

Abstract
Charcot-Marie-Tooth (CMT) disease is a clinically and genetically heterogeneous disorder. All mendelian patterns of inheritance have been described. We identified a homozygous p.A335V mutation in the MED25 gene in an extended Costa Rican family with autosomal recessively inherited Charcot-Marie-Tooth neuropathy linked to the CMT2B2 locus in chromosome 19q13.3. MED25, also known as ARC92 and ACID1, is a subunit of the human activator-recruited cofactor (ARC), a family of large transcriptional coactivator complexes related to the yeast Mediator. MED25 was identified by virtue of functional association with the activator domains of multiple cellular and viral transcriptional activators. Its exact physiological function in transcriptional regulation remains obscure. The CMT2B2-associated missense amino acid substitution p.A335V is located in a proline-rich region with high affinity for SH3 domains of the Abelson type. The mutation causes a decrease in binding specificity leading to the recognition of a broader range of SH3 domain proteins. Furthermore, Med25 is coordinately expressed with Pmp22 gene dosage and expression in transgenic mice and rats. These results suggest a potential role of this protein in the molecular etiology of CMT2B2 and suggest a potential, more general role of MED25 in gene dosage sensitive peripheral neuropathy pathogenesis.
Keywords: CMT, HMSN, MED25, PMP22, CMT2B2, ACID1, ARC92
Introduction
Charcot-Marie-Tooth disease (CMT) or hereditary motor and sensory neuropathy (HMSN) represents a group of frequent, clinically and genetically heterogenous disorders of peripheral nerves [1–3]. Two major CMT forms are distinguished by electrophysiological criteria: the demyelinating CMT (HMSN) type 1 and the axonal CMT type 2 [3]. Autosomal dominant CMT type 1 is most frequently caused by a 1.4-Mb tandem duplication including the PMP22 gene (CMT1A [MIM #118220]) [4, 5]. For the dominantly inherited axonal CMT type 2A mutations in the Mitofusin 2 gene (MFN2 [MIM *608507]) [6] are the most frequently observed variations. Intermediate types of CMT have also been described [7, 8]. To create animal models of the most frequently observed form of CMT, CMT1A, transgenic mice and rats that carry altered copy numbers of the PMP22 gene have been generated [9–11]. These animals recapitulate many pathological hallmarks of the human disease [12].
Both the autosomal recessive demyelinating (CMT4 [MIM #214400]) and the autosomal recessive axonal forms (ARCMT2 [MIM #605588, %605589]) are less frequently observed. For the autosomal recessive axonal ARCMT2, the first locus was mapped on chromosome 1q21.2–q21.3 [13]. Subsequently, De Sandre-Giovannoli et al. identified a homozygous R298C mutation in the LMNA gene in three consanguineous Algerian families with severe and early onset [14]. Furthermore, mutations in the ganglioside-induced differentiation-associated protein 1 (GDAP1) gene on chromosome 8q21.1 have been identified in three families of Spanish ancestry with ARCMT2 [15]. These patients were characterized by childhood onset, distal muscle weakness, sensory deficits, and vocal chord paresis in some of the patients [16].
We investigated an extended Costa Rican family (CR-P) with Spanish and Amerindian ancestors that segregated an autosomal recessive CMT type 2 neuropathy that mapped to chromosome 19q13.3 (ARCMT2 [MIM %605589]) [17, 18]. Here we provide genetic and functional data to support the contention that the homozygous p.A335V mutation in the gene for Mediator 25 (MED25) is responsible for the neuropathy segregating in the family CR-P from Costa Rica.
Materials and methods
Patients
The clinical and electrophysiological investigations took place in the city hospital of Palmares in Costa Rica, 31 family members underwent genetic analysis; clinical and electrophysiological evaluations were performed on 11 subjects. Eight of these studied subjects—three males and five females—were clinically diagnosed with motor and sensory neuropathy. Informed consent was obtained from all study participants. The age at onset of the chronic symmetric sensory-motor neuropathy was 28 to 42 years (mean 33.8 ± 5.3), the electrophysiologic data reflected a primary axonal degenerative process with mild myelin impairment in the course of the disease [18]. The electrophysiological investigations were described in detail elsewhere [18].
Genetic analysis
Genomic DNA was extracted from peripheral blood samples by the salting-out method. DNA was diluted in HPLC–H20 to a concentration of approximately 50 ng/ml and stored at 4°C. The critical interval in chromosome 19q13.3 could be refined to 1 Mb between marker D19S604 and a SNP (c.901 T>C) identified in the nuclear receptor subfamily 1, group H, member 2 (NR1H2) gene by homozygosity mapping.
Primers flanking all exons of 53 genes corresponding to 487 amplicons in the critical interval were designed using the Primer3 program. All 19 exons of MED25 have also been sequenced. Oligonucleotide sequences and PCR conditions are available on request. Bidirectional direct sequencing was implemented using the BigDye Terminator Cycle Sequencing Kit (Applied Biosystems) on an ABI 3730 capillary sequencer (Applied Biosystems) for all variations found with unidirectional sequencing. Sequence traces were evaluated using the DNAStar software package.
A BAC-contig on the critical interval was constructed using public databases and confirmed with Celera scaffolds. This interval, included on the public contig NT_011109 and the Celera-Scaffold GA_x2KMHMQE7RG (Celera Publication Site) of chromosome 19q, was analysed with several programs including NIX (http://www.hgmp.mrc.ac.uk/Registered/Webapp/nix/) and GrailEXP (http://compbio.ornl.gov/grailexp/). All genomic sequences included in the critical interval were also compared with the GenBank database of the National Institutes of Health of USA. (www.ncbi.nlh.gov/), using the simple-BLAST program against the “nr” database, for finding homologous sequences and expressed sequenced tags (ESTs) that correspond to the respective gene.
To identify the most related protein sequences with MED25, ”BLAST” and ”Advanced BLAST2 & Orthologue” (http://dove.embl-heidelberg.de/Blast2e/) programs were used. For selection of candidate genes, we also used the Build 35 of the public human genome assembly at UCSC. The UCSC map provided known proteins and genes as curated by SWISS-PROT and NCBI RefSeq project, aligned mRNAs and ESTs, Ensembl gene builds, Genscan and FGENESH gene predictions, and alignments of other genomes by similarity. Expression of candidate genes and anonymous transcripts in nervous system, and especially in spinal cord and peripheral nerve, was checked by querying public data repositories for EST matches and serial analysis of gene expression (SAGE) tags. Expression of candidates was also tested by RT-PCR.
Globular regions and elements of secondary structure in MED25 were predicted using the servers Globplot2.3 (http://globplot.embl.de/) and PsiPred (http://bioinf.cs.ucl.ac.uk/psipred/) with standard settings.
