

Abstract
The early endocytic patch protein Sla2 is important for morphogenesis and growth rates in Saccharomyces cerevisiae and Candida albicans, but the mechanism that connects these processes is not clear. Here we report that growth defects in cells lacking CaSLA2 or ScSLA2 are associated with a cell cycle delay that is influenced by Swe1, a morphogenesis checkpoint kinase. To establish how Swe1 monitors Sla2 function, we compared actin organization and cell cycle dynamics in strains lacking other components of early endocytic patches (Sla1 and Abp1) with those in strains lacking Sla2. Only sla2 strains had defects in actin cables, a known trigger of the morphogenesis checkpoint, yet all three strains exhibited Swe1-dependent phenotypes. Thus, Swe1 appears to monitor actin patch in addition to actin cable function. Furthermore, Swe1 contributed to virulence in a mouse model of disseminated candidiasis, implying a role for the morphogenesis checkpoint during the pathogenesis of C. albicans infections.
INTRODUCTION
Candida albicans is a multimorphic human pathogen that forms filamentous true hyphae as well as yeast cells and pseudohyphae. The ability to grow with yeast and hyphal morphologies contributes to virulence (Mitchell, 1998), and the ability to form hyphae appears necessary for maximal virulence (Banerjee et al., 2008; Carlisle et al., 2009), although the exact role of morphogenesis in virulence remains to be elucidated (Gow et al., 2002). Morphogenesis is tightly coupled to cell cycle progression (Berman, 2006). In yeast and subapical hyphal cells, a G1 delay occurs prior to daughter cell formation (Kron & Gow, 1995). In contrast, in pseudohyphal cells, no G1 delay occurs, resulting in synchronous cell cycles and symmetrical cell sizes in mother and daughter cells (Barelle et al., 2003; Kron & Gow, 1995).
Polarization of the actin cytoskeleton is required for morphogenesis of all C. albicans cell types (Akashi et al., 1994; Anderson & Soll, 1986). Just before evagination of either a bud or hypha, actin patches cluster at the site of evagination and then remain polarized within the daughter cell. With continued yeast bud growth, actin patches are redistributed isotropically throughout the bud cortex, whereas in hyphal daughters the majority of actin patches remain polarized to the hyphal tip. Cortical actin patches are concentrated at sites of polarized growth, where they appear to function in endocytosis (Moseley & Goode, 2006), acting with actin cables to recycle cortically localized polarity proteins before they can diffuse too far from the cell front and ensuring the maintenance of their polarized distrubution at daughter cell tips (Irazoqui et al., 2005). The actin cytoskeleton also includes actin cables, long bundles of actin filaments that converge at the tips of yeast and hyphal daughter cells during polarized growth. Actin cables serve as polarized tracks for the secretion of cargos needed to maintain polarity and build the daughter cell. Thus, polarized growth and cell shape are dependent upon the interplay between secretion and endocytosis, achieved by the activities of actin cables and patches, respectively.
In Saccharomyces cerevisiae, the integrity of the actin cytoskeleton is monitored during bud morphogenesis, such that defects cause delay of the nuclear cell cycle (McMillan et al., 1998). This morphogenesis checkpoint response, mediated by the Swe1 kinase, results in inhibitory phosphorylation of the cyclin-dependent kinase (Clb2/Cdc28) (Lew & Reed, 1995). Although the precise molecular defect(s) in the actin cytoskeleton that triggers Swe1 is not known, only actin cable mutants have been reported to activate the morphogenesis checkpoint (McMillan et al., 1998), suggesting that the function of actin cables is the cytoskeletal feature that is being monitored by Swe1. Swe1 also mediates a morphogenesis checkpoint in response to septin ring perturbations in C. albicans and S. cerevisiae (Barral et al., 1999; Longtine et al., 2000; Shulewitz et al., 1999; Wightman et al., 2004), presumably by suppressing the targeting, and subsequent degradation, of Swe1 that normally occurs at the mother–bud neck (Longtine et al., 2000).
The endocytic patch component Sla2 has been studied extensively in S. cerevisiae, where it is important for endocytosis by linking actin to clathrin, and is necessary for polarized growth (Baggett et al., 2003; Bidlingmaier & Snyder, 2002; Gourlay et al., 2003). In C. albicans, independent isolates containing a mutant allele lacking the C-terminal one-third of Sla2 were unable to form true hyphae in response to serum and other stimuli (Asleson et al., 2001). Consistent with this, a transposon-mediated disruption allele of Sla2 did not form hyphae in response to alkaline pH conditions (Davis et al., 2002). Polarization defects of the sla2 mutant strain were associated with delocalization of Rvs167, Gsc1 and membrane sterols, which normally localize to hyphal tips (Oberholzer et al., 2006). In addition, Sla2 is important for endocytosis: sla2 mutant strains were unable to internalize and utilize haemoglobin via a receptor (Rbt5)-mediated endocytic mechanism (Weissman et al., 2008) and have abnormal deposition of chitin and intolerance to cell wall stress (Oberholzer et al., 2006), possibly due to the lack of endocytic recycling/remodelling of the cell wall. Thus, similar to ScSla2, C. albicans Sla2 has roles in polarized growth and endocytosis.
ScSla2 is also important for normal rates of cell growth, especially at temperatures above 30 °C (Holtzman et al., 1993). Importantly for C. albicans, elevated temperature is a feature of the host niche and an inducer of hyphal morphogenesis. In this study, we investigated the role of Sla2 in C. albicans growth and found an association between endocytosis and the morphogenesis checkpoint kinase Swe1. Furthermore, Swe1 was important for C. albicans virulence in a mouse model of systemic candidiasis, consistent with the ideas that cells encounter cell cycle delays during growth in the host and that activation of morphogenesis/cycle checkpoints plays a role in the pathogenesis of C. albicans infections.
METHODS
Strains, media and growth conditions.
The C. albicans and S. cerevisiae strains used are listed in Table 1. Unless noted otherwise, strains were grown at 30 °C in rich medium (YPD) or synthetic complete (SDC) medium lacking uridine, histidine or arginine for selection during transformation (Sherman, 1991). Otherwise, all yeast media included uridine at 80 μg ml−1. YPAD medium is YPD with the addition of adenine (50 mg l−1). Hyphae were induced by growing cells in the presence of 10 % bovine serum at 37 °C. Pseudohyphae were enriched by growing cells at 37 °C without serum. To enrich for semi-synchronous cell growth in some experiments, yeast cells were grown to stationary phase prior to subculture.
Table 1.
