

Abstract
Mutations in the leucine-rich repeat kinase 2 (LRRK2) gene are the most common known cause of Parkinson's disease (PD). Whether loss of LRRK2 function accounts for neurodegeneration of dopamine neurons in PD is not known, nor is it known whether LRRK2 kinase activity modulates the susceptibility of dopamine (DA) neurons to the selective dopaminergic toxin, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). To better understand the role of LRRK2 in DA neuronal survival and its role in the susceptibility of DA neurons to MPTP, we generated LRRK2 knock-out (KO) mice lacking the kinase domain of LRRK2. Here, we show that LRRK2 KO mice are viable and have no major abnormalities and live to adulthood. The dopaminergic system is normal in LRRK2 KO mice as assessed via HPLC for DA and its metabolites and via stereologic assessment of DA neuron number in young and aged mice. Importantly, there is no significant difference in the susceptibility of LRRK2 KO and wild-type mice to MPTP. These results suggest that LRRK2 plays little if any role in the development and survival of DA neurons under physiologic conditions. Thus, PD due to LRRK2 mutations are likely not due to a loss of function. Moreover, LRRK2 is not required for the susceptibility of DA neurons to MPTP.
Introduction
Progressive and selective loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) is one of the main neuropathological findings in Parkinson's disease (PD). Although PD is mostly a sporadic disorder and the etiology is not known, its pathogenesis may involve genetic susceptibility and environmental factors, that will cause oxidative stress, impairments in mitochondria and the proteosome, or aggregation of misfolded proteins (Dawson and Dawson, 2003; Vila and Przedborski, 2004; Moore et al., 2005).
Only a 10% of the PD cases are linked to Mendelian genetic defects, and mutations in Leucine-rich repeat kinase 2 (LRRK2) are the most frequent of the familial forms of PD (Gasser, 2007; Lees et al., 2009). Several missense mutations distributed along the protein have been reported, but the role of LRRK2 in the pathophysiology of PD is still not well known. The kinase activity may be a link between LRRK2 and PD (Smith et al., 2005; West et al., 2005, 2007) as attenuation of LRRK2 kinase activity prevents LRRK2 toxicity in cellular models of PD. LRRK2 also has independent GTPase activity that can regulate its kinase activity (Greggio et al., 2008). It is assumed that LRRK2 mutations linked to PD are gain of function mutations in which the kinase domain is critical for the toxic actions of mutant LRRK2 (Biskup and West, 2009).
Previous studies showed that LRRK2 is associated with the mitochondria (Biskup et al., 2006), and that the toxicity mediated by LRRK2 mutants could be due to mitochondria-dependent apoptosis (Iaccarino et al., 2007). Wild-type (WT) LRRK2, but not the mutants, attenuate hydrogen peroxide (H2O2)-induced oxidative stress suggesting a protective role for LRRK2 (Liou et al., 2008). Moreover, data generated in lines of Caenorhabditis elegans expressing human wild-type and mutant LRRK2 suggest that LRRK2 plays a role modulating the response of the mitochondria to different stressors like rotenone and paraquat (Saha et al., 2009), and work in Drosophila indicate that LRRK2 mutant flies display increased sensitivity to rotenone, a mitochondrial complex I inhibitor (Ng et al., 2009). Thus, it seems that LRRK2 may play important roles in mitochondrial function.
Here, we show for the first time that the absence of LRRK2 in mice does not lead to major progressive behavioral, neurochemical, or anatomical deficits in the dopaminergic system. In addition, ablation of LRRK2 unexpectedly does not exacerbate the dopaminergic neurodegeneration caused by the parkinsonian neurotoxin 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine (MPTP). Thus, LRRK2 appears to play little if any role in the maintenance or the survival of dopamine (DA) neurons or the susceptibility of DA neurons to MPTP.
Materials and Methods
Gene targeting and generation of LRRK2-null mice.
