

Abstract
Background
Atherosclerosis is a chronic inflammatory disease associated with the accumulation of oxidized lipids in arterial lesions. Recently we studied the degradation of peroxidized linoleic acid and suggested that oxidation is an essential process that results in the generation of terminal products, namely mono- and dicarboxylic acids that may lack the pro-atherogenic effects of peroxidized lipids. In continuation of that study, we tested the effects of azelaic acid (AzA), one of the end products of linoleic acid peroxidation, on the development of atherosclerosis using low density lipoprotein receptor knockout (LDLr−/−) mice.
Methods and results
LDLr−/− mice were fed with a high fat and high cholesterol Western diet (WD group). Another group of animals were fed the same diet with AzA supplementation (WD+AzA group). After four months of feeding, mice were sacrificed and atherosclerotic lesions were measured. The results showed that the average lesion area in WD+AzA group was 38% (p<0.001) less as compared to WD group. The athero-protective effect of AzA was not related to changes in plasma lipid content. AzA supplementation decreased the level of CD68 macrophage marker by 34% (p<0.05).
Conclusions
The finding that AzA exhibits an anti-atherogenic effect suggests that oxidation of lipid peroxidation-derived aldehydes into carboxylic acids could be an important step in the body’s defense against oxidative damage.
Keywords: atherosclerosis, LDL receptor knockout mouse, lipid peroxidation, dicarboxylic acid, CD68, macrophages
1. Introduction
Development of atherosclerosis is thought to be associated with the oxidation of low density lipoprotein (LDL) [1–3]. The peroxidized lipids associated with oxidized LDL contribute to atherosclerosis in many ways, influencing various cellular pathways directly and indirectly through secondary products derived after lipid peroxide degradation and oxidation [4–6]. The aldehydes formed from the decomposition of peroxidized lipids may play a key role in atherosclerosis by modifying apolipoproteins leading to their recognition by macrophage scavenger receptors [7–9], generating antigenic responses [10–12], and affecting many cell processes [13,14]. Aldehydes are readily oxidized enzymatically and non-enzymatically to carboxylic acids [15], or become detoxified by GSH-coupling enzymatic systems [16]. Currently there is no data on whether lipid peroxidation-derived carboxylic acids, particularly dicarboxylic acids, affect the development of atherosclerosis.
Azelaic acid (AzA), a nine carbon saturated aliphatic dicarboxylic acid, is an end product of linoleic acid peroxide decomposition [15] and is derived from oxo-nonanoic acid (Fig. 1). Since linoleic acid represents the major polyunsaturated fatty acid in human plasma and LDL, we tested the effects of AzA on atherosclerosis development in cholesterol-fed LDL receptor knockout (LDLr−/−) mice. We present evidence that AzA inhibits atherosclerosis development without any significant effect on plasma lipid content.
Fig. 1.

The decomposition of peroxidized linoleic acid to AzA.
2. Materials and methods
2.1. Reagents
AzA, heparin, and other chemicals and reagents were purchased from Sigma-Aldrich Chemical Co (St. Louis, MO).
2.2. Animals, diets, and feeding timeline
All animal studies were performed in agreement with Public Health Service policy on use of laboratory animals, and in conformity with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. Animal use protocol was approved by the Institutional Animal Care and Use Committee of the Ohio State University.
Fifty four 4 week old female LDLr−/− mice on C57BL/6J background (B6.129S7-Ldlrtm1Her/J strain) were obtained from Jackson Laboratory (Bar Harbor, ME). Female animals were chosen purely for convenience and availability, as the focus of the study was not to find out potential gender differences in atherosclerotic responses to AzA. Two separate and independent studies, 27 animals each, were performed one after another designated as study AzA#1 and AzA#2. Both studies were similar in design with differences only in the content of cholesterol in the Western diet and the duration of mice feeding: 0.2% cholesterol for14 weeks in AzA#1 study and 0.125% cholesterol for 16 weeks in AzA#2 study.
