

Abstract
Given the highly infiltrative growth pattern of malignant glioma and the lack of specificity associated with currently available treatment regimens, alternative strategies designed to eradicate cancer cells while limiting collateral toxicity in normal tissues remain a high priority. To this end, the development of specific immunotherapies against targeted neoplastic cells represents a promising approach.
The epidermal growth factor receptor class III variant (EGFRvIII), a constitutively activated mutant of the wild‐type tyrosine kinase, is present in a substantial proportion of malignant gliomas and other human cancers, yet completely absent from normal tissues. This receptor variant consists of an in‐frame deletion, the translation of which produces an extracellular junction with a novel glycine residue, flanked by amino acid sequences that are not typically adjacent in the normal protein.
In this review, both preclinical and early clinical development of a peptide vaccine directed against this portion of the EGFRvIII antigenic domain are recapitulated. Following vaccination, our group has demonstrated potent, redirected cellular and humoral immunity against cancer cells expressing the mutant receptor without significant toxicity. Additionally, the corresponding therapeutic outcomes observed in these studies lend credence to the potential role of peptide‐based vaccination strategies among emerging antitumor immunotherapies in patients with malignant glioma.
Keywords: antigens, central nervous system neoplasms, epidermal growth factor receptor, immunotherapy
INTRODUCTION
Glioblastoma multiforme (GBM) is the most common primary malignant brain neoplasm, representing over 50% of all tumors in this category diagnosed each year (15). It is also one of the most aggressive and difficult cancers to treat; despite standard multimodal therapy, including surgical resection and radiotherapy plus temozolomide (TMZ), GBM remains uniformly lethal, with a median survival of less than 15 months from the time of diagnosis (96). Recurrent tumors exhibit an even poorer prognosis with a progression‐free survival of less than 20 weeks following currently available salvage therapy (102). Furthermore, although these outcomes represent remarkable advancements in the treatment of GBM, conventional strategies are ultimately limited by significant morbidity associated with nonspecific damage to normal cells and tissues (47). As a result, there is a clear need for more effective therapies that enable precise targeting of tumor cells while preserving an otherwise healthy milieu.
To this end, the immune system has emerged as a particularly promising approach. Over a century ago, Ehrlich first proposed that the body's natural immune system, with its inherent specificity and biologic efficiency, could be redirected to eradicate targeted neoplastic cells while reducing collateral toxicity (27). It has since been established in numerous studies that proper immunological manipulation, namely via vaccination against tumor antigens, can result in the regression of even bulky and invasive human cancers (80). While this type of manipulation has been shown to take many forms—including cellular, humoral and myriad passive, active and adoptive strategies—the discussion continues with regard to the precise interplay necessary to achieve the greatest antitumor response. This uncertainty notwithstanding, the prospect of harnessing the discriminatingly potent nature of the human immune system remains a high priority, and the understanding of its potential role in the treatment of GBM grows accordingly.
CNS IMMUNOPRIVILEGE
Antitumor immunotherapies, in the context of intracerebral tumors, encounter a distinct set of challenges, one of the most prominent being that of central nervous system (CNS) immune privilege. The first studies to suggest the concept of limited immune surveillance in the CNS and other select tissues were first reported in 1948 by Sir Peter Medawar, who showed that allogeneic tissue grafts transplanted into the brains of experimental animals were not rejected (64). Later research in the area of neuro‐immunology would support this finding based on unique characteristics that are now generally associated with the CNS: the presence of a specialized blood‐brain barrier (BBB) and the absence of conventional draining lymph nodes as well as resident antigen‐presenting cells (APCs) within the brain 30, 40.
While the CNS certainly exhibits immune privilege to some degree, a growing body of data suggests that its isolation from the immune system is not as complete as once believed. For instance, despite the BBB, immune cells have been shown to traffic to the brain relatively frequently 29, 44, 74, and, contrary to what was previously thought, antigen egress via cerebrospinal fluid (CSF) compartments and cervical lymphatics also appears to occur 19, 38. Furthermore, it has been proposed that specialized microglia (35) along with astrocytes (1) and certain cells of the choroid plexus epithelium (92) are able to mediate human leukocyte antigen (HLA) presentation, thereby functioning as surrogate APCs within the CNS.
CROSSING THE BBB
As previously mentioned, immune cells, specifically activated T lymphocytes, have the ability to penetrate the BBB under normal physiological conditions. This was first appreciated when experimental animals were injected intravenously with radioactively labeled T cell blasts, which were subsequently tracked to the CNS (29). Naïve T lymphocytes, however, are significantly restricted from entering the CNS, suggesting that penetration past the BBB is possible only after activation takes place (65). When it does occur, lymphocyte extravasation into the brain parenchyma is a highly regulated process mediated by several well‐characterized adhesion molecules and chemotactic factors (29). Once inside the CNS compartment, whether T lymphocytes proliferate and differentiate within the brain microenvironment has yet to be established, as previous studies differ on this point; nevertheless, it has been shown that these cells do remain in the CNS for longer periods of time if given the opportunity to interact with their cognate antigen 25, 62.
Central memory T lymphocytes that alternatively enter the CNS via the choroid plexus (79) flux continuously throughout subarachnoid spaces, and have purportedly significant roles in routine CNS immunosurveillance. Subarachnoid‐space macrophages and pericytes associated with CNS microvasculature are both considered to be critical in the presentation of recall antigens to this T cell population (29). At any given time, T lymphocytes represent over 80% of the approximately 150 000 cells normally found in the CSF of healthy individuals (29). As an absolute number, this somewhat diminutive quantity of cells may not be particularly relevant to immune responses within the brain parenchyma; however, it seems that these cells are relatively CSF enriched, given that lymphocytes typically compose less than 5% of all leukocytes present in circulating blood.
In its intact state, the BBB is thought to be poorly permeable to antibodies. This assumption stems from the observation that CSF titers in normal individuals are relatively low, especially in comparison to those measured in peripheral blood. Generally, the rate of immunoglobulin diffusion into the CNS varies depending on the molecular weight of a given protein (75); as an example, the physiological CSF/serum ratios for IgM and IgG have been quoted to range from 0.005% to 0.025% and 0.16% to 0.32%, respectively, reflecting the difference in size between these molecules (7). Although these limited ratios undoubtedly evidence CNS immune privilege to some degree, classic animal experiments have verified that, after both active and passive immunizations, corresponding antibodies can be detected within the CNS, specifically the brain, spinal cord and CSF (33). However, the reported fraction eventually isolated from these areas was again notably small—0.1% to 1% of that found in serum.
The theoretical possibility that even small amounts of antibody can cross the BBB and have physiologically relevant effector functions in the CNS is supported by the recent development of promising vaccines for patients with Alzheimer's disease (AD). These vaccines target amyloid‐β (AB), the cleavage product of amyloid precursor protein (APP); mutations in which have been shown to lead to parenchymal amyloid plaque accumulation 39, 91 in addition to other pathological features and clinical manifestations of AD 53, 60. Initial experiments using transgenic mice expressing mutant APP have shown that active immunization with the AB peptide reduces plaque burden and improves behavioral end points 13, 51, 88. This provided the first evidence that an immune response can be used as a potential treatment for AD, in theory by preventing formation of amyloid deposits and mediating clearance of preexisting plaques. Subsequent studies confirmed that the therapeutic effects of the vaccine are, at least in part, due to an antibody‐mediated mechanism. This was primarily demonstrated by animal experiments showing that peripherally administered AB‐specific antibody enters the CNS, localizes to plaques and achieves amyloid clearance mimicking that observed in previous mouse studies employing active immunization strategies 3, 4. Several hypotheses have been offered regarding the mechanism behind antibody‐mediated plaque clearance in AD 26, 94. Of these, one prominent theory states that passively administered antibody sequesters AB peptide in the periphery without crossing the BBB, thereby generating a concentration gradient favoring efflux out of the brain (24). Other suggested antibody mechanisms rely on passage of antibody across the BBB; these include direct plaque disaggregation (93) and Fc receptor‐mediated microglial phagocytosis (4).