The LRRK2 gene contains 51 exons and the target sequences for generating LRRK2 knock-out mice include partial exon 39 and complete exon 40. The map of designed targeting construct for generating LRRK2 knock-out mice is shown in Figure 1A. Restriction enzyme site BamHI was used for inserting the long arm into the targeting construct. ClaI and Aat II were used for inserting the short arm, and AscI and SalI were used for inserting a stop codon and a loxP flanked neomycin gene into the targeting construct. Rsr II was used for inserting the negative selection gene thymidine kinase (TK) into the targeting construct and XhoI was used to linearize the targeting construct. The expected mutant allele is also shown in Figure 1A. The expression of LRRK2 is disrupted by the deletion of partial exon 39 and complete exon 40 as well as by the introduction of a stop codon into the coding sequences. Embryonic stem cells carrying the mutant allele were injected into blastocysts and the resulting male chimeric mice were bred to C57BL/6 female mice to obtain heterozygous LRRK2 mutant male and female mice, which were subsequently bred to generate LRRK2-null mice.
Figure 1.

Targeted disruption of LRRK2 in LRRK2 KO mice. A, Schematic representation of the targeting strategy. B, Southern blot analysis of genomic DNA from WT (+/+), heterozygous (+/−) and homozygous LRRK2 KO (−/−) mice. C, PCR analysis of genomic DNA from heterozygous (+/−), WT (+/+), and homozygous LRRK2 KO (−/−) mice. D, A cDNA fragment of LRRK2 mRNA open reading frame was used as the probe for Northern blot analysis of total RNA from WT (+/+), homozygous LRRK2 KO (−/−), and heterozygous (+/−) mice. E, Immunoblot analysis of whole-brain lysate from WT, heterozygous (Het), and homozygous LRRK2 KO mice with affinity-purified rabbit polyclonal antibodies and monoclonal antibody 6.1 generated to LRRK2 protein using different synthetic peptides against human LRRK2. The blot was reprobed with anti-actin antibody as loading control.
Genotyping LRRK2 mice by PCR.
LRRK2 mice genomic DNA was purified from mouse tail tissue using standard protocols. Primer pair (forward: 5′CCCAGGGCTGAGAACGATTAAGTC3′; reverse: 5′CTGGAGTGGACTCAGGGTTAC AGC3′) was used to amplify a 590-bp DNA fragment from wild-type LRRK2 allele, and primer pair (forward: 5′GGCCTACCCGCTTCCATTGCTCAGCGG3′; reverse: 5′CCGAACAAACGACCCAACACCCGTGCG3′) was used to amplify a 328-bp DNA fragment from mutant LRRK2 allele. The amplification products were separated on a 1% agarose gel.
Southern and Northern blot analysis.
Southern blot analysis was performed by using DNA extracted from liver after proteinase K digestion. DNA (20 μg) was digested with SphI, separated on a 1% agarose gel, denatured, and neutralized by 0.5 m NaOH/1.5 m NaCl and 1 m Tris-HCl (pH 8.0)/1.5 m NaCl, respectively, and transferred onto a nylon membrane (Nytran SuperCharge, Schleicher & Schuell) in the presence of 10 × SSC. A region downstream of exon 40 of the mouse LRRK2 gene was amplified by PCR using a pair of primers (sense: TGCAGACAGGACATCACACCGTTT, antisense: AGGCTCAAACCCGGACATGTGA, see Fig. 1A, S-probe) from the target construct. This fragment was labeled in the presence of [32P]-dATP and used as probe for hybridization at 65°C for 16 h. After hybridization, the membrane was washed in 2 × SSC/0.1% SDS (5 min, room temperature) and 0.2 × SSC/0.1% SDS (2 × 10 min, 68°C), and the result was visualized using a Phospho-Imager system (Cyclone, Packard). For Northern blot analysis, total RNA was extracted from mouse brain using the acid-phenol-guanidine isothiocyanate method (TRIzol, Invitrogen). Total RNA (5 μg) was applied to a formaldehyde-denatured agarose gel and transferred onto a nylon membrane. A region of mouse LRRK2 cDNA (1730 to 2310 nt) was amplified by PCR using a plasmid containing the full-length mouse LRRK2 cDNA as template (kindly provided by Dr. Bettina Lorenz-Deperieux, Institut f̈ur Humangenetik, Neuherberg, Germany). The resultant fragment was labeled in the presence of [32P]-dATP and used to probe the membrane, and the result was visualized using a phospho-imager system.
