

Abstract
Tau is a microtubule-associated protein implicated in neurodegenerative tauopathies. Alternative splicing of the tau gene (MAPT) generates six tau isoforms, distinguishable by the exclusion or inclusion of a repeat region of exon 10, that are referred to as 3-repeat (3R) and 4-repeat (4R) tau, respectively. We developed transgenic mouse models that express the entire human MAPT gene in the presence and absence of the mouse Mapt gene and compared the expression and regulation of mouse and human tau isoforms during development and in the young adult. We found differences between mouse and human tau in the regulation of exon 10 inclusion. Despite these differences, the isoform splicing pattern seen in normal human brain is replicated in our mouse models. In addition, we found that all tau, both in the neonate and young adult, is phosphorylated. We also examined the normal anatomic distribution of mouse and human tau isoforms in mouse brain. We observed developmental and species-specific variations in the expression of 3R and 4R-tau within the frontal cortex and hippocampus. In addition, there were differences in the cellular distribution of the isoforms. Mice transgenic for the human MAPT gene exhibited higher levels of neuronal cell body expression of tau compared to wild-type mice. This neuronal cell body expression of tau was limited to the 3R isoform, whereas expression of 4R tau was more “synaptic like”, with granular staining of neuropil rather than in neuronal cell bodies. These developmental and species-specific differences in the regulation and distribution of tau isoforms may be important to the understanding of normal and pathologic tau isoform expression.
Keywords: splicing, phosphorylation, Alzheimer’s disease, frontotemporal dementia, 3-repeat and 4-repeat tau, tauopathy
Introduction
Tau is a normal, highly abundant protein in brain that is also part of the neuropathology of a number of neurodegenerative diseases including Alzheimer’s disease (AD) and frontotemporal dementia. In these diseases, tau aggregates form abnormal fibrillar tangles in neurons, and in some cases in oligodendrocytes and astrocytes (Buee et al., 2000; Lee et al., 2001; Mandelkow et al., 2007; Reed et al., 2001). The normal function of tau is to bind to microtubules and regulate the stability of these cytoskeletal structures. In diseases where fibrillar tau is found (tauopathies), tau, either in small aggregates, or as mature tangles is thought to be toxic and to contribute to cellular dysfunction.
Human tau is encoded by a single gene (MAPT) that produces a variety of isoforms by alternative splicing (Andreadis, 2005). In normal adult human brain, there are 6 isoforms that differ by sequences from exons 2 and 3 that encode N-terminal sequences, and exon 10 that encodes a microtubule binding repeat sequence (Goode and Feinstein, 1994; Gustke et al., 1994). When this latter exon is present, there are 4 microtubule repeats (4R tau) and when absent there are 3 microtubule-binding repeats (3R tau). In human and rodent fetal brain, there is predominantly a single tau isoform (0N3R) that lacks sequences from these 3 brain-relevant exons (Goedert et al., 1989a; Janke et al., 1999; Kosik et al., 1989; Takuma et al., 2003). In addition to this developmental regulation of alternative splicing, in adult human brain and rodent brain, there are region and cell-specific patterns of isoform expression, particularly with respect to sequences encoded by exon 10 (Bullmann et al., 2007; Goedert et al., 1989b). These spatial regulation patterns and the regulatory mechanisms that control the isoform complement in specific cells are not well understood.
In tauopathies, the isoform content of aggregated tau varies, depending on the disorder. In AD, all 6 brain isoforms are present in neurofibrillary tangles (Goedert et al., 1992), and the ratio of 4R to 3R is approximately 1 (Hong et al., 1998). This is the same ratio as non-pathogenic tau in bulk brain preparations, and thus in AD, the tangle isoform content appears to reflect the average synthesis ratio of 4R/3R tau. Both 3R and 4R tau isoforms are also observed in insoluble tau from Guam amyotrophic lateral sclerosis/parkinsonism dementia complex (ALS/PDC)(Winton et al., 2006). In frontotemporal dementia with parkinsonism - chromosome 17 type with MAPT mutations (FTDP-17T), the 3R/4R synthesis ratio, the isoform content of aggregated tau, and the regional and cellular localization of aggregated tau varies depending on the mutation (Clark et al., 1998; Hong et al., 1998; Poorkaj et al., 1998; van Swieten et al., 2007). In other tauopathies without MAPT mutations, 4R/3R ratios in pathologic structures can be skewed either towards increased 4R [PSP(Arai et al., 2001; Zhukareva et al., 2006) and corticobasal degeneration (CBD)(Fujino et al., 2005)] or increased 3R tau (Pick’s disease (Zhukareva et al., 2002)) with distinct localization of aggregated tau structures for each disorder. While the cause of these disease-specific differences is unknown, genetic variation in non-coding regions in and near MAPT influence susceptibility to PSP (Conrad et al., 1997; Pastor et al., 2004; Pittman et al., 2004; Rademakers et al., 2005), CBD (Di Maria et al., 2000; Houlden et al., 2001), FTD (Baker et al., 1999; Higgins et al., 2000; Hughes et al., 2003), Guam ALS/PDC (Poorkaj et al., 2001a; Sundar et al., 2007), PD (Goris et al., 2007; Zabetian et al., 2007), and possibly AD (Myers et al., 2005). The presumed consequence of these high-risk genetic variants is to alter MAPT gene regulation. These genetic findings along with the spectrum of neuropathology observed in tauopathies with and without MAPT mutations suggest that there is a complex relationship between the regulation of tau alternative splicing, tau expression levels, and the selective vulnerability of different brain regions and cell types. Unfortunately the cellular and regional pattern of tau alternative splicing and the factors regulating these patterns are not well understood for normal or disease brains.
Several approaches have been used to model tauopathies in mice. Initially, intronless cDNA sequences encoding a single isoform with expression driven by heterologous promoters were used as transgenes (Gotz et al., 1995; Ishihara et al., 1999). Promoters were selected to give robust pan-neuronal expression (Lewis et al., 2000) or glial expression (Forman et al., 2005), and FTDP-17T mutations were included to drive pathology [for reviews see (Gotz, 2001; Denk and Wade-Martins, 2007)]). These transgenic animals successfully model tau aggregation and tangle formation and the resulting inflammatory response. Also behavior deficits are seen and neuronal loss does occur. However, the caveats of these models are that only one isoform is produced, tau is grossly over-expressed relative to normal tau levels, and the expression pattern is determined by a heterologous promoter and is not the same as the MAPT promoter. Thus the selective vulnerability that differentiates the clinical and neuropathologic spectrum seen for different tauopathies is not modeled. Other approaches include using the endogenous mouse MAPT promoter to drive expression of a single isoform (Sennvik et al., 2007) and using a heterologous promoter to drive expression of a minigene that contains some of the sequences around relevant alternatively spliced exons 2, 3, and 10 (Mirra et al., 1993). Another approach is to use a genomic clone that contains the entire human MAPT gene as the transgene. In this model, expression of the normal tau protein is driven by the MAPT promoter and regulatory sequences (Andorfer et al., 2003, Andorfer et al., 2005). This approach has the potential to generate mice with expression patterns mirroring the human brain and FTDP-17T can be modeled by introducing mutations into the genomic clone.
Given the importance of tau isoforms in multiple neurodegenerative diseases, and the paucity of studies examining tau isoform regulation and neuroanatomic distribution in model systems, we conducted the current study to begin to address these issues. Specifically, we examined the levels and anatomic distribution of tau isoforms in embryonic and post-natal wildtype (WT) and transgenic mouse brain. We developed several transgenic mouse models that express the entire human MAPT gene (both normal and mutated sequences) in the presence and absence of the mouse Mapt gene. We found evidence for developmental and species-specific distribution of tau isoforms that may be important to the understanding of normal and pathologic tau isoform regulation. In addition, we demonstrate that FTDP-17T mutations introduced into MAPT genomic clones can recapitulate the isoform splicing pattern seen in normal human brain, providing possible mouse models for future studies of tauopathies.
Materials and Methods
Antibody Characterization
The 3R and 4R-specific tau antibodies were developed by de Silva and colleagues (de Silva et al., 2003). The 3R-specific mouse monoclonal antibody (RD3, clone 8E6/C11, catalog # 05-803, Upstate Cell Signaling) is raised against bovine thyroglobulin conjugated synthetic peptide corresponding to amino acids 209-224 of human tau (numbering based on the 0N3R isoform). The region brackets the exon 9/exon 10 junction at amino acids 216-217 (KHQPGGGKVQIVYKPV). This antibody detects 3 bands on western blots of rodent brain tissue representing 0N3R, 1N3R and 2N3R (manufacturer’s technical information and figure 2). The 4R-specific mouse monoclonal antibody (RD4, clone 1E1/A6, catalog # 05-804, Upstate Cell Signaling) is raised against bovine thyroglobulin conjugated synthetic peptide corresponding to amino acids 275-291 of human tau (numbering based on 2N4R tau) which are the first 17 amino acids in exon 10 (VQIINKKLDLSNVQSKC). The region is flanking junction coded by adjacent exons 9 and 11 with the inclusion of exon 10. This antibody detects 3 bands on western blots of rodent brain tissue representing 0N4R, 1N4R and 2N4R (manufacturer’s technical information and figure 2). When RD3 and RD4 are used together on the same western blot, all six brain tau isoforms are detected. The RD3 and RD4 antibodies did not stain brain tissue from tau knockout mice (figure 6), further demonstrating the specificity of these antibodies for tau. Rb17025, a rabbit polyclonal antibody raised against 2N4R recombinant tau that recognizes mouse and human tau, is from V.-M. Lee (Ishihara et al., 1999). T14 is a mouse monoclonal antibody that recognizes human tau residues 83-120 (based on 2N4R tau numbering)(Kosik et al., 1988) but not mouse tau and is from V.-M. Lee. Both T14 and Rb17025 specifically recognize all six tau isoforms on western blots of mouse brain tissue (figure 2) and recognize both phosphorylated and non-phosphorylated tau. Doublecortin (DCX) C-18 (catalog # sc-8066, Santa Cruz Biotechnology) is an affinity purified goat polyclonal antibody raised against an 18 amino acid peptide representing amino acids 384-401 of human doublecortin. This antibody detects a single 40-kDa band on western blot (Donovan et al., 2006). In addition, the pattern of staining we observed using this antibody for immunohistochemistry is similar to that previously reported (Donovan et al., 2006), with most immunoreactive cells in the hippocampus restricted to the subgranular zone.
Figure 2.