Given these conclusions, the concept of BBB permeability in the absence of frank inflammation appears to be garnering support. However, it has long been asserted and widely accepted that in the presence of neuro‐inflammatory disease states—including experimental autoimmune encephalitis, meningitis and cancer—the BBB undergoes changes that alter its ability to block the migration of leukocytes and serum proteins into the CNS (23). Furthermore, by virtue of their existence, paraneoplastic syndromes clearly demonstrate that such changes in the BBB occur, and that these changes are in fact clinically significant. Most paraneoplastic neurological disorders (PND) are likely immune mediated (21), as suggested by the demonstration of antineural antibodies in the CNS of patients with peripheral tumors. These antibodies represent the body's natural immune reactivity against systemic tumor antigens, and cross‐reactivity with neurological structures has been found to result in significant morbidity. A number of paraneoplastic antibodies have been cited in the involvement of PND pathogenesis; these include anti‐Hu, anti‐Yo, anti‐Ri, anti‐CV2/CRMP5, anti‐Ma and anti‐amphiphysin antibodies (21).
Because pathological antibodies have been shown to cross the BBB in the context of malignancy, it follows that peripherally administered therapeutic antibodies should also have access to the intracerebral environment with physiologically relevant outcomes. The development of radiolabeled monoclonal antibodies (MAbs) for the diagnosis and treatment of brain tumors was first explored by Day and coworkers in 1965 (22), and since then, numerous studies have supported that MAbs are capable of localizing to intracerebral malignancies. Using radioiodinated antitenascin MAb 81C6, our group has shown that not only does 81C6 exhibit therapeutic activity in mice with intracranial human glioma xenografts, but that selective tumor localization also occurs in patients with a variety of intracranial malignancies following peripheral administration of the antibody (109). However, tumor‐specific uptake of 81C6 remained quite low at less than 5 × 10−3% of the injected dose per gram, and nonspecific antibody accumulation also took place in other tissues besides the brain including the liver, spleen and bone marrow. In contrast to what was observed with 81C6, results from later human studies using radiolabeled chimeric ch806, a MAb specific for the epidermal growth factor receptor class III variant (EGFRvIII) tumor antigen, suggest that higher‐percentage BBB penetration may be achieved in the absence of cross‐reactivity with systemic antigens, effectively creating an intracerebral antibody sink at the tumor site (90). Using single‐photon emission computed tomography, this potential effect was observed given the physiological localization of Indium‐111‐labeled ch806, which was noted to accumulate within intracranial target lesions without visual evidence of nonspecific, residual binding in normal tissues (Figure 1).
Figure 1.
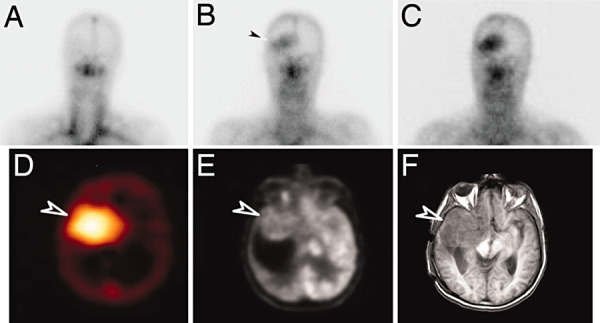
Targeting of glioma by radiolabeled chimeric monoclonal antibody directed against the EGFRvIII tumor antigen. (A–C) Planar images of the head and neck obtained on day 0 (A), day 3 (B), and day 7 (C) after infusion of 111In‐ch806. Initial blood pool activity is seen on day 0, and uptake of 111In‐ch806 in an anaplastic astrocytoma in the right frontal lobe is evident by day 3 (arrow) and increases by day 7. (D–F) Tumor‐specific uptake of 111In‐ch806 (arrow) is demonstrated in a SPECT image of the brain (D), 18F‐FDG (FDG, Fluorodeoxyglucose) positron emission tomography (E) and MRI (F). Figure reproduced with permission from reference (90).
TUMOR‐SPECIFIC REJECTION ANTIGENS
The landmark paper published over two decades ago by van Pel and Boon rekindled the then waning interest in cancer vaccine development when it suggested that even nonimmunogenic tumors display sufficiently “foreign,” and therefore immunologically susceptible antigen profiles (101). Since that time, a great deal of effort has gone toward characterizing a variety of human tumor antigens, the majority of which can now be placed into one of two main categories: those consisting of overexpressed normal gene products or, alternatively, those derived from mutations in somatic genes 36, 37.
Most well‐characterized targeted tumor antigens isolated to date correspond to overexpressed proteins that are also present in normal cells, two examples of which include CD20 and erbB2, proteins associated with lymphoma and breast cancer, respectively. The ability of antigens in this category to mediate optimal tumor rejection, however, is often compromised by the fact that proteins that are also found on normal cells have the potential to trigger immunologic tolerance to varying degrees. Notable exceptions to this limitation include antigens associated with fetal gene products, such as carcinoembryonic antigen (50), or those expressed solely in immunoprivileged, tissue‐specific sites like the testes. The latter group includes the melanoma MAGE, GAGE(100), and BAGE (11) family antigens, all of which, due to their limited expression, trigger little to no tolerance and should therefore make ideal tumor rejection antigens.
Cancer vaccination protocols that effectively target normal gene products invariably pose the risk of autoimmune toxicity (37). This untoward effect can be avoided to some extent by directing the immune response against a mutated protein specific only to tumor cells. As targets, these antigens have the advantage of avoiding central tolerance mechanisms, in theory, making them more suitable for tumor rejection. However, a limitation of these antigens is that they are generally patient specific as they often reflect random mutations associated with the inherent genetic instability of tumors 56, 59. Thus, to the extent that mutated gene products are incidental to the oncogenic process, they are conceivably restricted in their use as practical targets of widely applicable cancer vaccines. Conversely, although the majority of somatic mutations in tumors does appear to be sporadic (57), recent studies using high‐throughput screening have suggested that several functional mutations associated with, rather than incidental to, the oncogenic process are, in fact, not random, and that these variants are consistently shared among patients (98). The challenge, then, is to isolate and target these ideal antigens: frequent, highly specific, oncogenic mutations that are also absent from normal tissues, thereby avoiding the risk of autoimmunity. To date, few such antigens are known, although their discovery represents a potential boon for the further development of effective antitumor immunotherapies.
EGFRVIII TUMOR‐SPECIFIC ANTIGEN
Among the many antigens that have been shown to be overexpressed on tumor cells, the type I epidermal growth factor receptor (EGFR) represents one of the most frequently implicated cell‐surface markers for a wide range of human malignancies. Functionally, the EGFR has well‐characterized roles in oncogenesis and tumor progression, and as such, amplification and overexpression of the EGFR gene is considered a poor prognostic indicator (72). Regarding intracerebral cancers in particular, the EGFR gene is amplified in up to 50% and overexpressed in over 90% of GBM specimens 28, 49, suggesting significantly augmented cellular activity of this receptor in these tumors.
The EGFR is a 170‐kDa transmembrane glycoprotein, consisting of an extracellular ligand‐binding domain and an intracellular region with tyrosine kinase functionality (95). Activation via stimulatory interactions with growth factors—including epidermal growth factor (EGF) and transforming growth factor‐α—results in receptor dimerization and subsequent intracellular autophosphorylation on tyrosine residues, in turn leading to the activation of downstream molecules associated with cellular mitogenesis and survival (Figure 2) (14). Given the nature of these potentially oncogenic pathways, it was originally believed that the impact of EGFR on neoplastic processes was exclusively due to amplification of its corresponding gene. However, it is now clear that many tumors, including GBM, also express rearranged, aberrant forms of the EGFR gene that have significant physiological relevance 28, 32. Several of these mutations have been reported in the literature and are typically associated with tumors that also exhibit extensive wild‐type gene amplification 58, 107.
