol esters modulate the phosphorylation of human T-cell leukemia virus type I Tax. J Virol. 1993;67:4436–4441. doi: 10.1128/jvi.67.7.4436-4441.1993. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=237821. [DOI] [PMC free article] [PubMed]
- 253.Lindholm PF, Marriott SJ, Gitlin SD, Bohan CA, Brady JN. Induction of nuclear NF-kappa B DNA binding activity after exposure of lymphoid cells to soluble tax1 protein. New Biol. 1990;2:1034–1043. http://www.ncbi.nlm.nih.gov/pubmed/2101630. [PubMed]
- 254.Alefantis T, Mostoller K, Jain P, Harhaj E, Grant C, Wigdahl B. Secretion of the human T cell leukemia virus type I transactivator protein tax. J Biol Chem. 2005;280:17353–17362. doi: 10.1074/jbc.M409851200. http://www.ncbi.nlm.nih.gov/pubmed/15659397. [DOI] [PubMed]
- 255.Lippincott-Schwartz J, Smith CL. Insights into secretory and endocytic membrane traffic using green fluorescent protein chimeras. Curr Opin Neurobiol. 1997;7:631–639. doi: 10.1016/s0959-4388(97)80082-7. http://www.ncbi.nlm.nih.gov/pubmed/9384543. [DOI] [PubMed]
- 256.Lippincott-Schwartz J, Roberts TH, Hirschberg K. Secretory protein trafficking and organelle dynamics in living cells. Ann Rev Cell Dev Biol. 2000;16:557–589. doi: 10.1146/annurev.cellbio.16.1.557. http://www.ncbi.nlm.nih.gov/pubmed/11031247. [DOI] [PMC free article] [PubMed]
- 257.Bannykh SI, Balch WE. Selective transport of cargo between the endoplasmic reticulum and Golgi compartments. Histochem Cell Biol. 1998;109:463–475. doi: 10.1007/s004180050248. http://www.ncbi.nlm.nih.gov/pubmed/9681628. [DOI] [PubMed]
- 258.Bannykh SI, Nishimura N, Balch WE. Getting into the Golgi. Trend Cell Biol. 1998;8:21–25. doi: 10.1016/s0962-8924(97)01184-7. http://www.ncbi.nlm.nih.gov/pubmed/9695803. [DOI] [PubMed]
- 259.Nishimura N, Bannykh S, Slabough S, Matteson J, Altschuler Y, Hahn K, Balch WE. A di-acidic (DXE) code directs concentration of cargo during export from the endoplasmic reticulum. J Biol Chem. 1999;274:15937–15946. doi: 10.1074/jbc.274.22.15937. http://www.ncbi.nlm.nih.gov/pubmed/10336500. [DOI] [PubMed]
- 260.Sevier CS, Weisz OA, Davis M, Machamer CE. Efficient export of the vesicular stomatitis virus G protein from the endoplasmic reticulum requires a signal in the cytoplasmic tail that includes both tyrosine-based and di-acidic motifs. Mol Biol Cell. 2000;11:13–22. doi: 10.1091/mbc.11.1.13. http://www.molbiolcell.org/cgi/content/abstract/11/1/13. [DOI] [PMC free article] [PubMed]
- 261.Jain P, Mostoller K, Flaig KE, Ahuja J, Lepoutre V, Wigdahl B, et al. Identification of human T cell leukemia virus type 1 tax amino acid signals and cellular factors involved in secretion of the viral oncoprotein. J Biol Chem. 2007;282:34581–34593. doi: 10.1074/jbc.M707317200. http://www.jbc.org/cgi/content/abstract/282/47/34581. [DOI] [PubMed]
- 262.Alefantis T, Flaig KE, Wigdahl B, Jain P. Interaction of HTLV-1 Tax protein with calreticulin: Implications for Tax nuclear export and secretion. Biomed Pharmacother. 2007;61:194–200. doi: 10.1016/j.biopha.2007.02.005. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2043122. [DOI] [PMC free article] [PubMed]
- 263.Cowan EP, Alexander RK, Daniel S, Kashanchi F, Brady JN. Induction of tumor necrosis factor alpha in human neuronal cells by extracellular human T-cell lymphotropic virus type 1 Tax. J Virol. 1997;71:6982–6989. doi: 10.1128/jvi.71.9.6982-6989.1997. http://jvi.asm.org/cgi/content/abstract/71/9/6982. [DOI] [PMC free article] [PubMed]
- 264.Brady JN. Extracellular Tax1 protein stimulates NF-kB and expression of NF-kB-responsive Ig kappa and TNF beta genes in lymphoid cells. AIDS Res Hum Retroviruses. 1992;8:724–727. http://www.ncbi.nlm.nih.gov/pubmed/1515222?dopt=Abstract. [PubMed]
- 265.Selmaj KW, Raine CS. Tumor necrosis factor mediates myelin and oligodendrocyte damage in vitro. Ann Neurol. 1988;23:339–346. doi: 10.1002/ana.410230405. http://www.ncbi.nlm.nih.gov/pubmed/3132891. [DOI] [PubMed]