In each study, mice were evenly distributed into 3 groups, 9 animals in each, and fed with three different diets for 14 or 16 weeks. First group (Normal) was fed normal mice chow, second group (WD) was fed Western diet (21% milk fat with 0.2% or 0.125% cholesterol), and third group (WD+AzA) was fed Western diet supplemented with 0.5% AzA. The chow and diets were made by Harlan Teklad (Indianapolis, IN). At the end of feeding for14 or 16 weeks, the animals were fasted overnight and sacrificed according to the animal protocol. Blood was collected immediately by syringe aspiration from the heart using heparin as an anticoagulant. After centrifugation, the plasma was snap frozen for analysis of lipid and cytokine profiles. The aorta and liver were flushed with ice-cold phosphate buffered saline (PBS), cleaned from surrounding tissue, photo documented, and collected for extraction of total RNA.
2.3. Assessment of atherosclerotic lesions
The aortic trunk was washed with cold PBS through the left ventricle. The dissection of the aorta was performed under a stereomicroscope from the iliac bifurcation up to the heart, including the beginning of the brachiocephalic, carotid, and subclavian arteries. Detailed en face pictures of the aorta along with a ruler were obtained using a digital camera. Lesion areas were marked on the pictures under direct microscopic observations. Accordingly, lesion area size was quantified using ImageJ software [17]. The ruler in the pictures was used to determine the pixel to mm2 conversion factor.
2.4. Analysis of plasma lipids
All lipid analyses were performed by enzymatic methods on the Beckman CX7 (Fullerton, CA) automated clinical chemistry analyzer in Cardiovascular Specialty Labs (Atlanta, GA). Total cholesterol and triglycerides were determined using reagents from Beckman Coulter Diagnostics (Fullerton, CA). Cholesterol in LDL (LDLc) and in HDL (HDLc) was determined using homogeneous assays from Genzyme Diagnostics (Cambridge, MA).
2.5. Quantitative real-time PCR (qPCR)
After the mice were euthanized and blood was collected, the cremasteric artery was cut and the aorta and liver were flushed by injecting PBS into the left ventricle of the heart. Approximately 100 μg section from the large lobe of the liver was snap frozen in TRIzol (Invitrogen; Carlsbad, CA). The aorta was exposed, treated with RNALater (Ambion; Austin, TX) and cleaned from the ascending aorta to the renal artery bifurcation. The aorta was photographed, cut into two pieces: aortic arch half (including ascending and descending aorta) and abdominal aorta half, and snap frozen in Trizol. Total RNA from the aorta and liver were isolated using TRIzol reagent according to the manufacturer’s protocol. The concentration and quality of RNA was assessed by NanoDrop (Thermo Fisher Scientific, Waltham, MA). cDNA was generated from 10–100 ng of total RNA and 1/20th of the sample was taken for qPCR. cDNA syntheses and qPCRs were performed with SYBR GreenER Two-Step qRT-PCR Kit for iCycler (Invitrogen) according to the manufacturer’s protocol. qPCR was run in 20 μl of reaction mixture in sealed 96-well plates on iCycler (Bio-Rad; Hercules, CA) in duplicates. The melting curve and efficiency were assessed for all primer pairs. All primers were purchased from Invitrogen and are listed in the supplementary appendix. Threshold cycle (CT) was determined by Bio-Rad iQ5 v.2 software. The level of mRNA was calculated using the 2−ΔΔCT method and GAPDH (glyceraldehyde 3-phosphate dehydrogenase) as an internal control gene [18]. Data are expressed as fold induction of mRNA level in one group compared to another.
2.6. Mice plasma cytokines profile
Plasma cytokine levels were determined by RayBiotech, Inc. (Norcross, GA) using RayBio® Mouse G Series 1000 Array glass chip.
2.7. In vitro LDL oxidation assay
The oxidation of LDL was performed using copper [19]. 100 μg of LDL was incubated in quartz cuvette in 1ml PBS with 5 μM copper. AzA was added as 50 mM solution adjusted to pH 7.4 by sodium hydroxide. In some control experiments the same volume of 200 mM solution of acetic acid adjusted to pH 7.4 by sodium hydroxide was added. Time course of oxidation of LDL samples was followed by measuring the formation of conjugated diene at 234 nm in a UVIKON XL spectrophotometer (Bio-Tek Instruments, Winooski, VT) equipped with a 12-chamber cuvette changer. Samples were monitored continuously for periods of up to 8 h.