Figure 2.
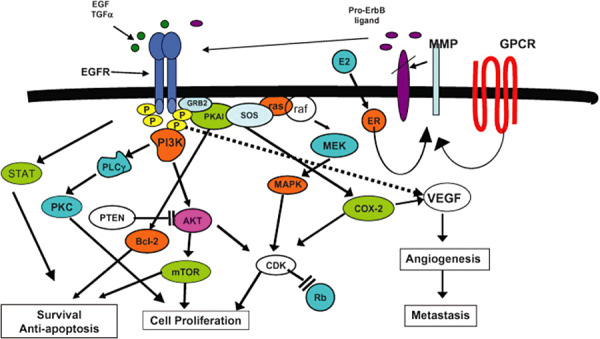
EGFR downstream signaling in cancer cells. Figure reproduced with permission from reference (6).
The most common and well‐characterized EGFR mutant was first identified in primary human GBM tumors and is commonly referred to as the EGFR class III variant (EGFRvIII). EGFRvIII is a constitutively active, ligand‐independent form of the EGF wild‐type receptor 5, 45, the expression of which has been shown to have tumorigenic effects, both augmenting proliferation and inhibiting apoptosis 5, 73. Specifically, EGFRvIII has also been shown to promote greater cellular motility 12, 76 as well as resistance to radiation and chemotherapy 54, 55, 68, characteristics often associated with highly malignant tumors.
A number of molecular mechanisms have been implicated in the oncogenic pathways coupled with EGFRvIII downstream signaling. In the absence of ligand binding and dimerization, for example, EGFRvIII has been observed to constitutively interact with adaptor proteins central to the Ras cascade 17, 77. Similarly, growth advantage in cells expressing EGFRvIII has been attributed at least in part to elevated phosphatidylinositol (PI) 3‐kinase levels and consequent activation of the c‐Jun N‐terminal kinase pathway 2, 70. The respective involvement of, and interplay among, these signals in neoplastic processes have yet to be fully described; however, it has been shown that malignant cells become dependent on these pathways to some extent, and that removal of such stimulation results in reduced cell survival (103).
Structurally, EGFRvIII is an 801 base pair in‐frame deletion of the wild‐type receptor that corresponds to mRNA exons 2–7, the absence of which leads to the translation of a truncated extracellular domain (Figure 3). A consequence of this deletion–mutation is the fusion of two otherwise distant portions of the molecule, which in turn creates an antigenic junction characterized by a novel glycine residue, flanked by amino acid sequences that are not typically adjacent in the wild‐type receptor 10, 58. This tumor‐specific epitope has been shown to be present on the surface tumor cells, yet completely absent from any normal adult tissues (46).
Figure 3.
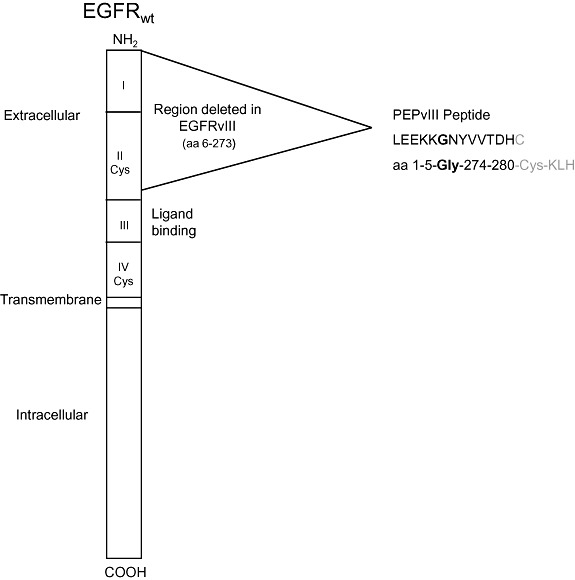
Schematic diagram of the EGFR wild‐type protein showing the area of in‐frame deletion which forms EGFRvIII. During the deletion, amino acids 6 and 273 are split forming a novel glycine at the junction of amino acids 5 and 274. PEPvIII is a 13 amino acid peptide with a terminal cysteine added to facilitate conjugation to KLH. Figure reproduced with permission from reference (85).
Immunohistochemical (IHC) analysis represents one of the most common assays used to identify the EGFRvIII mutant along with a number of second messenger molecules also expressed in malignant cells (Figure 4). Alternative approaches to IHC which employ molecular techniques such as Western blotting and reverse transcription‐polymerase chain reaction assays are currently being explored and have confirmed the specific expression of EGFRvIII in human GBM specimens; to date, data derived from IHC studies have been shown to be consistent with results obtained using other methods (31). As evidenced by IHC, EGFRvIII is consistently expressed in a wide variety of cancers, and can be found in approximately 20% of GBM specimens (69). Within such tumor samples, the proportion of EGFRvIII‐expressing cells has been shown to range from 37% to 86% (105), suggesting that cells within EGFRvIII‐positive tumors may translate the variant receptor with at least some level of homogeneity. Thus, given its oncogenic properties, inherent tumor specificity and frequent expression in malignancy, the EGFRvIII mutation represents a particularly attractive tumor‐specific target for the development of anticancer immunotherapies 52, 106.
Figure 4.
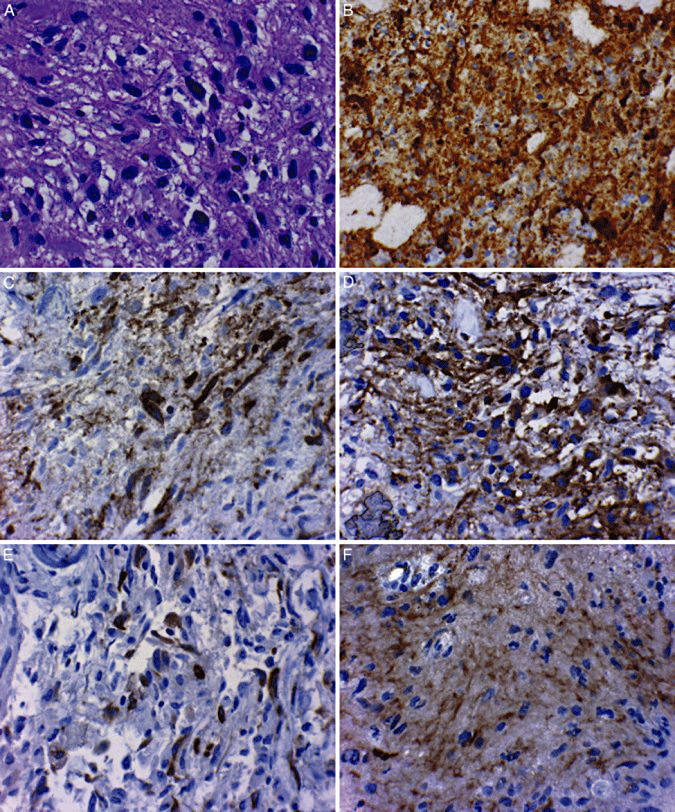
A. High‐grade astrocytoma used in subsequent immunohistochemical assays (hematoxylin–eosin, original magnification ×400). B. Antiepidermal growth factor receptor (anti‐EGFR) wild‐type immunohistochemistry showing strong diffuse cytoplasmic immunoreactivity, a pattern that is frequently associated with EGFR genetic amplification (original magnification ×400). C. Anti‐EGFRvIII immunohistochemical reactivity exhibiting strong cytoplasmic localization. EGFRvIII immunoreactivity is most commonly encountered in tumors also exhibiting amplification of the EGFR locus (original magnification ×400). D. Antiphosphatase and tensin homolog (anti‐PTEN) immunoreactivity demonstrating 80% of tumor cells with cytoplasmic reactivity, a pattern associated with an intact PTEN status in the tumor (original magnification ×400). E. Anti‐phospho‐S6 immunohistochemistry revealing approximately 20% of tumor cells labeling, indicating this messenger is activated in this tumor (original magnification ×400). F. Anti‐phospho‐Akt immunohistochemistry demonstrating approximately 80% of tumor cells labeling, indicating this messenger is activated in this tumor (original magnification ×400). Figure reproduced with permission from reference (63).
