

Summary
Epithelial–mesenchymal transition (EMT) is a phenotypic conversion that facilitates organ morphogenesis and tissue remodeling in physiological processes, such as embryonic development and wound healing. A similar phenotypic conversion is also detected in fibrotic diseases and neoplasia, and is associated with disease progression. EMT in cancer epithelial cells often seems to be an incomplete and bidirectional process. In this Review, we discuss the phenomenon of EMT as it pertains to tumor development, focusing on exceptions to the commonly held rule that EMT promotes invasion and metastasis. We also highlight the role of RAS-controlled signaling mediators, ERK1, ERK2 and phosphatidylinositol 3-kinase, as microenvironmental responsive regulators of EMT.
Keywords: epithelial, mesenchymal transition, extracellular matrix, microenvironment, tumor, ERK1, ERK2
Introduction
Simple epithelia are composed of cohesive sheets of cells connected by tight junctions and polarized in an apical–basal orientation relative to an underlying basement membrane (Figure 1). The surrounding mesenchymal cells are embedded within the interstitial extracellular matrix (ECM); lacking intercellular junctions, mesenchymal cells manifest primarily anterior–posterior polarity.1,2 These structural differences are reflected in the characteristic genes each cell type expresses: epithelial cells express distinct junctional proteins (e.g. E-cadherin, also known as cadherin 1, type 1) and epithelial-specific cytoskeletal proteins (e.g. cytokeratins), whereas mesenchymal cells express N-cadherin (also known as cadherin 2 type 1) and mesenchymal-specific vimentin3 (Box 1).
Figure 1.

Common morphologic characteristics of epithelial and mesenchymal cells. Epithelial morphology is characterized by an apical–basal polarity, contact with a basal basement membrane and formation of extensive cell–cell contacts, including tight junctions. An anterior–posterior polarity is lost if any cell–cell junctions and residency within a more unstructured interstitial matrix characterize mesenchymal morphology.
Box 1. Commonly used markers of epithelial–mesenchymal transition in tissue culture.
Increased expression of mesenchymal markers
α-Smooth muscle actin
Vimentin
Thrombospondin
Fibronectin
N-cadherin
Tenascin C
MMP3
CXCL1
Vitronectin
Collagen I
Collagen III
PAI1 (also known as serpin peptidase inhibitor, clade E)
TGF-β1
FGF1, FGF2 and FGF8
MMP2
MMP9
Decreased expression of epithelial markers
Epithelial CKs (e.g. CK8, CK18 and CK19)
E-cadherin
Occludin
Desmoplakin
Mucin1
TJP1
Altered location/activity of signaling factors
β-Catenin
Starch
Snail
Slug
Twist
Goosecoid
FOXC2
Sox 10
NFκB
SMAD2 and SMAD3
ERK1/ERK2
PI3K/AKT
Abbreviations: AKT, protein kinase B; CK, cytokeratin; ERK, extracellular signal-regulated kinase; FGF, fibroblast growth factor; FOXC2, forkhead box C2; FSP1, fibroblast-specific protein 1; MMP, matrix metalloproteinase; NFκB, nuclear factor κ B; PAI1, plasminogen activator inhibitor 1; PI3K, phosphatidylinositol 3-kinase; SMAD, mothers against decapentaplegic homologs; Sox 10, SRY (sex determining region Y)-box 10; TGF-β1, transforming growth factor β1; TJP1, tight junction protein 1.
Desmoplasia, the appearance of fibrous, mesenchymal-like tissue in the peritumor stroma, is associated with poor clinical outcome.4 Recent gene-profiling experiments suggest that the presence of a mesenchymal gene signature in tumors is predictive of poor clinical outcome in colorectal, breast and ovarian cancers.5–10 The principal cell types that contribute to the desmoplastic stromal reaction and mesenchymal gene signature are fibroblasts, which reside in the stroma and produce interstitial ECM molecules, and myofibroblasts, which produce growth factors, cytokines and ECM and also function to contract the ECM. Myofibroblasts have long been thought to be derived from fibroblasts, but recent data have shown that a substantial proportion of these cells are derived from epithelial–mesenchymal transition (EMT) associated with tumor progression, tissue fibrosis and other pathologies.8
EMT involves fundamental changes in gene expression that disrupt epithelial polarity and establish a mesenchymal phenotype, with concomitant alterations in cytoskeletal organization, cell adhesion and production of ECM (Figure 2).1,8,11 This process of phenotypic conversion is well conserved throughout the Vertebrata, having emerged more than 500 million years ago.11,12 More recent observations have led to suggestions that EMT contributes to the phenotypic conversions observed in tissue fibrosis,3,13–17 chronic inflammation18 and rheumatic diseases, which are similar to conversions that occur in cancer progression.1,8,19–23 Several recent reviews have summarized key signaling pathways involved in EMT and probed the link between the tumor microenvironment, fibrosis, EMT and cancer progression.8,24,25 In this Review, we expand on these ideas by analyzing the mechanistic processes involved in this EMT conversion as part of the broader function of epithelial plasticity in tumor progression. We focus on the role of RAS signaling in epithelial tissue plasticity, because EMT in cancer is a dynamic and often incomplete process regulated by the microenvironment and RAS. In addition, we review the role of RAS signaling effector pathways, most notably ERK1, ERK2 and phosphatidylinositol 3-kinase (PI3K)/AKT. Surprisingly, these pathways are responsive to the microenvironment even when RAS is mutated into an activated form.
Figure 2.
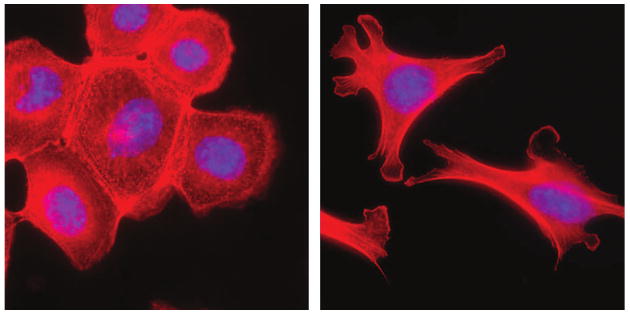
EMT of mammary epithelial cells. Treatment of mouse mammary epithelial cells with MMP3 stimulates breakdown of the epithelial structure and acquisition of a mesenchymal morphology. Red staining, f-actin; blue staining, DAPI. Abbreviations: DAPI, 4′-6-diamidino-2-phenylindole; EMT, epithelial–mesenchymal transition; MMP, matrix metalloproteinase.