- 266.Dhib-Jalbut S, Hoffman PM, Yamabe T, Sun D, Xia J, Ruscetti FW, et al. Extracellular human T-cell lymphotropic virus type I Tax protein induces cytokine production in adult human microglial cells. Ann Neurol. 1994;36:787–790. doi: 10.1002/ana.410360516. http://www.ncbi.nlm.nih.gov/pubmed/7979225. [DOI] [PubMed]
- 267.Ali A, Patterson S, Cruickshank K, Rudge P, Dalgleish AG, Knight SC. Dendritic cells infected in vitro with human T cell leukaemia/lymphoma virus type-1 (HTLV-1); enhanced lymphocytic proliferation and tropical spastic paraparesis. Clin Exp Immunol. 1993;94:32–37. doi: 10.1111/j.1365-2249.1993.tb05973.x. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1534377. [DOI] [PMC free article] [PubMed]
- 268.Makino M, Wakamatsu S, Shimokubo S, Arima N, Baba M. Production of functionally deficient dendritic cells from HTLV-I-infected monocytes: Implications for the dendritic cell defect in adult T cell leukemia. Virology. 2000;274:140–148. doi: 10.1006/viro.2000.0445. http://linkinghub.elsevier.com/retrieve/pii/S0042682200904458. [DOI] [PubMed]
- 269.Macatonia SE, Cruickshank JK, Rudge P, Knight SC. Dendritic cells from patients with tropical spastic paraparesis are infected with HTLV-1 and stimulate autologous lymphocyte proliferation. AIDS, Res Hum Retroviruses. 1992;8:1699–1706. doi: 10.1089/aid.1992.8.1699. http://cat.inist.fr/?aModele=afficheN&cpsidt=4437342. [DOI] [PubMed]
- 270.Makino M, Shimokubo S, Wakamatsu SI, Izumo S, Baba M. The role of human T-lymphotropic virus type 1 (HTLV-1)-infected dendritic cells in the development of HTLV-1-associated myelopathy/tropical spastic paraparesis. J Virol. 1999;73:4575–4581. doi: 10.1128/jvi.73.6.4575-4581.1999. http://jvi.asm.org/cgi/content/abstract/73/6/4575?maxtoshow=&HITS=10&hits=10&RESULTFORMAT=&fulltext=%C2%B5g&searchid=1&FIRSTINDEX=760&resourcetype=HWFIG. [DOI] [PMC free article] [PubMed]
- 271.Mostoller K, Norbury CC, Jain P, Wigdahl B. Human T-cell leukemia virus type I Tax induces the expression of dendritic cell markers associated with maturation and activation. J Neurovirol. 2004;10:358–371. doi: 10.1080/13550280490521104. http://www.ncbi.nlm.nih.gov/pubmed/15765807. [DOI] [PubMed]
- 272.Ahuja J, Kampani K, Datta S, Wigdahl B, Flaig KE, Jain P. Use of human antigen presenting cell gene array profiling to examine the effect of human T-cell leukemia virus type 1 Tax on primary human dendritic cells. J Neurovirol. 2006;12:47–59. doi: 10.1080/13550280600614981. http://cat.inist.fr/?aModele=afficheN&cpsidt=17824988. [DOI] [PubMed]
- 273.Ahuja J, Lepoutre V, Wigdahl B, Khan ZK, Jain P. Induction of pro-inflammatory cytokines by human T-cell leukemia virus type-1 Tax protein as determined by multiplexed cytokine protein array analyses of human dendritic cells. Biomed Pharmacother. 2007;61:1–8. doi: 10.1016/j.biopha.2007.02.006. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2043123. [DOI] [PMC free article] [PubMed]
- 274.Jain P, Ahuja J, Khan ZK, Shimizu S, Meucci O, Jennings SR, Wigdahl B. Modulation of dendritic cell maturation and function by the Tax protein of human T cell leukemia virus type 1. J Leukoc Biol. 2007;82:44–56. doi: 10.1189/jlb.1006641. http://www.ncbi.nlm.nih.gov/pubmed/17442856. [DOI] [PMC free article] [PubMed]
- 275.Sacktor N, McDermott MP, Marder K, Schifitto G, Selnes OA, Epstein L, et al. HIV-associated cognitive impairment before and after the advent of combination therapy. J Neurovirol. 2002;8:136–142. doi: 10.1080/13550280290049615. http://www.ncbi.nlm.nih.gov/pubmed/11935465. [DOI] [PubMed]
- 276.Letendre SL, McCutchan JA, Childers ME, Woods SP, Lazzaretto D, Ellis RJ, et al. Enhancing antiretroviral therapy for human immunodeficiency virus cognitive disorders. Ann Neurol. 2004;56:416–423. doi: 10.1002/ana.20198. http://www.ncbi.nlm.nih.gov/pubmed/15349869. [DOI] [PubMed]