

Abstract
The primary method for neuronal communication involves the release of chemical messengers that are packaged intracellularly in vesicles. Although experiments measuring release at single cells have classically been thought to assess the entire content of vesicles, there is evidence in the literature that suggests that the total transmitter stored in vesicles is not expelled during exocytosis. In this work, we introduce a novel technology using a microfluidic-based platform to electrochemically probe individual PC12 cell vesicles isolated from the cell environment. We measure the total vesicular content using methodology that circumvents the biophysical processes of the cell associated with exocytosis. Direct comparisons of amperometric data from release experiments at single PC12 cells versus our cell-free model reveal that on average vesicles release only 40% of their total transmitter load. The data support the intriguing hypothesis that the average vesicle does not open all the way during the normal exocytosis process, resulting in incomplete distention of the vesicular contents. In addition, we have shown that vesicular catecholamine levels can be altered with pharmacological manipulation and variances observed from these treatments can be resolved at the single vesicle level in a high-throughput manner, a process that we have termed electrochemical cytometry. Upon establishing that release in exocytotic processes proceeds in an incomplete manner, we related electrochemical data quantified from both single cell release experiments and electrochemical cytometry of vesicles to vesicular volume from electron microscopy measurements to investigate the location of intravesicular catecholamine stores retained postfusion.
Keywords: Vesicle, exocytosis, partial release, catecholamine, electrochemical cytometry, amperometry
Neurons communicate through the release of chemical messengers (e.g., neurotransmitters, neurohormones, and neuropeptides), which are packaged intracellularly in submicrometer sized structures called secretory vesicles (1−3). During exocytosis, the fundamental method for neurotransmission, vesicles migrate to the plasma membrane of a cell, fuse, and release their contents into the extracellular space. These messengers can then bind to receptors on a target cell, thus inducing a cascade of signaling events in a complex network (3,4). Researchers have focused on unraveling the elements that govern the exocytotic pathway, and direct measurements of chemical messengers released during exocytosis have allowed vesicle fusion dynamics and presynaptic regulatory processes of neurotransmission to be examined (5−10). Specific efforts have revolved around answering questions such as how is vesicle fusion regulated and how are chemical messengers extruded from vesicles into the extracellular space?
Monoamines (e.g., dopamine, serotonin, histamine, epinephrine, and norepinephrine) are a redox-active family of chemical messengers that have been examined in both the CNS (9−11) and periphery (12−20) of various animal model systems. The release of monoamines at the single vesicle level can be probed on a millisecond time scale using electrochemical methods, making these molecules an attractive target for quantitative mechanistic investigations of exocytosis. Constant potential amperometry performed at carbon-fiber microelectrodes has been used to monitor vesicular monoamine release from single cells (Figure 1A). In this measurement, Faraday’s law (Q = nNF) is used to quantify the mole amount of released monoamine, N, from the time integral of current transients (Q) on the amperometric trace, where n is the number of electrons exchanged in the oxidation reaction (2e− for most monoamines) and F is Faraday’s constant (96 485 C/mol). Long-standing assumptions made in amperometric analyses of exocytosis at single cells are that (i) each current transient is attributed to a discrete vesicle fusion event and (ii) each vesicle releases its entire contents during fusion.
Figure 1.
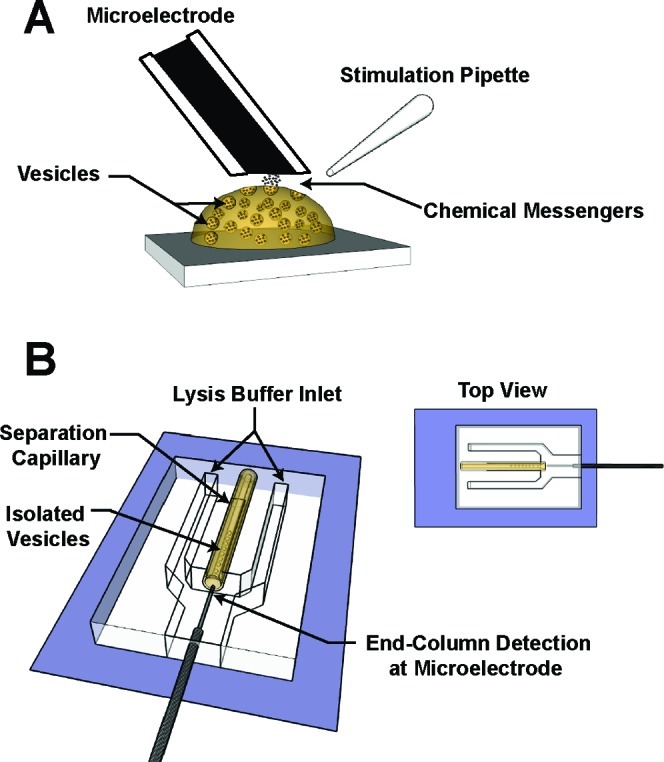
Experimental approaches to measure vesicular transmitter content (not drawn to scale): (A) Stimulated exocytosis at single cells measured using amperometric detection at a carbon-fiber disk microelectrode. The carbon-fiber disk microelectrode is placed onto the cell and held at a potential above the formal oxidation potential of the monoamine investigated (typically ∼0.7 V versus Ag/AgCl; reference electrode placed in outer buffer reservoir). A chemical secretatogue (e.g., elevated K+ solution) is puffed onto a single adherent cell using a micropipet to induce exocytosis. As vesicles undergo exocytosis, they fuse to the plasma membrane and release their contents into the extracellular space where they can be detected at the microelectrode sensor. (B) Isolated vesicle investigation on the microfluidic-based platform. Vesicles are isolated from cells off-line using differential centrifugation. A suspension of vesicles is then electrokinetically injected onto a fused-silica capillary that terminates onto a PDMS-based microfluidic platform. As individual vesicles exit the separation capillary, they are flushed with lysis solution from neighboring channels in a sheath-flow format. Their membranes are instantaneously lysed when they interact with this solution and their contents subsequently detected using end-column amperometry at a cylindrical carbon-fiber microelectrode.