Fluorescence spectroscopy and calculation of the binding constant
All fluorescence spectra were measured in a F-4500 fluorescence spectrophotometer (Hitachi, Tokyo, Japan) at an excitation wavelength of 280 nm and an emission wavelength of 340 nm at 294 K. A semi-micro quartz fluorescence cell (light path 10 × 4 mm) with magnetic stirrer was used. To obtain the titration curves for calculation of the binding constants (Kd), synthetic peptides according to the MED25 wildtype (Ac-QLPPGPPGAPKPPPAS-CONH2) and the Ala335Val mutation (Ac-QLPPGPPGVPKPPPAS-CONH2) were purchased from Jerini (Berlin, Germany). Stock solutions of up to 5 mM were added in small increments to samples containing 0.5 µM SH3 domains in 50 mM Tris/HCl, 150 mM NaCl, pH 7.4 Samples were stirred for 2 min, fluorescence was recorded for 30 s and averaged. Ligand binding was monitored by detecting the changes of intrinsic fluorescence for a tryptophane residue located in the binding site of the SH3 domains. Since the concentration of the SH3 domain protein was low compared to the peptide concentration, the experimental data were fitted to the standard equation [19]
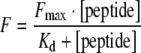 |
where [peptide] gives the final peptide concentration at each measurement point, F is the measured protein fluorescence intensity at the particular peptide concentration, and Fmax is the observed maximal fluorescence intensity of the protein when saturated with the peptide. Nonlinear regression curve fitting was carried out to fit the experimental data to the equation, with Fmax and Kd as fitted parameters. The change in protein concentration that occurred as a result of peptide addition was properly corrected.
Molecular modelling
Models of the complexes of wild-type and mutant MED25 with Abl and Lck were generated by homology modelling using previously published complexes of the respective SH3 domains with proline-rich ligands as templates [20, 21]. Proper alignment of the ligand sequences in complex with the SH3 domains was derived using the iSPOT server [22]. Models were generated using SwissModel [23] and the quality was verified by PROCHECK [24] and WHAT_CHECK [25] analyses. Visualization of the structures was performed using MolMol [26] and DS Viewer (Accelrys Inc.).
Transcription activation
Plasmid hARC92/ACID1 expression vectors were constructed by cloning of PCR products in the multiple cloning site of a previously modified pCIneo Vector (Promega), which contains a GAL4 DNA-binding domain (amino acids 1–147) fused to a Myc epitope positioned between the NheI and the EcoRI sites. ARC92/ACID1 derivatives correspond to the human factor. QuikChange site directed mutagenesis kit (Stratagene) was used to generate the A335V mutants. All clones were verified by sequencing. The GAL4 reporter pGLMRG5 is derived from pMRG5 [27] but contains a luciferase gene instead of a G-free cassette. Jurkat T cells (plasmid amounts refer to 1.0 × 107 cells) were transfected by electroporation with 10 µg of pGLMRG5 reporter and increasing amounts of GAL4-fused constructs. Total amounts of DNA (20 µg) were adjusted by supplementation with the respective empty expression vectors. A ß-Gal expression vector was co-transfected in order to normalize luciferase levels.
Animals
Wild-type, trembler, and transgenic mice
Transgenic Pmp22tg and Pmp22−/− mice have been described [28, 29]. For quantitative and RT-PCR analysis, RNA derived from sciatic nerves of four animals has been pooled. RNA was extracted with Trizol (Invitrogen), according to manufacturer’s recommendations, from rat sciatic nerve at the indicated developmental time points and adult tissues. Nerve cut and crush experiments were performed in strain C57/Bl6 mice 2 [30]. For each time point, the nerves of three animals were pooled, the contralateral site was used as control. Quantitative RT-PCR was carried out on an ABI Prism 7000 Sequence Detection System using TaqMan technology (Applied Biosystems). PCR was performed with the unlabelled primers 047Qfor (5′-CTCCGCAAGCACTACCTGCT-3′) and 047Qref (5′-CCCAAAGTCCGTCTCTGCA-3′) and the 5′ FAM labelled primer 047QP (5′-CCATAGAGTACTTCAACGGG-3′). The reactions were carried out in triplicate using TaqMan Universal PCR Master Mix was according to the manufacturer′s recommendations. The amount of 18S rRNA was determined in parallel reactions as an internal control.
Transgenic rats
Pmp22++/+ rats have been described [28]. RNA was derived from sciatic nerve sections and analysed via RT-PCR and quantitative PCR for Pmp22 and Med25 expression. Progesterone and onapristone supplementation and also the behavior tests using the bar holding time as phenotypic indicator were as described, progesterone diet was at 20 mg/kg [31]. RNA was extracted from rat sciatic nerve with Trizol (Invitrogen) according to manufacturer’s recommendations. Quantitative RT-PCR was carried out on an ABI Prism 7000 Sequence Detection System using TaqMan technology (Applied Biosystems). PCR was performed with the unlabelled primers 047Qfor (5′-CTCCGCAAGCACTACCTGCT-3′) and rat_047_Qrev (5′-CCCAAAGTCCGTCTCTGCG-3′) and the 5′ FAM labelled primer rat_047_QP (5′-CCATAGAGTACTTCAACGGA-3′). The reactions were performed in triplicate using TaqMan as described above with mice. Additionally the experiments have been replicated using GAPDH as an internal control.
Results
Identification of the neuropathy causing homozygous MED25 mutation p.A335V
Family CR-P originates from a single town in the province of Alajuela, Republic of Costa Rica in Central America. This family is genealogically related and the affected members are descendants of consanguineous matings (Fig. 1a). The critical interval in chromosome 19q13.3 could be refined to 1 Mb between marker D19S604 and a SNP (c.901 T>C) identified in the nuclear receptor subfamily 1, group H, member 2 (NR1H2) gene (Fig. 1a). This refinement could be done since individual B-5.3 [17] agreed with his participation on this study.
Fig. 1.
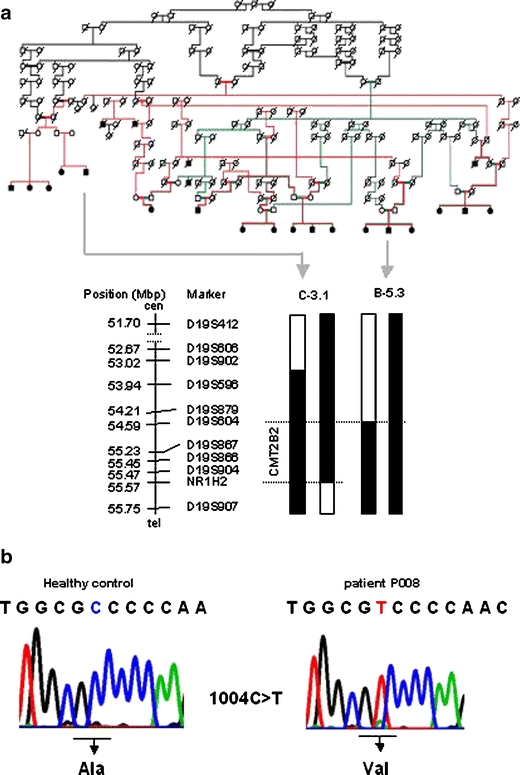
Pedigree of the CR-P family that carries the CMT2B2 mutation. Colored lines (red, green, black) were used to visualize the relationship within family CR-P. The redefined critical interval of 1 Mb between D19S604 and the c.901C>T SNP in NR1H2 is indicated (a). Sequence analysis of MED25 revealed the homozygous mutation A335V (b)
To identify the disease causing allele DNA sequence analysis of the entire critical region was implemented. Overall, 53 genes corresponding to 487 amplicons were analysed. Three genes, Pronapsin A (NAPSA), MED25 and the prostate tumor overexpressed gene 1 (PTOV1) showed variations (A310T, A335V, and I252V, respectively) that cosegregated with the recessive neuropathy trait in family CR-P. A p.A335V missense amino acid substitution in MED25 appeared to be the disease causing variation for CMT2B2 based on the following observations (Fig. 1b). The alanine at position 335 is evolutionarily conserved, indicating a biological function suggested also by its central position in a proline-rich motif (Fig. 2a). The population carrier frequency (0.68 %; allele frequency, 0.34 %) is below that usually observed for a polymorphism and in accordance with autosomal recessive inheritance (Table 1). The incidence of 1:88,000 (Hardy–Weinberg, q2 = 1.1391−5) is concordant with other recessive types of CMT [2]. We analyzed 2,064 chromosomes of Central American, European, and US American ancestry and found seven heterozygous p.A335V carriers; five were healthy controls from Costa Rica (632 Costa Rican controls analyzed), one was a healthy control from Germany (23 German controls analyzed), and one was a female CMT1 patient with unknown genetic defect from Germany (377 patients analyzed). Based on these data the incidence in Costa Rica is 1:66,358 (Hardy–Weinberg, q2 = 1.13914−5) at a population carrier frequency of 0.76%, combined for the USA and Germany in 1:154,449 (Hardy–Weinberg, q2 = 6.4746−6). In contrast, the p.A310T variation in NAPSA was observed at a population frequency of 5% (251 control chromosomes tested) consistent with a common polymorphism. The p.I252V (c.754 A>G) variation in PTOV1 was found homozygous in a healthy control, thus likely excluding PTOV1 as a candidate gene. Neither the p.A310T nor the p.I252V fall within conserved amino acids.