EMT is an Intrinsic Plasticity of Tumor Cells
When Boyer and colleagues studied cultured cells,26 they first described EMT as a morphologic change from epithelial-like sheets of tumor cells to scattered, fibroblast-like cells capable of invading the basement membrane (Figure 3). EMT has more recently been shown to occur during normal morphogenesis of the mammary gland and seems to be required for the formation of ducts.27 Since the initial observations, EMT in cultured cancer cells has been characterized on the molecular level; altered gene-expression profiles, subcellular localization of protein and activity levels are now commonly used to identify EMT in cell culture (Table 1).3 EMT in cell culture can be either stable, that is the mesenchymal phenotype is sustained after the stimulus provoking the conversion is removed, or reversible, that is the cells revert, or undergo a mesenchymal–epithelial transition (MET), if the stimulus is removed. Experiments that quantitatively define the transient and incomplete phenotypic changes often observed in cultured tumor cells provide insight into the dynamic role EMT might have in neoplastic processes.
Figure 3.

The dynamic role of EMT in mammary gland neoplastic processes. EMT during mammary tumor progression is postulated to facilitate invasion and colonization of distant tissues by tumor cells. EMT enables efficient penetration of vessels and escape into distant tissues, such as the lung or bone. A mesenchymal phenotype might be retained or revert to an epithelial phenotype (MET), depending on the tissue microenvironment. For example, some microenvironments, such as those provided by bones, can offer selective growth for a mesenchymal phenotype, whereas others (e.g. lung) might favor growth of an epithelial phenotype. Abbreviations: EMT, epithelial–mesenchymal transition; MET, mesenchymal–epithelial transition.
Table 1.
Comparison of the gene signature of epithelial–mesenchymal transition in EpH4 mammary cells and cancer-related gene signatures.
| Gene signature and site of origin | Gene unicode in EpH4 cells | Reference |
|---|---|---|
| EMT signatures | ||
| HNSCC | None | Chung et al. (2006)43 |
| Stromal signatures | ||
| Fibroblast serum response (human) | Fln, Mt1 and Top2a | Chang et al. (2004)6 |
| Stromal signature of HNSCC (human) | Pcolce | Roepman et al. (2006)30 |
| Stromal response of prostate cancer (mouse) | Mt2 | Bacac et al. (2006)31 |
| Signatures of metastasis–invasion in breast cancer | ||
| 70-gene prognostic signature | CCL2, MT1X and MT2A | van‘t Veer et al. (2002)46 |
| ‘Invasiveness’ gene signature | Ier5 and Fln | Liu et al. (2007)92 |
Abbreviations: EMT, epithelial–mesenchymal transition; HNSCC, head and neck squamous cell carcinoma.
Insight into the complexity of EMT has been provided by studies that have used gene transcription and proteomic microarrays to assess EMT and MET.28 Additional data suggest that the gene signatures of EMT and, more generally, plasticity of epithelial tumor cells are controlled by tissue and microenvironmental factors.8 A comparison of transcriptional analyses of transforming growth factor (TGF) β1-induced EMT in EpH4 mouse mammary epithelial cells (transient EMT associated with a scattering phenotype) and EMT induced in EpH4 derivatives, such as c-Fos–ER–EpH4 (stable EMT without induction of malignancy) and RAS–EpH4 (stable EMT with induction of malignancy), revealed a common gene signature for EMT distinct from that associated with scattering, metastasis or oncogene expression (Figure 4A). Furthermore, a number of the genes in this signature have been linked to poor outcome in breast cancer.28 Intriguingly, some changes in the gene-expression profile overlap with those documented to occur during epithelial–mesenchymal conversion of the medial edge epithelial seam in the embryonic palate.29
Figure 4.

Overlap between the EMT gene signature of EpH4 mammary cells and that of the embryonic palate. (A) A Venn diagram illustrating the overlap between the gene signature of EpH4 metastasis and the gene signature of EMT in EpH4. Both upregulated and downregulated genes are represented. These results show that EMT can be distinguished from metastasis as a molecular process. (B) A Venn diagram showing the number of upregulated EMT-specific genes in EpH4 cells that are also upregulated or downregulated by at least twofold in the embryonic palate undergoing EMT. Both EMT processes are in response to TGF-β1. Approximately 50% of EMT-specific genes upregulated in EpH4 cells are also upregulated during EMT associated with embryonic palate morphogenesis. Based on data from Jechlinger M et al. (2003) Oncogene 22: 7155–7169, and LaGamba D et al. (2005) Dev Dyn 234: 132–142. Abbreviation: EMT, epithelial–mesenchymal transition.
Figure 4B and Table 2 show the extent of this overlap and compares the expression of genes that are upregulated by at least twofold during EMT of EpH4 mammary cells with those altered during EMT in the embryonic palate.29 There are similar, but not identical, alterations in the expression of gene sets in EpH4 mammary cells and the embryonic palate. There is also an overlap between genes that are downregulated during EMT of EpH4 cells and alterations in the gene profile during palatogenesis: 70% of genes downregulated during EMT in EpH4 cells are altered by more than twofold during palatogenesis (Table 3). The similarity between these two EMT signatures might result from the important role of TGF-β1 in driving mesenchymal conversion of both EpH4 cells28 and the medial ridge epithelium of the embryonic palate.29 Even though an EMT involves acquisition of at least some mesenchymal properties, the EMT-specific gene signature of EpH4 cells has surprisingly few gene-expression changes in common with stromal signatures. Such gene-expression changes include the fibroblast serum response that encompasses genes that are commonly upregulated in fibroblasts following serum stimulation in cell culture and which predicts poor outcome in breast and other cancers and enhances the prognostic value of an ‘invasiveness’ gene signature that is predictive of poor outcome in breast cancer.6,17 A similarly limited overlap in altered gene expression is observed between the EMT-specific gene signature of EpH4 cells and the signatures of stromal tumors, such as head and neck squamous cell carcinoma30 and prostate cancer31 (Table 1). Furthermore, the EMT-specific gene signature of EpH4 mammary cells bears little resemblance to an EMT-specific signature of head and neck squamous cell carcinoma32 (Table 1). Although these comparisons are limited, gene-signature analysis indicates that EMT is a process that is distinct from metastasis or tumorigenesis per se and could be tissue-specific and microenvironment-specific but has some resemblance to the embryonic process, at least when driven by a common factor, such as TGF-β1.