Immunoblot analysis of RAB soluble human MAPT and mouse Mapt isoforms in brains from wildtype and transgenic mice. Whole brain homogenates were prepared as described in the Materials and Methods and subjected to electrophoresis and immunoblot analysis using the following antibodies: RD4 (1-1000 dilution, top panels, A and B); RD3 (1-3000 dilution, middle panels, A and B); Rb17025 (diluted 1-6000, bottom panel, A and B) which recognizes both mouse and human tau (all six isoforms); T14 (1-1000) (C, all panels), an antibody specific for human tau (all six isoforms) that does not recognize mouse tau. Protein loads/lane for A and B are: 1.2 ug, top panel; 0.1 ug, middle panel; 0.2 ug, bottom panel. Lanes labeled with “-” are untreated soluble tau and lanes labeled “+” were treated with λ-phosphatase. A. Wildtype mice produce predominantly 3R tau at P6, both 3R and 4R tau at P24 and predominantly 4R tau at P90. Note that for all isoforms, the mouse protein migrates faster than the human equivalent protein. However, spacing is not altered. B. The human gene in a Mapt-/- mouse produces predominantly 3R tau and no 4R tau at P6. By P24, other tau isoforms were observed including some that are 4R, although the predominant isoform is still 3R even in the adult mice (P90). C. The human gene in the adult mouse brain produces all 6 tau isoforms and FTDP-17 mutations that alter isoform ratios in man also affect splicing of E10 in mouse. RAB soluble whole brain homogenates were prepared from mice transgenic for either the normal MAPT gene (hT-PAC-N) or MAPT with the FTDP-17 mutations V337M, R5L, S305S, E10+14, P301L, or N279K on a mouse Mapt+ background. Samples in the top panel were untreated and samples in the bottom panel were treated with λ-phosphatase. Since different transgenic lines produce different amounts of human tau, loading was adjusted so that approximately the same amount of human tau was present in each lane. The protein loaded for phosphatase treated samples was as follows: 0.25 ug, hT-PAC-N; 0.17 ug, hT-PAC-337M; 0.43 ug, hT-PAC-5L; 0.9 ug, hT-PAC-305S; 0.18 ug, hT-PAC-E10+14; 0.2 ug, hT-PAC-301L; and 0.2 ug, hT-PAC-279K. The protein loaded for untreated samples was 0.5 ug, except for hT-PAC-337M, which was 0.1 ug. The standards are recombinant human tau.
Figure 6.

3R and 4R tau are undetectable in brain tissue of tau knockout mice. No immunostaining is observed in the hippocampus of P24 Mapt-/- mice using antibodies specific for 3R tau (RD3, A) and 4R tau (RD4, B). Scale bars A and B, 100 μm.
Transgenic mice
All experimental procedures were carried out under a protocol approved by Veterans Affairs Puget Sound Health Care System’s Institutional Animal Care and Use Committee and were in accordance with NIH guidelines for the care and use of laboratory animals. The following transgenic mouse lines were developed: transgenic mice expressing the normal human tau gene (MAPT) on a mouse tau gene (Mapt) background (hT-PAC-N, Mapt+/+), human MAPT on a Mapt null background (hT-PAC-N, Mapt-/- (Dawson et al., 2001), or mutated human MAPT on a Mapt background (see below). All transgenic mice have been backcrossed 10 or more generations into C57Bl/6J. All transgenic mice have the complete sequence of human MAPT as contained in the P1 artificial chromosome (PAC) 61D6 (Research Genetics). This PAC is 201 kb and contains approximately 5 kb sequence upstream of the MAPT promoter and an additional 62 kb downstream of the 3′ end of MAPT (Poorkaj et al., 2001b). This downstream segment contains the 3′ end of KIAA1267, and MAPT is the only complete gene on this PAC. For some lines (hT-YAC-301L), the 61D6 PAC was converted into a yeast artificial chromosome (YAC) and homologous recombination was used to introduce mutations as previously described (Poorkaj et al., 2000). For other transgenic lines (hT-PAC-337M, hT-PAC-5L, hT-PAC-305S, hT-PAC-E10+14, hT-PAC-279K), mutations were introduced into this PAC using a 2-step recombineering process. This method utilizes inducible recombination (red) genes from the bacteriophage lambda to carry out recombination between short terminal segments on linear DNA and corresponding target regions of homology on large insert clones such as PACs and BACs (Muyrers et al., 1999; Wang et al., 2006; Zhang et al., 1998; Zhang et al., 2003). We modified the procedures developed for bacterial artificial chromosomes (BACs) for use with the PACs. In the first step of the recombineering process, E.coli containing PAC 61D6 were transformed with vector pSC101-BAD-gbaA (Gene Bridges, Germany) and the gam, redβ, redα, and recA genes induced with arabinose. These induced cells were made electroporation-competent cells and transformed with a linear DNA construct that targets MAPT E10. This construct contained an rpsL-Zeo fusion DNA cassette flanked by 50 nt sequences that are homologous to MAPT E10. The rpsL-zeo gene encodes a fusion protein that when retained, makes cells resistant to Zeocin and sensitive to streptomycin. Typically we analyzed 100-200 colonies that had been screened for both Zeocin resistance and streptomycin sensitivity, and by colony PCR found ∼80% contained the rpsL-Zeo cassette integrated in the correct site. In the second step, the rpsL-Zeo cassette was replaced by a linear oligonucleotide containing one of the FTDP-17 point mutations. Streptomycin resistance was used to select recombinants. Typically 20-40% of the streptomycin resistant colonies contained the correct recombinant product as determined by DNA sequencing. Sequences of all the constructs and primers, and detailed protocols are available on request.
RNA analyses
Total RNA was isolated from mouse hemi-brains using the TRIzol (Invitrogen) procedure. Briefly, hemi-brains were weighed and 1 ml TRIzol/0.1g brain tissue was dounce-homogenized with a motorized hand-held pestle. Extracted RNA were treated with DNaseI and subjected to RT-PCR to separately detect mouse- and human-specific transcripts using mouse-specific primers ME9F2 (5′-CCCCCTAAGTCACCATCAGCTAGT-3′) and ME11R (5′-CACTTTGCTCAGGTCCACCGGC-3′) as well as human-specific primers HE9F2 (5′-ACCCAAGTCGCCGTCTTCCGCC-3′) and HE11R (5′-CACCTTGCTCAGGTCAACTGGT -3′) primers in exons 9 and 11, respectively. PCR reactions were performed for 18 - 22 cycles to obtain linear amplification and contained 1 ng of 32P-labelled mouse or human forward primer to facilitate detection and quantitation on a PhosphorImager (BioRad). RT-PCR product sizes for mouse were 172 bp (E10-) and 265 bp (E10+) and for human were 172 bp (E10-) and 265 bp (E10+). Each bar represents the average value from multiple brain samples as explained in the figure legends.
Immunoblot analysis
Protein for immunoblot experiments was prepared by homogenizing mouse brains that had been frozen immediately at the time of dissection. The brains were homogenized in RAB high-salt buffer (0.1 M MES, 1 mM EGTA, 0.5 mM MgSO4, 0.75 M NaCl, 0.02M NaF, 1 mM PMSF, and 0.1% protease inhibitor cocktail (complete Mini, Roche Applied Science). The homogenate is centrifuged at 40,000 × G for 40 minutes at 4° C. The resulting supernatant is boiled for 5 min., centrifuged at 13,000 × g at 4° C for 20 minutes, and the supernatant used for denaturing gel electrophoresis and immunoblotting. Some samples were dephosphorylated using λ-phosphatase (New England Biolabs) as described by others (Hanger et al., 2002). Samples were subjected to sodium dodecyl sulfate gel electrophoresis using Centurion 7.5% pre-cast gels (BioRad) and proteins transferred to membranes by conventional electroblotting. Antibody signals were detected using ECL Plus from Amersham. The tau standard is a mixture of recombinant tau for 6 isoforms from V. M.-Y. Lee.
Immunohistochemistry
For all genotypes (wildtype, hT-PAC-N, Mapt-/-, tau knockout) at all ages, 3 mice were examined by immunohistochemistry and the images presented are representative of each age/genotype group. While there was some variability in the intensity of the staining within a group of 3, the 3R and 4R distribution did not vary significantly. Postnatal day 3 (P3) day 6 (P6) and day 24 (P24) mice were anesthetized and fixed by transcardial perfusion with 4% paraformaldehyde. Brains were removed, paraffin embedded, and coronal sections from the frontal cortex and hippocampus were cut at 10 μm thickness and stored at 4°C until use. Sections were immunostained with RD3 (1:800), RD4 (1:80), and DCX (1:1000). For monoclonal antibodies, a Vector M.O.M. Immunodetection Kit (Vector, Burlingame, CA) was used to eliminate possible background staining resulting from use of an anti-mouse secondary antibody on mouse tissue. Sections were deparaffinized and rehydrated through alcohols, and an antigen retrieval step consisting of heat pretreatment by microwave (RD3, DCX) or pressure cooker (RD4) in DakoCytomation Target Retrieval Solution (Vector, Burlingame, CA) was used. Sections were treated for endogenous peroxidases with 3% hydrogen peroxide in PBS (pH 7.4) for 30 minutes and blocked in M.O.M. block (RD3 and RD4) or 5% non-fat milk in PBS (DCX) for 1 hour. Sections were incubated with primary antibody overnight at 4°C followed by biotinylated secondary antibody for 45 minutes at room temperature. Finally, sections were incubated for 1 hour in an avidin-biotin complex (Vector’s Vectastain Elite ABC kit, Burlingame, CA) and the reaction product was visualized with 0.05% diaminobenzidine (DAB)/0.01% hydrogen peroxide in PBS. Negative controls with secondary antibody alone did not immunostain tissue sections (data not shown). In addition, the RD3 and RD4 antibodies did not stain brain tissue from tau knockout mice (figure 6), further demonstrating the specificity of these antibodies for use in immunohistochemistry. Two methods were used to detect double labeling of RD3 and DCX in the hippocampus. To demonstrate that 3R-tau and DCX were expressed in the same population of subgranular zone cells in the dentate gyrus, we used a Vector M.O.M. Immunodetection Kit with an enzyme-based visualization method. Sections were incubated overnight at 4°C with both primary antibodies concurrently. Sections were then incubated with the biotinylated M.O.M. secondary antibody, followed by an alkaline phosphatase avidin-biotin complex (Vector, Burlingame, CA) and the RD3 reaction product was visualized with Vector Red (Vector, Burlingame, CA). Sections were then incubated with a biotinylated goat secondary antibody followed by an avidin-biotin complex and the DCX reaction product was visualized with DAB. To demonstrate that 3R-tau and DCX were co-expressed in the same cell, fluorescent immunohistochemistry was performed. Sections were incubated overnight at 4°C with both primary antibodies concurrently. Sections were then incubated with a Cy2-conjugated donkey anti-goat secondary antibody (Jackson ImmunoResearch, Westgrove, PA) and a biotinylated M.O.M. mouse secondary antibody. Sections were then incubated with the fluorescently tagged avidin system, Texas Red avidin D (Vector, Burlingame, CA).