NATURE OF THE PEPTIDE VACCINE
Generally, immunotherapy can be divided into either active or passive approaches (81). Active immunizations rely on the natural immune system to mount physiological responses against specific antigens that are either inoculated directly into the body or are instead presented on autologous APCs. Prior to vaccination, APCs are pulsed with the appropriate antigen, cancer cells or lysates thereof, and are thereby loaded with the immunogenic material of interest. Passive vaccination strategies involve either the direct infusion of antibodies or, alternatively, the adoptive transfer of antigen‐specific T lymphocytes.
Currently, a number of immunotherapeutic approaches targeting the unique EGFRvIII antigen are under investigation. Given the technical difficulty and relatively high cost of dendritic cell (DC) vaccination therapy, the most promising and practical active vaccination format to date is a peptide derived from the novel fusion junction amino acid sequence. PEPvIII (H‐Leu‐Glu‐Glu‐Lys‐Lys‐Gln‐Asn‐Tyr‐Val‐Val‐Thr‐Asp‐His‐Cys‐OH) (71) is a well‐characterized, EGFRvIII‐specific, 14‐mer peptide that has been shown, when coupled to keyhole limpet hemocyanin (KLH), to elicit both humoral and cellular immune responses. Our group has extensive experience with PEPvIII‐KLH, and we have clearly demonstrated the induction of considerable EGFRvIII‐specific immune responses in both murine tumor models and early clinical trials.
EGFRVIII: PRECLINICAL STUDIES
Our group has shown that passive administration of EGFRvIII‐specific antibodies Y10 and L8A4 (unarmed murine IgG2a and IgG1, respectively) leads to significant tumor growth inhibition in subcutaneous murine melanoma models. These studies, which use syngeneic tumors transfected with a murine homolog of the variant receptor (msEGFRvIII), have shown that while these two MAbs achieve therapeutic efficacy when given intraperitoneally, only those mice treated with Y10 exhibit lasting tumor‐free survival after treatment is discontinued (83). Evidence from in vitro studies suggests that Y10 has the ability to mediate a wide range of effector functions when incubated with cells expressing msEGFRvIII. These functions include the inhibition of DNA synthesis and cellular proliferation, as well as the activation of autologous, complement‐mediated and antibody‐dependent cell‐mediated cytotoxicity.
Active vaccination strategies targeted against msEGFRvIII in syngeneic murine tumor models have also been proven to be effective. Following intraperitoneal injection with DCs pulsed with PEPvIII‐KLH, C3H mice that had previously been challenged with intracerebral tumors demonstrated a significant increase in median survival. Furthermore, all the mice in this study survived rechallenge with tumor, suggesting that immunization was sufficient to create long‐term immunological memory against the msEGFRvIII antigen in this model system (42). Following this experiment, we conducted a similar study in which C3H mice were treated using a one‐time vaccination, this time with PEPvIII‐KLH in complete Freund's adjuvant as opposed to the DC vaccine. This vaccine protocol also resulted in increased median survival and, ultimately, long‐term survival in nearly half of the mice (43). Notably, mice with tumors that failed to exhibit responses to the PEPvIII‐KLH vaccine were found to have IHC evidence of down‐regulated or completely absent EGFRvIII expression, suggesting that antigen escape variants may be associated with failure to adequately treat some tumors.
EGFRVIII: CLINICAL STUDIES
Our group has also demonstrated, in clinical trials, induction of EGFRvIII‐specific immunity with vaccines targeted against the EGFRvIII tumor‐specific antigen. A number of EGFRvIII‐derived cytotoxic T lymphocyte epitopes have been characterized to date (108), and previous data have shown that EGFRvIII‐specific antibody titers, while absent in normal volunteers, may be detectable in patients with tumors expressing the mutant receptor (78). It is still unclear, however, whether cellular or humoral responses will ultimately provide the critical mediators for specific antitumor eradication using our approach.
Our first clinical study evaluating the toxicity and potential efficacy of EGFRvIII‐based vaccinations began with a Phase I trial (VICTORI) (86) conducted at Duke University Medical Center (PI: John H. Sampson).
Fifteen adults with newly diagnosed GBM (WHO grade III or IV) were enrolled in the study; criteria for eligibility did not include EGFRvIII expression. Of the 15 patients, three did not ultimately receive vaccine due to progression of their tumors during external beam radiotherapy (EBRT). Following gross‐total tumor resection and completion of EBRT, 12 patients underwent leukapheresis to obtain peripheral blood mononuclear cells in preparation for DC generation and immunologic monitoring. Prior to inoculation, DCs were pulsed for 2 hours with 500 µg PEPvIII peptide (Anaspec, San Jose, CA, USA) conjugated to KLH (Biosyn, Carlsbad, CA, USA). In total, patients received up to 1.1 × 108 DCs in three equal doses, injected intradermally every 2 weeks into the upper thigh, 10 cm below the inguinal ligament. Patients were followed for toxicity and evidence of radiographic or clinical progression.
Patients in the VICTORI trial did not suffer serious adverse events exceeding Grade II toxicity at any DC dose tested (National Cancer Institute Common Toxicity Criteria). Blood drawn from patients following vaccination showed ex vivo evidence of antigen‐specific cellular and humoral immune responses. Median survival for the 12 patients was 18.7 months after vaccination (CI95 14.5, 25.6) and 22.8 months after histological diagnosis (CI95 17.5, 29). These outcomes improve on what would have been expected by chance, according to Curran's recursive partition analysis (20). Eight of the 12 patients in this study belonged to group III, and the remaining four belonged to group IV, which have estimated survivals of 17.9 and 11.1 months, respectively. While nine of the 12 patients in our study surpassed these estimates, the increase in survival was not statistically significant (P = 0.083; binomial proportions), although these results may be negatively biased by the fact that EGFRvIII expression was not a criterion for inclusion in this Phase I toxicity trial.
Nevertheless, the outcomes associated with our DC‐based, PEPvIII‐specific vaccine were encouraging and warranted further testing at different centers; however, the inherent cost and variability associated with autologous DC manufacturing made this approach impractical on a large scale. Thus, given the success of our preclinical studies, we decided to proceed with a Phase II multicenter trial (ACTIVATE) (41) without the use of DCs, instead administering PEPvIII‐KLH directly in combination with granulocyte macrophage‐colony stimulating factor (GM‐CSF).
ACTIVATE, a Phase II, multicenter clinical trial conducted at Duke (PI: John H. Sampson) and University of Texas, M.D. Anderson Cancer Center (PI: Amy B. Heimberger), enrolled 19 adults who all had EGFRvIII‐expressing, newly diagnosed primary GBM (WHO Grade IV). Prior to receiving the KLH‐conjugated peptide vaccines, patients underwent >95% volumetric tumor resection, along with standard of care radiation therapy with concurrent TMZ. Vaccinations consisted of intradermal injections with 500 µg PEPvIII‐KLH (Anaspec) and GM‐CSF, administered near the inguinal region in the upper thigh, on alternating sides. The first three vaccines were given biweekly, followed by monthly injections until radiographic evidence of tumor progression or death.