Table 2.
Genes commonly altered during EMT in EpH4 mammary cells and the embryonic palate.
| EMT gene unicode | EpH4 cells | Embryonic palate |
|---|---|---|
| Atf1 | + | + |
| Creg | + | + |
| F2r | + | ↓ |
| Dpys13 | + | − |
| Dab2ip | + | ↓ |
| Eng | + | + |
| Gas1 | + | + |
| Hmox1 | + | − |
| Rpl7a | + | + |
| Hexb | + | + |
| Mt1 | + | − |
| Mt2 | + | + |
| Ppic | + | + |
| Pxmp3 | + | + |
| Pcolce | + | − |
| Col6a1 | + | + |
| Col6a2 | + | ↓ |
| Raew07 | + | − |
| Mcp1 | + | − |
| Vdlr | + | − |
| Zfhxla | + | − |
Abbreviations: +, increased expression; –, not altered; ↓, downregulated; EMT, epithelial–mesenchymal transition.
Based on data from Jechlinger M et al. (2003) Oncogene 22: 7155–7169, and LaGamba D et al. (2005) Dev Dyn 234: 132–142.
Table 3.
Downregulated EMT-specific genes in EpH4 mammary cells commonly altered during EMT in the embryonic palate.
| Unicode of downregulated EMT genes in EpH4 cells | Embryonic palate |
|---|---|
| Ap1b1 | ↓ |
| Actn4 | ↓ |
| Abcf2 | ↓ |
| Chka | ↓ |
| Cldn4 | ↓ |
| Ddb1 | + |
| Fln | + |
| Flii | − |
| Ier5 | + |
| Gspt1 | ↓ |
| Hsp110 | − |
| Hnrpdl | + |
| Irf3 | ↓ |
| Junb | ↓ |
| Jup | ↓ |
| Klf5 | − |
| Lamb3 | + |
| Lisch7 | ↓ |
| Mkrn3 | + |
| Msh2 | + |
| Atp1a3 | − |
| Nasp | − |
| Pctp | + |
| Pkp1 | + |
| Pou2fl | − |
| Arhgef1 | ↓ |
| Spint2 | + |
| Slc9a9 | − |
| Supt6h | ↓ |
| Top2a | − |
| Sgtb | − |
| Tgm2 | + |
| Usp5 | ↓ |
| Hiplr | − |
Abbreviations: +, increased expression; –, not altered; ↓, downregulated; EMT, epithelial–mesenchymal transition.
These studies illustrate that EMT in cancer is a complex process that seems to be a subset of an extensive phenotypic conversion program.1,33–37 The dynamic nature of interconversion of tumor phenotypes is more difficult to capture in vivo and has rarely been documented. Conversion of ductal epithelial cells of breast tumors into myoepithelial cells and myofibroblasts in vivo, however, is suggested by both the residual expression of epithelial keratin markers in myoepithelial and myofibroblast cells and the simultaneous expression of myoepithelial (e.g. K14, K17 and vimentin) and myofibroblast (e.g. vimentin and α-smooth muscle actin) markers.38 Retention of some epithelial and myoepithelial markers in ‘transdifferentiated’ myofibroblasts and evidence of nonrandom X-chromosome inactivation patterns38 also demonstrate epithelial plasticity towards the fibroblast phenotype. These results suggest that adult epithelial cells have a capacity to acquire aspects of a mesenchymal phenotype, and vice versa, in cell culture and breast cancer. The apparent rarity of an EMT in samples of human tumors probably reflects its transient nature and possible function as a brief proinvasion conversion program that is required for the colonization of distant tissues.
The Clinical Significance of EMT
Although EMT has been clearly documented in cultured human cancer cell lines and some human tumors, its prevalence in aggressive tumors and role in clinical progression are still controversial.33,39 A clear demonstration of EMT in most human neoplastic disease has been compromised by the cellular heterogeneity of most human tumors and by the lack of markers for mesenchymal and epithelial cells in solid tumor biopsies. Evidence that EMT might be highly localized and transient or limited to specific steps in metastatic colonization32,40 further complicates clinical analysis of this process. Uncertainty regarding a clinical role for EMT in tumor progression is fueled by the rarity of morphologic changes in primary tumors observed by pathologists. Nonetheless, a number of studies have shown that expression of EMT-related genes (Box 1)1,41 is associated with the metastatic/invasive phenotype. Furthermore, a recent study that compared the gene signature of metaplastic breast cancer with that of breast ductal carcinoma showed unique downregulation of epithelial genes and upregulation of mesenchymal genes in the metaplastic sample.42 EMT might, therefore, be a feature of subtypes of breast carcinoma. These studies justify further assessment of EMT as an essential component of malignancy.
Nevertheless, increased expression of markers of EMT has been detected at the invasive fronts of aggressive tumors.1 Numerous data illustrate an association between known regulators of EMT (e.g. Snail, Twist and Slug transcription factors) and both aggressive behavior of tumors in animal models and poor clinical outcome in patients with cancer, suggesting a role for EMT in tumor progression.1,23,43 The pleiotropic nature of regulators of EMT, such as Snail, make it difficult to determine the extent to which they are causative of EMT in human cancer.44 One approach used to detect diagnostic, tissue-specific markers of EMT is to identify alterations in gene expression associated with conversion in human tumors, animal models of cancer or cultured cells and then assess whether these gene signatures are correlated with clinical outcome. For example, such transcriptional profiling experiments resulted in the identification of a ‘wound-responsive’ gene signature7 that predicts poor outcome in patients with breast cancer and increases the predictive value of other gene signatures associated with poor outcome in this disease. A similar approach identified key transcriptional alterations associated with the response of human breast epithelial cells to organotypic three-dimensional (3D) culture conditions that were also predictive of outcome in patients with breast cancer.45
Although the prognostic value of EMT in breast cancer progression per se has to our knowledge not been reported, a number of genes in the EMT–metastatic gene signature of EpH4 mammary cells and gene signature of mammary tumor cells are associated with poor prognosis in breast cancer.28 The EMT-specific gene signature of EpH4 mammary cells has limited overlap with two gene signatures of metastasis–invasion that predict poor clinical outcome in breast cancer (Table 1).10,46 More in-depth studies could help to clarify the clinical significance of EMT in tumor progression.