Fig. 2.

Conservation of the amino acid alanine in position 335 (a) and the SH3-binding motif in MED25 (b). Genomic structure, domains, and peptide motifs present in MED25. The von Willebrandt factor type A (vWA) domain and an activator-interacting domain (ACID) were previously verified experimentally [35, 36]. The SH3-binding site result from a computational prediction and its SH3-binding ability has been proven in the present study
Table 1.
Frequency of the A335V mutation in controls and other neuropathy patients
| Healthy controls | Neuropathy patients without known mutation | Total | |
|---|---|---|---|
| Individuals | 655 | 377 | 1,032 |
| Ala335Val carrier | 6 | 1 | 7 |
The A335V mutation was analyzed via restriction analysis of PCR products of exon 9 using the enzyme HgaI and subsequent sequence analysis for confirmation. For the neuropathy patients without known mutation all coding exons of MED25 have been sequenced
Another 98 variations comprising translationally silent changes, splice site variants and 13 amino acid substitutions were identified, but could be excluded as causes of the axonal neuropathy mostly due to the existence of homozygous healthy carriers. The p.A335V mutation in MED25 is the only variation in the critical interval in 19q13.3 that fulfills all formal genetic criteria to be the disease causing variation for CMT2B2. We performed further experiments in order to assign a biological function to this region of MED25 and characterize the effects of this mutation.
Functional properties of the MED25 p.A335V mutation
The p.A335V mutation resides in a proline-rich region (Fig. 2a,b). This region was predicted to be a potential SH3 ligand by iSPOT (http://cbm.bio.uniroma2.it/ispot/) [22]. According to these predictions, the wildtype should represent an interaction partner of the SH3 domain of the Abelson tyrosine kinase family (AblSH3). The mutant MED25 may retain this affinity but is furthermore predicted to have a strong affinity for the SH3 domains of the Src-family of kinases. Thus, the mutant MED25 should bind a broader variety of SH3 groups.
To test this hypothesis, fluorescence binding experiments were performed using the SH3 domains from Abl (clone pET3d, a kind gift of Luis Serrano [32]) and from the Src-family kinase Lck [21] (Fig. 3a,b). Computational investigations of MED25 predict that the SH3 binding region is nonglobular and lacks any regular secondary structure. Therefore, synthetic MED25 peptides are expected to adequately reflect the binding properties of full-length MED25 Therefore, synthetic MED25 peptides that cover the binding site were used for the titration experiments.
Fig. 3.
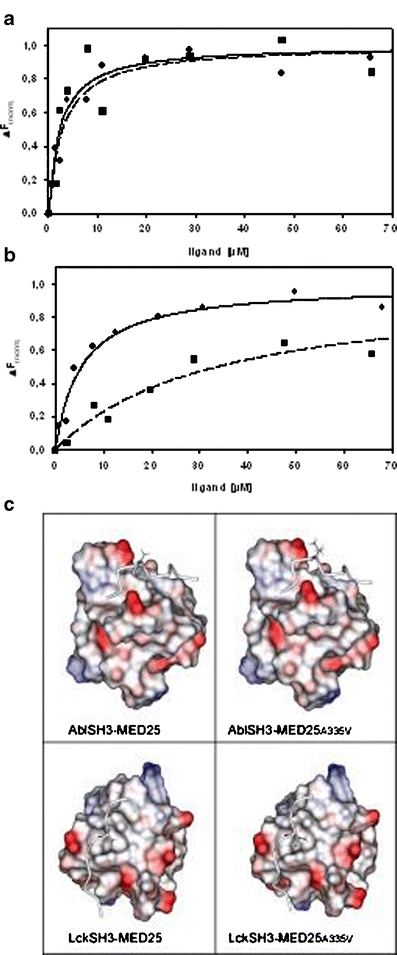
Affinity test results for MED25 binding to Abl SH3 (a) and Lck SH3 (b). Fluorescence titration experiments were performed for wild-type (filled square, dotted line) and mutant MED25 peptides carrying an A335V point mutation (filled circle, solid line). Curves were fitted according to the equation given in methods. c Models of MED25 in complex with Abl SH3 (top) and Lck SH3 (bottom) visualizing the effect of the A335V mutation. The SH3 domains are shown in space-filled representation and the acidic and basic surface patches are colored in red and blue, respectively. White surface patches correspond to nonpolar regions. The MED25 binding region is shown as tube and residue 335 is shown in stick presentation
Whereas the affinity of synthetic MED25 peptides for Abl is almost unaltered by the mutation (Kd(wt) = 2.37 ± 0.93 µM; Kd(mutant) = 2.91 ± 0.66 µM), the mutant binds one order of magnitude stronger to the Lck SH3 domain than wild-type MED25 (Kd(wt) = 33.47 ± 15.01 µM; Kd(mutant) = 5.38 ± 1.05 µM).
A quantitative comparison of the values for the binding affinity shows that the A335V mutant binds to AblSH3 and LckSH3 with a very similar affinity, while wild-type MED25 binds significantly stronger to Abl than to Lck. Thus, the increase in affinity for Lck observed in the mutant leads to a loss of binding specificity for Abelson SH3 domain.
To rationalize the effects of the mutation on its binding properties, we modelled the structure of the complexes formed by MED25 and the Abl or Lck SH3 domains (Fig. 3c). In the MED25–AblSH3 complex (Fig. 3c, top panel) residue 335 is oriented away from the SH3 domain explaining why the alanine to valine mutation does not affect affinity. In the MED25–LckSH3 complex (Fig. 3c, lower panel), Val335 packs into a hydrophobic pocket of the LckSH3 surface. The respective contacts cannot be formed by the shorter sidechain of wild-type alanine potentially explaining the observed increased affinity of the mutant. Src-family kinases generally exhibit a binding preference for ligands with long aliphatic sidechains at the respective sequence position [33, 34]. Thus, it can be anticipated that the mutation in MED25 may increase the affinity for a broad range of members of this family which will block the binding site and in effect may decrease the dosage of free mutant MED25 protein compared to the wildtype because of this sequestration.