Photomicrography and figure preparation
Photomicrographs were taken with a digital camera and imported into Adobe Photoshop for mounting. To optimize visualization of staining, photomicrographs were modified, when necessary, by adjusting brightness and contrast.
Results
The following work was performed to determine whether 3R and 4R isoforms are differentially regulated with respect to regional localization during development and in the adult animal. We examined 3R and 4R expression in both neonatal mouse where 3R tau isoforms are predominant and in young adult animals where 4R isoforms are predominant. We also compared expression of the endogenous mouse tau gene (Mapt) to the human gene (MAPT) expressed in mouse. Human gene expression was examined in mice transgenic for a P1 artificial chromosome (PAC) clone 61d6 that is 201 kb. This clone contains the entire MAPT gene, and includes 5kb upstream of the MAPT promoter, and 62 kb past the 3′ end of MAPT (Poorkaj et al., 2000, Poorkaj et al., 2001b). Regional isoform expression patterns were determined using antibodies specific for either 3R or 4R tau (de Silva et al., 2003). Since these antibodies recognize both mouse and human tau, expression of the PAC-encoded human gene was examined on a mouse Mapt null background (Mapt-/-)(Dawson et al., 2001).
RNA analysis of isoform regulation during development
To determine when 3R and 4R expression changes from the fetal neonatal pattern to the adult pattern, RNA expression was analyzed in WT mice and in transgenic mice expressing normal human MAPT on a mouse Mapt background (hT-PAC-N, Mapt+/+). These experiments were also performed to determine if expression of the human gene affected expression of the mouse gene and visa versa. RNA was prepared from the brains of mice ranging in ages from embryonic day 14 (E14) to postnatal day 36 (P36). RT-PCR primers were designed that specifically detected exon 10 minus (Ex10-) 3R-tau and exon 10 plus (Ex10+) 4R-tau and that were specific for mouse or human transcripts. As previously reported (Janke et al., 1999; Kampers et al., 1999; Takuma et al., 2003), in fetal and new-born WT mouse brain, the predominant transcript is Ex10- (figure 1 A and C). In adult mice, the predominant transcript changes to Ex10+. When Mapt expression was examined in embryonic and neonatal mice, 90% of Mapt transcripts from E14 to P3 were Ex10- and approximately 10% Ex10+ (figure 1 C). Ex10+ expression increased to 20% by P6, 80% by P12, and 100% (no detectable Ex10-) by P24.
Figure 1.

Human MAPT and mouse Mapt RNA levels in wildtype mice and in mice transgenic for human MAPT (hT-PAC-N Mapt+/+). RNA from either mouse or human brain was isolated as described in the Materials and Methods section and RT-PCR was performed using either mouse or human-specific primers that amplified transcripts from mid-E9 to mid-E11. The short fragment (165 bp) is E10- and the long fragment (265 bp) is E10+. A. Mouse-specific RT-PCR primers amplify mouse Mapt RNA but not human MAPT RNA. Using mouse-specific primers (ME9F2 and ME11R), no transcript was detected in human embryonic (E14) or adult brain RNA. In fetal mice (E14) the predominant product is E10- although some E10+ RNA is detected. In RNA from adult (90 days) hT-PAC-N, Mapt+/+ lines that have an intact mouse Mapt gene, the predominant transcript from the mouse gene is E10+. B. Human-specific RT-PCR primers amplify only transcripts from the human gene but not the mouse gene. Samples are the same as in A and the primers used were HE9F2 and HE11R. C. Quantitation of E10+ transcripts using mouse-specific and human-specific primers to compare the appearance of E10+ mouse Mapt and human MAPT transcripts in developing transgenic mouse brains. For WT mice (black bars), each bar represents values from 2 to 5 mice. For hT-PAC-N, Mapt+/+, each bar for mouse (gray) and human (open) is the average combined E10+ value from mouse lines 9, 12 and 13 and represents a minimum of 6 to a maximum of 14 mice. Mouse Mapt transcripts are predominantly E10- until P6 when the amount of E10+ begins to increase (WT 79.7% vs. transgenic 75.8%) until by day P24, all Mapt transcripts are E10+. The presence of the human MAPT gene in hT-PAC-N, Mapt+/+ does not influence the endogenous pattern of E10 splicing of transcripts from the mouse Mapt gene compared with non-transgenic mice. The human gene in the mouse produces no detectable E10+ transcripts until P12 (14.01%), at which stage endogenous mouse E10+ levels are significantly higher at 76% than human E10+ (p < 10-16). D. In adult human brain, transcripts from MAPT are 49 % E10+ (SD 0.03). For the human gene in mice (hT-PAC-N, mapt +/+), 21 % of transcripts were E10+ (SD 1.2, n = 13). This is the average from 4 different hT-PAC-N Mapt+/+ lines in adult (P90) mice with an intact Mapt gene. Error bars for individual lines represent values from at least 3 different brain samples with standard deviations as follows: adult human brain (SD = 3.0, n = 3), transgenic mice Line 12 (SD = 1.0, n = 3), Line 9 (SD = 1.0, n = 3), Line 4 (SD = 0.6, n = 4) and Line 13 (SD = 0.1, n = 13).
Developmental switching from Ex10- to Ex10+ expression also occurs in humans. Like mouse, in the human fetus, Ex10- is the predominant transcript (figure 1 B), as previously reported (Goedert et al., 1989a; Takuma et al., 2003). In adult human brain, 49% (SD = 3%, n = 3) of MAPT transcripts are Ex10+ (figure 1 B and D). When expression of the human MAPT gene in mice (hT-PAC-N lines, Mapt+/+) was examined and compared to regulation of the endogenous mouse, the following differences were noted. First, in adult mice, the transcripts from the human gene are 21% (SD = 1.2%, n = 13) E10+ compared to 100% E10+ from the mouse gene (no detectable E10-RNA, figure 1 C). This was observed in 4 different transgenic lines and in work by others (Grover et al., 1999). Second, as observed in whole brain preparations from newborn mice, the human gene produces almost no E10+ message whereas approximately 10% of transcripts from the mouse gene are E10+ (figure 1 C). Third, the change from the fetal/newborn pattern to the adult pattern occurs later for the human gene at approximately day 12, compared to the mouse gene where the change begins by P6. Expression of the human gene does not influence Ex10 expression pattern from the mouse gene (figure 1 C). Similar results were obtained in an hT-PAC transgenic mouse where the FTDP-17 mutation P301L had been introduced into the PAC by homologous recombination (hT-PAC-301L, data not shown).
Protein analysis of isoform regulation during development
Isoform expression was also examined by immunoblots using antibodies specific for 3R, 4R, or total tau (all isoforms). Since tau is phosphorylated, and this affects migration in electrophoresis experiments, we analyzed tau digested with λ-phosphatase and compared it to untreated tau (figure 2). The 3R and 4R antibodies were specific for the respective 3R and 4R isoforms, while the antibodies recognizing total tau (Rb17025 and T14) detected all six isoforms. Also, when RD3 and RD4 were used on the same immunoblot, all 6 isoforms were detected. In the WT mouse, at P6, most of the tau is 3R though some 4R is present (figure 2 A), a result consistent with the RNA analysis (figure 1). Note that phosphatase treatment altered the mobility of tau indicating that all tau is phosphorylated in normal mouse both in the new-born and in the young adult animals (figure 2 A). For example, without phosphatase treatment, there is no tau that co-migrates with the predominant isoform (0N4R) in adult mouse encoded by Mapt (figure 2 A, bottom panel). (Tau isoforms without exons 2 or 3 are 0N, isoforms with exon 2 but lacking exon 3 are 1N, and isoforms with both exons 2 and 3 are 2N.) Tau expressed from the human gene was examined on a Mapt null background (hT-PAC-N, Mapt-/-) because the 3R and 4R antibodies react with both human and mouse tau (figure 2 B). All human tau in the P6 animal was 3R with no 4R tau detectable until P24. This result is consistent with the absence of 4R RNA in the youngest animals (figure 1 C). By P24, all 6 human tau isoforms are present. Again, most if not all tau encoded by MAPT is phosphorylated.
While the human gene does undergo developmentally regulated isoform switching, Ex10 inclusion is much lower from the human gene compared to the mouse gene. Previous work demonstrated that Ex10 inclusion is regulated both by exon splicing enhancer and silencer sequences and intronic sequences adjacent to both ends of Ex10 (D’Souza et al., 1999; D’Souza and Schellenberg, 2000; D’Souza and Schellenberg, 2002). To determine if some of these sequences also regulate MAPT Ex10 in the mouse, we generated hT-PAC mice with point mutations that control Ex10 inclusion. We introduced FTDP-17 mutations that increased Ex10 inclusion (N279K, S305S, E10+14) and mutations that have no effect on Ex10 inclusion (V337M, R5L, P301L) (D’Souza et al., 1999; D’Souza and Schellenberg, 2002; Hong et al., 1998; Hutton et al., 1998; Poorkaj et al., 2002; Stanford et al., 2000). Previous work showed that the mutations that increase E10 inclusion act either by weakening an intron splicing silencer (ISS, the E10+14 mutation), or by strengthening the weak E10 5′ splice site (the S305S mutation), or by strengthening an exon splicing enhancer (ESE) located in Ex10 (N279K mutation) (D’Souza et al., 1999; D’Souza and Schellenberg, 2000; D’Souza and Schellenberg, 2002). Transgenes were expressed on a mouse Mapt background and tau isoforms expressed from the mutated human gene were compared to tau isoforms expressed from the normal human gene using a human specific tau antibody (T14). For the hT-PAC-N, Mapt+/+ (normal human sequence), all 6 isoforms are seen in over-exposed immunoblots with 0N3R as the predominant isoform and 0N4R being the second most abundant isoform (figure 2 C). Mice transgenic for mutations that do not change Ex10 inclusion in vivo and in ex vivo experiments (hT-PAC-R5L, hT-YAC-301L, and hT-PAC-337M), exhibit the same isoform pattern as hT-PAC-N mice (figure 2 C). In contrast, for mice transgenic for hT-PAC-305S ,and hT-PAC-E10+14, both FTDP-17 mutations that increase Ex10 inclusion in humans, the predominant isoform is changed from 0N3R to 0N4R (figure 2 C). These results indicate that despite the differences in the regulation of the mouse and human genes, the 5′-splice site and ISS targeted by these 2 mutations behave the same in mouse and in humans.