C. albicans and S. cerevisiae strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| C. albicans | ||
| CAF2 | URA3/ura3Δ : : imm434 | Fonzi & Irwin (1993) |
| CAI4 | ura3Δ : : imm434/ura3Δ : : imm434 | Fonzi & Irwin (1993) |
| BWP17 | ura3Δ : : imm434/ura3Δ : : imm434 his1 : : hisG/his1 : : hisG arg4 : : hisG/arg4 : : hisG | Wilson et al. (1999) |
| YJB3400 | CAI4 SLA2/sla2-1 : : hisG-URA3-hisG | Asleson et al. (2001) |
| YJB3401 | CAI4 SLA2/sla2-1 : : hisG | Asleson et al. (2001) |
| YJB3402, 3612 | CAI4 sla2-1 : : hisG/sla2-1 : : hisG-URA3-hisG | Asleson et al. (2001) |
| YJB3403 | CAI4 sla2-1 : : hisG/sla2-1 : : hisG | Asleson et al. (2001) |
| YJB5929, 5930 | CAI4 URA3:PMET3-SLA2 : : sla2-1/sla2-1 : : hisG | This study |
| YCG6655, 6658, 6659, 4994 | BWP17 swe1 : : HIS1/swe1 : : ARG4 | This study |
| YCG6792 | BWP17 swe1 : : HIS1/swe1 : : ARG4 arg4 : : hisG/ARG4-URA3 : : arg4:hisG | This study |
| YJB7435 | BWP17 SLA2/sla2Δ : : ARG4 | This study |
| YJB7662, 7663 | BWP17 URA3:PMET3-SLA2 : : sla2Δ/sla2Δ : : ARG4 | This study |
| YJB8658 | BWP17 URA3:PMET3-SLA2 : : sla2Δ/sla2Δ : : ARG4 CDC3/CDC3 : : GFP:HIS1 | This study |
| YJB7674, 7675 | BWP17 sla2Δ : : ARG4/sla2Δ : : HIS1 | This study |
| YCG7853, 7855, 7857 | BWP17 URA3:SWE1 : : swe1 : : HIS1/swe1 : : ARG4 | This study |
| YJB7904, 7905 | BWP17 swe1 : : HIS1/swe1 : : ARG4 sla2Δ : : dpl200/sla2Δ : : dpl200:URA3 | This study |
| YCG8190, 8193, 8195 | BWP17 URA3:swe1Δ : : swe1 : : HIS1/swe1 : : ARG4 | This study |
| YJB8417, 8418 | CAI4 SLA2/sla2-1-HA:URA3 | This study |
| YJB8457, 8458 | CAI4 SLA2-HA:URA3/sla2-1 | This study |
| YJB9105 | BWP17 sla2Δ : : ARG4/sla2Δ : : HIS1 arg4 : : hisG/ARG4-URA3 : : arg4 : : hisG | This study |
| YJB9955 (6284) | BWP17 HIS1 : : his1 : : hisG/his1 : : hisGARG4-URA3 : : arg4 : : hisG/arg4 : : hisG | Bensen et al. (2002) |
| S. cerevisiae | ||
| YJB2709 | MATa abp1 : : LEU2 ura3-52 lys2-801 leu2-3,112 | D. Drubin, University of California, Berkeley |
| YJB2713 | MATαsla2 : : URA3 ura3-52 leu2-3,112 | D. Drubin |
| YJB2756 | MATa ura3-52 leu2-Δ2 his3-Δ200 trp1-Δ63 lys2-801 | J. Pringle, Stanford University |
| YJB3046 | MATa ura3-52 leu2-3,112 | D. Drubin |
| YJB3047 | MATa sla1-Δ1 : : URA3 ura3-52 leu2-3,112 | D. Drubin |
| YJB4786 | MATa sla2 : : HIS3 ura3-52 leu2-Δ2 his3-Δ200 trp1-Δ63 lys2-801 | This study |
| YJB4788 | MATa swe1 : : LEU2 ura3-52 leu2-Δ2 his3-Δ200 trp1-Δ63 lys2-801 | J. Pringle |
| YJB2759 | MATαswe1 : : LEU2 ura3-52 leu2-Δ2 his3-Δ200 trp1-Δ63 lys2-801 swe1 : : LEU2 | J. Pringle |
| YJB4791 | MATa swe1 : : LEU2 sla2 : : HIS3 ura3-52 leu2-Δ2 his3-Δ200 trp1-Δ63 lys2-801 | This study |
| YJB11509* | MAT swe1 : : LEU2 sla1 : : URA3 trp1-Δ63 | This study |
| YJB11511† | MAT swe1 : : LEU2 abp1 : : LEU2 ura3-52 lys2-801 | This study |
*Derived from cross between YJB3047 and YJB2759.
†Derived from cross between YJB2709 and YJB2759.
Strain constructions.
The SLA2 and SWE1 ORFs were sequentially deleted from C. albicans strain BWP17 by PCR-mediated gene disruption (Wilson et al., 1999, 2000). Deletion cassettes were amplified from plasmids pRS-ARGΔSpeI, pGEM-HIS1, pGEM-URA3 or pDDB57 using primers 671F (5′-gcccaatgagtcgtgctgaagttgatttacaaacaagtgttaaaaaagcttgtaatgctgatgaagtaccgttttcccagtcacgacgtt-3′; plain text refers to gene-specific sequence and bold text refers to plasmid-specific sequence throughout) and 672R (5′-caactaagtattcaatcaaaaacagtttctggagttttatattcttgttttctaatttctgcaagtcttttgtggaattgtgagcggata-3′) for SLA2 and primers 523F (5′- ggattcaaacccgtgtcaagacgtatcgggtgataccagtagcacaccaatggccgttttcccagtcacgacgtt-3′) and 524R (5′-tataatatcgaagctatcgacttttttggatttcaagatattcaaagttggggtgtggaattgtgagcggata-3′) for SWE1. Strains YJB7662 and YJB5929, each containing a single allele of SLA2 regulated by the MET3 promoter (Care et al., 1999), were generated by digesting plasmid pMG1724 with BglII and transforming the SLA2/sla2Δ heterozygotes YJB7435 and YJB3401, respectively. Plasmid pMG1724 was constructed by digesting pYES-Sla2 (Asleson et al., 2001) with BamHI and NsiI. The desired 930 bp fragment of the SLA2 ORF was subcloned into pCaDis (Care et al., 1999) digested with BamHI and PstI to generate pMG1724. To reintegrate SWE1 into the swe1/swe1 strains, SWE1 (and 615 bp of 5′-upstream promoter sequence) was amplified from BWP17 by PCR using primers 1217 (5′-aaggaaaaaagcggccgctgtctgactttattaattcaatcag-3′; NotI site, underlined) and 1218 (5′-cgcgacgcgtgaattattccttccaacttcaatag-3′; MluI site, underlined) and Elongase (BRL Life Technologies). This SWE1 product was cloned into NotI/MluI-digested pGEM-URA3 (Wilson et al., 1999) to generate pMG2050 (SWE1-URA3). This plasmid was digested with HindIII and transformed into independently constructed ura3/ura3 swe1/swe1 strains YCG6655, YCG6658 and YCG6659 to generate YCG7853, YCG7855 and YCG7857, respectively. To generate isogenic control swe1/swe1 strains containing URA3 at the SWE1 locus as in the above SWE1-reintegrant strains, pMG2050 was digested with PstI and religated to remove 1400 bp of the SWE1 ORF, resulting in pMG2084 (swe1Δ-URA3). This reintegrant plasmid was also digested with HindIII and transformed into YCG6655, YCG6658 and YCG6659 to generate strains YCG8190, YCG8193 and YCG8195, respectively. URA3-containing reintegrant strains were verified by PCR to confirm that SWE1-URA3 or swe1Δ-URA3 had reintegrated into a disrupted swe1 locus. C. albicans swe1/swe1 and sla2/sla2 strains were made prototrophic for URA3 by transforming them with NotI-linearized pRS-ARG-URA-BN (Davis et al., 2000). Strain constructions were initially verified by PCR using primers outside the regions of integration and then by Southern blotting, to verify that the deletion cassette had integrated into the predicted locus, and by Northern blotting, to verify the expression or loss of expression of the message of interest.
S. cerevisiae strains lacking SLA2 were constructed by transforming strains YJB2756 and YJB4788 with a sla2 : : HIS3 disruption plasmid (pWA9, provided by H. Riezman, University of Geneva, Switzerland), digested with EcoRI to target integration to the SLA2 locus. Transformants were selected on SDC medium lacking histidine and analysed by PCR to confirm that HIS3 had replaced SLA2.
C. albicans strains carrying haemagglutinin (HA) and fluorescent protein fusions were constructed by PCR-mediated gene modification (Gerami-Nejad et al., 2001). YJB8417, YJB8418, YJB8457 and YJB8458, each expressing an HA-tagged version of Sla2, were generated using oligonucleotides 1245F (5′-tactgaatttgctacaattttcaatagttttattgctgaagatagaaatgaagatgaatcgagtttttctggtggtggtcggatccccgggttaattaa-3′) and 1246R (5′-caatattttttgtcatccctttggcatttaatgttaaatcattaatacttgaagttaaagatgatgtacttctagaaggaccacctttgattg-3′) to amplify a SLA2-specific 3-HA cassette from plasmid pMG1874 (Gerami-Nejad et al., 2009). YJB6674, expressing a yellow fluorescent protein (YFP)-tagged version of Sla2, was generated using oligonucleotides 863F (5′-tttagcagctcgtaaaagacttgcagaaattagaaaacaagaatataaaactccagaaactgtttttgatggtggtggtggtggtggtggttctaaaggtgaagaattatt-3′) and 694R (5′-tcttcttttcttctttttttttatatatatatatatttgtttaacaacaaacactaaaactatataacatcta gaaggaccacctttgattg-3′) to amplify a SLA2-specific YFP cassette from plasmid pYFP-URA3 (Gerami-Nejad et al., 2001). YJB8658 expressing a green fluorescent protein (GFP)-tagged version of Cdc3 was generated using oligonucleotides 620F (5′-acaaaaattattaccacaagacccaccagcacaaccagctccacaaaagagtcgtaaaggatttttacgtggtggtggttctaaaggtgaagaattatt-3′) and 641R (5′-aaaacgctattagactaaaagataatacaaaatgggcatatatttgccaattaaaattaaaaaaatcaaaagtaggaattccggaatatttatgagaaac-3′) to amplify a CDC3-specific GFP cassette from plasmid pGFP-URA3 (Gerami-Nejad et al., 2001). Genetic fusions were verified by PCR, using primers outside the area of integration.
Western blot analysis.