Possible Functions of EMT during Tumor Progression
Numerous studies have shown that inhibiting the expression or impairing the function of EMT-regulating factors blocks tumor migration and invasion in cultured epithelial cells. Invasion and metastasis of epithelial tumors, however, can occur in the absence of any detectible EMT and might not occur even in the presence of EMT.1 For example, metastasis of some bladder cancer cell lines was associated with conversion to an epithelial phenotype (MET) rather than retention of the mesenchymal phenotype,47 whereas desmoid tumors are mesenchymal and locally invasive but do not metastasize.48 EMT can be functionally uncoupled from the processes of invasion and metastasis. Conditional expression of TGF-β1 in mouse keratinocytes in the presence of a functional TGF-β1 receptor promoted EMT and metastasis in chemically induced papilloma; however, expression of a dominant-negative TGF-β1 receptor blocked the induction of EMT but did not influence the ability of TGF-β1 to promote metastasis.49 Such observations indicate that it might be an oversimplification to suggest that EMT is responsible only for increased migratory and invasive capacities.
A broader perspective on the role of EMT in cancer can be gleaned from the study of EMT in normal processes. Growing evidence suggests that EMT is integral to repair and renewal processes in normal tissue50–53 and might contribute to fibrosis if these processes are sustained or otherwise aberrant.1,8,15,18,54 As predicted by analysis of EMT gene signatures, EMT has been documented in migration of keratinocytes to wound sites55 and in response to ultraviolet irradiation. EMT also occurs transiently at the tips of growing mouse mammary gland branches as they invade the fat pad during branching morphogenesis,27 a process that resembles tumor invasion into adjacent tissues. In such instances, EMT-related processes coordinate epithelial cell movement rather than dissemination.
Transient EMT in cancer can provide fibroblast-like properties to tumor cells, even in the absence of complete morphologic alteration. Several reports suggest that conversion of non-small cell lung carcinoma cells to a mesenchymal phenotype affects their sensitivity to mitogens and antiproliferative drugs. For example, EMT in non-small-cell lung carcinoma, which was detected by a mesenchymal gene signature, predicted a loss of response to activation of the EGFR and insensitivity to the EGFR inhibitor erlotinib.56,57 Studies of experimental models have shown that acquisition of resistance to drugs, such as tamoxifen, is associated with, and might even promote, EMT.58 These studies demonstrated that EMT can be associated with changes in responsiveness to mitogens and antihormone therapy. It is unclear whether these effects are a direct or indirect consequence of phenotypic conversion. EMT could directly affect responses to mitogens and hormones as a result of altered expression of specific growth factor receptors, such as EGFR. Mesenchymal cells also differ from epithelial cells in their expression of transporters, and this could affect sensitivity to specific drugs.59,60
EMT might perform key immunomodulatory functions during tumor progression. For instance, fibroblasts possess immunomodulatory activities: they can permit leukocyte infiltration and retention within tissues at wound sites by presenting antigens to the immune system and by modifying T-cell responses. Fibroblasts could, therefore, function to mask tumor antigens and protect tumors from immune surveillance.61 Fibroblasts produce and respond to a different set of cytokines and growth factors than epithelial cells and are more responsive to mitogenic and motogenic effects of platelet-derived growth factor (PDGF) and fibroblast growth factor than epithelial cells. These growth factors are abundant in the microenvironment of tissues undergoing extensive remodeling, as well as in tumors, and transiently or reversibly, EMT might facilitate growth of epithelial tumor cells in these types of dynamic microenvironments.62 Transient EMT enables epithelial cells to temporarily evade the effects of growth-inhibitory factors. Using a tissue micropatterning approach, Nelson and coauthors showed that a transient EMT occurred in regions of the lowest concentration of the branching inhibitor TGF-β1.27 Transient EMT might provide the ductal cells with a temporary release from the inhibitory growth effects of TGF-β1 and enable response to other mitogens in the microenvironment.63 In summary, the probable transient nature of EMT and paucity of mesenchymal markers for EMT have limited assessment of the extent to which this phenotypic conversion contributes to tumor progression. Microdissection techniques that enable sampling of the tumor edge and improved imaging resolution in vivo will also contribute to clarification of the role of EMT in tumor metastasis.
Molecular Pathways that Regulate EMT
Identification of the molecular pathways that regulate EMT in cancer cells has been the subject of intense investigation.1,19,64–67 Although the processes involved in EMT have distinct characteristics in different tissues, RAS-regulated ERK1/ERK2 and PI3K signaling pathways are increasingly recognized as key mediators of tumor cell plasticity. Gene signatures of deregulated RAS pathways are common in human tumors and, in addition to signatures of other oncogenic pathways, have enabled risk stratification in many types of human cancer.68,69
RAS proteins function as switches that are triggered by microenvironmental factors, such as growth factors and ECM molecules, and control many downstream signaling pathways.70–72 Increased expression and/or mutation of RAS is a common early event in human tumors.66,71 In breast cancer, mutations that result in increased expression of RAS are more common than mutations that result in constitutive activation of RAS,73 and increased activity of RAS-regulated downstream mediators, PI3K, ERK1/ERK2, is prognostic of a poor outcome.74–79 RAS-regulated pathways can induce autonomous, stable EMT in mammary epithelial cells. For example, exposing EpH4 cells with activating RAS mutations to TGF-β1 stimulates autocrine production of mesenchymal factors, such as PDGFA, PDGFB and PDGF receptors α and β, which maintain EMT even if exogenous TGF-β1 is withheld. In the absence of constitutively activated RAS signaling pathways in parental EpH4 mammary cells, TGF-β1 induces an incomplete and transient mesenchymal conversion that is reversible if TGF-β1 is removed.80,81
ERK1/ERK2 and PI3K-regulated pathways have central roles in EMT of tumor cells. PI3K stimulates proliferation, blocks apoptosis and promotes switching of cadherin isotypes on exposure to interstitial collagens;65,82 ERK1/ERK2 disassemble adherens junctions and induce expression of mesenchymal ECM components, such as tenascin C, in addition to matrix metalloproteinases.83,84 Both pathways regulate the transcription factors Slug and Snail, which, in turn, promote EMT by suppressing expression of E-cadherin, genes encoding epithelial tight junction components and epithelial-specific cytokeratins; loss of E-cadherin induced by extracellular matrix metalloproteinases can induce EMT as well.85–87 Activation of the ERK1/ERK2 and PI3K pathways regulates the tumor-suppressive effects of environmental factors, such as TGF-β1, promoting growth and stabilization of EMT,88 an effect that is achieved by linking TGF-β1 receptor activity to PI3K and ERK1/ERK2 signaling pathways.28,49,80 RAS, PI3K and ERK1/ERK2 mediators are controlled through alteration of integrin-responsive signaling pathways: β1-integrins modulate the activity of growth factor receptors, such as the EGFR and PDGF receptor, which activate ERK1/ERK2 and PI3K signaling pathways.89,90 Additionally, the nuclear localization of activated ERK1/ERK2, which is required for its effects on gene transcription, is regulated by hyaluronan.91 Hence, ECM molecules and growth factors present in the tumor microenvironment control the localization and activation status of these RAS effectors, thereby determining the precise effect of these pathways on the behavior and differentiation or plasticity of tumor cells (Figure 5).