For constructs used to generate hT-PAC-E10+14 and hT-PAC-305S, the normal Ex10 is replaced by a single copy of E10 with the respective mutations. For the hT-PAC-279K construct, two copies of E10 with the 279K change were inserted in tandem upstream of the normal E10 (figure 3 A). Analysis of RNA from fetal hT-PAC-279K mice showed about 10% inclusion of E10-279K compared to no Ex10 inclusion in hT-PAC-N, Mapt+/+ mice (figure 3 B, C). In adult animals, Ex10 inclusion increased to ∼ 50.44% (n = 2) for the mutant mice compared to 20.1% (SD = 0.1, n = 3) for hT-PAC-N, Mapt+/+ mice. The increase in E10 was entirely due to increased inclusion of the mutant exons (figure 3 C). Immunoblot analysis of the hT-PAC-279K mice (figure 2 C, right panel), showed that the mutation resulted in an increase in protein bands with multiple copies of E10. Note that the 279K mutation increases fetal E10 inclusion suggesting the ESE targeted by this mutation is part of the regulation of fetal E10 inclusion.
Figure 3.
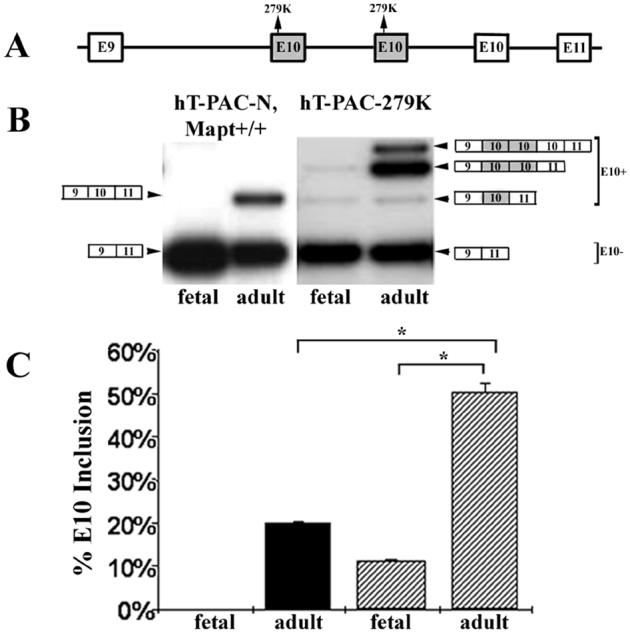
The hT-PAC-279K transgene has 3 copies of E10 and the 279K mutation increases inclusion of E10. A. The structure of the E10 region of MAPT in mouse line hT-PAC-279K retaining 2 mutant E10 copies (shaded box) upstream of a normal E10 copy. B. RT-PCR products from fetal (E14) and adult (P90) hT-PAC-N Mapt+/+ and hT-PAC-279K mice. The structure for the RT-PCR products with multiple E10 copies was determined by DNA sequencing. Note that the normal E10 copy is spliced only in adult mouse brain and, in hT-PAC-279K, is detected as a minor species in the presence of both mutant E10 copies. C. Quantitation of E10 inclusion. Each bar represents values from 3 different brain samples. The 279K mutation in mice having multiple copies of E10, stimulates inclusion of one mutant E10 copy in fetal brain at 11.2 % (SD 4.8, n = 3) and predominantly enhances mutant E10+ transcripts in adult brain to 50.4 % (SD = 1.8, n = 2). The E10+ bar for hT-PAC-279K mice is the sum of all E10+ species as observed in B. A corrected significance criteria of p < 0.025 was used in E10 splicing comparisons between adult WT and mutant 279K mice as well as between fetal and adult 279K mice, p < 1 × 10-4. Solid bars, hT-PAC-N mice; hatched bars, hT-PAC-279K mice.
Immunohistochemistry of WT mice
The results of the RNA experiments described in figure 1 indicate that P3-P6 animals express predominantly 3R tau mRNA and P24 animals express predominately 4R tau mRNA. Thus, for the immunohistochemistry work described below, P3 and P6 animals were examined for the fetal-neonatal expression pattern, and P24 animals for the adult expression pattern. We first examined the localization of 3R and 4R tau expression by immunohistochemistry in the frontal cortex and hippocampus of WT mice (figure 4). Mouse 3R-tau is expressed throughout all the layers of the cortex with the most intense staining in the outer layers of the cortex compared to the deeper layers in both developing and young adult mice (figure 4 A-C). In addition, at P3, 3R tau was primarily localized to neuronal cell bodies and fibers (figure 4 A and A’), while at P6 and P24, localization appeared more “synaptic-like”, with granular staining of neuropil similar to what is observed using antibodies against synaptic markers. Mouse 4R-tau expression was not detectable at P3 (figure 4 D), but by P6, 4R-tau was expressed in all cortical layers (figure 4 E). In the P24 animals (figure 4 F), 4R-tau immunostaining was most intense in the outer layers and weaker in the deeper layers, similar to what was observed for 3R-tau expression. In addition, 4R tau expression was not localized to neuronal cell bodies at any age but rather appeared primarily “synaptic-like”.
Figure 4.

Expression of mouse 3R and 4R tau in the frontal cortex and hippocampus of wildtype mice at P3 (A, D, G, J), P6 (B, E, H, K), and P24 (C, F, I, L). In the cortex, 3R tau is expressed in all layers at all ages examined (A-C) while 4R tau is undetectable in P3 mice (D) but is observed beginning at P6 (E). Higher magnification (A’) of boxed region in A showing neuronal cell body staining of 3R tau at P3. In the hippocampus, little specific staining is seen at P3 for either 3R or 4R tau (G and J, respectively). At P6, 4R staining is seen in the hilus, stratum oriens and stratum radiatum (K), while 3R staining is still faint (H). By P24, both 3R and 4R are abundant throughout the hippocampus (I and L). Panels A-C and G-I were stained with the RD3 antibody and panels D-F and J-L were stained with the RD4 antibody. Scale bars A-L, 100 μm; A’, 25 μm. Abbreviations: Or (stratum oriens layer); Rad (stratum radiatum layer); MF (mossy fibers).
In the hippocampus, in contrast to the cortex, there was no detectable 3R-tau in P3 and P6 animals (figure 4 G, H). However, in P24 animals, 3R expression is substantial (figure 4 I). 4R-tau is also undetectable in the hippocampus at P3, but is detected by day 6, and is highly expressed in P24 mice (figure 4 J-L). At P24 both 3R and 4R-tau are highly expressed in the CA3 mossy fiber projections but the staining pattern for the 2 isoforms in other hippocampal regions is strikingly different (figure 4 I, L). 3R-tau is highly expressed in the stratum oriens and stratum radiatum layers, whereas 4R-tau staining is much weaker in these regions. Differences between 3R and 4R-tau within the dentate gyrus of the WT animals are especially evident. Whereas 3R-tau is highly expressed in the subgranular zone of the dentate gyrus (figure 5 A), 4R-tau is highly expressed in the inner dentate molecular layer and hilus (figure 5 B). We also examined 3R-tau immunostaining in the hippocampus of postnatal day 90 mice and found that expression was extensively downregulated compared to P24, and restricted to cells in the subgranular zone (supplemental figure 1). As expected, 3R and 4R-tau are not expressed in the hippocampus of postnatal day 24 Mapt-/- control mice (figure 6 A, B). Tau was also undetectable in the hippocampus of the younger tau-knockout animals. Tau expression in the cortex was undetectable as well (data not shown).
Figure 5.

Expression of mouse tau isoforms and doublecortin (DCX) in the hippocampus of P24 wildtype mice. In the hippocampus of P24 wildtype mice, 3R tau (A, RD3 antibody) is expressed in the subgranular zone (SGZ) and 4R tau (B, RD4 antibody) is extensively expressed in the hilus and the inner dentate molecular layer (iDML). Mouse 3R tau and DCX are co-expressed in the same population of SGZ cells in the dentate gyrus of P24 wildtype mice (C, D). DCX was detected using an avidin-biotin complex and visualized with DAB under light microscopy (C). In the same tissue section, 3R tau was detected with an alkaline phosphatase avidin-biotin complex and visualized with Vector Red under immunofluoresence microscopy (D). Dcx and 3R tau are co-expressed in the same cells (E-G). DCX was detected using a Cy2-conjugated donkey anti-goat secondary antibody (E). 3R-tau was detected using a biotinylated M.O.M. mouse secondary antibody and the fluorescently tagged avidin system, Texas Red avidin D (F). Merge of the Cy-2 and Texas Red avidin D fluorescent signals (G). Arrowheads show individual cells within the SGZ that are co-labeled with both DCX and 3R-tau. Scale bars A-D, 100 μm; E-G, 25 μm. A magenta-green copy is available online (supplemental figure 2). Abbreviations: granular cell layer (GCL), dentate molecular layer (DML).
The SGZ of the dentate gyrus is a site of adult neurogenesis (Gould and Gross, 2002). High expression of 3R-tau in this region suggests that 3R-tau might be expressed in newly developing neurons in the adult dentate gyrus. Thus, we determined whether 3R-tau was co-expressed in these cells with doublecortin (DCX), a marker of adult neurogenesis (Rao and Shetty, 2004). To demonstrate that 3R-tau and DCX were expressed in the same population of subgranular zone cells in the dentate gyrus, double-labeling experiments were performed in which 3R-tau was detected with Vector Red and visualized by immunofluoresence microscopy (figure 5 D), while DCX was detected with DAB and visualized by light microscopy (figure 5 C). Our results clearly demonstrate that WT mice express 3R-tau and DCX within the same population of SGZ cells in the dentate gyrus. To demonstrate that 3R-tau and DCX were co-expressed within the same cell, double-labeling fluorescent immunohistochemistry was performed (figure 5 E-G). DCX was detected using a Cy2-conjugated secondary antibody (E), while 3R-tau was detected using a biotinylated secondary antibody and the fluorescently tagged avidin system, Texas Red avidin D (F). Our results show that 3R-tau and DCX are co-expressed within SGZ cells in the dentate gyrus.