Strains were cultured in YPAD for 6 h at 30 °C, harvested by centrifugation, resuspended in 1 ml buffer T (40 mM Tris/HCl pH 6.8, 5 % SDS, 8 M urea, 100 μM EDTA) with 1 ml of 500 μm glass beads, heated at 95 °C for 10 min, and lysed on a Mini Bead Beater (BioSpec Products) for 5 min at 4 °C. The resulting lysates were diluted 1 : 4 into electrophoresis sample buffer, heated for 5 min at 95 °C, and insoluble cell debris was removed by centrifugation for 6 min at 4 °C. Samples were separated on a 7.5 % acrylamide gel, and blotted onto PVDF membrane. The top half of the blot was incubated with a 1 : 10 000 dilution of HRP-conjugated mouse anti-HA antibody (Roche) for 1 h at room temperature. The bottom half was incubated with a 1 : 5000 dilution of rabbit anti-PSTAIRE (anti-Cdc28) primary antibody for 1 h at room temperature, and HRP-conjugated anti-rabbit secondary antibody for 1 h at room temperature (Santa Cruz Biotech). Both blot halves were developed using chemiluminescence (Pierce SuperSignal Fempto, Pierce Chemical). The blots were exposed on an Alpha Innotech ChemiImager 5500 digital imaging system, and chemiluminescent signals on the blots were quantified with ImageJ software (http://rsb.info.nih.gov/ij/).
Flow cytometry.
Strains were grown to exponential phase in YPD and fixed by adding 95 % ethanol to a final concentration of 70 %. Cells were washed twice with 50 mM Tris/HCl pH 8.0, 5 mM EDTA, and sonicated during the second wash for 5 s. Cell pellets were resuspended in 500 μl of a 2 mg ml−1 RNase A solution and incubated overnight at 37 °C. Cells were harvested by centrifugation, resuspended in 250 μl of 5 mg pepsin ml−1 in 55 mM HCl, incubated for 1 h at 37 °C, washed twice with 50 mM Tris/HCl pH 7.5, 5 mM EDTA, and resuspended in 250 μl of the same solution. Fifty-microlitre samples of the cell preparations were then stained with 500 μl of 1 μM SYBR green I (Invitrogen) in 50 mM Tris/HCl pH 7.5, 5 mM EDTA and incubated in the dark overnight at 4 °C. Cells were then analysed by flow cytometry using a FACSCalibur with HTS option (Becton Dickinson). FACS is optimized to remove cells from the analysis that have not separated. G1/G2 ratios (average of two independent experiments) were determined by measuring the height, in pixels, of the G1 peak and dividing by the height, in pixels, of the G2 peak.
Microscopy and cell staining.
All micrographs were obtained using a Nikon E600 microscope fitted for epifluorescence and differential interference contrast (DIC) microscopy. YFP and GFP filter cubes (#41028 and #41017, Chroma) were used for visualization of fluorescent fusion proteins. Images were captured with a CoolSnap HQ cooled CCD camera (Photometrics). Image processing and data collection from 16-bit images were accomplished with Metamorph imaging software, v6.2 (Universal Imaging Corp.). For figure assembly, images were converted to 8-bit and exported to Adobe Photoshop v7.0 and/or Microsoft Powerpoint.
To assess cell viability, strains were grown to stationary phase in SDC at 30 °C, diluted 1 : 20 in fresh medium and cultured at 30 °C, 37 °C or 42 °C for 4 h. Cells were collected by centrifugation and resuspended in fresh medium containing 10 mM FUN1 (Molecular Probes), incubated (in the dark at the same temperatures) for 1 h, and then analysed with the Endow GFP filter. Bright green staining with internal red dots or bars indicated live, metabolizing cells. To visualize nuclei, yeast cells were grown to exponential phase in YPD and fixed in 70 % ethanol for 10 min, washed twice in 1 ml 1× PBS (130 mM NaCl, 10 mM Na2HPO4, 10 mM NaH2PO4, pH 7.2), resuspended in 1× PBS containing 10 μg 4′,6-diamidino-2-phenylindole (DAPI) ml−1 (final concentration), and incubated for at least 30 min at room temperature, protected from light. Cells were washed twice with 1× PBS, and viewed with a UV-1A filter cube (Nikon). To detect actin, yeast cells were fixed in 3.7 % formaldehyde, stained with Alexa Fluor 568 phalloidin (Molecular Probes) as previously described (Hausauer et al., 2005) and viewed with a Texas red filter cube (Nikon).
Mouse virulence studies.
C. albicans strains were grown to stationary phase in 50 ml YPD. The doubling times of all strains used were identical. Strains were harvested, washed twice with sterile water, counted on a haemacytometer, and cell suspensions were prepared at 1×106 cells ml−1. A 100 μl aliquot of cell suspension was injected via the lateral tail vein into ICR male mice weighing 22–25 g at the time of infection. Each infection group consisted of at least five mice, and the experiment was performed six times. In all cases, the experimenter was blinded to the genotype of the tested strains. Survival curve statistics were calculated by the Kaplan–Meier method using the Mantel–Haenszel test (Mantel & Haenszel, 1959) with GraphPad Prism v3.00.
RESULTS
C. albicans sla2 disruption and deletion strains have similar morphogenetic defects
We previously generated two independent C. albicans strains (YJB3402 and YJB3612) homozygous for a truncated disruption allele (sla2-1) in which the URA-blaster cassette was inserted between codons 713 and 714 of the SLA2 gene in C. albicans strain CAI4. Since ScSLA2 is essential for viability in some S. cerevisiae strain backgrounds (Na et al., 1995), and since the sla2-1 disruption allele had the potential to produce a truncated protein with some residual function, we constructed independent strains lacking both copies of the entire SLA2 coding sequence (sla2Δ) as well as strains in which the expression of Sla2 could be regulated (PMET3-SLA2/sla2Δ) (see Methods for details).
Like the sla2-1/sla2-1 strains (Asleson et al., 2001), the sla2Δ/sla2Δ cells (and the PMET3-SLA2/sla2Δ cells grown in repressing conditions) were larger and rounder than the SLA2 parental strain (discussed in detail below), suggesting that the sla2-1 allele behaved like a null allele. To ask if the truncated Sla2-1 protein was present in cells, we compared the levels of C-terminally epitope-tagged versions of both wild-type Sla2 and Sla2-1 (Sla2-HA and Sla2-1-HA). The truncated Sla2-1-HA protein band migrated faster than the full-length Sla2-HA protein (Fig. 1), indicating that the disruption allele produces a truncated protein of the expected size (713 aa protein plus the HA tag). Furthermore, similar to results in S. cerevisiae, where a nonsense mutation in the C-terminal portion of Sla2 resulted in reduced protein amounts as compared to wild-type Sla2 (Baggett et al., 2003), levels of Sla2-1 were ∼20-fold lower than the levels of wild-type Sla2 (Fig. 1). Thus, sla2-1 is not a null allele; it produces a truncated protein that is present at very low levels, presumably because it is less stable than the full-length protein.
Fig. 1.
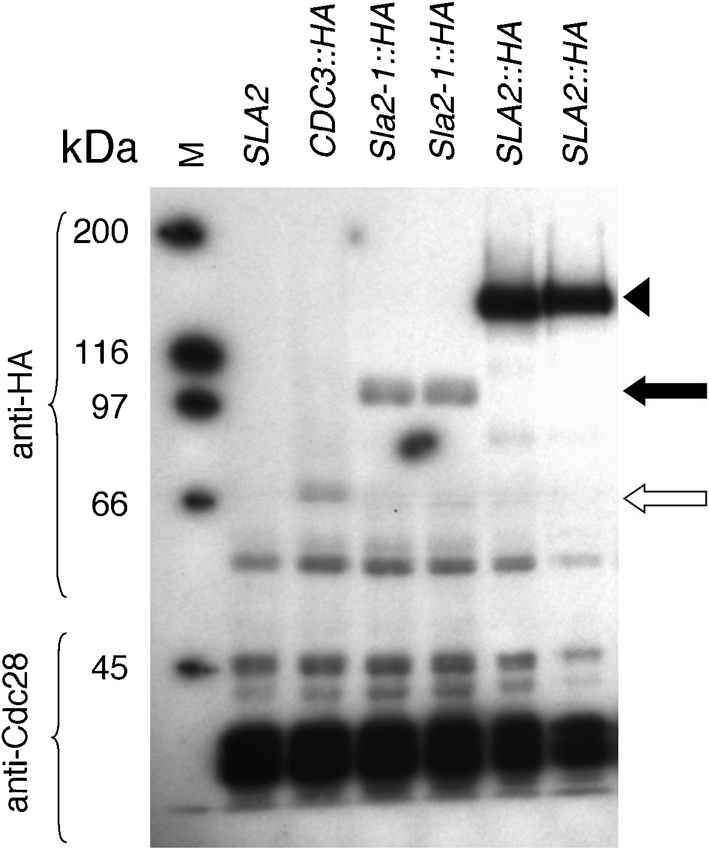
Sla2-1 is a truncated protein present at low levels: immunoblot analysis of cell lysates from strains expressing wild-type and mutant Sla2-HA detected with anti-HA antibody. Lanes listed left to right: M, molecular mass markers; SLA2 (BWP17); CDC3 : : HA (Gerami-Nejad et al., 2009) (white arrow); Sla2-1 : : HA (YJB8417 and YJB8418); SLA2 : : HA (YJB8457 and YJB8458). The upper panel was probed with anti-HA antibody; the lower panel was probed with anti-Cdc28 as a loading control. The wild-type Sla2-HA runs at approximately 126 kDa (arrowhead), and Sla2-1-HA (black arrow) at 87.9 kDa.