2.8. Cytochrome C reduction assay
Cytochrome C reduction was run in quartz cuvette in 1ml PBS containing 100 μM xanthine and 10 μM cytochrome C from equine heart. The AzA was added as 50 mM solution adjusted to pH 7.4 by sodium hydroxide and was added up to a final concentration of 1 mM. In some control experiments the same volume of 100 mM solution of acetic acid adjusted to pH 7.4 by sodium hydroxide was added. Reduction of cytochrome C was started by adding 0.02 units of xanthine oxidase and was followed by monitoring the optical density at 550 nm for 30 min in a UVIKON XL spectrophotometer (Bio-Tek Instruments, Winooski, VT).
2.9. LC-MS/MS analysis of AzA level in plasma
AzA was extracted and measured for 6 samples from each diet group from AzA#2 study using 75 μl of mouse plasma. For normalization of the extraction yield 100 μM sebacic acid was added to the plasma samples. After dilution to 500 μl and acidification to 400 mM HCl, samples were extracted with 3 ml of ether, dried, dissolved in 150 μl of water, centrifuged 12,000 g for 20 minutes, and 50 μl aliquots were subjected to LC-MS/MS analysis.
The high-performance liquid chromatography was run on Shimadzu (Columbia, MD) system consisted of two LC-20AD pumps, SIL-20A autosampler, and CBM-20A controller. Separation was performed on μBondapak C18 125A 10 μm 3.9 × 300 mm column (Waters; Milford, MA) using 50 minute linear gradient from 40% acetonitrile in water to 50% methanol in water followed by 10 minutes of 50% methanol in water at 0.5 ml/min.
3200 Q TRAP triple quadrupole mass spectrometer (Applied Biosystems; Foster City, CA) was used to carry out negative ion electrospray tandem mass spectrometric analysis in multiple reactions monitoring mode. The instrument was controlled by Analyst software v. 1.2 (Applied Biosystems). The source temperature was 350°C, ion spray voltage was −4200 V, declustering potential was −37 V, entrance potential was −2 V, collision energy was −10 V and collision cell exit potential was −3 V. The MRM transitions of 187.2 amu to 125.2 amu and 202.3 amu to 140.3 amu were monitored for the detection of AzA and sebacic acid respectively.
2.10. Statistical analysis
All data are presented as mean ± SD. Statistical significance for differences in lesion areas and plasma lipid concentrations were evaluated using Student’s t-test. Statistical significance for relative gene expression was evaluated using one-way ANOVA with Tukey post-hoc test using 95% confidence intervals. Differences were considered significant at P < 0.05.
3. Results
3.1. Effect of AzA on atherosclerosis lesion development in mice
LDLr knockout mice were randomly divided into 3 groups, 9 animals in each group, and were fed with normal chow (“Normal”), Western diet (“WD” group) and Western diet with AzA supplementation (“WD+AzA” group) for 4 months. No adverse effects of AzA were noted and the animals grew normally and looked healthy. There were no statistically significant differences in food consumption or in weight gain between the groups (data not shown).
At the end of the feeding period the mice were sacrificed. AzA plasma levels were 0.2–0.7 μM for WD animals, and 0.9–21 μM for WD+AzA animals. Animals fed Western diet (WD and WD+AzA groups) developed atherosclerotic lesions while no lesions were seen in normal chow fed mice. Analysis of lesion area size revealed differences between mice which were fed the Western diet with and without AzA supplementation. In AzA#1 study, the average area was 0.63±0.24 mm2 for WD animals and 0.36±0.10 mm2 for WD+AzA animals, revealing a decrease of 44% (p<0.01). In AzA#2 study, the average lesion size was 0.70±0.19 mm2 for WD animals, and 0.47±0.20 mm2 for WD+AzA animals, showing a decrease of 33% (p<0.05). When the lesion area size was expressed as fold change compared to the average size for each study, aggregated data from 18 animals in each group suggested a 39% reduction in lesion size (p=0.001) in the WD+AzA group as compared to the WD group (Figure 2)
Fig. 2.