To further investigate the in vivo function of this proline-rich motif as part of the Mediator complex within RNA polymerase II, clones comprising the protein coding region of the MED25 p.A335V mutation were tested for transcriptional activation. The mutation has been analyzed in the context of a GAL4 fusion protein using a transient reporter assay. GAL4-MED25 proteins have previously been shown to activate transcription that correlated with recruitment of a functional Mediator complex [35]. In this assay, the p.A335V mutation has little influence on transcription activation (Fig. 4). As expected from the previous characterization of the Mediator binding domain located in the N-terminal domain (NTD, aa 1–226) wild-type and mutant proteins bound indistinguishably to Mediator upon transfection into HeLa cells. Thus, a more significant influence of the mutation on specific genes may be possible.
Fig. 4.
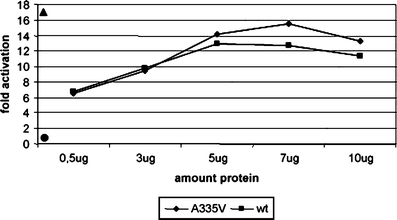
Transcription activation-Gal4 reporter assay using full-length clones (1–754) for the wild-type (filled square) and A335V (filled diamond) mutation at different protein concentrations. The highest activation at 5 µg was found for a Gal-(1–715) clone (filled triangle, left side top). The empty vector pGLMRG5 was used as control (filled circle, left side down)
In summary, the p.A335V mutation results in an interaction with an extended range of SH3 domains. We hypothesized that this may affect the specificity and degree of activation of the downstream target genes in the peripheral nervous system (PNS).
Refined expression analysis of Med25 in wild-type rats
The findings described above suggest that the p.A335V mutation in MED25 causes CMT2B2 by a "loss-of-specific-function". A quantitative RT-PCR expression analysis for Med25 using RNA derived from several tissues of adult rats was performed to establish a highly differentiated gene expression profile. This analysis revealed an ubiquitous expression of Med25 (Fig. 5a) confirming and extending previous Northern blot studies [35, 36] to dorsal root ganglia and sciatic nerve, tissues affected in peripheral neuropathies. The highest expression levels were found in dorsal root ganglia, cerebellum, cortex, and optic nerve indicating an important function in the nervous system, whereas the lowest expression was in the skin.
Fig. 5.
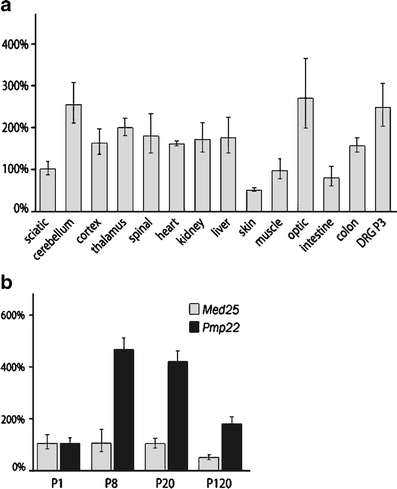
Expression profile and refined expression analysis for Med25. RNA extracts derived from rat tissue have been analyzed via qRTPCR (a). An ubiquitous expression was demonstrated, for peripheral nerves the expression in DRG and sciatic nerve is shown. The time course of Med25 expression during developing sciatic nerve of rats is constant till day 20, but reduced late in development at day 120 (b). In comparison, Pmp22 expression represents a typical time course of myelination
An analysis reflecting the time course of sciatic nerve development and maintenance in rat-derived tissue revealed a relatively constant expression from day 1 to day 20 after birth for Med25 compared to Pmp22. However, at day 120 an approximately 50% reduction was found for Med25 (Fig. 5b).
Med25 expression in Pmp22-transgenic mice and rats
The CR-P patients were diagnosed as having primary axonal peripheral neuropathy; however, a mild myelin impairment in the late disease course indicated by slightly reduced nerve conduction velocities was also observed [18]. This points to complex effects on the peripheral nervous system. On the other hand, axonal damage is also a consequence of prolonged loss of glial support observed for most demyelinating neuropathies including CMT1A [37, 38]. We hypothesized that MED25 may represent a transcription activator with a certain role for both types of peripheral neuropathies. The currently best characterized animal models for demyelinating peripheral neuropathies are transgenic mice and rats with altered Pmp22 gene dosage. Mice with increased or reduced copy numbers of Pmp22, or altered transcription levels of Pmp22 using a tet promotor [39], develop a demyelinating as well as a pronounced distal axonopathy that correlates with the clinical progression of the respective disease [40–42]. Thus, it was of basic interest to investigate if Med25 might be involved in transcriptional regulation of Pmp22. We performed quantitative expression analysis of Med25 in sciatic nerve extracts derived from mice carrying either the Trembler (Tr) point mutation in Pmp22 [43], a null mutant (Pmp22−/−) [29] and overexpressing mutants (Pmp22tg, [28]. The respective phenotypes resemble DSN, hereditary neuropathy with liability to pressure palsies (HNPP, Pmp22−/−), or CMT1A. Surprisingly, Med25 was coordinately expressed with Pmp22 gene dosage and expression in Pmp22−/− and Pmp22tg transgenic mice (Fig. 6a). High Pmp22 transcript levels are associated with high Med25 levels and a reduced Pmp22 expression with a low Med25 expression compared to wildtype. In contrast, the Pmp22Tr mutation had almost no effect on Med25 expression. This may indicate a different molecular pathology for point mutations in Pmp22 compared to the gene dosage related peripheral nerve disorders [44]. For further confirmation of this result, we analyzed Med25 expression in a second animal model, transgenic rats carrying three to five Pmp22 copies [28]. Almost identical to the results in Pmp22tg mice, the Med25 expression correlated also with the increased Pmp22 dosage and expression (Figs. 6b and 7a). Thus, the correlation of Med25 and Pmp22 expression was observed in independent animal models.
Fig. 6.
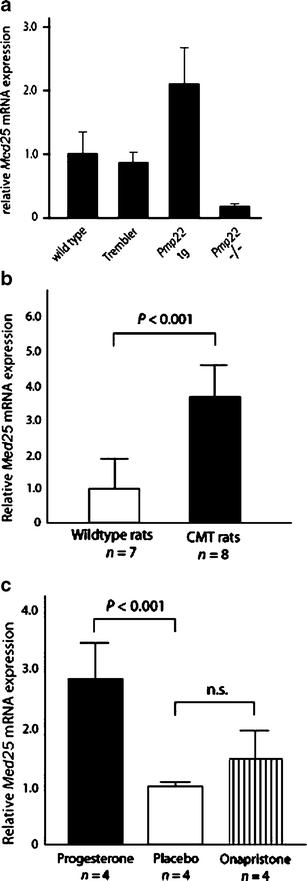
Med25 qRTPCR expression analysis in Pmp22 transgenic animals. Wild-type and trembler mice showed similar Med25 expression, whereas Pmp22tg mice showed an increased and Pmp22−/− mice a reduced expression (a). CMT (Pmp22++/+) rats revealed also a significantly increased expression compared to wildtype (b). After progesterone supplementation, similar significant results were obtained, whereas onapristone had no significant effect (c). Med25 dosage is strictly correlated with Pmp22 dosage in Pmp22−/− and Pmp22tg mice and Pmp22++/+ rats (n = number of animals examined)
Fig. 7.