Similar to what was observed in VICTORI, patients participating in ACTIVATE did not experience serious adverse events aside from local reactions at the injection site. We demonstrated that this vaccine formulation elicits both humoral (89) and delayed‐type hypersensitivity immune responses specific for PEPvIII and EGFRvIII in a number of patients, and that detection of these responses predicts greater median overall survival (OS). Median time‐to‐progression (TTP) following surgery in patients who received the vaccine is 12 months (n = 12), exceeding a median TTP of 7.1 months (n = 29) calculated from a historical matched unvaccinated control group (P = 0.0058). If and when tumors recurred, pathological samples were obtained and evaluated by IHC to determine EGFRvIII expression. Of the specimens examined in this trial, none were found to contain cells that display positive staining for EGFRvIII.
Following ACTIVATE, our lab initiated the ACT II trial (84), which enrolled a total of 21 patients who essentially followed the same treatment scheme as those in ACTIVATE, except for the addition of two different TMZ dosing schedules concurrent with vaccination cycles; patients either received 200 mg/m2 TMZ × 5/28 days [ACT IIA (n = 13)] or 100 mg/m2 TMZ × 21/28 days [ACT IIB (n = 8)]. While grade 2 TMZ‐associated lymphopenia was observed in the majority of ACT II patients, we found that all immune responses were unexpectedly either sustained or enhanced with successive TMZ treatments. The seemingly paradoxical relationship between TMZ‐induced lymphopenia and improved PEPvIII‐KLH‐specific immunogenicity is currently under further investigation.
In summary, these trials to date collectively show that vaccination with a peptide containing the PEPvIII tumor epitope safely elicits a specific immune response against EGFRvIII, and that this approach might be effective against cancers bearing the variant antigen. While our group has demonstrated significantly greater TTP and OS in GBM patients who have received the PEPvIII‐KLH vaccine, definitive evidence for this promising effect will require confirmation from our ongoing randomized Phase III clinical trials.
DISCUSSION
While the mechanisms underlying the beneficial effects of our vaccine in patients with GBM are still unclear, it is our hope that additional experience with PEPvIII‐KLH will elucidate our general understanding of various peptide vaccination strategies and their potential role in eliciting effective antitumor responses. Previous trials employing peptide vaccines have targeted a wide range of cancers including those of the colon, prostate, breast, cervix, pancreas and ovaries. The most convincing evidence in favor of peptide vaccine efficacy comes from the melanoma literature, in which a number of tumor‐associated antigens have been specifically targeted with relative success, some of which include MART‐1 (16), MAGE‐3 (61), tyrosinase (87), and NY‐ESO‐1 (48). Data from many early clinical vaccine trials for melanoma and other neoplasms have been fairly encouraging, although some have claimed that these results have been overly optimistic due to a reliance on subjective or “soft” end points. In 2004, Rosenberg et al suggested that criteria for clinical responses to cancer vaccines should only include objective measurements such as those denoting tumor size and volume. Taking these new parameters into account, he reassessed 35 National Cancer Institute trials involving a wide range of cancers and concluded that overall, there were only seven objective tumor responses out of a total of 175 patients (4%) who had received some form of peptide vaccine. At that time, the implications of these results could not be understated, and thus represented a turning point for the field of cancer immunotherapy. However, the conclusions drawn by Rosenberg and colleagues have since been challenged as excessively pessimistic and potentially misleading 67, 99. Opposing views suggest that the literature instead supports more favorable tumor response rates (10%) following treatment with peptide vaccines, and that this frequency may even be higher when employing the objective criteria as mandated by Response Evaluation Criteria in Solid Tumors (97). In addition, it was brought to attention that many patients receive vaccinations only after completion of standard chemotherapeutic regimens, conceivably making their tumors more aggressive and resistant to treatment when compared with those who had not received therapy. Finally, it has been suggested that a number of immunological variables (eg, adjuvants, HLA haplotypes) have yet to be explored in the context of peptide vaccines, knowledge of which might ultimately improve efficacy of existing therapeutic regimens. Regardless of the ongoing discussion on the future of active cancer vaccination strategies, Rosenberg et al and other authors agree that, at this stage, it would be unwise to interpret any lack of clinical evidence as an “investigational dead end,” insofar as present shortcomings may simply reflect the need for further exploration (82).
As the field of tumor immunology moves forward, in addition to the standardization of and adherence to objective end points, there are still a number of issues regarding the specific targeting of cancer epitopes by peptide vaccines that must be addressed. It is known, for example, that although a variety of tumors including malignant glioma have been found to express the EGFRvIII mutation, cells within these cancers often exhibit significant antigenic heterogeneity 8, 9, 104, 105. This confounds immunotherapeutic approaches designed to target single tumor‐specific antigens, as even effective vaccines will fail to target those cells in the tumor that do not happen to express a given epitope. We observed the potential consequences of this issue in both murine studies (43) and clinical trials, in which the majority of recurrent tumors from vaccinated patients no longer expressed EGFRvIII. Thus, in the context of cancer vaccines, greater antitumor effects may be achieved with the development of multiantigenic vaccines that target the various aberrant biological processes often present in GBM and other tumors (34); the recently characterized expression of human cytomegalovirus antigens in GBM, for instance, may provide multiple targets for such an approach 18, 66. Alternatively, strategies that enrich EGFRvIII‐positive cell populations prior to treatment may also prove to increase antitumor efficacy.
GBM is a devastating disease, and despite recent advances in antitumor therapy, there is still a pressing need for novel and effective approaches designed to potently eradicate tumor cells while minimizing toxicity to neighboring tissues. The potential ability to harness and redirect the cytotoxic power and inherent specificity of the immune system against neoplastic cells provides one such approach. As reviewed herein, our lab has shown that peptide vaccines can be employed to safely generate both B and T cell‐mediated immune responses against tumor‐specific epitopes such as the EGFRvIII antigen. Moreover, we have demonstrated that these responses are efficacious in murine models, and that similar therapeutic outcomes may be expected in humans with malignant glioma, even in the context of severe immunosuppression.