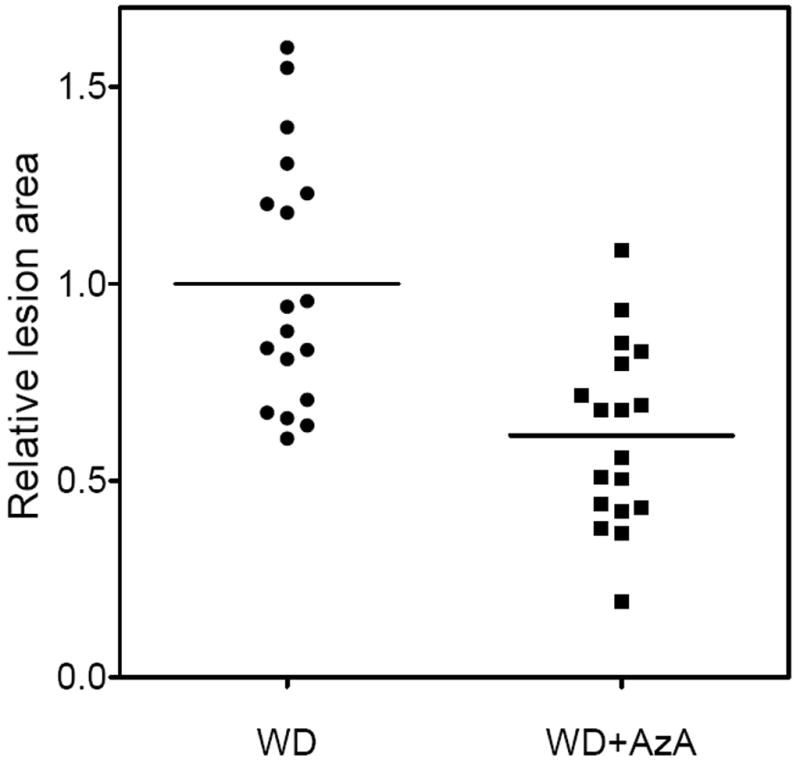
Relative lesion area in 5 months old LDLr−/− mice after 4 months of feeding with Western-type diet (WD), and the same diet with 0.5% AzA supplement (WD+AzA). The aggregated data from AzA#1 and AzA#2 studies are shown. Lesion area size is expressed as fold change relative to an average lesion size for WD group in corresponding study. Averages for WD and WD+AzA groups are shown as lines. P<0.001 assessed by Student’s t-test
AzA supplemented animals demonstrated a decrease not only in lesion area size, but also in the number of lesions. In the first study the number dropped from 4.0±1.4 in WD group to 2.7±1.0 in WD+AzA group (decrease of 33%, p<0.05). In the second study the average number of lesions per animal was 4.9±1.8 for WD group and 3.0±1.3 for WD+AzA group (decrease of 39%, p<0.05). Both studies aggregated suggest a decrease of 36% (p=0.001).
3.2. Plasma lipid profile
AzA did not change the majority of parameters in both studies, such as total cholesterol (CHOL), triglyceride level (TG), LDLc, and very low density lipoprotein cholesterol (VLDLc) (Figure 3A,B,C). The only difference we observed was increased HDL cholesterol in WD+AzA group compared to WD group in AzA#1 study: 65 ±10 and 46±9 mg/100ml respectively (increase of 42%, p<0.001). However, we did not observe a similar increase in AzA#2 study; WD+AzA and WD groups had almost identical concentrations of HDL cholesterol: 42.7±6.9 and 42.6±9.1 mg/100ml, respectively.
Fig. 3.

Lipid profiles of 5 months old LDLr−/− mice after 4 months of feeding with Western-type diet (WD), and the same diet with 0.5% AzA supplement (WD+AzA). A: HDL cholesterol profile for AzA#1 and AzA#2 studies; B: Lipid profile for AzA#1 study; C: Lipid profile for AzA#2 study
3.3. Gene expression in mouse aorta
We assessed mRNA levels of several inflammatory marker genes in the animal aortas. Each aorta was cut into two halves: aortic arch and abdominal parts. qPCR revealed that the aortic arch of WD and WD+AzA animals had a 3–5 fold increase in the level of mRNA of MCP-1, CD4, and P-selectin (p < 0.01) compared to Normal group. However, those markers could not distinguish the extent of inflammation between WD and WD+AzA groups (Figure 4A). Also, these markers were not significantly different in the abdominal segments of the aortas between the three diet groups (data not shown).
Fig. 4.

mRNA level of several marker genes in aorta of 5 months old LDLr−/− mice after 4 months of feeding with Western-type diet (WD), and the same diet with 0.5% AzA supplement (WD+AzA). A: MCP-1, CD4 and P-selectin mRNA level in aortic arch. P < 0.01 for differences in Norm and WD or WD+AzA groups; B: Relative level of mRNA of CD68 in aortic arch and abdominal aorta. P < 0.001 for differences in Norm and WD or WD+AzA groups.