sla2 mutants exhibit a delay in nuclear cell cycle progression
The growth phenotypes of the C. albicans sla2Δ/sla2Δ strains were similar to the phenotypes of the sla2-1/sla2-1 strains. Both sla2 strains exhibited a slight reduction in growth at 30 °C and similar, more dramatic growth defects at 37 °C and 42 °C as compared to the wild-type SLA2 and heterozygous SLA2/sla2 strains (Fig. 2a). For example, at 37 °C, population doubling times were 205 min for wild-type SLA2 strains, 314 min for sla2-1 strains and 396 min for sla2Δ strains. Strains growing under conditions that repress PMET3-SLA2 were also temperature-sensitive at 37 °C and 42 °C, although they grew somewhat better than the sla2Δ mutants, possibly due to incomplete repression of the MET3 promoter (Fig. 2a). Importantly, at 37 °C and 42 °C both sla2 mutant strains exhibited a marked reduction in growth, yet cell viability was not decreased to the same extent (Fig. 2b). This suggests that at least a proportion of cells undergo a growth delay rather than cell death at elevated temperatures.
Fig. 2.

(a) sla2 mutants have temperature-sensitive growth defects. Strains streaked on SDC plates containing 10 mM methionine and 2 mM cysteine (to repress SLA2 expression from the MET3 promoter in the relevant strains; phenocopy of the sla2 null morphology was confirmed by microscopy of the cells) were incubated at the indicated temperatures for 6 days. Strain order was the same on all plates. +/+, CAF2; +/−, YJB3400; −1/−1, YJB3402; −1/−1, YJB3612; M/−, YJB5929; M/−, YJB5930; +/+, BWP17; +/Δ, YJB7435; Δ/Δ, YJB7674; Δ/Δ, YJB7675; M/Δ, YJB7662; M/Δ, YJB7663. Black diagonal lines separate strains constructed from strain BWP17 (left) from those constructed from strain CAI4 (right). (b) sla2 mutants remain viable at high temperatures. Exponential-phase cells were incubated with FUN1 for 1 h and metabolizing cells were counted (at least 200 cells per strain; see Methods). sla2-1/sla2-1, combined data for YJB3402 and YJB3612; sla2Δ/sla2Δ, combined data for YJB7674 and YJB7675; SLA2/sla2Δ, data for YJB7435.
To ask if loss of CaSla2 caused a specific delay in the cell cycle, we analysed the sla2 mutants for steady-state DNA content by flow cytometry at 30 °C. This temperature decreases growth rate without inducing morphogenesis (pseudohyphae and hyphae) as seen at higher temperatures. Wild-type cells, as well as SLA2/sla2Δ heterozygotes, had a normal distribution of cells with 2N (G1) and 4N (G2) DNA (Fig. 3a). In contrast, strains lacking Sla2 (sla2Δ/sla2Δ), as well as strains carrying only the sla2-1 allele, were enriched in 4N DNA, suggesting a delay in the G2 stage of the cell cycle even at 30 °C (Fig. 3a).
Fig. 3.

(a) C. albicans sla2 strains exhibit a cell cycle delay at 30 °C: flow cytometry of exponential-phase cultures. Top to bottom: wild-type (WT), BWP17; SLA2/sla2Δ, YJB7435; sla2-1/sla2-1, YJB3402; sla2Δ/sla2Δ, YJB7674. (b) Nuclear division is delayed in sla2 mutants. In wild-type (CAI4) cells, medium-sized buds have received a nucleus while in sla2 cells (YJB3403), many medium and large buds remain anucleate (arrows). Bar, 10 μm. (c) S. cerevisiae sla2 and sla1 strains exhibit a cell cycle delay at 30 °C: flow cytometry of exponential-phase cultures. Top to bottom: WT, YJB2756; sla2, YJB4786; sla1, YJB3047; abp1, YJB2709. The G1/G2 ratio for each strain is indicated.
Cell cycle synchrony is difficult to achieve with C. albicans; alpha factor does not arrest a majority of cells and mutants that give clean cell cycle arrest and release are not available. In classic studies of cell cycle dynamics in S. cerevisiae, the timing of mitosis correlates with attainment of certain mother and bud cell sizes (Hartwell et al., 1970). To examine the cell cycle characteristics of C. albicans sla2 mutants, we compared the range of mother and bud cell sizes and the size of buds that had received a nucleus in populations of exponential-phase wild-type and sla2 cells. Consistent with what has been seen in S. cerevisiae sla2 mutants (Holtzman et al., 1993), C. albicans sla2 mother cells were significantly larger than wild-type mother cells (6.83 μm vs 6.18 μm in diameter, respectively; P<0.001 by Student's t test, n=200 mother–daughter pairs analysed for each strain). In contrast, bud size was not significantly different between sla2 and wild-type cultures (3.56 μm vs 3.44 μm in diameter, respectively; P>0.2). This suggests that mother cells become larger due to inappropriate growth in subsequent cell cycles, rather than during the time they are buds. More importantly, 40 % of wild-type buds contained nuclei by the time they reached ∼3–4 μm in diameter, while sla2 buds reached 5–6 μm in diameter before 40 % of the buds had received a nucleus. In other words, there is a delay in nuclear division/mitosis relative to bud growth in sla2 mutant cells (Fig. 3b). Taken together, the DNA content and nuclear distribution data support the idea that sla2 mutant cells exhibit a cell-cycle delay at the G2/M stage of the cell cycle.
The cell cycle delay in sla2 cells is SWE1-dependent
In S. cerevisiae yeast cells, Swe1 mediates a morphogenesis checkpoint that links bud growth and mitosis (Lew, 2003). Abnormalities in the actin cytoskeleton or the septin ring trigger inhibitory phosphorylation of the cyclin-dependent kinase (Clb2/Cdc28) by Swe1, resulting in a delay in cell cycle progression prior to mitosis (reviewed by Lew, 2003). We hypothesized that a Swe1-mediated mechanism delays cell cycle progression in sla2 mutant strains. If this is the case, then deletion of Swe1 in these cells should uncouple nuclear cell cycle progression from bud morphogenesis. To test this prediction, we first analysed growth of sla2 and swe1 sla2 double mutant strains. Because a relationship between Swe1 and Sla2 has not been described in S. cerevisiae, we performed the same analyses in both C. albicans and S. cerevisiae (Fig. 4a). As previously reported in both yeasts, strains lacking Swe1 grew to a similar extent as the wild-type strain (Fig. 4a; McMillan et al., 1998; Wightman et al., 2004). In both C. albicans and S. cerevisiae, swe1 sla2 double mutant strains grew less well than sla2 single mutant strains (Fig. 4a). This is consistent with the idea that Swe1 enables sla2 cells to maintain viability despite morphogenetic defects.
Fig. 4.

Swe1 effects on endocytosis mutants. (a) Dilutions (left to right) of YPAD cultures grown overnight at 24 °C were spotted onto SDC medium and incubated for 2 days (C. albicans) or 3 days (S. cerevisiae) at the indicated temperatures. C. albicans strains (twofold dilutions, starting with 5×105 cells, top panels): wild-type (WT), YJB9955; swe1Δ, YJB6792; sla2Δ, YJB9105; swe1Δ sla2Δ, YJB7904. S. cerevisiae strains (twofold dilutions, starting with 5×105 cells, middle panels): WT, YJB2756; swe1, YJB4788; sla2, YJB4786; swe1 sla2, YJB4791. S. cerevisiae strains (tenfold dilutions, starting with 2×107 cells, lower panels): WT2, YJB2756; abp1, YJB2709; swe1, YJB2759; sla1, YJB3047; swe1Δ/sla1Δ, YJB11509; and swe1/abp1, YJB11511. (b) Strains were cultured at 30 °C overnight in YPAD, diluted 1 : 20 into fresh YPAD, grown at 30 °C for 5 h and stained with DAPI as described in Methods. Micrographs of cell morphology (top panels in each set, DIC) and nuclear localization (bottom panel in each set, DAPI) of C. albicans strains (WT, YJB9955; swe1, YJB6792; sla2, YJB9105; swe1 sla2, YJB7904) and S. cerevisiae strains (WT, YJB2756; swe1, YJB4788; sla2, YJB4786; swe1 sla2, YJB4791). Bars, 10 μm.