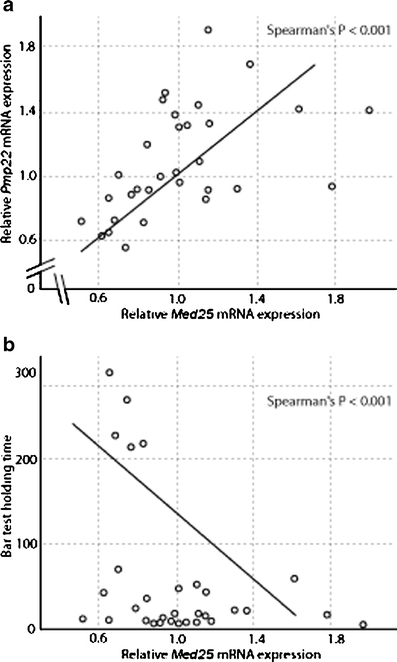
Correlation of Pmp22 and Med25 in expression and behavior of CMT rats. A significant correlation of Pmp22 and Med25 expression in sciatic nerve extracts was found by qRTPCR analysis of 32 animals (12 onapristone-, 8 placebo-, and 12 progesterone-treated (a). A significant correlation of holding time on the bar and Med25 expression was found, high Med25 expression results in short bar holding time. Low Med25 expression results in an extended bar holding time (b)
The effects of progesterone and a progesterone-antagonist, onapristone, on epigenetic modification of Pmp22 expression in peripheral nerves of Pmp22 transgenic rats were reported recently [31]. Progesterone increases Pmp22 expression and the progesterone antagonist decreases its expression accompanied by improvement of the phenotype in rats. The correlation of Med25 dosage with Pmp22 dosage in peripheral nerves of transgenic animals was an intriguing finding that stimulated us to perform additional experiments with regard to progesterone and onapristone treatment of these transgenic rats. Comparable to the observation for Myelin Protein Zero (MPZ, P0) expression, progesterone-treated (20 mg/kg) male rats revealed a further increase of the Med25 expression (Fig. 6c). However, onapristone treatment had no significant reducing effect (Fig. 6c). This may indicate that Med25 expression is not regulated directly by the progesterone receptor but is linked via an unknown pathway to Pmp22 dosage and expression.
Med25 expression was also correlated with the behavioral phenotype of these animals measured in the bar holding test [28, 31]. Med25 and Pmp22 transcript levels vary in a wide range in these Pmp22++/+ CMT rats (Fig. 7a). The Pmp22++/+ (progesterone-treated) rats at age of 5 weeks with relatively low Med25 expression showed an increased bar holding time, whereas the animals with high Med25 expression had a short time on the bar (Fig. 7b).
Med25 expression and nerve injury
We have further investigated Med25 and Pmp22 expression after injury to peripheral nerves using qRT-PCR analysis. After permanent sciatic nerve transsection, without allowing nerve regeneration, Pmp22 transcript levels were strongly reduced while Med25 expression was only moderately decreased (Fig. 8a). If a crush injury was applied, similar results were obtained 12 days after lesion (Fig. 8b). Sixty days after crush lesion, when nerve fibers have mostly regenerated and remyelinated, Pmp22 levels are almost back to controls of unlesioned nerves while Med25 levels have only partially recovered. We conclude that both Med25 and Pmp22 expression are regulated by neuron–Schwann cell interactions in lesioned peripheral nerves although this phenomenon is more pronounced for Pmp22.
Fig. 8.
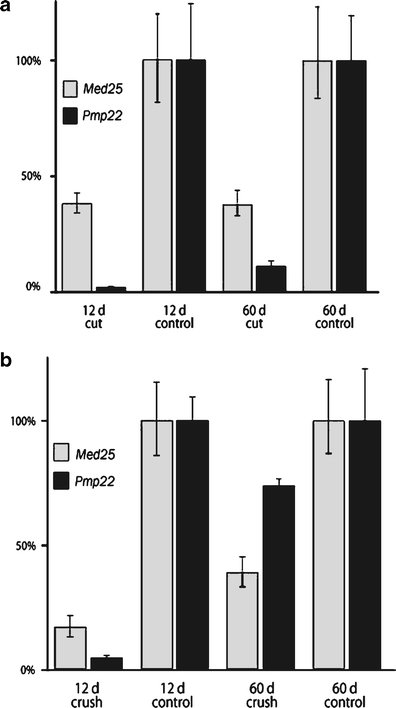
Med25 and PMP22 expression levels analyzed by qRTPCR after sciatic nerve transsection (a) or crush injury (b). Transcript levels are given as percentage of control (unlesioned) nerves, 12 days or 60 days after lesions
Discussion
We identified a missense mutation p.A335V in a yet unassigned transcription activation domain of MED25 cosegregating with a peripheral neuropathy in a Costa Rican kindred. Heterozygous p.A335V was found in five out of 632 healthy Costa Rican controls and furthermore in one German healthy control supporting the recessive nature of this mutation. These observations suggest that A335V is not a private mutation, but a very rare variant, until now, only in homozygous state in CMT2B2 patients [15, 45]. The incidence for CMT in Costa Rica is 1:66,358 (Hardy–Weinberg, q2 = 1.507−5), whereas that combined for the USA and Germany is 1:154,449 (Hardy–Weinberg, q2 = 6.4746−6). The difference may be explained by a potential founder effect for the p.A335V MED25 mutation due to the Spanish ancestry of immigrants in the sixteenth century of the extended Costa Rican family. Interestingly, for a recessive GDAP1 mutation (p.Q163X) a similar common haplotype shared by nonrelated patients and families with Spanish origin was described [46, 47].
There are two variants in exon 9 of MED25 reported in public databases (rs34889830 and rs4290565), nevertheless these polymorphisms were not found in additional ESTs or genomic sequences in databases and were not observed in the 377 neuropathy patients via sequence analysis. For MED25 several splice variants are known, especially for the 3′ region [35, 36, 48]. One of them is the P78 cDNA expressed in prostate cancer cells [48]. Some conserved domains, among them the von Willebrandt factor type A (vWA) domain- and Synapsin-related regions, were assigned according to homology data [35, 36]. An interaction with Mediator of the vWA domain was recently shown by immunoprecipitation [35, 36]. It was also shown that MED25 contains an activator-interacting domain (ACID) which is a functionally important target of the VP16 transcriptional activator [35, 36]. The MED25 VP16-binding domain was later assigned to a discrete region between amino acids 402 and 590 [35, 36].
We describe the proline-rich SH3 recognition motif comprising the p.A335V mutation. Affinity experiments using synthetic MED25 peptides showed a loss of binding specificity of the mutant as shown for the interaction with the Abl and Lck SH3 domains (Fig. 3). Although both types of kinases have been associated with the nervous system [49, 50], their interconnection with transcription activation as suggested by the MED25 mutation in family CR-P has not yet been shown. This raises the question whether additional cofactors, mediated by their SH3 domains, might act as interaction partners of MED25 in the nervous system. At present, 839 different human SH3 domains [51] of mostly uncharacterized binding preferences are known, which renders the existence of additional MED25 interaction partners feasible. Based on the extension of affinity to a broader range of SH3 domains detected for the MED25 mutant, however, we anticipate that the number of binding partners will be larger for the mutant than for the wildtype. Such interactions include an increased number of nonphysiological interactions and a concomitant "loss-of-specific-function". As a further consequence, the MED25 physiological function will be reduced in a dose-dependent manner by increased competitive binding to additional interaction partners. This situation could be compared to recessively inherited deletions, e. g. spinal muscular atrophy with two distinct survival motor neuron genes responsible for the resulting phenotype.