Histopathology and clinical pathology.
For histopathology assessment, tissues were fixed in 10% neutral buffered formalin, processed and embedded in paraffin. Five micron sections were mounted on glass slides and stained with hematoxylin and eosin for microscopic examination. Automated CBCs (HemaVet 950FS; Drew Scientific) and serum chemistry panels (Vet Ace; Diagnostic Technologies) were performed at an in-house core laboratory according to manufacturer's instructions.
Immunohistochemistry and stereological cell counts.
Mice were perfused with ice-cold PBS and 4% paraformaldehyde/PBS, pH 7.4. Brains were removed and postfixed 4 h in the same fixative. After cryoprotection in 20% and 30% sucrose/PBS, pH 7.4, brains were frozen, and serial coronal sections (30 μm sections) were cut with a microtome. Free-floating sections were blocked with 4% goat serum/PBS plus 0.2% Triton X-100 and incubated with primary antibodies against tyrosine hydroxylase (TH) (rabbit polyclonal; Novus Biologicals) or α-synuclein (mouse monoclonal; BD Transduction Laboratories), followed by incubation with biotin-conjugated anti-rabbit or anti-mouse (goat polyclonal; Jackson ImmunoResearch), ABC reagents (Vector Laboratories), and SigmaFast DAB Peroxidase Substrate (Sigma). The sections stained for TH were then counterstained with Nissl (0.09% thionin) for 6 min, followed by destaining with 1% formalin acetic acid for 40 s. Cell counts of TH- and Nissl-positive neurons of the SNpc were counted using the optical fractionator STEREO INVESTIGATOR software (MicroBrightField).
HPLC analysis of biogenic amines.
Biogenic amines in discrete brain regions were measured by sonicating the respective tissues in chilled 0.1 m perchloric acid (100 μl/mg). After centrifugation (15,000 × g, 20 min, 4°C), 10 μl of the supernatant was isocratically eluted through a 80 × 4.6 mm C18 column (ESA) with a mobile phase containing 75 mm NaH2PO4, 1.5 mm octane sulfonic acid, 5% acetonitrile, pH 3.0, and detected by a two-channel Coulochem II electrochemical detector (ESA) kept at a potential of 0.7 mv and flow rate of 1 ml/min. Data were collected and processed using external standards for respective amines on a EZChrome Elite Client Workstation (ESA). Data were normalized by protein concentration or tissue weight.
MPTP injections.
All procedures were approved by and conformed to the guidelines of the Institutional Animal Care Committee of Johns Hopkins University. LRRK2 KO animals were backcrossed to C57B6 mice for 10 generations and LRRK2 KO and WT littermates from F9 and F10 heterozygote crosses were used for the MPTP studies. Animals received four intraperitoneal injections of MPTP-HCl (20 mg/kg free base, Sigma-Aldrich) or saline control every 2 h. Animals were decapitated or perfused 7 d after the last MPTP injection and brains were removed for analysis.
Immunoblot analysis.