Figure 5.

Microenvironmental and spatial regulation of signaling pathways controlling EMT. The RAS–ERK1/ERK2 pathway is an example of a signaling module that is responsive to microenvironmental cues and requires specific subcellular localization to determine the consequences of gene expression and tumor phenotype. A simplified version of this complex process is illustrated in the diagram. ECM components interact with integrin receptors at the cell surface (step 1) and the affinity of this interaction is modified by growth-factor-regulated signaling, in a process known as ‘inside-out signaling’ (steps 1–3). Conversely, the affinity of the integrin–ECM interaction affects growth-factor-regulated signaling, in a process known as ‘outside-in signaling’ (steps 2–3). The collective interactions between integrins and growth factor receptors promote the localization and activation of lipid-modified RAS (step 2) at the inner cell membrane leaflet (step 4). Activated RAS then selectively activates kinases, such as ERK1/ERK2 and other pathways, such as the PI3K/AKT pathway. RAS also blocks the tumor-suppressing activity of TGF-β1 by linking this pathway to ERK1/ERK2 signaling pathways (step 5). This linkage promotes the proinvasion properties of TGF-β1. Activated ERK1/ERK2 must translocate to the nucleus or cell-adhesion sites (known as ‘focal contacts’) to have access to target proteins that regulate EMT, motility and/or invasion. ERK1-regulated and ERK2-regulated gene expression (e.g. MMP9) further modifies the microenvironment of the tumor cell, thereby affecting the activation status of the integrin–growth factor receptor signaling pathway. This downstream consequence of RAS–ERK1–ERK2 activation and the reversal of steps 1–5 have profound effects on the phenotype even if other pathways are mutated. Abbreviations: AKT, protein kinase B; ECM, extracellular matrix; EMT, epithelial–mesenchymal transition; ERK1, extracellular signal-regulated kinase 1; ERK2, extracellular signal-regulated kinase 2; MMP, matrix metalloproteinase; PI3K, phosphatidylinoinositol 3-kinase; TGF, transforming growth factor.
Conclusions
A more complete definition of how EMT contributes to cancer progression requires analysis of EMT during normal tissue renewal, development of mechanistic assays for in situ detection of EMT and continued identification of effectors of EMT that are prognostic for tumor outcome. RAS-regulated (ERK1/ERK2 and PI3K) signaling pathways are modulated by elements of the tumor microenvironment, suggesting functions beyond their well-studied roles in motility and proliferation. Another active field involves identification of regulatory pathways of EMT in the context of epithelial plasticity, to identify potential targets for therapy. In parallel, technical advances in accurate sampling and visualization of individual cells will enable isolation and analysis of the key regulators of EMT in tumors.
Key Points.
Fibroblast activity (e.g. desmoplasia and stromal transcriptomes) are associated with poor outcome in cancers
Epithelial–mesenchymal transition (EMT) of epithelial cancer cells contributes to this activity in the peritumor stroma, and EMT is a subset of the more generalized cell phenotype plasticity exhibited by aggressive tumor cells
Clear evidence for EMT of tumor cells in human tumors is rare, and this might be owing to its transient and dynamic nature
The clinical significance of EMT is still under study but has been associated with the metastatic/invasive phenotype and specific breast carcinoma subtypes
Study of EMT has focused on its role in active dissemination of tumor cells (e.g. migration and invasion), but emerging evidence suggests a broader functional role in tumor progression, including drug resistance and immune modulation
RAS-regulated pathways connect regulation of EMT to the tumor microenvironment
Footnotes
Review Criteria: The information for this Review was compiled by searching the PubMed and MEDLINE databases for articles published until 31 March 2007. Only articles published in English were considered. The search terms used included “DNA microarray” in association with the following search terms: “transcriptome”, “gene signature”, “epithelial to mesenchymal phenotype conversion”, “gene expression profiling”, “tumor markers”, “prognosis”, “clinical outcome” and “morphogenesis”. When possible, primary sources have been quoted. Full articles were obtained and references were checked for additional material, as appropriate.
Competing interests: The authors declared no competing interests.
Contributor Information
Eva A Turley, London Health Sciences Centre and University of Western Ontario, London, ON, Canada.
Mandana Veiseh, Life Sciences Division, at the Lawrence Berkeley National Laboratory, Berkeley, CA, USA.
Derek C Radisky, Mayo Clinic Cancer Center, Jacksonville, FL.
Mina J Bissell, Life Sciences Division, at the Lawrence Berkeley National Laboratory, Berkeley, CA, USA.