The level of mRNA of monocyte/macrophage marker CD68 was 12.6±3.9 fold higher in the aortic arch of WD animals compared to Normal group (p < 0.001) and about 3 fold higher in the abdominal aorta of WD animals compared to Normal group (p<0.05). There was a difference in the level of CD68 between WD and WD+AzA animals in the aortic arch segments and lesion-free abdominal segments (Figure 4B). AzA supplementation decreased CD68 levels in the aortic arch by 34% (p<0.05) and by 62% (p<0.05) in abdominal segments of the aorta
3.4. Gene expression in mouse liver
Because of the discrepancy in AzA’s effect on HDL cholesterol levels in AzA#1 and AzA#2 studies, we analyzed the expression of ApoA1 in the liver of animals from AzA#1 study. In this study, WD+AzA mice had elevated levels of HDLc when compared to WD mice; this is in contrast to our AzA#2 study where both WD and WD+AzA animals had similar levels of HDLc. No difference in ApoA1 gene expression in the liver between WD and WD+AzA groups was observed (data not shown). We assume that the HDL cholesterol values were an anomaly, perhaps due to very high plasma cholesterol values.
The expression of several other genes that could be athero-protective, such as paraoxonase 1 (PON1) and catalase were also assessed. There was a decrease in ApoA1 and PON1 expression in the liver of WD animals compared to Normal animals. However, the changes between WD and WD+AzA groups were not significant (data not shown).
3.5. Cytokine protein array
Significant changes in the levels of seven inflammatory mediators were noted in WD animals as compared to Normal animals (Table 1). Supplementation of Western diet with AzA caused minimal changes in the profile of 96 inflammation-related proteins as measured by the cytokine array. The only statistically significant difference in WD+AzA group compared to WD group was in the level of insulin-like growth factor binding protein 3 (IGFBP-3). Additional cytokines with the lowest P-values are also listed in Table 1.
Table 1.
Changes in the level of inflammatory mediators in mouse plasma between different diet groups. Five samples from each of normal chow (Norm), western diet (WD) and western diet with AzA supplement (WD+AzA) groups were analyzed by a sandwich ELISA. Proteins with highest statistical significant different levels in WD compare to Norm, and in WD+AzA compare to WD are shown. The protein levels expressed as a fold change between two groups.
| Cytokines differently presented in Norm and WD groups | |||
|---|---|---|---|
| Protein | Relative protein level compare to Norm, fold difference | P-value | |
| Norm | WD | WD vs Norm | |
| Thymus CK-1 | 1 | 0.56 | 0.001 |
| G-CSF | 1 | 1.66 | 0.008 |
| L-Selectin | 1 | 1.54 | 0.011 |
| P-Selectin | 1 | 1.22 | 0.016 |
| Leptin R | 1 | 2.08 | 0.020 |
| IGFBP-2 | 1 | 0.65 | 0.025 |
| axl | 1 | 3.03 | 0.030 |
| Cytokines differently presented in WD+AzA and WD groups | |||
| Protein | Relative protein level compare to WD, fold difference | P-value | |
| WD | WD+AzA | WD+AzA vs. WD | |
| IGFBP-3 | 1 | 0.40 | 0.001 |
| VCAM-1 | 1 | 1.25 | 0.077 |
| Pro-MMP-9 | 1 | 1.58 | 0.096 |
| DPPIV/CD26 | 1 | 0.83 | 0.105 |
3.6. Assessment of antioxidant properties of AzA
Although no antioxidant properties can be inferred from the chemical structure of AzA, it can interfere with oxidation processes by chelating metal ions, inhibiting enzymes, and other indirect routes. We assessed whether AzA affected the oxidation of LDL by copper ions in vitro. Neither the lag time, the time course of oxidation, nor the extent of oxidation of LDL was affected by AzA (up to 0.5 mM). In addition, AzA did not affect the reduction of cytochrome C by superoxide generated by xanthine oxidase (data not shown).
4. Discussion
Oxidized LDL provides a large pool of lipid peroxides and products of its degradation in atherosclerotic plaques. In our previous study, we established that dicarboxylic acids, such as AzA, are normally generated from oxidized lipids [15]. AzA has been noted to inhibit the generation of reactive oxygen species by neutrophils [20]; however, the effect of the dicarboxylic acid on the development of atherosclerosis has not been elucidated. Also, AzA has been shown to have a positive therapeutic effect in the treatment of some skin conditions, such as rosacea, acne, erythema [21]. To evaluate the effect of AzA on the development of atherosclerotic plaques, we performed a study utilizing female LDLr−/− mice fed high fat, high cholesterol (“Western”) diet, a regular animal model of atherosclerosis.