Mouse brain tissue from 10-week old females was homogenized in 4 × volumes of ice-cold PBS containing 0.1% Triton X-100 with protease inhibitors [0.5 mm AEBSF (Sigma), 2 μg/ml Aprotinin (Roche), 5 μg/ml Bestatin (Sigma), 5 μg/ml E-64 (Roche), 5 μg/ml Leupeptin (Roche), and 1 μg/ml Pepstatin (Sigma)]. The tissue homogenate was centrifuged at 37,000 × g at 4°C for 20 min. The supernatant was used for the Western blot. Protein concentration was measured by BCA protein assay. Brain lysates (40 μg) were electrophoresed in 7% SDS-PAGE gels and transferred to nitrocellulose membranes. Membranes were incubated with primary antibody diluted in blocking solution (5% nonfat milk overnight at 4°C, and for 1 h with a secondary antibody labeled with horseradish peroxidase (1:5000–20,000). The signal was detected using chemiluminescent substrates. Primary polyclonal antibodies 5514, which recognizes amino acids 2500–2515 (Biskup et al., 2006), 1182E which recognizes amino acids 841–960 (Waxman et al., 2009), 5824 which recognizes amino acids 1–500, and monoclonal antibody 6.1, which recognizes amino acids 1–500 (X. Li, V.L. Dawson and T.M. Dawson, unpublished observation) were used for immunoblot against LRRK2. After extensive washing, the blot was incubated with the secondary anti-Rabbit-HRP antibody (GE Healthcare) (for antibody 5514, 1182, and 5824) or the secondary anti-Mouse-HRP antibody (GE Healthcare) (for antibody 6.1) at room temperature for 1 h using a 1:10,000 dilution. The immunoblot signal was detected by SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific).
Statistical analysis.
Significance was determined by one- or two-way ANOVA followed by the Student–Newman–Keuls test, or a two-tailed nonpaired Student's t test. Significance was set at p ≤ 0.05.
Results
Generation of LRRK2 KO mice
LRRK2 was disrupted by deletion of exon 39 and 40, which contains the kinase domain, and by the introduction of a stop codon and a neo selection cassette (Fig. 1A). Heterozygous male and female mice were bred to generate homozygous mice and WT littermates. Southern blot and PCR analysis (Fig. 1B,C) confirms disruption of LRRK2. Northern blot analysis of LRRK2 KO mouse brain shows the lack of endogenous LRRK2 transcript (Fig. 1D). Immunoblot analyses using four different antibodies spanning different regions of the LRRK2 protein, including C-terminal and N-terminal antibodies, indicate that there is a reduction of LRRK2 protein in heterozygous mice and no LRRK2 protein expression in LRRK2 KO mice (Fig. 1E).
Histopathology and clinical pathology
The offspring of the heterozygous crosses gave rise to all three genotypes and 7–10% of the homozygous mice are not viable (Table 1). The oldest homozygous mice currently have lived to 2 years of age. No general abnormalities are noted in newborn through young adult LRRK2 KO mice and they appear normal in their growth rates and fertility. A cohort that included 26 LRRK2 KO and 19 WT mice was aged and analyzed at 16–20 months of age. The brain and nonbrain tissues were analyzed separately. Alopecia of the skin is commonly observed in the LRRK2 KO mice (data not shown). The alopecia is usually centered on the muzzle and head and is interpreted as being due to excessive grooming. Splenomegaly is also common in KO mice. However, even though the average spleen size of KO mice was larger than in WT mice (KO = 263 ± 65 mg, n = 30; WT = 200 ± 49 mg, n = 19), the difference is not significantly different as some WT mice of the same age had splenomegaly and some KO mice had spleens of normal size. Although there was some mild individual variation, no consistent alterations in the automated CBC, serum chemistry, or histopathology screens distinguish LRRK2 KO from WT mice (data not shown).
Table 1.