Immunohistochemistry of transgenic mice
Human tau expression was examined in the frontal cortex and hippocampus of mice transgenic for the human MAPT gene and lacking mouse tau (hT-PAC-N Mapt-/-; figure 7). 3R-tau was detected in all cortical layers in P3, P6 and P24 animals, but the distribution and intensity differed with age. Strong immunostaining for human 3R-tau was detected in the younger animals (P3 and P6), with localization to neuronal cell bodies and fibers in all but the outermost layer (figure 7 A and B). At P3, this neuronal cytoplasmic staining was particularly robust (figure 7 A and A’) but decreased by P6 and was undetectable by P24. While cortical 3R immunostaining was still apparent at P24, the localization appeared more “synaptic-like”, similar to what we observed for mouse 3R-tau immunostaining in the WT animals. In contrast to mouse 4R-tau immunostaining in the WT animals, we were unable to detect human 4R-tau in any layers of the cortex in the hT-PAC-N Mapt-/- animals (figure 7 D-F). However, 4R tau was detected in the hippocampus (see below).
Figure 7.

Expression of human 3R and 4R tau in the frontal cortex and hippocampus of hT-PAC-N, Mapt-/- mice at P3 (A, D, G, J), P6 (B, E, H, K), and P24 (C, F, I, L). In the cortex, 3R tau is observed in all cortical layers at all ages (A-C), while 4R tau is not observed at any age (D-F). Higher magnification (A’) of boxed region in A showing neuronal cell body staining of 3R tau at P3. In the hippocampus, 3R tau was expressed at all ages (G-I) while 4R tau was not detected until P24 (L). The transgenic animals expressed 3R tau in CA3 pyramidal neurons of the hippocampus (N). This neuronal cell body staining is also evident in human CA3 neurons (O) but is undetectable in the CA3 neurons of wildtype mice (M). Panels A-C, G-I and M-O were stained with the RD3 antibody and panels D-F and J-L were stained with the RD4 antibody. Scale bars A-O, 100 μm; A’, 25 μm. Abbreviations: MF (mossy fibers), CA3 (pyramidal neurons of the CA3).
We also examined 3R and 4R expression in the hippocampus of hT-PAC-N Mapt-/- animals. 3R-tau is highly expressed throughout the hippocampus in both developing and adolescent mice (figure 7 G-I), whereas 4R-tau is undetectable until P24 and at that age, detectable 4R expression is limited to the CA3 mossy fiber projections and the hilus (figure 7 J-L). Interestingly, expression of human 3R-tau in the hT-PAC-N Mapt-/- mice appeared to be lower in the SGZ of the dentate gyrus compared to the high expression of mouse 3R-tau in the SGZ of WT animals (figure 4 I and 7 I). In addition, the human 3R-tau in the hT-PAC-N Mapt-/- mice was expressed in pyramidal neurons of the hippocampus. Neuronal expression of 3R tau was especially robust in the younger animals (P3 and P6), but was still evident in the CA3 pyramidal neurons at P24 (figure 7 N). In contrast, no 3R-tau immunostaining was detected in the CA3 pyramidal neurons of WT mice (figure 7 M). The neuronal expression of 3R-tau in the hT-PAC-N Mapt-/- mice is similar to the expression of 3R-tau in CA3 pyramidal neurons in the hippocampus of a normal adult human (figure 7 O).
Discussion
Above we show that tau 3R and 4R are differentially regulated in the developing and in the mature nervous system. At a given developmental stage, 3R and 4R can be expressed in different regions and cells. The expression pattern changes as the brain matures, still with different cells having different complements of 3R and 4R tau. Presumably, this pattern of expression reflects different functions that tau normally performs in different cells. Tau functions in the developing nervous system to establish neuronal cell polarity and to promote axonal elongation, and growth cone translocation. Tau participates in these functions by binding directly to microtubules (Feinstein and Wilson, 2005). Neuronal development requires dynamic microtubules and tau regulates these processes by controlling microtubule elongation and shortening (Drechsel et al., 1992; Goode et al., 1997; Panda et al., 1999; Panda et al., 2003; Trinczek et al., 1995). In contrast to developing neurons, in the mature brain in a fully differentiated neuron, microtubules must be relatively stable, though not necessarily static. Even in the same cell, some microtubules may be relatively stable while others are more dynamic (Feinstein and Wilson, 2005). To carry out these diverse functions, tau levels are controlled by transcriptional regulation, the isoforms present are controlled by alternative splicing (D’Souza et al., 1999; D’Souza and Schellenberg, 2000; D’Souza and Schellenberg, 2002), function is influenced by phosphorylation (Buee et al., 2000), and the location of translation by 3′ untranslated region sequences (Aronov et al., 2001). The regulation of alternative splicing that controls the cellular isoform content is particularly critical because mutations that cause changes in the 3R and 4R content of a cell result in severe neurodegeneration leading to cell death. In the work described above, we show that the neuronal tau expression levels and isoform content is highly cell and region specific both during development and in the mature brain. Thus the regulation of both expression and splicing is highly complex.
Previous studies have demonstrated that 3R-tau is the predominant isoform in fetal and new-born WT mouse brain (Janke et al., 1999; Kampers et al., 1999; Takuma et al., 2003), while in adult mice, the predominant isoform changes to 4R-tau. Our immunohistochemistry experiments also demonstrate these developmental variations in the expression of 3R and 4R-tau and provide additional information regarding some anatomical differences in this expression. In the youngest animals we examined (P3), 3R-tau immunostaining was abundant in the cortex of WT mice, but was undetectable in the hippocampus. Hippocampal expression of 3R-tau appeared sometime between P6 and P24. As expected, 4R-tau immunostaining was undetectable in our youngest animals (P3), but was apparent in both the cortex and hippocampus by P6. At P24, both 3R and 4R-tau were abundant in both the cortex and hippocampus, thus the switch to the 4R isoform must occur at an older age. In contrast to our results, Bullman et al report that 3R-tau immunostaining is detectable in the rat hippocampus from the time of birth. These differences may reflect species variations in the expression of tau isoforms or differences in experimental protocol. Given the strong immunostaining of 3R-tau in the hippocampus at P24 (figure 4 I) despite the paucity of exon 10-RNA (figure 1 C) and the low level of 3R-tau protein as determined by western blot analysis (figure 2 A) at this timepoint, we also immunostained the hippocampus of postnatal day 90 mice. At P90, there is no 3R-tau detectable by western blot analysis using whole brain extract (figure 2 A) and thus we would expect to see little or no 3R-tau immunostaining at this age. Indeed, we found an extensive decrease in 3R-tau immunostaining throughout the hippocampus at P90, with expression restricted to cells in the subgranular zone of the dentate gyrus (supplemental figure 1), a result similar to what has been previously reported in the rat (Bullmann et al., 2007). These results indicate that the robust immunostaining detected with the RD3 antibody in the hippocampus of P24 animals is specific for 3R-tau.
Developmental switching from the 3R to 4R-tau isoforms also occurs in humans. Like mouse, in the human fetus, 3R-tau is the predominant isoform (Goedert et al., 1989a; Takuma et al., 2003), while in adult human brain, 3R and 4R-tau isoforms are approximately equal. While our immunohistochemical results indicate that human 3R-tau is abundant in the cortex and hippocampus of transgenic mice at P3-P24, the appearance of the 4R-tau isoform does not occur until P24, is restricted to the hippocampus and expression levels are quite low. RNA analysis also indicated that human 4R-tau transcripts were undetectable until P12 and were sparse, even at P36. Low human 4R-tau expression in our mouse model may reflect species-specific regulatory mechanisms. It is possible that the mouse splicing machinery negatively affects human 4R-tau expression without impacting human 3R-tau expression. Despite this drawback, we believe that our mouse models provide important information regarding tau isoform regulation and expression, especially with respect to 3R-tau.
The complex pattern of tau regulation is consistent with the fact that 3R tau and 4R tau are not functionally equivalent with respect to interactions with microtubules. In vitro, 4R has an approximately 3-fold higher binding affinity for microtubules compared to 3R tau (Butner and Kirschner, 1991; Goode et al., 2000). Likewise, in cultured cells, 4R tau can displace 3R tau from microtubules (Lu and Kosik, 2001). In addition, 4R tau is better at initiating and promoting microtubule assembly than 3R tau (Goedert and Jakes, 1990; Gustke et al., 1994; Lee et al., 1989; Panda et al., 2003). In terms of microtubule dynamics, when the rate and extent of shortening and lengthening of microtubules is measured, both isoforms suppress microtubule dynamics (Feinstein and Wilson, 2005). However, quantitatively, 4R is an approximately 3-fold more potent inhibitor compared to 3R tau. Qualitatively, both isoforms inhibit microtubule lengthening, while 4R inhibits shortening but 3R does not (Feinstein and Wilson, 2005; Panda et al., 2003; Trinczek et al., 1995). The same differences in 4R versus 3R were observed in cultured cells. Both isoforms suppressed microtubule growth with 4R being more potent than 3R. As seen in vitro, 4R but not 3R inhibited microtubule shortening (Bunker et al., 2004). The differential effect of tau on microtubule dynamics implies that cells with predominantly 3R tau would have more dynamic microtubules as would be required during development where axonal extension and synaptogenesis occurs. In the mature nervous system, more stable microtubules may be required corresponding to an increase in 4R tau expression. Thus the 3R:4R ratio may be carefully regulated to match the required microtubule function for a given cell. In contrast to effects on microtubule stability, transport rates of tau along microtubules is not isoform dependent (Utton et al., 2002). In FTDP-17 cases where mutations alter the isoform ratio, subjects remain clinically normal for the first 3-4 decades of life. Thus, it appears that neurons can tolerate alterations of tau isoform ratios, at least for some period of time. However, it is possible that a shift in tau isoform ratio may be damaging over extended time and/or with aging.