The transmission of chemical messengers by exocytosis has classically been thought to be an all-or-none process that is quantal in nature (21−24). However, the data in several reports suggest that full fusion does not result in complete expulsion of the transmitter in a vesicle, a concept that has not been widely accepted. To experimentally address this hypothesis, we used electrochemistry to investigate vesicle content from the neuroendocrine secretory cell model rat adrenal pheochromocytoma cells (PC12 cells). This immortalized mammalian cell line has been well characterized by us and others for quantification of neurosecretion (25). PC12 cells contain large dense core vesicles that release catecholamines (dopamine and perhaps norepinephrine), a specific group of monoamine messengers implicated in physiological phenomena including neurological disease, learning and memory, and addiction.
In this study, we provide direct measurements of vesicular catecholamine content using electrochemical cytometry. This novel experimental approach is based on technology that we have developed to electrochemically interrogate individual vesicles using a microfluidic-based platform (26). In a previous report, we have demonstrated the chemical selectivity of this detection scheme using artificial vesicles both containing and lacking an electroactive analyte (26). Here, the total catecholamine content of vesicles extracted from secretory cells is directly compared with the more conventional amperometric release experiments performed on intact single cells from matched populations. We show that the entire catecholamine content of the vesicle is not released during fusion. This cell-free model simplifies the complexity of vesicular transmitter quantification measured from standard methods at single cells by eliminating elements of exocytosis governed by the cell machinery and membrane dynamics in the fusion processes (27).
Results and Discussion
Schematic illustrations showing the experimental approaches used here for single cell amperometry and the electrochemical cytometry of vesicles are presented in Figure 1A,B, respectively. The electrochemical cytometry platform is based on methodology used to separate subcellular components by capillary electrophoresis (28,29). First, vesicles from PC12 cells are isolated off-line using differential centrifugation (details provided in Methods). They are then injected onto a fused-silica capillary and separated by an applied electric field. The capillary terminates into a PDMS-based microfluidic device where individual vesicles undergo chemical lysis at the outlet. The vesicle contents are then quantitatively (>95% coulometric efficiency) detected using end-column amperometry at carbon-fiber microelectrodes.
Vesicular Release via Exocytosis Is Not All-or-None
Measurements resulting from the amperometric electrooxidation of catecholamine in vesicles extracted from the cell were compared with measurements from single cell release experiments in which exocytosis was induced by chemical secretagogues (Figure 2). Representative data for these measurements are shown in Figure 2A,B, where each current transient corresponds to the measurement of a single vesicle. Interestingly, as indicated by the amperometric traces for each of the two measurements, many more current transients are detected from the suspension of isolated vesicles (∼5000 events per ∼1 nL injection) compared with that from stimulated exocytosis at single cells (∼100 events with multiple cells and stimulations), making this measurement a high-throughput survey of vesicles within the cell.
Figure 2.