Reduced numbers of knockout animals
| Background | Total | +/+ | +/− | −/− | Percentage −/− |
|---|---|---|---|---|---|
| Mixed | 300 | 72 (24%) | 174 (58%) | 54 (18%) | 18% |
| 129 | 140 | 35 (25%) | 84 (60%) | 21 (15%) | 15% |
Nigrostriatal dopaminergic system in LRRK2 KO mice
LRRK2 KO animals show no differences in brain development and gross anatomy. Gross histological examination of the brain reveals normal morphology in LRRK2 KO mice of different ages (data not shown). Total content of DA, serotonin and its metabolites in different brain regions were analyzed in KO and WT age-matched littermate mice. No significant changes in the levels of neurotransmitters are observed (Table 2). We examined striatal TH immunoreactivity in LRRK2 KO and WT age-matched littermate mice. There is no difference in the dopaminergic innervation of the striatum in 2–3-month- (data no shown) and 18–24-month-old mice (Fig. 2A,B). The number of dopaminergic neurons in the substantia nigra pars compacta (SNpc) was assessed by TH immunoreactivity (Fig. 2C,D). We performed stereological quantification of TH- and Nissl-positive neurons in SNpc (Fig. 2E). We did not find significant differences in the number of dopaminergic neurons in SNpc of 2–3 month and 18–22 month LRRK2 KO mice compared with WT mice (Fig. 2E). In addition, we determined the concentration of DA and its metabolites in the striatum and we observed no change in the levels of DA and its metabolites, 3,4-dihydroxyphenylactic acid (DOPAC) and homovanillic acid (HVA) in KO mice compared with WT age-matched littermate (Fig. 2F,G).
Table 2.
Concentrations (ng/mg of tissue) of dopamine, serotonin, and their metabolites determined by HPLC in 2–3 month LRRK2 WT and KO mice (n = 6)
| Cortex |
Cerebellum |
Brainstem |
||||
|---|---|---|---|---|---|---|
| WT | KO | WT | KO | WT | KO | |
| DA | 1.22 ± 0.44 | 1.18 ± 0.31 | — | — | — | — |
| HVA | 0.21 ± 0.05 | 0.16 ± 0.03 | — | — | — | — |
| DOPAC | 0.34 ± 0.05 | 0.31 ± 0.06 | — | — | — | — |
| 5-HT | 0.34 ± 0.03 | 0.30 ± 0.01 | 0.14 ± 0.09 | 0.09 ± 0.04 | 0.86 ± 0.16 | 0.78 ± 0.11 |
| 5HIAA | 0.22 ± 0.04 | 0.23 ± 0.04 | 0.20 ± 0.04 | 0.14 ± 0.05 | 0.80 ± 0.21 | 0.69 ± 0.08 |
| NE | 0.24 ± 0.04 | 0.22 ± 0.03 | 0.44 ± 0.12 | 0.35 ± 0.10 | 0.8 ± 0.13 | 0.69 ± 0.07 |
Figure 2.

Characterization of the nigrostriatal dopaminergic system in LRRK2 KO mice. A–D, Immunohistochemical staining for TH and Nissl was performed on coronal brain sections from LRRK2 WT (A, C) and KO (B, D) mice. Striatum (A, B) and ventral midbrain (C, D); scale bars, 500 μm. E, Stereological analysis of the number of TH-positive and Nissl-positive neurons in substantia nigra of 2–3 month and 18–22 month mice (n = 5). F, G, Striatal concentrations of dopamine and its metabolites were determined by HPLC with electrochemical detection in young (F) and aged (G) LRRK2 WT and KO mice (n = 6). The data are the mean ± SEM.