In the work described above, the isoform expression pattern seen in the young adult hippocampus offers some clues to tau function. Co-expression of 3R tau and DCX in the hippocampal SGZ, a region of adult neurogenesis, in P24 WT and hT-PAC-N Mapt-/- mice suggests that 3R tau is expressed in newly generated neurons in the adult. In contrast, 4R tau is not expressed in this region. In addition, we determined that 3R-tau immunostaining in the hippocampus of postnatal day 90 mice is restricted to cells in the SGZ. Others have also reported the persistence of 3R tau expression in the SGZ in 3-month old rats despite the almost complete switch to 4R tau expression in other brain regions (Bullmann et al., 2007). Given the differential effects of 3R and 4R tau on microtubule stability discussed above, it is possible that continued expression of 3R tau in the adult must be maintained in discreet regions such as the SGZ in order to promote increased plasticity of microtubules. It is possible that alterations in the 3R:4R tau ratio could lead to abnormalities in adult neurogenesis that may have implications in the development of neurodegeneration. Increased neurogenesis has been reported in post-mortem AD brain (Jin et al., 2004) suggesting an attempt to replace dying neurons. In addition, several mouse models of AD exhibit decreased neurogenesis, which may contribute to the cognitive deficits reported in these mice (Donovan et al., 2006; Feng et al., 2001). However, there is no information regarding alterations in SGZ neurogenesis in other tauopathies such as PSP, FTD, CBD, Guam ALS/PDC, and the alterations in AD have not been directly linked to tau or tau isoforms. Further studies are needed to determine if such an association exists.
In the mature brain, the regulation of alternative splicing has profound effects on function. This is best illustrated by the class of FTDP-17T mutations that results in elevated 4R synthesis. In subjects with this type of mutation, the tangles formed are composed primarily of 4R tau, reflecting the change in synthesis ratios. Unlike AD, tangles are observed in both neurons and glia. Some mutations in this class are intronic (e.g. E10 +14) or silent (e.g. L284L), and the excess 4R tau that is produced has a normal amino acid sequence (D’Souza et al., 1999; Hutton et al., 1998). This altered synthesis ratio of non-mutant tau is sufficient to cause severe neurodegeneration by the 4th-5th decade of life. Other mutations such as R5L and V337M, do not alter the 3R/4R synthesis ratio, yet the former mutation produces tangles that are predominantly 4R, and the regional localization of aggregated tau is similar to progressive supra nuclear palsy (PSP) with tangles in both neurons and astrocytes (Poorkaj et al., 1998; Poorkaj et al., 2002). In contrast, the V337M-induced neuropathology is primarily cortical, tangles are only found in neurons, and the 4R/3R ratio in tangles is approximately 1 (Hong et al., 1998; Sumi et al., 1992). Finally, the Δ280K mutation reduces the 4R/3R ratio resulting in the formation of Pick bodies and neuronal inclusions that were predominantly 3R-tau (van Swieten et al., 2007). Whether mutations that alter alternative splicing and 3R versus 4R production act the same in all cells and all regions is not presently known. Our results using transgenic mice with point mutations that control exon splicing indicate that the splicing sequences that regulate the human MAPT exon 10 retain a similar function when expressed in mouse. Thus, these mice are attractive models for future studies of tauopathies. Current studies are underway to determine the pattern of expression of 3R and 4R isoforms, as well as any pathological changes (i.e. tau aggregation) that occur with aging in these mice.
The fact that in some diseases, pathologic tau is primarily 4R (PSP, CBD) and in others either primarily 3R (PiD) or a mixture of 3R and 4R suggests several hypotheses. One possibility is that the disease process causes specific isoforms (e.g. 4R in PSP) to selectively aggregate even though the affected cells make 3R and 4R in equal amounts. A second hypothesis is that not all cells make the same isoform complement and that for some diseases, cells making predominantly 3R (PiD) or 4R (PSP, CBD) are affected by the disease process resulting in selective aggregation of specific isoforms. A third hypothesis is that the disease process affected regulation of isoform production and that for PSP, 4R isoforms are up-regulated. The results of the current study support, in part, the second hypothesis, in that we observed regional variation in 3R and 4R tau expression. Of interest, we found 3R to be relatively over-expressed, in comparison to 4R, in the SGZ of the dentate granule cell layer; it is the dentate granule cell layer that develops 3R predominant Pick bodies in PiD. Also of interest is the higher level of neuronal cell body expression of tau in the human brain and in mice transgenic for the human MAPT gene, versus the wildtype mouse. This neuronal cell body expression of tau was limited to the 3R isoform, whereas localization of 4R tau appeared more “synaptic-like”. In addition, this neuronal cell body pattern of 3R expression was more prominent in younger animals. Although the biological significance of this localization is unknown, it may play a role in the different functions of tau with respect to microtubule dynamics or in the transportation of proteins and vesicles to and from the synapse. In addition, it is possible that abnormalities in this distribution could be important in the development of tau-specific pathologies such as neurofibrillary tangles.
The complex nature of MAPT regulation in normal and in tauopathy brains makes modeling these diseases in animals difficult. The approach we took, using a genomic clone as the transgene, has the advantage that all isoforms are produced and thus the regulation of the human gene in mouse should be closer to human MAPT expression patterns. Regulation of the human MAPT gene in mouse is similar but not identical to how it is regulated in the human brain. In mouse the human gene undergoes a developmental switch from producing exclusively 3R-tau to a mixture of 3R and 4R although only about 20% of the total tau is 4R. Another difference is that even though all 6 isoforms are produced from the human gene in the mouse context, only small amounts of protein with exons 2 and exons 2+3 sequences are produced while in humans the predominant isoform is 1N3R and 1N4R (Ex2+). The sequences tested that regulate exon 10 inclusion appear to function in the mouse as in humans as demonstrated by the mice generated using the PAC constructs with FTDP-17T mutations (figure 2 C, and figure 3). Note that these mutations do not appear to influence inclusion of exon 2 and exon 3 sequences (figure 2 C). Differences between the mouse and human gene regulation in the mouse are also evident. The mouse gene produces predominantly 4R tau in the adult (P90) while the human gene produces both 3R and 4R (figure 2 A and B). This expression profile more closely resembles tau expression in an adult human in which both 3R and 4R tau are expressed. Another difference is that the mouse gene produces more 4R tau prior to the developmental switch (P6) compared to the human gene. The expression levels of tau do not appear to be controlled by a feedback mechanism since when the tau synthesis is increased by adding the hT-PAC-N to the normal endogenous tau levels, transcription from the endogenous gene does not change (figure 1 C). Another observation from the above work is that all tau, both in the neonate and young adult, is phosphorylated. This is consistent with previous work (Matsuo et al., 1994) showing that when either human or rodent brain is frozen rapidly after harvesting, tau from normal brains is highly phosphorylated. When post-mortem intervals are extended, dephosphorylation of soluble tau occurs while insoluble tau remains highly phosphorylated. Thus the relationship between tau phosphorylation in tauopathies may be important for insoluble but not soluble tau.
In conclusion, we have demonstrated distinct regional regulation of tau isoforms in wild-type and transgenic mice. Further study of the regulation of tau isoform expression levels and distribution in human tauopathies and animal models could provide important insights into the pathophysiology of many neurodegenerative diseases.
Supplementary Material
Acknowledgements
We thank Darrin Bisset and Elaine Loomis for technical support.
This work was supported by NIA grants PO1 AG17586 and R37 AG 11762-10, the American Health Assistance Foundation and the Department of Veteran’s Affairs.
Literature Cited
- Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, Duff K, Davies P. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J. Neurochem. 2003;86:582–590. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J. Neurosci. 2005;25:5446–5454. doi: 10.1523/JNEUROSCI.4637-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreadis A. Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim. Biophys. Acta. 2005;1739:91–103. doi: 10.1016/j.bbadis.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Arai T, Ikeda K, Akiyama H, Shikamoto Y, Tsuchiya K, Yagishita S, Beach T, Rogers J, Schwab C, Mcgeer PL. Distinct isoforms of tau aggregated in neurons and glial cells in brains of patients with Pick’s disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropath. 2001;101:167–173. doi: 10.1007/s004010000283. [DOI] [PubMed] [Google Scholar]
- Aronov S, Aranda G, Behar L, Ginzburg I. Axonal tau mRNA localization coincides with tau protein in living neuronal cells and depends on axonal targeting signal. J. Neurosci. 2001;21:6577–6587. doi: 10.1523/JNEUROSCI.21-17-06577.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez-Tur J, Hardy J, Lynch T, Bigio E, Hutton M. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum. Mol. Genet. 1999;8:711–715. doi: 10.1093/hmg/8.4.711. [DOI] [PubMed] [Google Scholar]
- Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Rev. 2000;33:95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- Bullmann T, de Silva R, Holzer M, Mori H, Arendt T. Expression of embryonic tau protein isoforms persist during adult neurogenesis in the hippocampus. Hippocampus. 2007;17:98–102. doi: 10.1002/hipo.20255. [DOI] [PubMed] [Google Scholar]