The wild-type and mutated proline-rich motif in MED25 revealed in our study an intrinsic transcription activation property in a GAL4 reporter assay indicating a novel in vivo function. The "loss-of-specific-function" induced by the A335V mutation may activate gene transcription in peripheral nerves with remarkably reduced degree and specificity for the cognate downstream target genes. The reduced, but maintained wildtype function may potentially explain a mild peripheral neuropathy with late age of onset (mean 33.8 years) in an autosomal recessive trait.
The significant correlation of Med25 and Pmp22 dosage and disease phenotype in transgenic animals was also present in preliminary siRNA knockdown experiments (data not shown). This raises the question of how a structural membrane protein can influence the expression of a transcription activator in a yet unknown signal transduction pathway [9]. EGR2, a transcription factor associated with CMT1A, DSN, and CHN, plays an essential role during peripheral nerve myelination and differentiation of Schwann cells [52]. The recessive p.I268N mutation abrogates the binding of NAB repressors and causes, in homozygous state, a severe phenotype (CHN). This finding may reflect a threshold effect in which certain increased protein level of the downstream PNS target gene(s), e. g. PMP22 and P0, must be achieved for manifestation of phenotype [53, 54]. In Tr mice abnormal retention within the endoplasmic reticulum of myelinating Schwann cells was described earlier [55]. However, the pathway from increased PMP22 dosage to myelin impairment and axonal damage remains enigmatic. Furthermore, the PNS target genes regulated by MED25 are currently completely unknown.
The p.A335V mutation was identified in patients presenting with an axonally classical peripheral neuropathy accompanied by mild myelin impairment. With regard to our expression analysis results for MED25 in sciatic nerves of transgenic animals and after injury a primary, subtle molecular lesion in the CR-P patients could be occurring in Schwann cells. The pathway from Schwann cell defect to axonal damage is unknown. MED25 is expressed in reasonable amounts in the cerebellum, cortex, DRG, and optic nerve (Fig. 5a). Hence, a MED25 mutation could potentially affect the central nervous system. The absence of central nervous clinical findings like ataxia or impairment of the optic nerve in the CR-P patients points to differences in molecular pathways between PNS and CNS, may be because central axons are myelinated by oligodendrocytes. In this context it is of interest that a patient with partial trisomy 19q including the MED25 gene among several others had severe central nervous impairment [56]. Clinical and electrophysiological re-examination revealed, however, no impairment at all of the peripheral nervous system. This increased gene dosage for MED25 is in heterozygous state thus supporting the assumption of an autosomal recessive mode of inheritance for CMT2B2.
Our data demonstrate that functional effects of p.A335V include loss-of-specific-function for SH3 ligand binding that probably sequester MED25 to nonphysiological pathways. Such potential altering of the availability of MED25 may affect transcriptional activation or regulation of target genes in the peripheral nervous system. We further document that Pmp22 expression correlates with Med25 expression levels in: (1) transgenic animals, both mice and rats, wherein the Pmp22 expression is altered by effectively changing gene copy number, (2) in animals wherein the Pmp22 expression is altered by epigenetic means, and (3) in a nerve-crush/injury paradigm. These latter findings suggest that PMP22 may be at least one of the genes downstream of MED25 that is mediating the neuropathic effects of the p.A335V mutation.
Acknowledgement
We want to thank the Costa Rican patients for their cooperation. This research was funded by the DFG, the Boehringer Ingelheim foundation, the DGM, ELAN funds, and the Association Francaise contre les myopathies (AFN). Support from the University of Costa Rica is gratefully acknowledged (project 742-98-241). AL was a DAAD Ph. D. fellow. U.S. is supported by the Swiss National Science Foundation and NCCR Neural Plasticity and Repair. We thank Claudia Preller for excellent technical support and Heike Meiselbach for help with Fig. 3c.
Footnotes
An erratum to this article can be found at http://dx.doi.org/10.1007/s10048-009-0213-1
Electronic database information
Accession numbers NR1H2: BC007790.1; NAPSA: AF090386; MED25: NM_030973; PTOV1: NM_017432. Online Mendelian Inheritance in Man(OMIM), http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM.
References
- 1.Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin Genet. 1974;6:98–118. doi: 10.1111/j.1399-0004.1974.tb00638.x. [DOI] [PubMed] [Google Scholar]
- 2.Mostacciuolo ML, Muller E, Fardin P, Micaglio GF, Bardoni B, Guioli S, Camerino G, Danieli GA. X-linked Charcot-Marie-Tooth disease. A linkage study in a large family by using 12 probes of the pericentromeric region. Hum Genet. 1991;87:23–27. doi: 10.1007/BF01213086. [DOI] [PubMed] [Google Scholar]
- 3.Shy M, Dyck Peter J, Chance Philipp F, Lupski James R, Klein Christopher J. Hereditary motor and sensory neuropathies. 4. Philadelphia: Elsevier Saunders; 2005. [Google Scholar]
- 4.Lupski JR, Oca-Luna RM, Slaugenhaupt S, Pentao L, Guzzetta V, Trask BJ, Saucedo-Cardenas O, Barker DF, Killian JM, Garcia CA, et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;66:219–232. doi: 10.1016/0092-8674(91)90613-4. [DOI] [PubMed] [Google Scholar]
- 5.Raeymaekers P, Timmerman V, Nelis E, Jonghe P, Hoogendijk JE, Baas F, Barker DF, Martin JJ, Visser M, Bolhuis PA, et al. Duplication in chromosome 17p11.2 in Charcot-Marie-Tooth neuropathy type 1a (CMT 1a). The HMSN Collaborative Research Group. Neuromuscul Disord. 1991;1:93–97. doi: 10.1016/0960-8966(91)90055-W. [DOI] [PubMed] [Google Scholar]
- 6.Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- 7.Jordanova A, Thomas FP, Guergueltcheva V, Tournev I, Gondim FA, Ishpekova B, Vriendt E, Jacobs A, Litvinenko I, Ivanova N, et al. Dominant intermediate Charcot-Marie-Tooth type C maps to chromosome 1p34-p35. Am J Hum Genet. 2003;73:1423–1430. doi: 10.1086/379792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zuchner S, Noureddine M, Kennerson M, Verhoeven K, Claeys K, Jonghe PD, Merory J, Oliveira SA, Speer MC, Stenger JE, et al. Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease. Nat Genet. 2005;37:289–294. doi: 10.1038/ng1514. [DOI] [PubMed] [Google Scholar]