LRKK2 KO susceptibility to MPTP-induced nigrostriatal neuronal death
Since LRRK2 is localized, in part, to mitochondria (Biskup et al., 2006), and studies suggest that it may play an important role in mitochondrial function (Iaccarino et al., 2007; Ng et al., 2009; Saha et al., 2009), the vulnerability of nigral dopaminergic neurons to the neurotoxin, MPTP, was assessed (Fig. 3). Two- to 3-month-old WT and KO mice were injected with MPTP (20 mg/kg every 2 h, four times) or saline. The effects of MPTP 7 d after the MPTP treatment were examined histologically. Immunostaining in LRRK2 KO and WT SNpc sections showed decreased TH-positive neuron numbers (Fig. 3B,D) compared with saline-treated controls (Fig. 3A,C). Quantitation of TH- and Nissl-positive neuron numbers in MPTP-treated LRRK2 KO and WT mice shows a significant loss of dopaminergic neurons in mice treated with MPTP (Fig. 3E) and striatal dopaminergic nerve terminals (data not shown), but unexpectedly no significant differences are observed between KO and WT mice. HPLC analysis reveals a significant reduction in striatal dopamine and its metabolites (DOPAC and HVA) in WT and LRRK2 KO mice injected with MPTP compared to saline controls (Fig. 3F). However, no statistical differences in the levels of dopamine and its metabolites after MPTP administration are observed between WT and KO mice (Fig. 3F). Since LRRK2 and α-synuclein are found in a percentage of Lewy bodies, sections were immunostained for α-synuclein in KO and WT mice treated with saline or MPTP. No difference in staining was observed between groups (data not shown).
Figure 3.
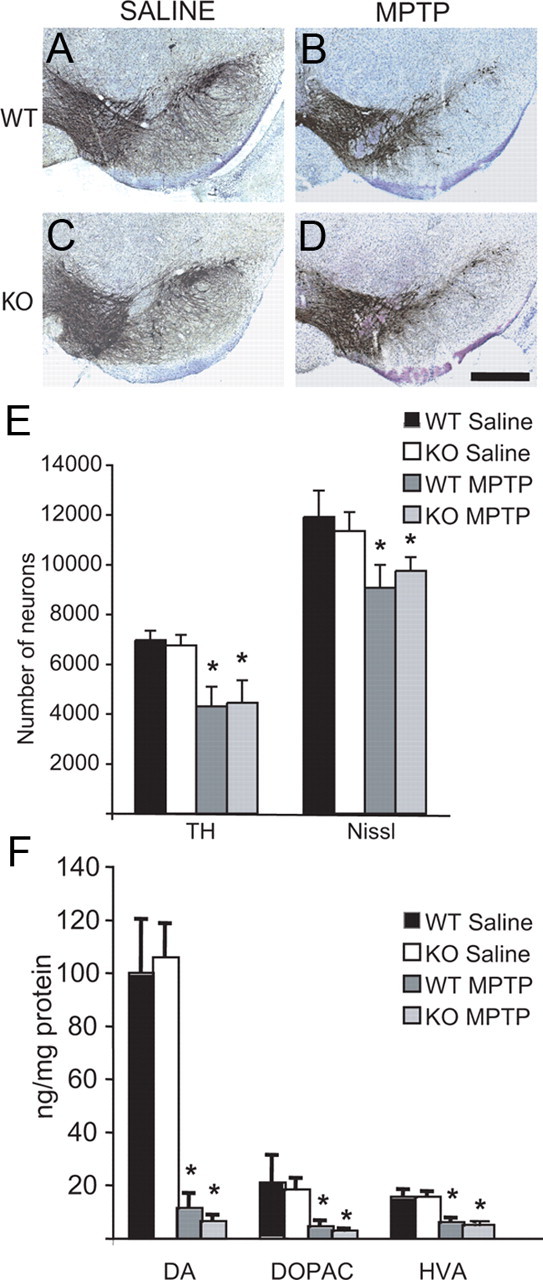
Evaluation of the effect of MPTP in LRRK2 KO mice. Stereological counting of dopaminergic neurons in the substantia nigra of mice treated with MPTP. TH immuno-staining in the substantia nigra of WT (A, B) and LRRK2 KO (C, D) mice treated (B, D) or nontreated (A, C) with MPTP; scale bars, 500 μm. E, Number of TH- and Nissl-positive neurons in the substantia nigra of WT and LRRK2 KO mice treated or nontreated (saline) with MPTP, determined by stereological counting. The data are the mean ± SEM (n = 6). F, Levels of DA and its metabolites (DOPAC and HVA) in the striatum of WT and LRRK2 KO mice treated or nontreated (saline) with MPTP. The data are the mean ± SEM (n = 7). *p ≤ 0.01.