Next, we compared nuclei in sla2 single mutants and in the swe1 sla2 double mutant strain. Importantly, swe1 sla2 double mutants produced a subpopulation of mother cells containing more than one nucleus (Fig. 4b, far right panels). This was true for both C. albicans and S. cerevisiae grown at 30 °C, conditions that are semi-permissive for the sla2 mutation and that promote growth with a yeast morphology in C. albicans. Furthermore, there were more binucleate mother cells in swe1 sla2 double mutant strains than in either the wild-type or single mutant strains (Table 2). Thus, it appears that in the absence of the checkpoint delay, swe1 sla2 cells exhibit mitotic defects. Together with the cell growth data, these results indicate that loss of Sla2 function causes a cell cycle delay that is mediated by Swe1 in both C. albicans and S. cerevisiae.
Table 2.
Quantitative nuclear analysis of yeast strains
Strains are described in the legend to Fig. 4(a). Values are given as percentages of total cells.
| C. albicans | Uni-nuclear* | Multi-nuclear† | Mitotic‡ |
|---|---|---|---|
| Wild-type (n=241) | 91 | 0 | 9 |
| sla2Δ/sla2Δ (n=226) | 91 | 4 | 5 |
| swe1Δ/swe1Δ (n=249) | 94 | 0 | 6 |
| swe1Δ/swe1Δ sla2Δ/sla2Δ (n=249) | 63 | 32 | 5 |
| S. cerevisiae | |||
| Wild-type (n=273) | 100 | 0 | 0 |
| sla2 (n=257) | 97 | 1 | 1 |
| swe1 (n=249 | 98 | 1 | 1 |
| swe1sla2 (n=216) | 87 | 12 | 1 |
| abp1 (n=309) | 99 | 0 | 1 |
| swe1abp1 (n=297) | 93 | 6 | 1 |
| sla1 (n=250) | 98 | 1 | 1 |
| swe1sla1 (n=266) | 90 | 9 | 1 |
*Includes budded and unbudded mother cells.
†Includes budded and unbudded mother cells; the multi-nuclear designation pertains to the mother cell.
‡Includes cells in which the nucleus is stretched between the mother cell and bud.
Sla2 influences the organization of actin patches and actin cables
In S. cerevisiae, the morphogenesis checkpoint is triggered by defects in the septin ring and the actin cytoskeleton, although the specific nature of the actin defects has not been identified. We investigated the role of CaSla2 in septin organization by analysing septin Cdc3, fused to GFP, in sla2 strains. Cdc3 localized in a ring pattern at the mother–bud necks of sla2 strains. This is similar to its localization in wild-type cells (Fig. 5a) and is consistent with the idea that sla2 does not trigger the checkpoint via disruption of the septin ring.
Fig. 5.

(a) Septin rings are not perturbed in cells lacking Sla2. C. albicans YJB8658 expressing Cdc3-GFP was grown overnight at 24 °C to repress expression of PMET3 : : SLA2 (SDC+Met +Cys) and then transferred to inducing medium (SDC −Met −Cys, PMET3 : : SLA2/sla2 (on)) or maintained in repressing conditions (PMET3 : : SLA2/sla2 (off); phenocopy of the sla2 null morphology was confirmed by microscopy of the cells) for 8 h at 30 °C. Representative images are shown. Bar, 10 μm. (b, c) Actin cable organization in C. albicans (b) and S. cerevisiae (c) strains by Alexa phalloidin staining. Bars, 10 μm; *, actin cables; arrows, actin comet tails (bar-like structures). (b) C. albicans: wild-type, BWP17; sla2/sla2, YJB7674. (c) S. cerevisiae: wild-type, YJB3046; sla2, YJB2713; sla1, YJB3047; abp1, YJB2709. (d) Sla2-YFP (YJB6674) localizes to cortical patches. Bar, 5 μm. (e) Higher magnification of panel (1) in (b) to highlight the bar-like actin structures (arrow).
To address the role of Sla2 in actin function, we analysed actin localization in sla2 strains. Consistent with classical studies (Anderson & Soll, 1986), cortical actin patches polarize almost exclusively to the daughter cell during bud growth in wild-type strains (Fig. 5b, wild-type panel). In contrast, in the sla2 strain, actin patches were delocalized, with an increase in the number of patches in mother yeast cells during budding (Fig. 5b, sla2 panels). These actin patches were larger and often exhibited a bar-like structure that radiated from the cell surface inward (Fig. 5b, arrow), similar to the actin ‘comet tail-like’ structure described in S. cerevisiae sla2 and clathrin heavy chain mutants (Fig. 5c, sla2 panel, and Fig. 5e, arrow; Kaksonen et al., 2003; Newpher & Lemmon, 2006). In addition, sla2 mutants had reduced rates of patch movement and patch stability (amount of time that Abp1-YFP-labelled patches remained visible) (Supplementary Movies S1 and S2 and Supplementary Fig. S1). These observations are consistent with the idea that CaSla2, like ScSla2, is involved in actin patch organization and dynamics.
In budding wild-type C. albicans and S. cerevisiae yeast cells, actin cables are oriented toward the growing bud (Fig. 5b, c, panels with asterisks). In contrast, in sla2 strains, actin cable localization and orientation was defective. First, actin cables were much less apparent and, second, those that were present were oriented randomly (Fig. 5b, c, sla2 panels), consistent with reported S. cerevisiae sla2 phenotypes (Holtzman et al., 1993). CaSla2-YFP, like ScSla2 (Yang et al., 1999), localized in cortical and bud neck patches (Fig. 5d), colocalizing with a subset of actin patches (Supplementary Fig. S2). Importantly, no localization of Sla2 to cables was evident. To ask if other mutants that affect endocytosis also affect actin cables, we analysed actin organization in S. cerevisiae strains lacking Sla1 and Abp1, components of cortical actin patches (Holtzman et al., 1993; Mulholland et al., 1994; Pruyne & Bretscher, 2000; Fig. 5c). Importantly, actin cables were readily detected and were polarized toward the buds (90 % of cells, n=30 for each), much like cable orientation in the wild-type strain (95 % of cells, n=30) and in contrast to the Scsla2 strain (25 % of cells, n=30). Thus, Sla2 is important for actin cable formation and polarization while the endocytic patch components Sla1 and Abp1 are not.
Mutations in endocytic patch components Sla1 and Abp1 also exhibit Swe1-dependent phenotypes
In S. cerevisiae, Swe1 has been reported to be activated by defects of actin cables (McMillan et al., 1998). Thus, we tested the hypothesis that it is the cable defect rather than the defect in patch organization that triggers a Swe1-mediated cell cycle delay in Scsla2 (and Casla2) mutants. If this hypothesis is correct, then Scabp1 and Scsla1 strains, which do not have obvious actin cable defects (Fig. 5c), should not trigger Swe1-dependent cell cycle phenotypes. Importantly, both Scsla2 and Scsla1 strains exhibited a cell cycle delay at G2/M (Fig. 3c). Furthermore, all three endocytosis mutants produced a subpopulation of mother cells containing more than one nucleus when combined with a swe1 mutation (Table 2). Together, these data indicate that defects in endocytic patch proteins cause phenotypes that are mediated by Swe1.
Swe1 contributes to polarized growth of sla2 strains grown in hyphal induction conditions
In S. cerevisiae, Swe1 is required for polarized growth in a number of strains carrying mutations that cause pseudohyphal-like morphologies (Ahn et al., 1999, 2001; Edgington et al., 1999; La Valle & Wittenberg, 2001), such as overexpression of the Clb2 mitotic cyclin or inactivation of Hsl1 and Hsl7, negative regulators of Swe1. These mutant strains are defective in the switch from polarized to isotropic bud growth that normally occurs during early mitosis and are considered ‘pseudo-pseudohyphae’ (Wightman et al., 2004). C. albicans sla2 strains exhibit dramatically reduced polarized growth but some residual polarization is detectable on pseudohyphal and hyphal induction media (Fig. 6). To ask if CaSwe1 is necessary for the polarized growth of sla2 strains under these conditions, we compared the morphology of swe1 sla2 double mutant strains to wild-type, sla2, and swe1 single mutant strains. As previously reported for swe1 mutant strains (Wightman et al., 2004), there were no obvious morphogenesis or growth defects in pseudohyphae or hyphae lacking Swe1 (Fig. 6). Importantly, swe1 sla2 double mutant cells were less polarized than either single mutant (Fig. 6). Thus, as in S. cerevisiae, the morphogenesis checkpoint is dispensable for normal pseudohyphal and true hyphal growth in C. albicans, while it is required for polarized growth in sla2 mutants.
Fig. 6.