- 9.Maier M, Berger P, Suter U. Understanding Schwann cell–neurone interactions: the key to Charcot-Marie-Tooth disease? J Anat. 2002;200:357–366. doi: 10.1046/j.1469-7580.2002.00044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muller HW, Suter U, Broeckhoven C, Hanemann CO, Nelis E, Timmerman V, Sancho S, Barrio L, Bolhuis P, Dermietzel R, et al. Advances in Charcot-Marie-Tooth disease research: cellular function of CMT-related proteins, transgenic animal models, and pathomechanisms. The European CMT Consortium. Neurobiol Dis. 1997;4:215–220. doi: 10.1006/nbdi.1997.0148. [DOI] [PubMed] [Google Scholar]
- 11.Nelis E, Timmerman V, Jonghe P, Broeckhoven C, Rautenstrauss B. Molecular genetics and biology of inherited peripheral neuropathies: a fast-moving field. Neurogenetics. 1999;2:137–148. doi: 10.1007/s100480050074. [DOI] [PubMed] [Google Scholar]
- 12.Berger P, Young P, Suter U. Molecular cell biology of Charcot-Marie-Tooth disease. Neurogenetics. 2002;4:1–15. doi: 10.1007/s10048-002-0130-z. [DOI] [PubMed] [Google Scholar]
- 13.Bouhouche A, Benomar A, Birouk N, Mularoni A, Meggouh F, Tassin J, Grid D, Vandenberghe A, Yahyaoui M, Chkili T, et al. A locus for an axonal form of autosomal recessive Charcot-Marie-Tooth disease maps to chromosome 1q21.2-q21.3. Am J Hum Genet. 1999;65:722–727. doi: 10.1086/302542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sandre-Giovannoli A, Chaouch M, Kozlov S, Vallat JM, Tazir M, Kassouri N, Szepetowski P, Hammadouche T, Vandenberghe A, Stewart CL, et al. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am J Hum Genet. 2002;70:726–736. doi: 10.1086/339274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cuesta A, Pedrola L, Sevilla T, Garcia-Planells J, Chumillas MJ, Mayordomo F, LeGuern E, Marin I, Vilchez JJ, Palau F. The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat Genet. 2002;30:22–25. doi: 10.1038/ng798. [DOI] [PubMed] [Google Scholar]
- 16.Sevilla T, Cuesta A, Chumillas MJ, Mayordomo F, Pedrola L, Palau F, Vilchez JJ. Clinical, electrophysiological and morphological findings of Charcot-Marie-Tooth neuropathy with vocal cord palsy and mutations in the GDAP1 gene. Brain. 2003;126:2023–2033. doi: 10.1093/brain/awg202. [DOI] [PubMed] [Google Scholar]
- 17.Leal A, Morera B, Del Valle G, Heuss D, Kayser C, Berghoff M, Villegas R, Hernandez E, Mendez M, Hennies HC, et al. A second locus for an axonal form of autosomal recessive Charcot-Marie-Tooth disease maps to chromosome 19q13.3. Am J Hum Genet. 2001;68:269–274. doi: 10.1086/316934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berghoff C, Berghoff M, Leal A, Morera B, Barrantes R, Reis A, Neundorfer B, Rautenstrauss B, Del Valle G, Heuss D. Clinical and electrophysiological characteristics of autosomal recessive axonal Charcot-Marie-Tooth disease (ARCMT2B) that maps to chromosome 19q13.3. Neuromuscul Disord. 2004;14:301–306. doi: 10.1016/j.nmd.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 19.Bauer F, Hofinger E, Hoffmann S, Rosch P, Schweimer K, Sticht H. Characterization of Lck-binding elements in the herpesviral regulatory Tip protein. Biochemistry. 2004;43:14932–14939. doi: 10.1021/bi0485068. [DOI] [PubMed] [Google Scholar]
- 20.Pisabarro MT, Serrano L, Wilmanns M. Crystal structure of the abl-SH3 domain complexed with a designed high-affinity peptide ligand: implications for SH3-ligand interactions. J Mol Biol. 1998;281:513–521. doi: 10.1006/jmbi.1998.1932. [DOI] [PubMed] [Google Scholar]
- 21.Schweimer K, Hoffmann S, Bauer F, Friedrich U, Kardinal C, Feller SM, Biesinger B, Sticht H. Structural investigation of the binding of a herpesviral protein to the SH3 domain of tyrosine kinase Lck. Biochemistry. 2002;41:5120–5130. doi: 10.1021/bi015986j. [DOI] [PubMed] [Google Scholar]
- 22.Brannetti B, Via A, Cestra G, Cesareni G, Helmer-Citterich M. SH3-SPOT: an algorithm to predict preferred ligands to different members of the SH3 gene family. J Mol Biol. 2000;298:313–328. doi: 10.1006/jmbi.2000.3670. [DOI] [PubMed] [Google Scholar]
- 23.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 24.Laskowski RA, Moss DS, Thornton JM. Main-chain bond lengths and bond angles in protein structures. J Mol Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- 25.Hooft RW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- 26.Koradi R., Billeter M., Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55, 29–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 27.Xie J, Collart M, Lemaire M, Stelzer G, Meisterernst M. A single point mutation in TFIIA suppresses NC2 requirement in vivo. EMBO J. 2000;19:672–682. doi: 10.1093/emboj/19.4.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sereda M, Griffiths I, Puhlhofer A, Stewart H, Rossner MJ, Zimmerman F, Magyar JP, Schneider A, Hund E, Meinck HM, et al. A transgenic rat model of Charcot-Marie-Tooth disease. Neuron. 1996;16:1049–1060. doi: 10.1016/S0896-6273(00)80128-2. [DOI] [PubMed] [Google Scholar]
- 29.Adlkofer K, Martini R, Aguzzi A, Zilasek J, Toyka KV, Suter U. Hypermyelination and demyelinating peripheral neuropathy in Pmp22-deficient mice. Nat Genet. 1995;11:274–286. doi: 10.1038/ng1195-274. [DOI] [PubMed] [Google Scholar]
- 30.Pot C, Simonen M, Weinmann O, Schnell L, Christ F, Stoeckle S, Berger P, Rulicke T, Suter U, Schwab ME. Nogo-A expressed in Schwann cells impairs axonal regeneration after peripheral nerve injury. J Cell Biol. 2002;159:29–35. doi: 10.1083/jcb.200206068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sereda MW, Meyer zu Horste G, Suter U, Uzma N, Nave KA. Therapeutic administration of progesterone antagonist in a model of Charcot-Marie-Tooth disease (CMT-1A) Nat Med. 2003;9:1533–1537. doi: 10.1038/nm957. [DOI] [PubMed] [Google Scholar]
- 32.Pisabarro MT, Serrano L. Rational design of specific high-affinity peptide ligands for the Abl-SH3 domain. Biochemistry. 1996;35:10634–10640. doi: 10.1021/bi960203t. [DOI] [PubMed] [Google Scholar]