Discussion
In the present study, we show for the first time that ablation of LRRK2 gene in mice does not lead to the loss of dopaminergic neurons. Moreover, LRRK2 KO mice are viable, and live a normal life span with clinical and pathologic feature indistinguishable from WT littermate mice. In addition, we provide evidence that the LRRK2 protein is dispensable for the development and maintenance of the nigrostriatal pathway. Our results agree with a previous report, which observed that the Drosophila ortholog of LRRK2 is dispensable for the survival of dopaminergic neurons (Wang et al., 2008). A different study performed in Drosophila showed opposite results, in which the loss of the LRRK2 ortholog induced degeneration of DA neurons (Lee et al., 2007). What factors account for the difference in the two Drosophila studies is not clear. However, in mice LRRK2 does not appear to be essential for the development and maintenance of DA neurons. It is somewhat surprising that Mendelian ratios obtained from offspring of heterozygote matings suggest that ablation of LRRK2 leads to a 7–10% loss of homozygotes during embryonic and perinatal development in mice. The reason for this is not clear at present.
The neurotoxin MPTP is an inhibitor of the mitochondrial complex I, and several lines of evidence have linked LRRK2 with the mitochondrial function (Biskup et al., 2006; Iaccarino et al., 2007; Ng et al., 2009; Saha et al., 2009). Surprisingly, when we challenged the nigrostriatal pathway with the neurotoxin MPTP, we did not find any statistically significant differences in the number of neurons of the SNpc or the DA content between WT and LRRK2 KO mice. These results could point to the involvement of LRRK2 in a pathway independent of the mitochondria. LRRK2 has been associated with the extrinsic cell death pathway that it is mitochondrial independent (Ho et al., 2009) and with Wnt signaling pathways (Sancho et al., 2009). Interestingly, a recent report in lrk-1 ablated C. elegans reported an enhanced vulnerability of dopaminergic neurons to rotenone, while WT LRRK2 overexpressing animals showed protection against this mitochondrial toxin (Saha et al., 2009). In addition, transgenic expression of mutant LRRK2 in Drosophila, but not WT LRRK2 renders dopaminergic neurons more vulnerable to rotenone (Ng et al., 2009). Thus, WT LRRK2 seems to play little if any role as a determinant of susceptibility of mitochondria to mitochondrial toxins, whereas mutant LRRK2 through as of yet unidentified gain of function mechanism could increase the vulnerability of mitochondria to mitochondrial toxins. Further study is required to understand the relationship of mutant LRRK2 to mitochondrial dysfunction, which will contribute to elucidating the role of LRRK2 in the pathogenesis of PD.
Footnotes
This work was supported by grants from National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke (NINDS) (P50 NS38377, NS054207). M.F.B. was supported by the Michael J. Fox Foundation, the Parkinson's Disease Foundation, and Department of Defense, and B.T. by NIH/NINDS Grant NS060885. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases. We thank Drs. Darren Moore and Andrew West for assistance with the antibody generation and initial characterization of the mice. E.A.-M. and R.M. had Postdoctoral Fellowship from Ministerio de Educación y Ciencia, Spain.