SLA2, but not SWE1, is required for pseudohyphal (PH) and hyphal (H) growth. Overnight cultures of WT (BWP17), swe1Δ/swe1Δ (YJB4994), sla2Δ/sla2Δ (YJB7674) and swe1Δ/swe1Δ sla2Δ/sla2Δ (YJB7905) strains were cultured in SDC at 37 °C for 2 h in the absence (PH) or presence (H) of 10 % serum. Bar, 10 μm.
The Swe1 checkpoint kinase is important for C. albicans virulence
Because C. albicans is an opportunistic pathogen, its virulence is closely tied to its ability to survive and grow in the human host; we therefore tested the hypothesis that the Swe1 checkpoint kinase is important for C. albicans to cause disease. A set of prototrophic isogenic swe1 mutants and SWE1 reintegrants were tested using a mouse model of systemic candidiasis (see Methods). To minimize the effects of differential URA3 expression between strains (Brand et al., 2004; Lay et al., 1998; Sundstrom et al., 2002), SWE1-URA3 was inserted into the disrupted SWE1 locus. Importantly, strains lacking Swe1 were attenuated in virulence relative to the isogenic reintegrant strains (Fig. 7). A composite of six separate virulence tests (see Methods) showed a significant difference (P≤0.0002). Thus, like other cell cycle checkpoint proteins (Bai et al., 2002), the Swe1 morphogenesis checkpoint kinase, which is not required for growth or morphogenesis (Figs 4a and 7; Wightman et al., 2004), contributes to virulence in a mouse model of systemic candidiasis.
Fig. 7.
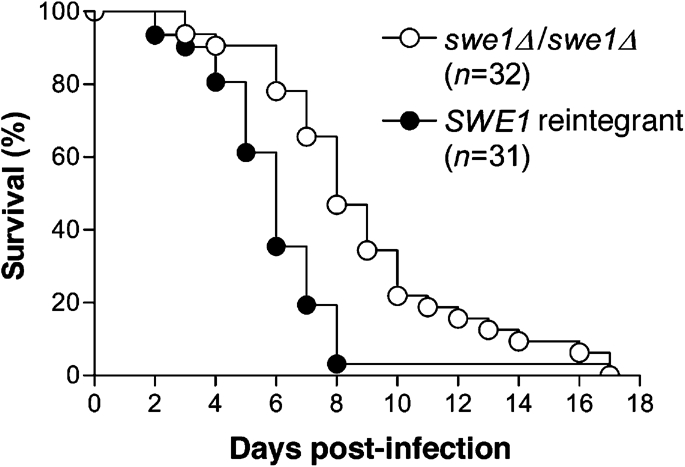
SWE1 contributes to virulence in a mouse model of systemic candidiasis. Survival as a function of time following inoculation of 106 cells per mouse (see Methods). The swe1Δ/swe1Δ curve (○) is a composite of data from strains YJB8190, 8193 and 8195. The SWE1-reintegrant curve (•) is a composite of data from strains YJB7853, 7855 and 7857. The two curves are significantly different (P≤0.0002).
DISCUSSION
Numerous C. albicans genes affect both cell cycle progression and morphogenesis, yet little is known about how these two processes are coordinated. Here, we found that CaSla2, a protein necessary for normal endocytosis, has a function that is monitored by the Swe1 checkpoint kinase. Cells lacking sla2 are delayed in G2, with a higher proportion of cells with 4N DNA and an increase in uninucleate, large-budded mother cells (Fig. 3). Furthermore, this cell cycle delay requires the Swe1 to coordinate nuclear division with bud growth, because swe1 sla2 cells are often bi- (or multi-)nucleate and exhibit a growth defect (Fig. 4b, Table 2). While it is clear that Swe1 contributes to sla2 phenotypes, it is also possible that other checkpoints, in particular a checkpoint that monitors the alignment of the spindle, also play a role in the cell cycle delay observed in sla2 mutant strains.
How do mutations in Sla2 interact with Swe1? In S. cerevisiae, Swe1 is activated by disruption of the septin ring or actin cytoskeleton. Because no defects in septin ring structure at the bud neck were evident (Fig. 5a), we propose that perturbations of the actin cytoskeleton trigger the morphogenesis checkpoint in sla2 mutants. While it is not yet clear how these defects promote Swe1-mediated cell cycle arrest (Keaton & Lew, 2006), S. cerevisiae actin mutants reported to trigger the morphogenesis checkpoint appear to affect actin cables rather than actin patches [Tpm1 (binds and stabilizes actin cables), Myo2 (transports cell components along actin cables), Sac6 (localizes to actin cables) and Pfy1 (Arp2/3-independent nucleation of actin filaments and actin cable formation)] (McMillan et al., 1998). We found that Sla2 has a major role in actin patch assembly and dynamics, as well as a role in actin cable polarization (Fig. 5b, c). Consistent with this, an Scsla2 mutation (sla2-82) interacts genetically with genes involved in actin cable assembly (Yoshiuchi et al., 2006). Thus, we tested the hypothesis that the Swe1-mediated delay in sla2 strains was due to actin cable, rather than actin patch, defects. However, two other endocytic patch components (Abp1 and Sla1), which do not affect actin cables (Fig. 5c), also interact genetically with Swe1 (Table 2). While we cannot rule out subtle defects in septin ring or cable function, these results suggest that the Swe1 morphogenesis checkpoint monitors defects in actin patch function. Nonetheless, Sla2 appears to be more important than Abp1 and Sla1 for the coordination of morphogenesis with the nuclear cell cycle because sla2 mutants have stronger cell cycle delays and Swe1-dependent phenotypes. This may be because Sla2 is more important than Abp1 or Sla1 in the endocytic function that is monitored by Swe1. Alternatively, Swe1 may monitor the integrity of both actin patches and cables, and Sla2 may trigger a stronger interaction with the morphogenesis checkpoint because it perturbs them both.
Sla2, Abp1 and Sla1 all function in the nucleation and early assembly of actin at cortical patches (Ayscough et al., 1999; Goode et al., 2001; Holtzman et al., 1993; Li et al., 1995; Wesp et al., 1997; Yang et al., 1999) and all interact by the yeast two-hybrid method with Ynl094w (Drees et al., 2001), a protein of unknown function that localizes to cortical actin patches (Drees et al., 2001) and interacts with Swe1 (Shulewitz et al., 1999). It is tempting to speculate that the morphogenesis checkpoint monitors defects in endocytosis through its interaction with Ynl094w.
Physiological stresses on the yeast cell were thought to induce the morphogenesis checkpoint by perturbation of the actin cytoskeleton. However, exposure of yeast cells to osmotic stress leads to a Swe1-dependent cell cycle delay that is independent of actin and is mediated by the stress-activated protein kinase Hog1 (Clotet et al., 2006). Interestingly, C. albicans sla2 strains have transcript profiles that are similar to those of wild-type cells exposed to osmotic shock (Oberholzer et al., 2006). However, no hyperphosphorylation of Hog1 was observed in these strains, implying that Hog1 was not activated and that sla2 does not activate Swe1 via the Hog1 MAPK cascade. Rather, we propose that defects in the actin cytoskeleton, and specifically in actin patch function, trigger the morphogenesis checkpoint in sla2 mutants.
In C. albicans, perturbation of cell cycle events often results in cells locked in filamentous growth states (Bachewich et al., 2005; Bensen et al., 2002, 2005; Berman, 2006; Chapa y Lazo et al., 2005; Finley et al., 2008; Wightman et al., 2004). For example, Bub2, a mitotic spindle checkpoint protein, is necessary for the polarized growth phenotype of cells arrested in mitosis due to either Cdc5 depletion (Bachewich et al., 2005) or dynein defects (Finley et al., 2008). Similarly, we find that Swe1, a morphogenesis checkpoint protein, contributes to the residual polarized growth in sla2 cells grown in hyphal conditions. Together, these results suggest that polarized growth can be modulated by cell cycle checkpoints and support the idea that polarized growth is a general response to cell cycle defects at most stages of the cell cycle (Berman, 2006). Interestingly, however, yeast-form sla2 cells do not exhibit the elongated cell phenotype of mitotic cell cycle delay. We propose that sla2 yeast cells are unable form hyperpolarized daughters because endocytic/membrane recycling defects and/or the polarized actin cable defects affect yeast-form growth more than hyphal-form growth. The observation that yeast and hyphal cell morphologies are affected differently by Sla2 and Swe1 highlights the concept that polarized growth mechanisms in the two forms are distinguishable.