- Bunker JM, Wilson L, Jordan MA, Feinstein SC. Modulation of microtubule dynamics by tau in living cells: implications for development and neurodegeneration. Mol. Biol. Cell. 2004;15:2720–2728. doi: 10.1091/mbc.E04-01-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butner KA, Kirschner MW. Tau protein binding to microtubules through a flexible array of distributed weak sites. J. Cell Biol. 1991;115:717–730. doi: 10.1083/jcb.115.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark LN, Poorkaj P, Wszolek Z, Geschwind DH, Nasreddine ZS, Miller B, Li D, Payami H, Awert F, Markopoulou K, Andreadis A, D’Souza I, Lee VMY, Reed L, Trojanowski JQ, Zhukareva V, Bird T, Schellenberg GD, Wilhelmsen KC. Pathogenic implications of mutations in the tau gene in pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc. Natl. Acad. Sci. U.S.A. 1998;95:13103–13107. doi: 10.1073/pnas.95.22.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad C, Andreadis A, Trojanowski J, Dickson D, Kang D, Chen X, Wiederholt W, Hansen L, Masliah E, Thal L, Katzman R, Xia Y, Saitoh T. Genetic evidence for the involvement of Tau in progressive supranuclear palsy. Ann. Neurol. 1997;41:277–281. doi: 10.1002/ana.410410222. [DOI] [PubMed] [Google Scholar]
- D’Souza I, Poorkaj P, Hong M, Nochlin D, Lee VMY, Bird TD, Schellenberg GD. Missense and silent tau gene mutations cause front temporal dementia with parkinsonism - chromosome 17 type by affecting multiple alternative RNA splicing regulatory elements. Proc. Natl. Acad. Sci. USA. 1999;96:5598–5603. doi: 10.1073/pnas.96.10.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza I, Schellenberg GD. Determinants of 4 repeat tau expression: Coordination between enhancing and inhibitory splicing sequences for exon 10 inclusion. J. Biol. Chem. 2000;275:17700–17709. doi: 10.1074/jbc.M909470199. [DOI] [PubMed] [Google Scholar]
- D’Souza I, Schellenberg GD. Tau exon 10 expression involves a bipartite intron 10 regulatory sequence and a weak 3′ splice site. J. Biol. Chem. 2002;277:26587–26599. doi: 10.1074/jbc.M203794200. [DOI] [PubMed] [Google Scholar]
- Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, Vitek MP. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J. Cell Sci. 2001;114:1179–1187. doi: 10.1242/jcs.114.6.1179. [DOI] [PubMed] [Google Scholar]
- de Silva R, Lashley T, Gibb G, Hanger D, Hope A, Reid A, Bandopadhyay R, Utton M, Strand C, Jowett T, Khan N, Anderton B, Wood N, Holton J, Revesz T, Lees A. Pathological inclusion bodies in tauopathies contain distinct complements of tau with three or four microtubule-binding repeat domains as demonstrated by new specific monoclonal antibodies. Neuropath. Appl. Neurobiol. 2003;29:288–302. doi: 10.1046/j.1365-2990.2003.00463.x. [DOI] [PubMed] [Google Scholar]
- Denk F, Wade-Martins R. Knock-out and transgenic mouse models of tauopathies. Neurobiol. Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.05.010. doi:10.1016/j.neurobiolaging.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Maria E, Tabaton M, Vigo T, Abbruzzese G, Bellone E, Donati C, Frasson E, Marchese R, Montagna P, Munoz DG, Pramstaller PP, Zanusso G, Ajmar F, Mandich P. Corticobasal degeneration shares a common genetic background with progressive supranuclear palsy. Ann. Neurol. 2000;47:374–377. doi: 10.1002/1531-8249(200003)47:3<374::aid-ana15>3.3.co;2-#. [DOI] [PubMed] [Google Scholar]
- Donovan MH, Yazdani U, Norris RD, Games D, German DC, Eisch AJ. Decreased adult hippocampal neurogenesis in the PDAPP mouse model of Alzheimer’s disease. J. Comp. Neurol. 2006;495:70–83. doi: 10.1002/cne.20840. [DOI] [PubMed] [Google Scholar]
- Drechsel DN, Hyman AA, Cobb MH, Kirschner MW. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol. Biol. Cell. 1992;3:1141–1154. doi: 10.1091/mbc.3.10.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein SC, Wilson L. Inability of tau to properly regulate neuronal microtubule dynamics: a loss-of-function mechanism by which tau might mediate neuronal cell death. Biochim. Biophys. Acta. 2005;1739:268–279. doi: 10.1016/j.bbadis.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Feng RB, Rampon C, Tang YP, Shrom D, Jin J, Kim M, Sopher B, Martin GM, Kim SH, Langdon RB, Sisodia SS, Tsien JZ. Deficient neurogenesis in forebrain-specific presenilin-1 knockout mice is associated with reduced clearance of hippocampal memory traces. Neuron. 2001;32:911–926. doi: 10.1016/s0896-6273(01)00523-2. [DOI] [PubMed] [Google Scholar]
- Forman MS, Lal D, Zhang B, Dabir DV, Swanson E, Lee VMY, Trojanowski JQ. Transgenic mouse model of tau pathology in astrocytes leading to nervous system degeneration. J. Neurosci. 2005;25:3539–3550. doi: 10.1523/JNEUROSCI.0081-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujino Y, Wang DS, Thomas N, Espinoza M, Davies P, Dickson DW. Increased frequency of argyrophilic grain disease in Alzheimer disease with 4R tau-specific immunohistochemistry. J. Neuropathol. Exp. Neurol. 2005;64:209–214. doi: 10.1093/jnen/64.3.209. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989a;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNA’s in human brain. EMBO J. 1989b;8:393–399. doi: 10.1002/j.1460-2075.1989.tb03390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Jakes R. Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990;9:4225–4230. doi: 10.1002/j.1460-2075.1990.tb07870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Crowther RA. Cloning of a big tau microtubule-associated protein characteristic of the peripheral nervous system. Proc. Natl. Acad. Sci. USA. 1992;89:1983–1987. doi: 10.1073/pnas.89.5.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode BL, Feinstein SC. Identification of a novel microtubule binding and assembly domain in the developmentally regulated inter-repeat region of tau. J. Cell Biol. 1994;124:769–782. doi: 10.1083/jcb.124.5.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode BL, Denis PE, Panda D, Radeke MJ, Miller HP, Wilson L, Feinstein SC. Functional interactions between the proline-rich and repeat regions of tau enhance microtubule binding and assembly. Mol. Biol. Cell. 1997;8:353–365. doi: 10.1091/mbc.8.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode BL, Chau M, Denis PE, Feinstein SC. Structural and functional differences between 3-repeat and 4-repeat tau isoforms. J. Biol. Chem. 2000;275:38182–38189. doi: 10.1074/jbc.M007489200. [DOI] [PubMed] [Google Scholar]
- Goris A, WilliamsGray CH, Clark GR, Foltynie T, Lewis SJG, Brown J, Ban M, Spillantini MG, Compston A, Burn DJ, Chinnery PF, Barker RA, Sawcer SJ. Tau and alpha-synuclein in susceptibility to, and dementia in, Parkinson’s disease. Ann. Neurol. 2007;62:145–153. doi: 10.1002/ana.21192. [DOI] [PubMed] [Google Scholar]
- Gotz J, Probst A, Spillantini MG, Schafer T, Jakes R, Burki K, Goedert M. Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. EMBO J. 1995;14:1304–1313. doi: 10.1002/j.1460-2075.1995.tb07116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz J. Tau and transgenic animal models. Brain Res. Rev. 2001;35:266–286. doi: 10.1016/s0165-0173(01)00055-8. [DOI] [PubMed] [Google Scholar]
- Gould E, Gross CG. Neurogenesis in Adult Mammals: Some Progress and Problems. J. Neurosci. 2002;22:619–623. doi: 10.1523/JNEUROSCI.22-03-00619.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover A, Houlden H, Baker M, Adamson J, Lewist J, Prihart G, Pickering-Brown S, Duff K, Hutton M. 5′ splice mutations in tau associated with the inherited dementia FTDP-17 affect a stem-loop structure that regulates alternative splicing of exon 10. J. Biol. Chem. 1999;274:15134–15143. doi: 10.1074/jbc.274.21.15134. [DOI] [PubMed] [Google Scholar]
- Gustke N, Trinczek B, Biernat J, Mandelkow EM, Mandelkow E. Domains of tau protein and interactions with microtubules. Biochemistry. 1994;33:9511–9522. doi: 10.1021/bi00198a017. [DOI] [PubMed] [Google Scholar]
- Hanger DP, Gibb GM, de Silva R, Boutajangout A, Brion JP, Revesz T, Lees AJ, Anderton BH. The complex relationship between soluble and insoluble tau in tauopathies revealed by efficient dephosphorylation and specific antibodies. FEBS Lett. 2002;531:538–542. doi: 10.1016/s0014-5793(02)03611-6. [DOI] [PubMed] [Google Scholar]
- Higgins JJ, Golbe LI, Debiase A, Jankovic J, Factor SA, Adler RL. An extended 5 ′-tau susceptibility haplotype in progressive supranuclear palsy. Neurology. 2000;55:1364–1367. doi: 10.1212/wnl.55.9.1364. [DOI] [PubMed] [Google Scholar]
- Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VMY. Mutation-specific functional impairments in distinct Tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–1917. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- Houlden H, Baker M, Morris HR, MacDonald N, Pickering-Brown S, Adamson J, Lees AJ, Rossor MN, Quinn NP, Kertesz A, Khan MN, Hardy J, Lantos PL, George-Hyslop PS, Munoz DG, Mann D, Lang AE, Bergeron C, Bigio EH, Litvan I, Bhatia KP, Dickson D, Wood NW, Hutton M. Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology. 2001;56:1702–1706. doi: 10.1212/wnl.56.12.1702. [DOI] [PubMed] [Google Scholar]
- Hughes A, Mann D, Pickering-Brown S. Tau haplotype frequency in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Exp. Neurol. 2003;181:12–16. doi: 10.1016/s0014-4886(03)00024-4. [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JBJ, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, Lee VMY. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 1999;24:751–762. doi: 10.1016/s0896-6273(00)81127-7. [DOI] [PubMed] [Google Scholar]
- Janke C, Beck M, Stahl T, Holzer M, Brauer K, Bigl V, Arendt T. Phylogenetic diversity of the expression of the microtubule-associated protein tau: implications for neurodegenerative disorders. Mol. Brain Res. 1999;68:119–128. doi: 10.1016/s0169-328x(99)00079-0. [DOI] [PubMed] [Google Scholar]
- Jin KL, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, Greenberg DA. Increased hippocampal neurogenesis in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA. 2004;101:343–347. doi: 10.1073/pnas.2634794100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampers T, Pangalos M, Geerts H, Wiech H, Mandelkow E. Assembly of paired helical filaments from mouse tau: implications for the neurofibrillary pathology in transgenic mouse models for Alzheimer’s disease. FEBA Lett. 1999;451:39–44. doi: 10.1016/s0014-5793(99)00522-0. [DOI] [PubMed] [Google Scholar]