AzA has been used as an energy source for parenteral nutrition [22]. Animal experiments did not reveal any toxic or teratogenic effects of AzA for single doses of up to 4g per 1 kg of body weight when fed orally [23]. A maximum single oral dose of 10 g or one hour intravenous infusion of AzA was tested on humans, and no adverse effects were found. AzA successfully reached the plasma when orally fed and was rapidly excreted in the urine [24]. Based on these data we chose to use 0.5% AzA supplementation to the Western diet. The average food consumption was 2.5–3 g per animal per day, providing up to 15 mg of AzA per animal per day, or less than 1g per 1 kg of body weight per day. Although we did not determine the steady-state level of AzA in mouse plasma, the level after overnight fasting was up to 21 μM for WD+AzA animals, while below 0.7μM for WD animals.
The WD fed animals showed predictable changes in lipid levels, inflammatory markers, and gene expression profiles compared to animals fed with normal chow. However, there was practically no difference between the WD and the WD+AzA fed animals suggesting that factors beyond the correction of dyslipidemia and inflammation should have played a role in the anti-atherosclerotic effects of AzA. Many antioxidants have been shown to reduce atherosclerosis in WD fed mice. We were unable to document any relevant antioxidant effects of AzA in the current study. The only known redox-related property of AzA is that it competitively inhibits thioredoxin reductase [25], an enzyme that has been observed to be elevated in the atherosclerotic artery [26]. It is unclear whether the inhibition of TrxR affected the development of atherosclerosis in WD+AzA animals. In addition, there were no effects of AzA on liver gene expression of PON 1, an antioxidant enzyme associated with HDL, or that of apolipoprotein A1. There was also no difference in the aortic expression of MCP-1, an oxidation responsive gene.
We observed a decreased level of plasma IGFBP-3 in animals with reduced atherosclerosis (WD+AzA animals as compared with WD animals), yet the plasma changes in IGF-I or IGF-II were not statistically significant (data not shown). It has been noted that an increased level of IGFBP-3 is associated with a higher degree of atherosclerosis in humans [27]. However, in most human studies, the opposite association was observed: a lower level of IGFBP-3 was associated with an increased extent of atherosclerosis, a higher risk of nonfatal myocardial infarction and ischemic stroke, and decreased plaque stability [28–31]. Based upon our current understanding it is unknown whether the decrease in IGFBP-3 is pro- or anti-atherogenic by itself.
How do we reconcile the decreased atherosclerosis with almost no changes in any of the known markers of the disease? Interestingly, we observed a decrease in the mRNA level of CD68, a marker for monocyte/macrophages, in WD+AzA animals compared to WD animals. The decrease of CD68 in the aortic arch area that contained lesions is a confirmation that the presence of foam cell forming macrophages in AzA animals was reduced. The decrease of CD68 mRNA was also observed in the abdominal aorta section, which did not contain any visible foam cell lesions which might suggest that AzA may have reduced the number of residential macrophages inside the arterial wall or on its surface. Although there was no difference in MCP-1 gene expression between WD and WD+AzA groups, the possibility that reduced expression of other chemotactic factors may have contributed to the reduced presence of macrophages cannot be ruled out.
Dicarboxylic acids bind divalent metals, particularly calcium. AzA is an amphiphilic dicarboxylic acid. It is possible that AzA could have affected many of the calcium-dependent processes that are involved in atherosclerosis.
Although the mechanism of the reduction in atherosclerosis by AzA is unclear, the main conclusion of this study is that generation of AzA can slow the progress of atherosclerosis. The rapid oxidation of lipid peroxidation-derived aldehydes may be a beneficial process that reduces the damage otherwise caused by decomposing lipid peroxides. In conclusion, it appears that our current study supports the hypothesis that terminal oxidation of fatty acids is an essential protective physiological process during atherosclerosis [4].