The ability of C. albicans to switch between filamentous and non-filamentous morphologies makes an important contribution to its virulence (Saville et al., 2008). For example, mutants locked in one growth form are avirulent (Braun & Johnson, 1997; Lo et al., 1997) and overexpression of UME6 increases both filamentous growth and virulence (Banerjee et al., 2008). However, many of the mutations that affect morphogenesis are transcription factors that likely affect other processes as well, making it difficult to definitively demonstrate a 1 : 1 relationship between the two processes. Our results indicate that, even though it is not required for growth or morphogenesis in vitro, Swe1 is important for virulence of C. albicans in a mouse model of systemic infection. Similar results were seen for the Mad2 cell cycle checkpoint (Bai et al., 2002). These connections between checkpoint proteins and virulence suggest that growth conditions in vivo affect cell cycle progression. Thus, an important theme emerging from this work and that of others is that the ability to appropriately delay cell cycle progression through checkpoint activation during growth in vivo is important for virulence, presumably by enabling the pathogen to survive environmental stresses present in the host.
Acknowledgments
We thank Maryam Gerami-Nejad, Sandy Vandoninck, Carter Myers, Carrie Ketel, Pete Jauert and John Asleson for technical assistance. We thank Duncan Clarke, Dana Davis, David Drubin, Aaron Mitchell, John Pringle, Howard Riezman and Peter Sudbery for providing strains and/or plasmids. This work was supported by NIH AI/DE 14666, NIH AI 0624273 and Burroughs Welcome Senior Scholar Award #0677 to J. B., by NIH F32 AI 10647 to E. B., by University of MN UROP funding to L. C., and by NIH AI 057440, University of MN Graduate School and MN Medical Foundation awards to C. A. G. K. R. F. was supported, in part, by NIH Biotechnology Training Grant GM08347.
Abbreviations
DAPI, 4′,6-diamidino-2-phenylindole
DIC, differential interference contrast
GFP, green fluorescent protein
HA, haemagglutinin
YFP, yellow fluorescent protein
Footnotes
Two supplementary movies and two supplementary figures are available with the online version of this paper.
References
- Ahn, S. H., Acurio, A. & Kron, S. J. (1999). Regulation of G2/M progression by the STE mitogen-activated protein kinase pathway in budding yeast filamentous growth. Mol Biol Cell 10, 3301–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn, S. H., Tobe, B. T., Fitz Gerald, J. N., Anderson, S. L., Acurio, A. & Kron, S. J. (2001). Enhanced cell polarity in mutants of the budding yeast cyclin-dependent kinase Cdc28p. Mol Biol Cell 12, 3589–3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi, T., Kanbe, T. & Tanaka, K. (1994). The role of the cytoskeleton in the polarized growth of the germ tube in Candida albicans. Microbiology 140, 271–280. [DOI] [PubMed] [Google Scholar]
- Anderson, J. M. & Soll, D. R. (1986). Differences in actin localization during bud and hypha formation in the yeast Candida albicans. J Gen Microbiol 132, 2035–2047. [DOI] [PubMed] [Google Scholar]
- Asleson, C. M., Bensen, E. S., Gale, C. A., Melms, A. S., Kurischko, C. & Berman, J. (2001). Candida albicans INT1-induced filamentation in Saccharomyces cerevisiae depends on Sla2p. Mol Cell Biol 21, 1272–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayscough, K. R., Eby, J. J., Lila, T., Dewar, H., Kozminski, K. G. & Drubin, D. G. (1999). Sla1p is a functionally modular component of the yeast cortical actin cytoskeleton required for correct localization of both Rho1p-GTPase and Sla2p, a protein with talin homology. Mol Biol Cell 10, 1061–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachewich, C., Nantel, A. & Whiteway, M. (2005). Cell cycle arrest during S or M phase generates polarized growth via distinct signals in Candida albicans. Mol Microbiol 57, 942–959. [DOI] [PubMed] [Google Scholar]
- Baggett, J. J., D'Aquino, K. E. & Wendland, B. (2003). The Sla2p talin domain plays a role in endocytosis in Saccharomyces cerevisiae. Genetics 165, 1661–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai, C., Ramanan, N., Wang, Y. M. & Wang, Y. (2002). Spindle assembly checkpoint component CaMad2p is indispensable for Candida albicans survival and virulence in mice. Mol Microbiol 45, 31–44. [DOI] [PubMed] [Google Scholar]
- Banerjee, M., Thompson, D. S., Lazzell, A., Carlisle, P. L., Pierce, C., Monteagudo, C., Lopez-Ribot, J. L. & Kadosh, D. (2008). UME6, a novel filament-specific regulator of Candida albicans hyphal extension and virulence. Mol Biol Cell 19, 1354–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barelle, C. J., Bohula, E. A., Kron, S. J., Wessels, D., Soll, D. R., Schafer, A., Brown, A. J. & Gow, N. A. (2003). Asynchronous cell cycle and asymmetric vacuolar inheritance in true hyphae of Candida albicans. Eukaryot Cell 2, 398–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barral, Y., Parra, M., Bidlingmaier, S. & Snyder, M. (1999). Nim1-related kinases coordinate cell cycle progression with the organization of the peripheral cytoskeleton in yeast. Genes Dev 13, 176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensen, E. S., Filler, S. G. & Berman, J. (2002). A forkhead transcription factor is important for true hyphal as well as yeast morphogenesis in Candida albicans. Eukaryot Cell 1, 787–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensen, E. S., Clemente-Blanco, A., Finley, K. R., Correa-Bordes, J. & Berman, J. (2005). The mitotic cyclins Clb2p and Clb4p affect morphogenesis in Candida albicans. Mol Biol Cell 16, 3387–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman, J. (2006). Morphogenesis and cell cycle progression in Candida albicans. Curr Opin Microbiol 9, 595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidlingmaier, S. & Snyder, M. (2002). Large-scale identification of genes important for apical growth in Saccharomyces cerevisiae by directed allele replacement technology (DART) screening. Funct Integr Genomics 1, 345–356. [DOI] [PubMed] [Google Scholar]
- Brand, A., MacCallum, D. M., Brown, A. J., Gow, N. A. & Odds, F. C. (2004). Ectopic expression of URA3 can influence the virulence phenotypes and proteome of Candida albicans but can be overcome by targeted reintegration of URA3 at the RPS10 locus. Eukaryot Cell 3, 900–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun, B. R. & Johnson, A. D. (1997). Control of filament formation in Candida albicans by the transcriptional repressor TUP1. Science 277, 105–109. [DOI] [PubMed] [Google Scholar]
- Care, R. S., Trevethick, J., Binley, K. M. & Sudbery, P. E. (1999). The MET3 promoter: a new tool for Candida albicans molecular genetics. Mol Microbiol 34, 792–798. [DOI] [PubMed] [Google Scholar]
- Carlisle, P. L., Banerjee, M., Lazzell, A., Monteagudo, C., Lopez-Ribot, J. L. & Kadosh, D. (2009). Expression levels of a filament-specific transcriptional regulator are sufficient to determine Candida albicans morphology and virulence. Proc Natl Acad Sci U S A 106, 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapa y Lazo, B., Bates, S. & Sudbery, P. (2005). The G1 cyclin Cln3 regulates morphogenesis in Candida albicans. Eukaryot Cell 4, 90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clotet, J., Escote, X., Adrover, M. A., Yaakov, G., Gari, E., Aldea, M., de Nadal, E. & Posas, F. (2006). Phosphorylation of Hsl1 by Hog1 leads to a G2 arrest essential for cell survival at high osmolarity. EMBO J 25, 2338–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, D., Edwards, J. E., Jr, Mitchell, A. P. & Ibrahim, A. S. (2000). Candida albicans RIM101 pH response pathway is required for host-pathogen interactions. Infect Immun 68, 5953–5959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, D. A., Bruno, V. M., Loza, L., Filler, S. G. & Mitchell, A. P. (2002). Candida albicans Mds3p, a conserved regulator of pH responses and virulence identified through insertional mutagenesis. Genetics 162, 1573–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drees, B. L., Sundin, B., Brazeau, E., Caviston, J. P., Chen, G. C., Guo, W., Kozminski, K. G., Lau, M. W., Moskow, J. J. & other authors (2001). A protein interaction map for cell polarity development. J Cell Biol 154, 549–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgington, N. P., Blacketer, M. J., Bierwagen, T. A. & Myers, A. M. (1999). Control of Saccharomyces cerevisiae filamentous growth by cyclin-dependent kinase Cdc28. Mol Cell Biol 19, 1369–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley, K. R., Bouchonville, K. J., Quick, A. & Berman, J. (2008). Dynein-dependent nuclear dynamics affect morphogenesis in Candida albicans by means of the Bub2p spindle checkpoint. J Cell Sci 121, 466–476. [DOI] [PubMed] [Google Scholar]