References
- 1.Lee JM, et al. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172:973–981. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Valles AM, et al. Acidic fibroblast growth factor is a modulator of epithelial plasticity in a rat bladder carcinoma cell line. Proc Natl Acad Sci USA. 1990;87:1124–1128. doi: 10.1073/pnas.87.3.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Desmouliere A, et al. The stroma reaction myofibroblast: a key player in the control of tumor cell behavior. Int J Dev Biol. 2004;48:509–517. doi: 10.1387/ijdb.041802ad. [DOI] [PubMed] [Google Scholar]
- 5.Nuyten DS, et al. Predicting a local recurrence after breast-conserving therapy by gene expression profiling. Breast Cancer Res. 2006;8:R62. doi: 10.1186/bcr1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang HY, et al. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol. 2004;2:E7. doi: 10.1371/journal.pbio.0020007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang HY, et al. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proc Natl Acad Sci USA. 2005;102:3738–3743. doi: 10.1073/pnas.0409462102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Radisky DC, et al. Fibrosis and cancer: do myofibroblasts come also from epithelial cells via EMT? J Cell Biochem. 2007;101:830–839. doi: 10.1002/jcb.21186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adler AS, Chang HY. From description to causality: mechanisms of gene expression signatures in cancer. Cell Cycle. 2006;5:1148–1151. doi: 10.4161/cc.5.11.2798. [DOI] [PubMed] [Google Scholar]
- 10.Liu ET, et al. In the pursuit of complexity: systems medicine in cancer biology. Cancer Cell. 2006;9:245–247. doi: 10.1016/j.ccr.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 11.Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–746. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 12.Hay ED. An overview of epithelio-mesenchymal transformation. Acta Anat (Basel) 1995;154:8–20. doi: 10.1159/000147748. [DOI] [PubMed] [Google Scholar]
- 13.McAnulty RJ. Fibroblasts and myofibroblasts: their source, function and role in disease. Int J Biochem Cell Biol. 2007;39:666–671. doi: 10.1016/j.biocel.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 14.Willis BC, et al. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc. 2006;3:377–382. doi: 10.1513/pats.200601-004TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neilson EG. Mechanisms of Disease: fibroblasts—a new look at an old problem. Nat Clin Pract Nephrol. 2006;2:101–108. doi: 10.1038/ncpneph0093. [DOI] [PubMed] [Google Scholar]
- 16.Faulkner JL, et al. Origin of interstitial fibroblasts in an accelerated model of angiotensin II-induced renal fibrosis. Am J Pathol. 2005;167:1193–1205. doi: 10.1016/S0002-9440(10)61208-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y. Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention. J Am Soc Nephrol. 2004;15:1–12. doi: 10.1097/01.asn.0000106015.29070.e7. [DOI] [PubMed] [Google Scholar]
- 18.Zvaifler NJ. Relevance of the stroma and epithelial-mesenchymal transition (EMT) for the rheumatic diseases. Arthritis Res Ther. 2006;8:210. doi: 10.1186/ar1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vincent-Salomon A, Thiery JP. Host microenvironment in breast cancer development: epithelial-mesenchymal transition in breast cancer development. Breast Cancer Res. 2003;5:101–106. doi: 10.1186/bcr578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Z, et al. Transdifferentiation of neoplastic cells. Med Hypotheses. 2001;57:655–666. doi: 10.1054/mehy.2001.1435. [DOI] [PubMed] [Google Scholar]
- 21.Katoh M. Epithelial-mesenchymal transition in gastric cancer. Int J Oncol. 2005;27:1677–1683. [PubMed] [Google Scholar]
- 22.Bates RC, Mercurio AM. The epithelial-mesenchymal transition (EMT) and colorectal cancer progression. Cancer Biol Ther. 2005;4:365–370. doi: 10.4161/cbt.4.4.1655. [DOI] [PubMed] [Google Scholar]
- 23.Yang J, et al. Exploring a new twist on tumor metastasis. Cancer Res. 2006;66:4549–4552. doi: 10.1158/0008-5472.CAN-05-3850. [DOI] [PubMed] [Google Scholar]
- 24.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 25.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 26.Boyer B, et al. Reversible transition towards a fibroblastic phenotype in a rat carcinoma cell line. Int J Cancer Suppl. 1989;4:69–75. doi: 10.1002/ijc.2910440719. [DOI] [PubMed] [Google Scholar]
- 27.Nelson CM, et al. Tissue geometry determines sites of mammary branching morphogenesis in organotypic cultures. Science. 2006;314:298–300. doi: 10.1126/science.1131000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jechlinger M, et al. Expression profiling of epithelial plasticity in tumor progression. Oncogene. 2003;22:7155–7169. doi: 10.1038/sj.onc.1206887. [DOI] [PubMed] [Google Scholar]
- 29.LaGamba D, et al. Microarray analysis of gene expression during epithelial-mesenchymal transformation. Dev Dyn. 2005;234:132–142. doi: 10.1002/dvdy.20489. [DOI] [PubMed] [Google Scholar]
- 30.Roepman P, et al. Maintenance of head and neck tumor gene expression profiles upon lymph node metastasis. Cancer Res. 2006;66:11110–11114. doi: 10.1158/0008-5472.CAN-06-3161. [DOI] [PubMed] [Google Scholar]
- 31.Bacac M, et al. A mouse stromal response to tumor invasion predicts prostate and breast cancer patient survival. PLoS ONE. 2006;1:e32. doi: 10.1371/journal.pone.0000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brabletz T, et al. Invasion and metastasis in colorectal cancer: epithelial-mesenchymal transition, mesenchymal-epithelial transition, stem cells and beta-catenin. Cells Tissues Organs. 2005;179:56–65. doi: 10.1159/000084509. [DOI] [PubMed] [Google Scholar]
- 33.Christiansen JJ, Rajasekaran AK. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006;66:8319–8326. doi: 10.1158/0008-5472.CAN-06-0410. [DOI] [PubMed] [Google Scholar]
- 34.Fridriksdottir AJ, et al. Maintenance of cell type diversification in the human breast. J Mammary Gland Biol Neoplasia. 2005;10:61–74. doi: 10.1007/s10911-005-2541-6. [DOI] [PubMed] [Google Scholar]