- Kosik KS, Orecchio LD, Binder L, Trojanowski JQ, Lee VML, Lee G. Epitopes that span the tau molecule are shared with paired helical filaments. Neuron. 1988;1:817–826. doi: 10.1016/0896-6273(88)90129-8. [DOI] [PubMed] [Google Scholar]
- Kosik KS, Orecchio LD, Bakalis S, Neve RL. Developmentally regulated expression of specific tau sequences. Neuron. 1989;2:1389–1397. doi: 10.1016/0896-6273(89)90077-9. [DOI] [PubMed] [Google Scholar]
- Lee G, Neve RL, Kosik KS. The microtubule binding domain of tau protein. Neuron. 1989;2:1615–1624. doi: 10.1016/0896-6273(89)90050-0. [DOI] [PubMed] [Google Scholar]
- Lee VMY, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Ann. Rev. Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van, Slegtenhorst M, Gwinn-Hardy K, Murphy MP, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin W-L, Yen S-H, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Lu M, Kosik KS. Competition for microtubule-binding with dual expression of tau missense and splice isoforms. Molec.Biol. Cell. 2001;12:171–184. doi: 10.1091/mbc.12.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow E, von Bergen M, Biernat J, Mandelkow EM. Structural principles of tau and the paired helical filaments of Alzheimer’s disease. Brain Pathol. 2007;17:83–90. doi: 10.1111/j.1750-3639.2007.00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo ES, Shin RW, Billingsley ML, vad deVoorde A, Oconnor M, Trojanowski JQ, Lee VMY. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer’s disease paired helical filament tau. Neuron. 1994;13:989–1002. doi: 10.1016/0896-6273(94)90264-x. [DOI] [PubMed] [Google Scholar]
- Mirra SM, Hart MN, Terry RD. Making the diagnosis of Alzheimer’s disease. A primer for practicing pathologists. Arch. Pathol. Lab. Med. 1993;117:132–144. [PubMed] [Google Scholar]
- Muyrers JPP, Zhang Y, Testa G, Stewart AF. Rapid modification of bacterial artificial chromosomes by ET-recombination. Nucleic Acids Res. 1999;27:1555–1557. doi: 10.1093/nar/27.6.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers AJ, Kaleem M, Marlowe L, Pittman AM, Lees AJ, Fung HC, Duckworth J, Leung D, Gibson A, Morris CM, deSilva R, Hardy J. The H1c haplotype at the MAPT locus is associated with Alzheimer’s disease. Hum. Mol. Genet. 2005;14:2399–2404. doi: 10.1093/hmg/ddi241. [DOI] [PubMed] [Google Scholar]
- Panda D, Miller HP, Wilson L. Rapid treadmilling of brain microtubules free of microtubule-associated proteins in vitro and its suppression by tau. Proc. Natl. Acad. Sci. USA. 1999;96:12459–12464. doi: 10.1073/pnas.96.22.12459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda D, Samuel JC, Massie M, Feinstein SC, Wilson L. Differential regulation of microtubule dynamics by three- and four-repeat tau: Implications for the onset of neurodegenerative disease. Proc. Natl. Acad. Sci. USA. 2003;100:9548–9553. doi: 10.1073/pnas.1633508100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor P, Ezquerra M, Perez JC, Chakraverty S, Norton J, Racette BA, McKeel D, Perlmutter JS, Tolosa E, Goate AM. Novel haplotypes in 17q21 are associated with progressive supranuclear palsy. Ann. Neurol. 2004;56:249–258. doi: 10.1002/ana.20178. [DOI] [PubMed] [Google Scholar]
- Pittman AM, Myers AJ, Duckworth J, Bryden L, Hanson M, Abou-Sleiman P, Wood NW, Hardy J, Lees A, de Silva R. The structure of the tau haplotype in controls and in progressive supranuclear palsy. Hum. Mol. Genet. 2004;13:1267–1274. doi: 10.1093/hmg/ddh138. [DOI] [PubMed] [Google Scholar]
- Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann. Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- Poorkaj P, Peterson KR, Schellenberg GD. Single-step conversion of P1 and P1 artificial chromosome clones into yeast artificial chromosomes. Genomics. 2000;68:106–110. doi: 10.1006/geno.2000.6267. [DOI] [PubMed] [Google Scholar]
- Poorkaj P, Tsuang D, Wijsman EM, Nemens E, Garruto RM, Craig U, Anderson L-J, Bird TD, Plato CC, Weiderholt W, Galasko D, Schellenberg GD. Tau is a susceptibility gene for amyotropic lateral sclerosis-parkinsonism dementia complex of Guam. Arch. Neurol. 2001a;58:1871–1878. doi: 10.1001/archneur.58.11.1871. [DOI] [PubMed] [Google Scholar]
- Poorkaj P, Kas A, D’Souza I, Zhou Y, Pham O, Olson MV, Schellenberg GD. A genomic sequence analysis of the mouse and human microtubule-associated protein tau. Mamm. Genome. 2001b;12:700–712. doi: 10.1007/s00335-001-2044-8. [DOI] [PubMed] [Google Scholar]
- Poorkaj P, Muma NA, Zhukareva V, Cochran EJ, Shannon KM, Hurtig H, Koller WC, Bird TD, Trojanowski JQ, Lee VMY, Schellenberg GD. A R5L tau mutation in a subject with a progressive supranuclear palsy phenotype. Ann. Neurol. 2002;52:511–516. doi: 10.1002/ana.10340. [DOI] [PubMed] [Google Scholar]
- Rademakers R, Melquist S, Cruts M, Theuns J, Del-Favero J, Poorkaj P, Baker M, Sleegers K, Crook R, De Pooter T, Kacem SB, Adamson J, Van den Bossche D, Van den Broeck M, Gass J, Corsmit E, DeRijk P, Thomas N, Engelborghs S, Heckman M, Litvan I, Crook J, De Deyn PP, Dickson D, Schellenberg GD, Van Broeckhoven C, Hutton ML. High-density SNP haplotyping suggests altered regulation of tau gene expression in progressive supranuclear palsy. Hum. Mol. Genet. 2005;14:3281–3292. doi: 10.1093/hmg/ddi361. [DOI] [PubMed] [Google Scholar]
- Rao MS, Shetty AK. Efficacy of doublecortin as a marker to analyse the absolute number anddendritic growth of newly generated neurons in the adult dentate gyrus. Eur. J. Neurosci. 2004;19:234–246. doi: 10.1111/j.0953-816x.2003.03123.x. [DOI] [PubMed] [Google Scholar]
- Reed LA, Wszolek ZK, Hutton M. Phenotypic correlations in FTDP-17. Neurobiol. Aging. 2001;22:89–107. doi: 10.1016/s0197-4580(00)00202-5. [DOI] [PubMed] [Google Scholar]
- Sennvik K, Boekhoorn K, Lasrado R, Terwel D, Verhaeghe S, Korr H, Schmitz C, Tomiyama T, Mori H, Krugers H, Joels M, Ramakers GJ, Lucassen PJ, Van Leuven F. Tau-4R suppresses proliferation and promotes neuronal differentiation in the hippocampus of tau knockin/knockout mice. FASEB J. 2007;21:2149–2161. doi: 10.1096/fj.06-7735com. [DOI] [PubMed] [Google Scholar]
- Stanford PM, Halliday GM, Brooks WS, Kwok BJ, Storet CE, Creasey H, Morris JGL, Fulham MJ, Schofield PRj. Progressive supranuclear palsy caused by a novel silent mutation in exon 10 of the tau gene. Brain. 2000;123:880–893. doi: 10.1093/brain/123.5.880. [DOI] [PubMed] [Google Scholar]
- Sumi SM, Bird TD, Nochlin D, Raskind MA. Familial presenile dementia with psychosis associated with cortical neurofibrillary tangles and neurodegeneration of the amygdala. Neurology. 1992;42:120–127. doi: 10.1212/wnl.42.1.120. [DOI] [PubMed] [Google Scholar]
- Sundar PD, Yu CE, Sieh W, Steinbart E, Garruto RM, Oyanagi K, Craig UK, Bird TD, Wijsman EM, Galasko DR, Schellenberg GD. Two sites in the MAPT region confer genetic risk for Guam ALS/PDC and dementia. Hum. Molec. Genet. 2007;16:295–306. doi: 10.1093/hmg/ddl463. [DOI] [PubMed] [Google Scholar]
- Takuma H, Arawaka S, Mori H. Isoforms changes of tau protein during development in various species. Develop. Brain Res. 2003;142:121–127. doi: 10.1016/s0165-3806(03)00056-7. [DOI] [PubMed] [Google Scholar]
- Trinczek B, Biernat J, Baumann K, Mandelkow EM, Mandelkow E. Domains of tau protein, differential phosphorylation, and dynamic instability of microtubules. Molec.Biol. Cell. 1995;6:1887–1902. doi: 10.1091/mbc.6.12.1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utton MA, Connell J, Asuni AA, van Slegtenhorst M, Hutton M, de Silva R, Lees AJ, Miller CCJ, Anderton BH. The Slow Axonal Transport of the Microtubule-Associated Protein Tau and the Transport Rates of Different Isoforms and Mutants in Cultured Neurons. J. Neurosci. 2002;22:6394–6400. doi: 10.1523/JNEUROSCI.22-15-06394.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Swieten JC, Bronner IF, Azmani A, Severijnen LA, Kamphorst W, Ravid R, Rizzu P, Willemsen R, Heutink P. The DeltaK280 mutation in MAP tau favors exon 10 skipping in vivo. J. Neuropathol. Exp. Neurol. 2007;66:17–25. doi: 10.1097/nen.0b013e31802c39a4. [DOI] [PubMed] [Google Scholar]
- Wang J, Sarov M, Rientjes J, Fu J, Hollak H, Kranz H, Xie W, Stewart AF, Zhang Y. An improved recombineering approach by adding RecA to lambda Red recombination. Mol. Biotechnol. 2006;32:43–53. doi: 10.1385/mb:32:1:043. [DOI] [PubMed] [Google Scholar]
- Winton MJ, Joyce S, Zhukareva V, Practico D, Perl DP, Galasko D, Craig U, Trojanowski JQ, Lee VMY. Characterization of tau pathologies in gray and white matter of Guam parkinsonism-dementia complex. Acta Neuropathol. 2006;111:401–412. doi: 10.1007/s00401-006-0053-0. [DOI] [PubMed] [Google Scholar]
- Zabetian CP, Hutter CM, Factor SA, Nutt JG, Higgins DS, Griffith A, Roberts JW, Leis BC, Kay DM, Yearout D, Montimurro JS, Edwards KL, Samii A, Payami H. Association analysis of MAPT H1 haplotype and subhaplotypes in Parkinson’s disease. Ann. Neurol. 2007;62:137–144. doi: 10.1002/ana.21157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Buchholz F, Muyrers JPP, Stewart AF. A new logic for DNA engineering using recombination in Escherichia coli. Nat. Genet. 1998;20:123–128. doi: 10.1038/2417. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Muyrers JPP, Rientjes J, Stewart AF. Phage annealing proteins promote oligonucleotide-directed mutagenesis in Escherichia coli and mouse ES cells. BMC Mol Biol. 2003;4:1–14. doi: 10.1186/1471-2199-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhukareva V, Mann D, Pickering-Brown S, Uryu K, Shuck T, Shah K, Grossman M, Miller BL, Hulette CM, Feinstein SC, Trojanowski JQ, Lee VMY. Sporadic Pick’s disease: A tauopathy characterized by a spectrum of pathological tau isoforms in gray and white matter. 2002 doi: 10.1002/ana.10222. [DOI] [PubMed] [Google Scholar]
- Zhukareva V, Joyce S, Schuck T, Van Deerlin V, Hurtig H, Albin R, Gilman S, Chin S, Miller B, Trojanowski JQ, Lee VMY. Unexpected abundance of pathological tau in progressive supranuclear palsy white matter. Ann. Neurol. 2006;60:335–345. doi: 10.1002/ana.20916. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.