- Fonzi, W. A. & Irwin, M. Y. (1993). Isogenic strain construction and gene mapping in Candida albicans. Genetics 134, 717–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerami-Nejad, M., Berman, J. & Gale, C. A. (2001). Cassettes for PCR-mediated construction of green, yellow, and cyan fluorescent protein fusions in Candida albicans. Yeast 18, 859–864. [DOI] [PubMed] [Google Scholar]
- Gerami-Nejad, M., Dulmage, K. & Berman, J. (2009). Additional cassettes for epitope and fluorescent fusion proteins in Candida albicans. Yeast 26, 399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode, B. L., Rodal, A. A., Barnes, G. & Drubin, D. G. (2001). Activation of the Arp2/3 complex by the actin filament binding protein Abp1p. J Cell Biol 153, 627–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourlay, C. W., Dewar, H., Warren, D. T., Costa, R., Satish, N. & Ayscough, K. R. (2003). An interaction between Sla1p and Sla2p plays a role in regulating actin dynamics and endocytosis in budding yeast. J Cell Sci 116, 2551–2564. [DOI] [PubMed] [Google Scholar]
- Gow, N. A., Brown, A. J. & Odds, F. C. (2002). Fungal morphogenesis and host invasion. Curr Opin Microbiol 5, 366–371. [DOI] [PubMed] [Google Scholar]
- Hartwell, L. H., Culotti, J. & Reid, B. (1970). Genetic control of the cell-division cycle in yeast. I. Detection of mutants. Proc Natl Acad Sci U S A 66, 352–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausauer, D. L., Gerami-Nejad, M., Kistler-Anderson, C. & Gale, C. A. (2005). Hyphal guidance and invasive growth in Candida albicans require the Ras-like GTPase Rsr1p and its GTPase-activating protein Bud2p. Eukaryot Cell 4, 1273–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman, D. A., Yang, S. & Drubin, D. G. (1993). Synthetic-lethal interactions identify two novel genes, SLA1 and SLA2, that control membrane cytoskeleton assembly in Saccharomyces cerevisiae. J Cell Biol 122, 635–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irazoqui, J. E., Howell, A. S., Theesfeld, C. L. & Lew, D. J. (2005). Opposing roles for actin in Cdc42p polarization. Mol Biol Cell 16, 1296–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaksonen, M., Sun, Y. & Drubin, D. G. (2003). A pathway for association of receptors, adaptors, and actin during endocytic internalization. Cell 115, 475–487. [DOI] [PubMed] [Google Scholar]
- Keaton, M. A. & Lew, D. J. (2006). Eavesdropping on the cytoskeleton: progress and controversy in the yeast morphogenesis checkpoint. Curr Opin Microbiol 9, 540–546. [DOI] [PubMed] [Google Scholar]
- Kron, S. J. & Gow, N. A. (1995). Budding yeast morphogenesis: signalling, cytoskeleton and cell cycle. Curr Opin Cell Biol 7, 845–855. [DOI] [PubMed] [Google Scholar]
- La Valle, R. & Wittenberg, C. (2001). A role for the Swe1 checkpoint kinase during filamentous growth of Saccharomyces cerevisiae. Genetics 158, 549–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lay, J., Henry, L. K., Clifford, J., Koltin, Y., Bulawa, C. E. & Becker, J. M. (1998). Altered expression of selectable marker URA3 in gene-disrupted Candida albicans strains complicates interpretation of virulence studies. Infect Immun 66, 5301–5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew, D. J. (2003). The morphogenesis checkpoint: how yeast cells watch their figures. Curr Opin Cell Biol 15, 648–653. [DOI] [PubMed] [Google Scholar]
- Lew, D. J. & Reed, S. I. (1995). A cell cycle checkpoint monitors cell morphogenesis in budding yeast. J Cell Biol 129, 739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, R., Zheng, Y. & Drubin, D. G. (1995). Regulation of cortical actin cytoskeleton assembly during polarized cell growth in budding yeast. J Cell Biol 128, 599–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo, H. J., Kohler, J. R., DiDomenico, B., Loebenberg, D., Cacciapuoti, A. & Fink, G. R. (1997). Nonfilamentous C. albicans mutants are avirulent. Cell 90, 939–949. [DOI] [PubMed] [Google Scholar]
- Longtine, M. S., Theesfeld, C. L., McMillan, J. N., Weaver, E., Pringle, J. R. & Lew, D. J. (2000). Septin-dependent assembly of a cell cycle-regulatory module in Saccharomyces cerevisiae. Mol Cell Biol 20, 4049–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantel, N. & Haenszel, W. (1959). Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst 22, 719–748. [PubMed] [Google Scholar]
- McMillan, J. N., Sia, R. A. L. & Lew, D. J. (1998). A morphogenesis checkpoint monitors the actin cytoskeleton in yeast. J Cell Biol 142, 1487–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, A. P. (1998). Dimorphism and virulence in Candida albicans. Curr Opin Microbiol 1, 687–692. [DOI] [PubMed] [Google Scholar]
- Moseley, J. B. & Goode, B. L. (2006). The yeast actin cytoskeleton: from cellular function to biochemical mechanism. Microbiol Mol Biol Rev 70, 605–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland, J., Preuss, D., Moon, A., Wong, A., Drubin, D. & Botstein, D. (1994). Ultrastructure of the yeast actin cytoskeleton and its association with the plasma membrane. J Cell Biol 125, 381–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na, S., Hincapie, M., McCusker, J. H. & Haber, J. E. (1995). MOP2 (SLA2) affects the abundance of the plasma membrane H+-ATPase of Saccharomyces cerevisiae. J Biol Chem 270, 6815–6823. [DOI] [PubMed] [Google Scholar]
- Newpher, T. M. & Lemmon, S. K. (2006). Clathrin is important for normal actin dynamics and progression of Sla2p-containing patches during endocytosis in yeast. Traffic 7, 574–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberholzer, U., Nantel, A., Berman, J. & Whiteway, M. (2006). Transcript profiles of Candida albicans cortical actin patch mutants reflect their cellular defects: contribution of the Hog1p and Mkc1p signaling pathways. Eukaryot Cell 5, 1252–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruyne, D. & Bretscher, A. (2000). Polarization of cell growth in yeast. J Cell Sci 113, 571–585. [DOI] [PubMed] [Google Scholar]
- Saville, S. P., Lazzell, A. L., Chaturvedi, A. K., Monteagudo, C. & Lopez-Ribot, J. L. (2008). Use of a genetically engineered strain to evaluate the pathogenic potential of yeast cell and filamentous forms during Candida albicans systemic infection in immunodeficient mice. Infect Immun 76, 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman, F. (1991). Getting started with yeast. Methods Enzymol 194, 3–20. [DOI] [PubMed] [Google Scholar]
- Shulewitz, M. J., Inouye, C. J. & Thorner, J. (1999). Hsl7 localizes to a septin ring and serves as an adapter in a regulatory pathway that relieves tyrosine phosphorylation of Cdc28 protein kinase in Saccharomyces cerevisiae. Mol Cell Biol 19, 7123–7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundstrom, P., Cutler, J. E. & Staab, J. F. (2002). Reevaluation of the role of HWP1 in systemic candidiasis by use of Candida albicans strains with selectable marker URA3 targeted to the ENO1 locus. Infect Immun 70, 3281–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman, Z., Shemer, R., Conibear, E. & Kornitzer, D. (2008). An endocytic mechanism for haemoglobin-iron acquisition in Candida albicans. Mol Microbiol 69, 201–217. [DOI] [PubMed] [Google Scholar]
- Wesp, A., Hicke, L., Palecek, J., Lombardi, R., Aust, T., Munn, A. L. & Riezman, H. (1997). End4p/Sla2p interacts with actin-associated proteins for endocytosis in Saccharomyces cerevisiae. Mol Biol Cell 8, 2291–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wightman, R., Bates, S., Amornrrattanapan, P. & Sudbery, P. (2004). In Candida albicans, the Nim1 kinases Gin4 and Hsl1 negatively regulate pseudohypha formation and Gin4 also controls septin organization. J Cell Biol 164, 581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, R. B., Davis, D. & Mitchell, A. P. (1999). Rapid hypothesis testing with Candida albicans through gene disruption with short homology regions. J Bacteriol 181, 1868–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, R. B., Davis, D., Enloe, B. M. & Mitchell, A. P. (2000). A recyclable Candida albicans URA3 cassette for PCR product-directed gene disruptions. Yeast 16, 65–70. [DOI] [PubMed] [Google Scholar]
- Yang, S., Cope, M. J. & Drubin, D. G. (1999). Sla2p is associated with the yeast cortical actin cytoskeleton via redundant localization signals. Mol Biol Cell 10, 2265–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiuchi, S., Yamamoto, T., Sakane, H., Kadota, J., Mochida, J., Asaka, M. & Tanaka, K. (2006). Identification of novel mutations in ACT1 and SLA2 that suppress the actin-cable-overproducing phenotype caused by overexpression of a dominant active form of Bni1p in Saccharomyces cerevisiae. Genetics 173, 527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]