REFERENCES
- 1. Aloisi F, Ria F, Adorini L (2000) Regulation of T‐cell responses by CNS antigen‐presenting cells: different roles for microglia and astrocytes. Immunol Today 21:141–147. [DOI] [PubMed] [Google Scholar]
- 2. Antonyak MA, Moscatello DK, Wong AJ (1998) Constitutive activation of c‐Jun N‐terminal kinase by a mutant epidermal growth factor receptor. J Biol Chem 273:2817–2822. [DOI] [PubMed] [Google Scholar]
- 3. Bacskai BJ, Kajdasz ST, Christie RH, Carter C, Games D, Seubert P et al (2001) Imaging of amyloid‐beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med 7:369–372. [DOI] [PubMed] [Google Scholar]
- 4. Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H et al (2000) Peripherally administered antibodies against amyloid beta‐peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 6:916–919. [DOI] [PubMed] [Google Scholar]
- 5. Batra SK, Castelino‐Prabhu S, Wikstrand CJ, Zhu X, Humphrey PA, Friedman HS, Bigner DD (1995) Epidermal growth factor ligand‐independent, unregulated, cell‐transforming potential of a naturally occurring human mutant EGFRvIII gene. Cell Growth Differ 6:1251–1259. [PubMed] [Google Scholar]
- 6. Bianco R, Gelardi T, Damiano V, Ciardiello F, Tortora G (2007) Rational bases for the development of EGFR inhibitors for cancer treatment. Int J Biochem Cell Biol 39:1416–1431. [DOI] [PubMed] [Google Scholar]
- 7. Bickel U (1995) Antibody delivery through the blood‐brain barrier. Adv Drug Deliv Rev 15:53–72. [PubMed] [Google Scholar]
- 8. Bigner DD (1981) Biology of gliomas: potential clinical implications of glioma cellular heterogeneity. Neurosurgery 9:320–326. [PubMed] [Google Scholar]
- 9. Bigner DD, Schold C, Bigner SH, Bullard DE, Wikstrand C (1981) How heterogeneous are gliomas? Cancer Treat Rep 65(Suppl. 2):45–49. [PubMed] [Google Scholar]
- 10. Bigner SH, Humphrey PA, Wong AJ, Vogelstein B, Mark J, Friedman HS, Bigner DD (1990) Characterization of the epidermal growth factor receptor in human glioma cell lines and xenografts. Cancer Res 50:8017–8022. [PubMed] [Google Scholar]
- 11. Boel P, Wildmann C, Sensi ML, Brasseur R, Renauld JC, Coulie P et al (1995) BAGE: a new gene encoding an antigen recognized on human melanomas by cytolytic T lymphocytes. Immunity 2:167–175. [DOI] [PubMed] [Google Scholar]
- 12. Boockvar JA, Kapitonov D, Kapoor G, Schouten J, Counelis GJ, Bogler O et al (2003) Constitutive EGFR signaling confers a motile phenotype to neural stem cells. Mol Cell Neurosci 24:1116–1130. [DOI] [PubMed] [Google Scholar]
- 13. Brendza RP, Holtzman DM (2006) Amyloid‐beta immunotherapies in mice and men. Alzheimer Dis Assoc Disord 20:118–123. [DOI] [PubMed] [Google Scholar]
- 14. Carpenter G, Cohen S (1990) Epidermal growth factor. J Biol Chem 265:7709–7712. [PubMed] [Google Scholar]
- 15. CBTRUS (2008) Statistical report: primary brain tumors in The United States, 2000–2004. Published by The Central Brain Tumor Registry of The United States. [Google Scholar]
- 16. Cebon J, Jager E, Shackleton MJ, Gibbs P, Davis ID, Hopkins W et al (2003) Two phase I studies of low dose recombinant human IL‐12 with Melan‐A and influenza peptides in subjects with advanced malignant melanoma. Cancer Immun 3:7. [PubMed] [Google Scholar]
- 17. Chu CT, Everiss KD, Wikstrand CJ, Batra SK, Kung HJ, Bigner DD (1997) Receptor dimerization is not a factor in the signalling activity of a transforming variant epidermal growth factor receptor (EGFRvIII). Biochem J 324:855–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH et al (2002) Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res 62:3347–3350. [PubMed] [Google Scholar]
- 19. Cserr HF, Knopf PM (1992) Cervical lymphatics, the blood‐brain barrier and the immunoreactivity of the brain: a new view. Immunol Today 13:507–512. [DOI] [PubMed] [Google Scholar]
- 20. Curran WJ Jr, Scott CB, Horton J, Nelson JS, Weinstein AS, Fischbach AJ et al (1993) Recursive partitioning analysis of prognostic factors in three Radiation Therapy Oncology Group malignant glioma trials. J Natl Cancer Inst 85:704–710. [DOI] [PubMed] [Google Scholar]
- 21. Dalmau J, Rosenfeld MR (2008) Paraneoplastic syndromes of the CNS. Lancet Neurol 7:327–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Day ED, Lassiter S, Woodhall B, Mahaley JL, Mahaley MS Jr (1965) The localization of radioantibodies in human brain tumors. I. Preliminary exploration. Cancer Res 25:773–778. [PubMed] [Google Scholar]
- 23. De Vries HE, Kuiper J, De Boer AG, Van Berkel TJ, Breimer DD (1997) The blood‐brain barrier in neuroinflammatory diseases. Pharmacol Rev 49:143–155. [PubMed] [Google Scholar]
- 24. DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM (2001) Peripheral anti‐A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 98:8850–8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Deshpande SS, Angkeow P, Huang J, Ozaki M, Irani K (2000) Rac1 inhibits TNF‐alpha‐induced endothelial cell apoptosis: dual regulation by reactive oxygen species. FASEB J 14:1705–1714. [DOI] [PubMed] [Google Scholar]
- 26. Duff K (1999) Curing amyloidosis: will it work in humans? Trends Neurosci 22:485–486. [DOI] [PubMed] [Google Scholar]
- 27. Ehrlich P, Bolduan C (1906) Collected Studies on Immunity, 1st edn. J. Wiley & Sons: New York. [Google Scholar]
- 28. Ekstrand AJ, James CD, Cavenee WK, Seliger B, Pettersson RF, Collins VP (1991) Genes for epidermal growth factor receptor, transforming growth factor alpha, and epidermal growth factor and their expression in human gliomas in vivo . Cancer Res 51:2164–2172. [PubMed] [Google Scholar]
- 29. Engelhardt B, Ransohoff RM (2005) The ins and outs of T‐lymphocyte trafficking to the CNS: anatomical sites and molecular mechanisms. Trends Immunol 26:485–495. [DOI] [PubMed] [Google Scholar]
- 30. Fabry Z, Raine CS, Hart MN (1994) Nervous tissue as an immune compartment: the dialect of the immune response in the CNS. Immunol Today 15:218–224. [DOI] [PubMed] [Google Scholar]
- 31. Feldkamp MM, Lala P, Lau N, Roncari L, Guha A (1999) Expression of activated epidermal growth factor receptors, Ras‐guanosine triphosphate, and mitogen‐activated protein kinase in human glioblastoma multiforme specimens. Neurosurgery 45:1442–1453. [DOI] [PubMed] [Google Scholar]
- 32. Frederick L, Wang XY, Eley G, James CD (2000) Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res 60:1383–1387. [PubMed] [Google Scholar]
- 33. Freund J (1930) Accumulation of antibodies in the central nervous system. J Exp Med 51:889–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A et al (2007) Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev 21:2683–2710. [DOI] [PubMed] [Google Scholar]
- 35. Gehrmann J, Matsumoto Y, Kreutzberg GW (1995) Microglia: intrinsic immuneffector cell of the brain. Brain Res Brain Res Rev 20:269–287. [DOI] [PubMed] [Google Scholar]
- 36. Gilboa E (1999) The makings of a tumor rejection antigen. Immunity 11:263–270. [DOI] [PubMed] [Google Scholar]
- 37. Gilboa E (2004) The promise of cancer vaccines. Nat Rev Cancer 4:401–411. [DOI] [PubMed] [Google Scholar]
- 38. Goldmann J, Kwidzinski E, Brandt C, Mahlo J, Richter D, Bechmann I (2006) T cells traffic from brain to cervical lymph nodes via the cribroid plate and the nasal mucosa. J Leukoc Biol 80:797–801. [DOI] [PubMed] [Google Scholar]
- 39. Hardy J (1997) Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci 20:154–159. [DOI] [PubMed] [Google Scholar]