References
- Biskup S, West AB. Zeroing in on LRRK2-linked pathogenic mechanisms in Parkinson's disease. Biochim Biophys Acta. 2009;1792:625–633. doi: 10.1016/j.bbadis.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biskup S, Moore DJ, Celsi F, Higashi S, West AB, Andrabi SA, Kurkinen K, Yu SW, Savitt JM, Waldvogel HJ, Faull RL, Emson PC, Torp R, Ottersen OP, Dawson TM, Dawson VL. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol. 2006;60:557–569. doi: 10.1002/ana.21019. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson's disease. Science. 2003;302:819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- Gasser T. Update on the genetics of Parkinson's disease. Mov Disord. 2007;22(Suppl 17):S343–S350. doi: 10.1002/mds.21676. [DOI] [PubMed] [Google Scholar]
- Greggio E, Zambrano I, Kaganovich A, Beilina A, Taymans JM, Daniëls V, Lewis P, Jain S, Ding J, Syed A, Thomas KJ, Baekelandt V, Cookson MR. The Parkinson disease-associated leucine-rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J Biol Chem. 2008;283:16906–16914. doi: 10.1074/jbc.M708718200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho CC, Rideout HJ, Ribe E, Troy CM, Dauer WT. The Parkinson disease protein leucine-rich repeat kinase 2 transduces death signals via Fas-associated protein with death domain and caspase-8 in a cellular model of neurodegeneration. J Neurosci. 2009;29:1011–1016. doi: 10.1523/JNEUROSCI.5175-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaccarino C, Crosio C, Vitale C, Sanna G, Carrì MT, Barone P. Apoptotic mechanisms in mutant LRRK2-mediated cell death. Hum Mol Genet. 2007;16:1319–1326. doi: 10.1093/hmg/ddm080. [DOI] [PubMed] [Google Scholar]
- Lee SB, Kim W, Lee S, Chung J. Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem Biophys Res Commun. 2007;358:534–539. doi: 10.1016/j.bbrc.2007.04.156. [DOI] [PubMed] [Google Scholar]
- Lees AJ, Hardy J, Revesz T. Parkinson's disease. Lancet. 2009;373:2055–2066. doi: 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- Liou AK, Leak RK, Li L, Zigmond MJ. Wild-type LRRK2 but not its mutant attenuates stress-induced cell death via ERK pathway. Neurobiol Dis. 2008;32:116–124. doi: 10.1016/j.nbd.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson's disease. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- Ng CH, Mok SZ, Koh C, Ouyang X, Fivaz ML, Tan EK, Dawson VL, Dawson TM, Yu F, Lim KL. Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. J Neurosci. 2009;29:11257–11262. doi: 10.1523/JNEUROSCI.2375-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha S, Guillily MD, Ferree A, Lanceta J, Chan D, Ghosh J, Hsu CH, Segal L, Raghavan K, Matsumoto K, Hisamoto N, Kuwahara T, Iwatsubo T, Moore L, Goldstein L, Cookson M, Wolozin B. LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans. J Neurosci. 2009;29:9210–9218. doi: 10.1523/JNEUROSCI.2281-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho RM, Law BM, Harvey K. Mutations in the LRRK2 Roc-COR tandem domain link Parkinson's disease to Wnt signalling pathways. Hum Mol Genet. 2009;18:3955–3968. doi: 10.1093/hmg/ddp337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Moore DJ, Liang Y, West AB, Dawson VL, Dawson TM, Ross CA. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc Natl Acad Sci U S A. 2005;102:18676–18681. doi: 10.1073/pnas.0508052102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila M, Przedborski S. Genetic clues to the pathogenesis of Parkinson's disease. Nat Med. 2004;10(Suppl):S58–S62. doi: 10.1038/nm1068. [DOI] [PubMed] [Google Scholar]
- Wang D, Tang B, Zhao G, Pan Q, Xia K, Bodmer R, Zhang Z. Dispensable role of Drosophila ortholog of LRRK2 kinase activity in survival of dopaminergic neurons. Mol Neurodegener. 2008;3:3. doi: 10.1186/1750-1326-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman EA, Covy JP, Bukh I, Li X, Dawson TM, Giasson BI. Leucine-rich repeat kinase 2 expression leads to aggresome formation that is not associated with alpha-synuclein inclusions. J Neuropathol Exp Neurol. 2009;68:785–796. doi: 10.1097/NEN.0b013e3181aaf4fd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet. 2007;16:223–232. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]