Supplementary Material
Acknowledgments
This study was supported by the National Institutes of Health grants HL69038 and DK056353.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Berliner JA, Watson AD. A role for oxidized phospholipids in atherosclerosis. N Engl J Med. 2005;353:9–11. doi: 10.1056/NEJMp058118. [DOI] [PubMed] [Google Scholar]
- 2.Parthasarathy S, Khan-Merchant N, Penumetcha M, Santanam N. Oxidative stress in cardiovascular disease. J Nucl Cardiol. 2001;8(3):379–89. doi: 10.1067/mnc.2001.114150. [DOI] [PubMed] [Google Scholar]
- 3.Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med. 1989;320(14):915–24. doi: 10.1056/NEJM198904063201407. [DOI] [PubMed] [Google Scholar]
- 4.Parthasarathy S, Litvinov D, Selvarajan K, Garelnabi M. Lipid peroxidation and decomposition--conflicting roles in plaque vulnerability and stability. Biochim Biophys Acta. 2008;1781:221–31. doi: 10.1016/j.bbalip.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parthasarathy S, Santanam N, Ramachandran S, Meilhac O. Oxidants and antioxidants in atherogenesis. An appraisal J Lipid Res. 1999;40:2143–57. [PubMed] [Google Scholar]
- 6.Leonarduzzi G, Arkan MC, Başağa H, Chiarpotto E, Sevanian A, Poli G. Lipid oxidation products in cell signaling. Free Radic Biol Med. 2000;28:1370–8. doi: 10.1016/s0891-5849(00)00216-1. [DOI] [PubMed] [Google Scholar]
- 7.Hoff HF, O’Neil J, Chisolm GM, 3rd, Cole TB, Quehenberger O, Esterbauer H, Jürgens G. Modification of low density lipoprotein with 4-hydroxynonenal induces uptake by macrophages. Arteriosclerosis. 1989;9:538–49. doi: 10.1161/01.atv.9.4.538. [DOI] [PubMed] [Google Scholar]
- 8.Steinbrecher UP, Lougheed M, Kwan WC, Dirks M. Recognition of oxidized low density lipoprotein by the scavenger receptor of macrophages results from derivatization of apolipoprotein B by products of fatty acid peroxidation. J Biol Chem. 1989;264:15216–23. [PubMed] [Google Scholar]
- 9.Leitinger N. Cholesteryl ester oxidation products in atherosclerosis. Mol Aspects Med. 2003;24:239–50. doi: 10.1016/s0098-2997(03)00019-0. [DOI] [PubMed] [Google Scholar]
- 10.Branch DW, Mitchell MD, Miller E, Palinski W, Witztum JL. Pre-eclampsia and serum antibodies to oxidised low-density lipoprotein. Lancet. 1994;343:645–6. doi: 10.1016/s0140-6736(94)92639-5. [DOI] [PubMed] [Google Scholar]
- 11.Romero FI, Khamashta MA, Hughes GR. Lipoprotein(a) oxidation and autoantibodies: a new path in atherothrombosis. Lupus. 2000;9:206–9. doi: 10.1191/096120300678828253. [DOI] [PubMed] [Google Scholar]
- 12.Sjögren P, Fredrikson GN, Rosell M, de Faire U, Hamsten A, Nilsson J, Hellenius ML, Fisher RM. Autoantibodies against modified apolipoprotein B-100 in relation to low-density lipoprotein size and the metabolic syndrome in otherwise healthy men. Metabolism. 2008;57:362–6. doi: 10.1016/j.metabol.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 13.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 14.Dianzani MU, Barrera G, Parola M. 4-Hydroxy-2,3-nonenal as a signal for cell function and differentiation. Acta Biochim Pol. 1999;46:61–75. [PubMed] [Google Scholar]
- 15.Raghavamenon AC, Garelnabi M, Babu S, Aldrich A, Litvinov D, Parthasarathy S. α-Tocopherol is ineffective in preventing the decomposition of pre-formed lipid peroxides and may promote the accumulation of toxic aldehydes: A potential explanation for the failure of antioxidants to affect human atherosclerosis. Antioxid Redox Signal. 2009;11:1237–1248. doi: 10.1089/ars.2008.2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conklin D, Prough R, Bhatanagar A. Aldehyde metabolism in the cardiovascular system. Mol Biosyst. 2007;3:136–50. doi: 10.1039/b612702a. [DOI] [PubMed] [Google Scholar]