- 40. Hart DN, Fabre JW (1981) Demonstration and characterization of Ia‐positive dendritic cells in the interstitial connective tissues of rat heart and other tissues, but not brain. J Exp Med 154:347–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Heimberger A, Hussain SF, Aldape K, Sawaya R, Archer GA, Friedman H et al (2006) Tumor‐specific peptide vaccination in newly‐diagnosed patients with GBM. J Clin Oncol 24: abstr 2529. [Google Scholar]
- 42. Heimberger AB, Archer GE, Crotty LE, McLendon RE, Friedman AH, Friedman HS et al (2002) Dendritic cells pulsed with a tumor‐specific peptide induce long‐lasting immunity and are effective against murine intracerebral melanoma. Neurosurgery 50:158–164; discussion 64–66. [DOI] [PubMed] [Google Scholar]
- 43. Heimberger AB, Crotty LE, Archer GE, Hess KR, Wikstrand CJ, Friedman AH et al (2003) Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clin Cancer Res 9:4247–4254. [PubMed] [Google Scholar]
- 44. Hickey WF (1991) Migration of hematogenous cells through the blood‐brain barrier and the initiation of CNS inflammation. Brain Pathol 1:97–105. [DOI] [PubMed] [Google Scholar]
- 45. Hou ST, Wang CC, Chu ML (1997) Ribotyping and random amplification of polymorphic DNA for nosocomial Enterobacter cloacae isolates in a pediatric intensive‐care unit. Infect Control Hosp Epidemiol 18:769–771. [DOI] [PubMed] [Google Scholar]
- 46. Humphrey PA, Wong AJ, Vogelstein B, Zalutsky MR, Fuller GN, Archer GE et al (1990) Anti‐synthetic peptide antibody reacting at the fusion junction of deletion‐mutant epidermal growth factor receptors in human glioblastoma. Proc Natl Acad Sci USA 87:4207–4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Imperato JP, Paleologos NA, Vick NA (1990) Effects of treatment on long‐term survivors with malignant astrocytomas. Ann Neurol 28:818–822. [DOI] [PubMed] [Google Scholar]
- 48. Jager E, Gnjatic S, Nagata Y, Stockert E, Jager D, Karbach J et al (2000) Induction of primary NY‐ESO‐1 immunity: CD8+ T lymphocyte and antibody responses in peptide‐vaccinated patients with NY‐ESO‐1+ cancers. Proc Natl Acad Sci USA 97:12198–12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jaros E, Perry RH, Adam L, Kelly PJ, Crawford PJ, Kalbag RM et al (1992) Prognostic implications of p53 protein, epidermal growth factor receptor, and Ki‐67 labelling in brain tumours. Br J Cancer 66:373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kass E, Schlom J, Thompson J, Guadagni F, Graziano P, Greiner JW (1999) Induction of protective host immunity to carcinoembryonic antigen (CEA), a self‐antigen in CEA transgenic mice, by immunizing with a recombinant vaccinia‐CEA virus. Cancer Res 59:676–683. [PubMed] [Google Scholar]
- 51. Koller MF, Mohajeri MH, Huber M, Wollmer MA, Roth Z'graggen BV, Sandmeier E et al (2004) Active immunization of mice with an Abeta‐Hsp70 vaccine. Neurodegener Dis 1:20–28. [DOI] [PubMed] [Google Scholar]
- 52. Kuan CT, Wikstrand CJ, Bigner DD (2001) EGF mutant receptor vIII as a molecular target in cancer therapy. Endocr Relat Cancer 8:83–96. [DOI] [PubMed] [Google Scholar]
- 53. Lahiri DK, Chen DM, Lahiri P, Bondy S, Greig NH (2005) Amyloid, cholinesterase, melatonin, and metals and their roles in aging and neurodegenerative diseases. Ann N Y Acad Sci 1056:430–449. [DOI] [PubMed] [Google Scholar]
- 54. Lammering G, Hewit TH, Holmes M, Valerie K, Hawkins W, Lin PS et al (2004) Inhibition of the type III epidermal growth factor receptor variant mutant receptor by dominant‐negative EGFR‐CD533 enhances malignant glioma cell radiosensitivity. Clin Cancer Res 10:6732–6743. [DOI] [PubMed] [Google Scholar]
- 55. Lammering G, Valerie K, Lin PS, Hewit TH, Schmidt‐Ullrich RK (2004) Radiation‐induced activation of a common variant of EGFR confers enhanced radioresistance. Radiother Oncol 72:267–273. [DOI] [PubMed] [Google Scholar]
- 56. Lengauer C, Kinzler KW, Vogelstein B (1998) Genetic instabilities in human cancers. Nature 396:643–649. [DOI] [PubMed] [Google Scholar]
- 57. Lennerz V, Fatho M, Gentilini C, Frye RA, Lifke A, Ferel D et al (2005) The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc Natl Acad Sci USA 102:16013–16018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Libermann TA, Nusbaum HR, Razon N, Kris R, Lax I, Soreq H et al (1985) Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature 313:144–147. [DOI] [PubMed] [Google Scholar]
- 59. Loeb LA (2001) A mutator phenotype in cancer. Cancer Res 61:3230–3239. [PubMed] [Google Scholar]
- 60. Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L et al (1999) Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol 155:853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Marchand M, Van Baren N, Weynants P, Brichard V, Dreno B, Tessier MH et al (1999) Tumor regressions observed in patients with metastatic melanoma treated with an antigenic peptide encoded by gene MAGE‐3 and presented by HLA‐A1. Int J Cancer 80:219–230. [DOI] [PubMed] [Google Scholar]
- 62. Masson F, Calzascia T, Di Berardino‐Besson W, De Tribolet N, Dietrich PY, Walker PR (2007) Brain microenvironment promotes the final functional maturation of tumor‐specific effector CD8+ T cells. J Immunol 179:845–853. [DOI] [PubMed] [Google Scholar]
- 63. McLendon RE, Turner K, Perkinson K, Rich J (2007) Second messenger systems in human gliomas. Arch Pathol Lab Med 131:1585–1590. [DOI] [PubMed] [Google Scholar]
- 64. Medawar PB (1948) Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br J Exp Pathol 29:58–69. [PMC free article] [PubMed] [Google Scholar]
- 65. Mitchell DA, Fecci PE, Sampson JH (2008) Immunotherapy of malignant brain tumors. Immunol Rev 222:70–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mitchell DA, Xie W, Schmittling R, Learn C, Friedman A, McLendon RE, Sampson JH (2008) Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neurooncol 10:10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mocellin S, Mandruzzato S, Bronte V, Marincola FM (2004) Cancer vaccines: pessimism in check. Nat Med 10:1278–1279; author reply 9–80. [DOI] [PubMed] [Google Scholar]
- 68. Montgomery RB, Guzman J, O'Rourke DM, Stahl WL (2000) Expression of oncogenic epidermal growth factor receptor family kinases induces paclitaxel resistance and alters beta‐tubulin isotype expression. J Biol Chem 275:17358–17363. [DOI] [PubMed] [Google Scholar]
- 69. Moscatello DK, Holgado‐Madruga M, Godwin AK, Ramirez G, Gunn G, Zoltick PW et al (1995) Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res 55:5536–5539. [PubMed] [Google Scholar]
- 70. Moscatello DK, Holgado‐Madruga M, Emlet DR, Montgomery RB, Wong AJ (1998) Constitutive activation of phosphatidylinositol 3‐kinase by a naturally occurring mutant epidermal growth factor receptor. J Biol Chem 273:200–206. [DOI] [PubMed] [Google Scholar]
- 71. Moscatello DK, Ramirez G, Wong AJ (1997) A naturally occurring mutant human epidermal growth factor receptor as a target for peptide vaccine immunotherapy of tumors. Cancer Res 57:1419–1424. [PubMed] [Google Scholar]
- 72. Neal DE, Sharples L, Smith K, Fennelly J, Hall RR, Harris AL (1990) The epidermal growth factor receptor and the prognosis of bladder cancer. Cancer 65:1619–1625. [DOI] [PubMed] [Google Scholar]
- 73. Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK, Huang HJ (1994) A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci USA 91:7727–7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Owens T, Renno T, Taupin V, Krakowski M (1994) Inflammatory cytokines in the brain: does the CNS shape immune responses? Immunol Today 15:566–571. [DOI] [PubMed] [Google Scholar]