- 35.Petersen OW, et al. The plasticity of human breast carcinoma cells is more than epithelial to mesenchymal conversion. Breast Cancer Res. 2001;3:213–217. doi: 10.1186/bcr298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gudjonsson T, et al. Myoepithelial cells: their origin and function in breast morphogenesis and neoplasia. J Mammary Gland Biol Neoplasia. 2005;10:261–272. doi: 10.1007/s10911-005-9586-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adriance MC, et al. Myoepithelial cells: good fences make good neighbors. Breast Cancer Res. 2005;7:190–197. doi: 10.1186/bcr1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petersen OW, et al. Epithelial to mesenchymal transition in human breast cancer can provide a nonmalignant stroma. Am J Pathol. 2003;162:391–402. doi: 10.1016/S0002-9440(10)63834-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tarin D, et al. The fallacy of epithelial mesenchymal transition in neoplasia. Cancer Res. 2005;65:5996–6000. doi: 10.1158/0008-5472.CAN-05-0699. [DOI] [PubMed] [Google Scholar]
- 40.Spaderna S, et al. A transient, EMT-linked loss of basement membranes indicates metastasis and poor survival in colorectal cancer. Gastroenterology. 2006;131:830–840. doi: 10.1053/j.gastro.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 41.Alonso SR, et al. A high-throughput study in melanoma identifies epithelial-mesenchymal transition as a major determinant of metastasis. Cancer Res. 2007;67:3450–3460. doi: 10.1158/0008-5472.CAN-06-3481. [DOI] [PubMed] [Google Scholar]
- 42.Lien HC, et al. Molecular signatures of metaplastic carcinoma of the breast by large-scale transcriptional profiling: identification of genes potentially related to epithelial-mesenchymal transition. Oncogene. 2007;26:7859–7871. doi: 10.1038/sj.onc.1210593. [DOI] [PubMed] [Google Scholar]
- 43.Chung CH, et al. Gene expression profiles identify epithelial-to-mesenchymal transition and activation of nuclear factor-κB signaling as characteristics of a high-risk head and neck squamous cell carcinoma. Cancer Res. 2006;66:8210–8218. doi: 10.1158/0008-5472.CAN-06-1213. [DOI] [PubMed] [Google Scholar]
- 44.Barrallo-Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005;132:3151–3161. doi: 10.1242/dev.01907. [DOI] [PubMed] [Google Scholar]
- 45.Fournier MV, et al. Gene expression signature in organized and growth-arrested mammary acini predicts good outcome in breast cancer. Cancer Res. 2006;66:7095–7102. doi: 10.1158/0008-5472.CAN-06-0515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van’t Veer LJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 47.Chaffer CL, et al. Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: role of fibroblast growth factor receptor-2. Cancer Res. 2006;66:11271–11278. doi: 10.1158/0008-5472.CAN-06-2044. [DOI] [PubMed] [Google Scholar]
- 48.Nieuwenhuis MH, et al. Genotype-phenotype correlations as a guide in the management of familial adenomatous polyposis. Clin Gastroenterol Hepatol. 2007;5:374–378. doi: 10.1016/j.cgh.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 49.Han G, et al. Distinct mechanisms of TGF-beta1-mediated epithelial-to-mesenchymal transition and metastasis during skin carcinogenesis. J Clin Invest. 2005;115:1714–1723. doi: 10.1172/JCI24399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zavadil J, et al. Genetic programs of epithelial cell plasticity directed by transforming growth factor-beta. Proc Natl Acad Sci USA. 2001;98:6686–6691. doi: 10.1073/pnas.111614398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Flanders KC. Smad3 as a mediator of the fibrotic response. Int J Exp Pathol. 2004;85:47–64. doi: 10.1111/j.0959-9673.2004.00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hudson LG, et al. Ultraviolet radiation stimulates expression of Snail family transcription factors in keratinocytes. Mol Carcinog. 2007;46:257–268. doi: 10.1002/mc.20257. [DOI] [PubMed] [Google Scholar]
- 53.Prindull G, Zipori D. Environmental guidance of normal and tumor cell plasticity: epithelial mesenchymal transitions as a paradigm. Blood. 2004;103:2892–2899. doi: 10.1182/blood-2003-08-2807. [DOI] [PubMed] [Google Scholar]
- 54.Rastaldi MP. Epithelial-mesenchymal transition and its implications for the development of renal tubulointerstitial fibrosis. J Nephrol. 2006;19:407–412. [PubMed] [Google Scholar]
- 55.Savagner P, et al. Developmental transcription factor slug is required for effective re-epithelialization by adult keratinocytes. J Cell Physiol. 2005;202:858–866. doi: 10.1002/jcp.20188. [DOI] [PubMed] [Google Scholar]
- 56.Yauch RL, et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res. 2005;11:8686–8698. doi: 10.1158/1078-0432.CCR-05-1492. [DOI] [PubMed] [Google Scholar]
- 57.Thomson S, et al. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 2005;65:9455–9462. doi: 10.1158/0008-5472.CAN-05-1058. [DOI] [PubMed] [Google Scholar]
- 58.Hiscox S, et al. Tamoxifen resistance in MCF7 cells promotes EMT-like behaviour and involves modulation of beta-catenin phosphorylation. Int J Cancer. 2006;118:290–301. doi: 10.1002/ijc.21355. [DOI] [PubMed] [Google Scholar]
- 59.Carrozzino F, et al. Inducible expression of Snail selectively increases paracellular ion permeability and differentially modulates tight junction proteins. Am J Physiol Cell Physiol. 2005;289:C1002–C1014. doi: 10.1152/ajpcell.00175.2005. [DOI] [PubMed] [Google Scholar]
- 60.Aroeira LS, et al. Mesenchymal conversion of mesothelial cells as a mechanism responsible for high solute transport rate in peritoneal dialysis: role of vascular endothelial growth factor. Am J Kidney Dis. 2005;46:938–948. doi: 10.1053/j.ajkd.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 61.Parsonage G, et al. A stromal address code defined by fibroblasts. Trends Immunol. 2005;26:150–156. doi: 10.1016/j.it.2004.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hogaboam CM, et al. Novel roles for chemokines and fibroblasts in interstitial fibrosis. Kidney Int. 1998;54:2152–2159. doi: 10.1046/j.1523-1755.1998.00176.x. [DOI] [PubMed] [Google Scholar]