- 17.Abramoff MD, Magelhaes PJ, Ram SJ. Image Processing with ImageJ. Biophotonics International. 2004;11:36–42. [Google Scholar]
- 18.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 19.Santanam N, Parthasarathy S. Paradoxical actions of antioxidants in the oxidation of low density lipoprotein by peroxidases. J Clin Invest. 1995;95(6):2594–600. doi: 10.1172/JCI117961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akamatsu H, Komura J, Asada Y, Miyachi Y, Niwa Y. Inhibitory effect of azelaic acid on neutrophil functions: a possible cause for its efficacy in treating pathogenetically unrelated diseases. Arch Dermatol Res. 1991;283:162–6. doi: 10.1007/BF00372056. [DOI] [PubMed] [Google Scholar]
- 21.Gooderham M. Rosacea and its topical management. Skin Therapy Lett. 2009;14:1–3. [PubMed] [Google Scholar]
- 22.Grego AV, Mingrone G. Dicarboxylic acids, an alternate fuel substrate in parenteral nutrition: an update. Clin Nutr. 1995;14:143–8. doi: 10.1016/s0261-5614(95)80011-5. [DOI] [PubMed] [Google Scholar]
- 23.Mingrone G, Greco AV, Nazzaro-Porro M, Passi S. Toxicity of azelaic acid. Drugs Exptl Clin Res. 1983;9:447–455. [Google Scholar]
- 24.Passi S. Azelaic acid: biochemistry and metabolism. Acta Derm Veneorol Suppl-Stockh. 1989;143:8–13. doi: 10.2340/00015555143813. [DOI] [PubMed] [Google Scholar]
- 25.Schallreuter KU, Wood JM. Azelaic acid as a competitive inhibitor of thioredoxin reductase in human melanoma cells. Cancer Lett. 1987;36:297–305. doi: 10.1016/0304-3835(87)90023-1. [DOI] [PubMed] [Google Scholar]
- 26.Furman C, Rundlöf AK, Larigauderie G, Jaye M, Bricca G, Copin C, Kandoussi M, Fruchart JC, Arnér ES, Rouis M. Thioredoxin reductase 1 is upregulated in atherosclerotic plaques: specific induction of the promoter in human macrophages by oxidized low-density lipoproteins. Free Radic Biol Med. 2004;37:71–85. doi: 10.1016/j.freeradbiomed.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 27.Kawachi S, Takeda N, Sasaki A, Kokubo Y, Takami K, Sarui H, Hayashi M, Yamakita N, Yasuda K. Circulating insulin-like growth factor-1 and insulin-like growth factor binding protein-3 are associated with early carotid atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:617–21. doi: 10.1161/01.ATV.0000154486.03017.35. [DOI] [PubMed] [Google Scholar]
- 28.Schuler-Luttmann S, Monnig G, Enbergs A, Schulte H, Breithardt G, Assmann G, Kerber S, von Eckardstein A. Insulin-like growth factor-binding protein-3 is associated with the presence and extent of coronary arteriosclerosis. Arterioscler Thromb Vasc Biol. 2000;20:E10–5. [PubMed] [Google Scholar]
- 29.Kaplan RC, McGinn AP, Pollak MN, Kuller LH, Strickler HD, Rohan TE, Cappola AR, Xue X, Psaty BM. Association of total insulin-like growth factor-I, insulin-like growth factor binding protein-1 (IGFBP-1), and IGFBP-3 levels with incident coronary events and ischemic stroke. J Clin Endocrinol Metab. 2007;92:1319–25. doi: 10.1210/jc.2006-1631. [DOI] [PubMed] [Google Scholar]
- 30.Johnsen SP, Hundborg HH, Sørensen HT, Orskov H, Tjønneland A, Overvad K, Jørgensen JO. Insulin-like growth factor (IGF) I, -II, and IGF binding protein-3 and risk of ischemic stroke. J Clin Endocrinol Metab. 2005;90:5937–41. doi: 10.1210/jc.2004-2088. [DOI] [PubMed] [Google Scholar]
- 31.Martin RM, Gunnell D, Whitley E, Nicolaides A, Griffin M, Georgiou N, Davey Smith G, Ebrahim S, Holly JM. Associations of insulin-like growth factor (IGF)-I, IGF-II, IGF binding protein (IGFBP)-2 and IGFBP-3 with ultrasound measures of atherosclerosis and plaque stability in an older adult population. J Clin Endocrinol Metab. 2008;93:1331–8. doi: 10.1210/jc.2007-2295. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.