- 75. Pardridge WM (1991) Peptide Drug Delivery to the Brain. Raven Press: New York. [Google Scholar]
- 76. Pedersen MW, Tkach V, Pedersen N, Berezin V, Poulsen HS (2004) Expression of a naturally occurring constitutively active variant of the epidermal growth factor receptor in mouse fibroblasts increases motility. Int J Cancer 108:643–653. [DOI] [PubMed] [Google Scholar]
- 77. Prigent SA, Nagane M, Lin H, Huvar I, Boss GR, Feramisco JR et al (1996) Enhanced tumorigenic behavior of glioblastoma cells expressing a truncated epidermal growth factor receptor is mediated through the Ras‐Shc‐Grb2 pathway. J Biol Chem 271:25639–25645. [DOI] [PubMed] [Google Scholar]
- 78. Purev E, Cai D, Miller E, Swoboda R, Mayer T, Klein‐Szanto A et al (2004) Immune responses of breast cancer patients to mutated epidermal growth factor receptor (EGF‐RvIII, Delta EGF‐R, and de2‐7 EGF‐R). J Immunol 173:6472–6480. [DOI] [PubMed] [Google Scholar]
- 79. Ransohoff RM, Kivisakk P, Kidd G (2003) Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol 3:569–581. [DOI] [PubMed] [Google Scholar]
- 80. Rosenberg SA (2001) Progress in human tumour immunology and immunotherapy. Nature 411:380–384. [DOI] [PubMed] [Google Scholar]
- 81. Rosenberg SA (2004) Shedding light on immunotherapy for cancer. N Engl J Med 350:1461–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Rosenberg SA, Yang JC, Restifo NP (2004) Cancer immunotherapy: moving beyond current vaccines. Nat Med 10:909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sampson JH, Crotty LE, Lee S, Archer GE, Ashley DM, Wikstrand CJ et al (2000) Unarmed, tumor‐specific monoclonal antibody effectively treats brain tumors. Proc Natl Acad Sci USA 97:7503–7508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sampson JH, Archer GE, Bigner D, Davis T, Friedman HS, Keler T et al (2008) Effect of EGFRvIII‐targeted vaccine (CDX‐110) on immune response and TTP when given with simultaneous standard and continuous temozolomide in patients with GBM. J Clin Oncol 26: abstr 2011. [Google Scholar]
- 85. Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Bigner DD (2008) Tumor‐specific immunotherapy targeting the EGFRvIII mutation in patients with malignant glioma. Semin Immunol 20(5):267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sampson JH, Archer GE, Mitchell DA, Heimberger A, Herndon JE 2nd, Lally‐Goss D et al (2009) An epidermal growth factor receptor variant III‐targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme (submitted for publication). [DOI] [PMC free article] [PubMed]
- 87. Scheibenbogen C, Schmittel A, Keilholz U, Allgauer T, Hofmann U, Max R et al (2000) Phase 2 trial of vaccination with tyrosinase peptides and granulocyte‐macrophage colony‐stimulating factor in patients with metastatic melanoma. J Immunother 23:275–281. [DOI] [PubMed] [Google Scholar]
- 88. Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T et al (1999) Immunization with amyloid‐beta attenuates Alzheimer‐disease‐like pathology in the PDAPP mouse. Nature 400:173–177. [DOI] [PubMed] [Google Scholar]
- 89. Schmittling RJ, Archer GE, Mitchell DA, Heimberger A, Pegram C, Herndon JE et al (2008) Detection of humoral response in patients with glioblastoma receiving EGFRvIII‐KLH vaccines. J Immunol Methods 339:74–81. [DOI] [PubMed] [Google Scholar]
- 90. Scott AM, Lee FT, Tebbutt N, Herbertson R, Gill SS, Liu Z et al (2007) A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors. Proc Natl Acad Sci USA 104:4071–4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Selkoe DJ (1999) Translating cell biology into therapeutic advances in Alzheimer's disease. Nature 399(6738 Suppl.):A23–A31. [DOI] [PubMed] [Google Scholar]
- 92. Serot JM, Foliguet B, Bene MC, Faure GC (1997) Ultrastructural and immunohistological evidence for dendritic‐like cells within human choroid plexus epithelium. Neuroreport 8:1995–1998. [DOI] [PubMed] [Google Scholar]
- 93. Solomon B, Koppel R, Frankel D, Hanan‐Aharon E (1997) Disaggregation of Alzheimer beta‐amyloid by site‐directed mAb. Proc Natl Acad Sci USA 94:4109–4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. St. George‐Hyslop PH, Westaway DA (1999) Alzheimer's disease. Antibody clears senile plaques. Nature 400:116–117. [DOI] [PubMed] [Google Scholar]
- 95. Stoscheck CM, King LE Jr (1986) Functional and structural characteristics of EGF and its receptor and their relationship to transforming proteins. J Cell Biochem 31:135–152. [DOI] [PubMed] [Google Scholar]
- 96. Stupp R, Mason WP, Van Den Bent MJ, Weller M, Fisher B, Taphoorn MJ et al (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996. [DOI] [PubMed] [Google Scholar]
- 97. Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L et al (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92:205–216. [DOI] [PubMed] [Google Scholar]
- 98. Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, Lin WM et al (2007) High‐throughput oncogene mutation profiling in human cancer. Nat Genet 39:347–351. [DOI] [PubMed] [Google Scholar]
- 99. Timmerman JM, Levy R (2004) Cancer vaccines: pessimism in check. Nat Med 10:1279; author reply 1279–80. [DOI] [PubMed] [Google Scholar]
- 100. Van den Eynde B, Peeters O, De Backer O, Gaugler B, Lucas S, Boon T (1995) A new family of genes coding for an antigen recognized by autologous cytolytic T lymphocytes on a human melanoma. J Exp Med 182:689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Van Pel A, Boon T (1982) Protection against a nonimmunogenic mouse leukemia by an immunogenic variant obtained by mutagenesis. Proc Natl Acad Sci USA 79:4718–4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Vredenburgh JJ, Desjardins A, Herndon JE 2nd, Marcello J, Reardon DA, Quinn JA et al (2007) Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol 25:4722–4729. [DOI] [PubMed] [Google Scholar]
- 103. Weinstein IB (2002) Cancer. Addiction to oncogenes—the Achilles heal of cancer. Science 297:63–64. [DOI] [PubMed] [Google Scholar]
- 104. Wikstrand CJ, Bigner SH, Bigner DD (1983) Demonstration of complex antigenic heterogeneity in a human glioma cell line and eight derived clones by specific monoclonal antibodies. Cancer Res 43:3327–3334. [PubMed] [Google Scholar]
- 105. Wikstrand CJ, McLendon RE, Friedman AH, Bigner DD (1997) Cell surface localization and density of the tumor‐associated variant of the epidermal growth factor receptor, EGFRvIII. Cancer Res 57:4130–4140. [PubMed] [Google Scholar]
- 106. Wikstrand CJ, Reist CJ, Archer GE, Zalutsky MR, Bigner DD (1998) The class III variant of the epidermal growth factor receptor (EGFRvIII): characterization and utilization as an immunotherapeutic target. J Neurovirol 4:148–158. [DOI] [PubMed] [Google Scholar]
- 107. Wong AJ, Ruppert JM, Bigner SH, Grzeschik CH, Humphrey PA, Bigner DS, Vogelstein B (1992) Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci USA 89:2965–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Wu AH, Xiao J, Anker L, Hall WA, Gregerson DS, Cavenee WK et al (2006) Identification of EGFRvIII‐derived CTL epitopes restricted by HLA A0201 for dendritic cell based immunotherapy of gliomas. J Neurooncol 76:23–30. [DOI] [PubMed] [Google Scholar]
- 109. Zalutsky MR, Moseley RP, Coakham HB, Coleman RE, Bigner DD (1989) Pharmacokinetics and tumor localization of 131I‐labeled anti‐tenascin monoclonal antibody 81C6 in patients with gliomas and other intracranial malignancies. Cancer Res 49:2807–2813. [PubMed] [Google Scholar]