- 63.Strutz F, et al. TGF-beta 1 induces proliferation in human renal fibroblasts via induction of basic fibroblast growth factor (FGF-2) Kidney Int. 2001;59:579–592. doi: 10.1046/j.1523-1755.2001.059002579.x. [DOI] [PubMed] [Google Scholar]
- 64.Wu WS. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006;25:695–705. doi: 10.1007/s10555-006-9037-8. [DOI] [PubMed] [Google Scholar]
- 65.Larue L, Bellacosa A. Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3[prime] kinase/AKT pathways. Oncogene. 2005;24:7443–7454. doi: 10.1038/sj.onc.1209091. [DOI] [PubMed] [Google Scholar]
- 66.Malaney S, Daly RJ. The ras signaling pathway in mammary tumorigenesis and metastasis. J Mammary Gland Biol Neoplasia. 2001;6:101–113. doi: 10.1023/a:1009572700317. [DOI] [PubMed] [Google Scholar]
- 67.Huber MA, et al. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17:548–558. doi: 10.1016/j.ceb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 68.Bild AH, et al. Linking oncogenic pathways with therapeutic opportunities. Nat Rev Cancer. 2006;6:735–741. doi: 10.1038/nrc1976. [DOI] [PubMed] [Google Scholar]
- 69.Massague J. Sorting out breast-cancer gene signatures. N Engl J Med. 2007;356:294–297. doi: 10.1056/NEJMe068292. [DOI] [PubMed] [Google Scholar]
- 70.Chambard JC, et al. ERK implication in cell cycle regulation. Biochim Biophys Acta. 2007;1773:1299–1310. doi: 10.1016/j.bbamcr.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 71.Giehl K. Oncogenic Ras in tumour progression and metastasis. Biol Chem. 2005;386:193–205. doi: 10.1515/BC.2005.025. [DOI] [PubMed] [Google Scholar]
- 72.Nottage M, Siu LL. Rationale for Ras and raf-kinase as a target for cancer therapeutics. Curr Pharm Des. 2002;8:2231–2242. doi: 10.2174/1381612023393107. [DOI] [PubMed] [Google Scholar]
- 73.Guerra E, et al. Prognostic value of mutations in TP53 and RAS genes in breast cancer. Int J Biol Markers. 2003;18:49–53. doi: 10.5301/jbm.2008.3870. [DOI] [PubMed] [Google Scholar]
- 74.Kim D, et al. Targeting the phosphatidylinositol-3 kinase/Akt pathway for the treatment of cancer. Curr Opin Investig Drugs. 2005;6:1250–1258. [PubMed] [Google Scholar]
- 75.Milde-Langosch K, et al. Expression and prognostic relevance of activated extracellular-regulated kinases (ERK1/2) in breast cancer. Br J Cancer. 2005;92:2206–2215. doi: 10.1038/sj.bjc.6602655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gee JM, et al. Biological and clinical associations of c-jun activation in human breast cancer. Int J Cancer. 2000;89:177–186. doi: 10.1002/(sici)1097-0215(20000320)89:2<177::aid-ijc13>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 77.Gee JM, et al. Phosphorylation of ERK1/2 mitogen-activated protein kinase is associated with poor response to anti-hormonal therapy and decreased patient survival in clinical breast cancer. Int J Cancer. 2001;95:247–254. doi: 10.1002/1097-0215(20010720)95:4<247::aid-ijc1042>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 78.Janes PW, et al. Activation of the Ras signalling pathway in human breast cancer cells overexpressing erbB-2. Oncogene. 1994;9:3601–3608. [PubMed] [Google Scholar]
- 79.Nakopoulou L, et al. Effect of different ERK2 protein localizations on prognosis of patients with invasive breast carcinoma. APMIS. 2005;113:693–701. doi: 10.1111/j.1600-0463.2005.apm_236.x. [DOI] [PubMed] [Google Scholar]
- 80.Janda E, et al. Ras and TGFβ cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol. 2002;156:299–313. doi: 10.1083/jcb.200109037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Grunert S, et al. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol. 2003;4:657–665. doi: 10.1038/nrm1175. [DOI] [PubMed] [Google Scholar]
- 82.Shintani Y, et al. Phosphoinositide-3 kinase-Rac1-c-Jun NH2-terminal kinase signaling mediates collagen I-induced cell scattering and up-regulation of N-cadherin expression in mouse mammary epithelial cells. Mol Biol Cell. 2006;17:2963–2975. doi: 10.1091/mbc.E05-12-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zavadil J, Bottinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–5774. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- 84.Nawshad A, et al. Transforming growth factor-beta signaling during epithelial-mesenchymal transformation: implications for embryogenesis and tumor metastasis. Cells Tissues Organs. 2005;179:11–23. doi: 10.1159/000084505. [DOI] [PubMed] [Google Scholar]
- 85.Lochter A, et al. Misregulation of stromelysin-1 expression in mouse mammary tumor cells accompanies acquisition of stromelysin-1-dependent invasive properties. J Biol Chem. 1997;272:5007–5015. doi: 10.1074/jbc.272.8.5007. [DOI] [PubMed] [Google Scholar]
- 86.Lochter A, et al. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J Cell Biol. 1997;139:1861–1872. doi: 10.1083/jcb.139.7.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Radisky DC, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–127. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Savagner P. Leaving the neighborhood: molecular mechanisms involved during epithelial-mesenchymal transition. Bioessays. 2001;23:912–923. doi: 10.1002/bies.1132. [DOI] [PubMed] [Google Scholar]
- 89.Tibbles LA, Woodgett JR. The stress-activated protein kinase pathways. Cell Mol Life Sci. 1999;55:1230–1254. doi: 10.1007/s000180050369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Scaltriti M, Baselga J. The epidermal growth factor receptor pathway: a model for targeted therapy. Clin Cancer Res. 2006;12:5268–5272. doi: 10.1158/1078-0432.CCR-05-1554. [DOI] [PubMed] [Google Scholar]
- 91.Tolg C, et al. Rhamm–/– fibroblasts are defective in CD44-mediated ERK1,2 motogenic signaling, leading to defective skin wound repair. J Cell Biol. 2006;175:1017–1028. doi: 10.1083/jcb.200511027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liu R, et al. The prognostic role of gene signature from tumorigenic breast cancer cells. N Engl J Med. 2007;356:217–226. doi: 10.1056/NEJMoa063994. [DOI] [PubMed] [Google Scholar]