- 33.Feng S, Kasahara C, Rickles RJ, Schreiber SL. Specific interactions outside the proline-rich core of two classes of Src homology 3 ligands. Proc Natl Acad Sci USA. 1995;92:12408–12415. doi: 10.1073/pnas.92.26.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sparks AB, Rider JE, Hoffman NG, Fowlkes DM, Quillam LA, Kay BK. Distinct ligand preferences of Src homology 3 domains from Src, Yes, Abl, Cortactin, p53bp2, PLCgamma, Crk, and Grb2. Proc Natl Acad Sci USA. 1996;93:1540–1544. doi: 10.1073/pnas.93.4.1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mittler G, Stuhler T, Santolin L, Uhlmann T, Kremmer E, Lottspeich F, Berti L, Meisterernst M. A novel docking site on Mediator is critical for activation by VP16 in mammalian cells. EMBO J. 2003;22:6494–6504. doi: 10.1093/emboj/cdg619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang F, DeBeaumont R, Zhou S, Naar AM. The activator-recruited cofactor/Mediator coactivator subunit ARC92 is a functionally important target of the VP16 transcriptional activator. Proc Natl Acad Sci USA. 2004;101:2339–2344. doi: 10.1073/pnas.0308676100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krajewski KM, Lewis RA, Fuerst DR, Turansky C, Hinderer SR, Garbern J, Kamholz J, Shy ME. Neurological dysfunction and axonal degeneration in Charcot-Marie-Tooth disease type 1A. Brain. 2000;123(Pt 7):1516–1527. doi: 10.1093/brain/123.7.1516. [DOI] [PubMed] [Google Scholar]
- 38.Kamholz J, Menichella D, Jani A, Garbern J, Lewis RA, Krajewski KM, Lilien J, Scherer SS, Shy ME. Charcot-Marie-Tooth disease type 1: molecular pathogenesis to gene therapy. Brain. 2000;123(Pt 2):222–233. doi: 10.1093/brain/123.2.222. [DOI] [PubMed] [Google Scholar]
- 39.Robertson A, Perea J, Tolmachova T, Thomas PK, Huxley C. Effects of mouse strain, position of integration and tetracycline analogue on the tetracycline conditional system in transgenic mice. Gene. 2002;282:65–74. doi: 10.1016/S0378-1119(01)00793-4. [DOI] [PubMed] [Google Scholar]
- 40.Sancho S, Magyar JP, Aguzzi A, Suter U. Distal axonopathy in peripheral nerves of PMP22-mutant mice. Brain. 1999;122(Pt 8):1563–1577. doi: 10.1093/brain/122.8.1563. [DOI] [PubMed] [Google Scholar]
- 41.Robertson AM, Perea J, McGuigan A, King RH, Muddle JR, Gabreels-Festen AA, Thomas PK, Huxley C. Comparison of a new pmp22 transgenic mouse line with other mouse models and human patients with CMT1A. J Anat. 2002;200:377–390. doi: 10.1046/j.1469-7580.2002.00039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suter U, Scherer SS. Disease mechanisms in inherited neuropathies. Nat Rev Neurosci. 2003;4:714–726. doi: 10.1038/nrn1196. [DOI] [PubMed] [Google Scholar]
- 43.Patel PI, Roa BB, Welcher AA, Schoener-Scott R, Trask BJ, Pentao L, Snipes GJ, Garcia CA, Francke U, Shooter EM, et al. The gene for the peripheral myelin protein PMP-22 is a candidate for Charcot-Marie-Tooth disease type 1A. Nat Genet. 1992;1:159–165. doi: 10.1038/ng0692-159. [DOI] [PubMed] [Google Scholar]
- 44.Giambonini-Brugnoli G, Buchstaller J, Sommer L, Suter U, Mantei N. Distinct disease mechanisms in peripheral neuropathies due to altered peripheral myelin protein 22 gene dosage or a Pmp22 point mutation. Neurobiol Dis. 2005;18:656–668. doi: 10.1016/j.nbd.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 45.Nelis E, Erdem S, Tan E, Lofgren A, Ceuterick C, Jonghe P, Broeckhoven C, Timmerman V, Topaloglu H. A novel homozygous missense mutation in the myotubularin-related protein 2 gene associated with recessive Charcot-Marie-Tooth disease with irregularly folded myelin sheaths. Neuromuscul Disord. 2002;12:869–873. doi: 10.1016/S0960-8966(02)00046-9. [DOI] [PubMed] [Google Scholar]
- 46.Jordanova A, Jonghe P, Boerkoel CF, Takashima H, Vriendt E, Ceuterick C, Martin JJ, Butler IJ, Mancias P, Papasozomenos S, et al. Mutations in the neurofilament light chain gene (NEFL) cause early onset severe Charcot-Marie-Tooth disease. Brain. 2003;126:590–597. doi: 10.1093/brain/awg059. [DOI] [PubMed] [Google Scholar]
- 47.Claramunt R, Pedrola L, Sevilla T, Lopez de Munain A, Berciano J, Cuesta A, Sanchez-Navarro B, Millan JM, Saifi GM, Lupski JR, et al. Genetics of Charcot-Marie-Tooth disease type 4A: mutations, inheritance, phenotypic variability, and founder effect. J Med Genet. 2005;42:358–365. doi: 10.1136/jmg.2004.022178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang C, McCarty IM, Balazs L, Li Y, Steiner MS. A prostate-derived cDNA that is mapped to human chromosome 19 encodes a novel protein. Biochem Biophys Res Commun. 2002;296:281–287. doi: 10.1016/S0006-291X(02)00872-0. [DOI] [PubMed] [Google Scholar]
- 49.Moresco EM, Koleske AJ. Regulation of neuronal morphogenesis and synaptic function by Abl family kinases. Curr Opin Neurobiol. 2003;13:535–544. doi: 10.1016/j.conb.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 50.Omri B, Crisanti P, Marty MC, Alliot F, Fagard R, Molina T, Pessac B. The Lck tyrosine kinase is expressed in brain neurons. J Neurochem. 1996;67:1360–1364. doi: 10.1046/j.1471-4159.1996.67041360.x. [DOI] [PubMed] [Google Scholar]
- 51.Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer EL, et al. The Pfam protein families database. Nucleic Acids Res. 2004;32:D138–D141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Topilko P, Schneider-Maunoury S, Levi G, Baron-Van Evercooren A, Chennoufi AB, Seitanidou T, Babinet C, Charnay P. Krox-20 controls myelination in the peripheral nervous system. Nature. 1994;371:796–799. doi: 10.1038/371796a0. [DOI] [PubMed] [Google Scholar]
- 53.Warner LE, Svaren J, Milbrandt J, Lupski JR. Functional consequences of mutations in the early growth response 2 gene (EGR2) correlate with severity of human myelinopathies. Hum Mol Genet. 1999;8:1245–1251. doi: 10.1093/hmg/8.7.1245. [DOI] [PubMed] [Google Scholar]
- 54.Warner LE, Mancias P, Butler IJ, McDonald CM, Keppen L, Koob KG, Lupski JR. Mutations in the early growth response 2 (EGR2) gene are associated with hereditary myelinopathies. Nat Genet. 1998;18:382–384. doi: 10.1038/ng0498-382. [DOI] [PubMed] [Google Scholar]
- 55.Shames I, Fraser A, Colby J, Orfali W, Snipes GJ. Phenotypic differences between peripheral myelin protein-22 (PMP22) and myelin protein zero (P0) mutations associated with Charcot-Marie-Tooth-related diseases. J Neuropathol Exp Neurol. 2003;62:751–764. doi: 10.1093/jnen/62.7.751. [DOI] [PubMed] [Google Scholar]
- 56.Dorn T, Riegel M, Schinzel A, Siegel AM, Kramer G. Epilepsy and trisomy 19q–different seizure patterns in a brother and a sister. Epilepsy Res. 2001;47:119–126. doi: 10.1016/S0920-1211(01)00303-5. [DOI] [PubMed] [Google Scholar]