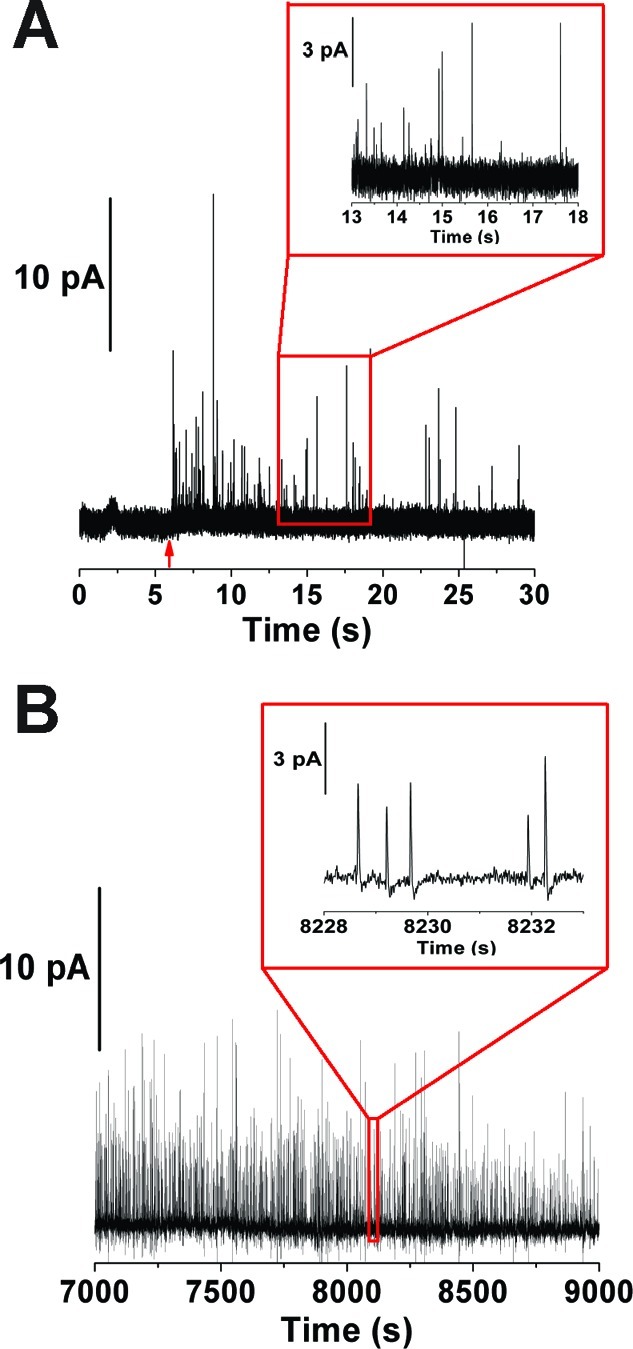
Amperometric quantification of catecholamine amounts in PC12 cells. (A) Representative amperometric trace resulting from exocytotic release at intact cells. Red arrows indicate elevated K+ application used to induce exocytosis. (B) Representative electropherogram for the end-column lysis and amperometric detection of individual vesicles. Vesicles were isolated off-line by differential centrifugation from a matched population of PC12 cells investigated in panel A. Vesicles in panel B were injected at 111 V/cm for 5 s and separated in an applied field of 333 V/cm. Individual events are shown in the expanded axes in panels A and B in order to illustrate typical peak characteristics observed for the analyses. The amperometric signals from single cell experiments were filtered at 2 kHz bandpass and at 15 Hz bandpass for the electrochemical cytometry experiments, providing different baselines for these different experimental approaches.
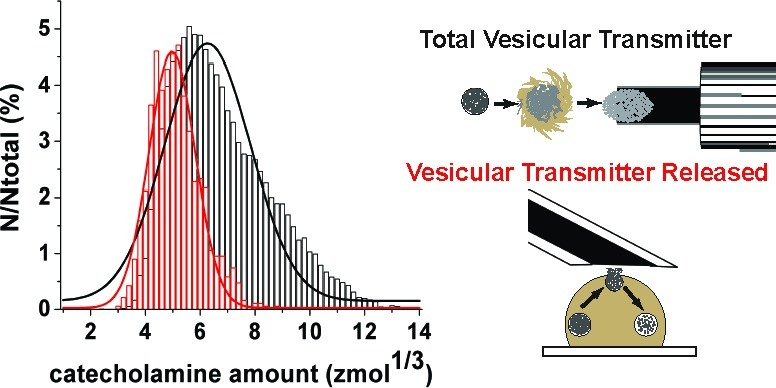
On the basis of Faraday’s law, the mole amount of catecholamine from individual current transients was quantified, binned, and plotted as a normalized frequency histogram of vesicle content (Figure 3). When the total vesicular catecholamine content (as measured in the cell-free model) is compared with the amount released (as measured from single cell amperometry experiments), the data reveal that, on average, approximately 40% of the catecholamine in a vesicle is released during exocytosis. Indeed, significantly less catecholamine was measured by stimulated exocytosis at single cells, 141 ± 3 zmol, versus 387 ± 2 zmol for isolated vesicles measured on the microfluidic platform (Mann−Whitney U-test, p < 0.0001; error is SEM). A Gaussian fit of the data reveals a shift in the distribution for the amount released from cells using stimulated exocytosis (red) compared with that from isolated vesicles investigated on the microfluidic platform (black). Interestingly, if the data from the isolated vesicle experiments is fit to two Gaussian distributions (Figure 3, inset), the correlation coefficient of the fit increases from 0.90 to 0.99, suggesting two populations of vesicles. The mean of each distribution is greater than that measured from exocytosis at single cells (4.97 zmol1/3 for single cell experiments versus 5.5 and 7.7 zmol1/3 for isolated vesicles). This result challenges classical assumptions of quantal release for the stimulated exocytosis of single cells (vide supra).
Figure 3.
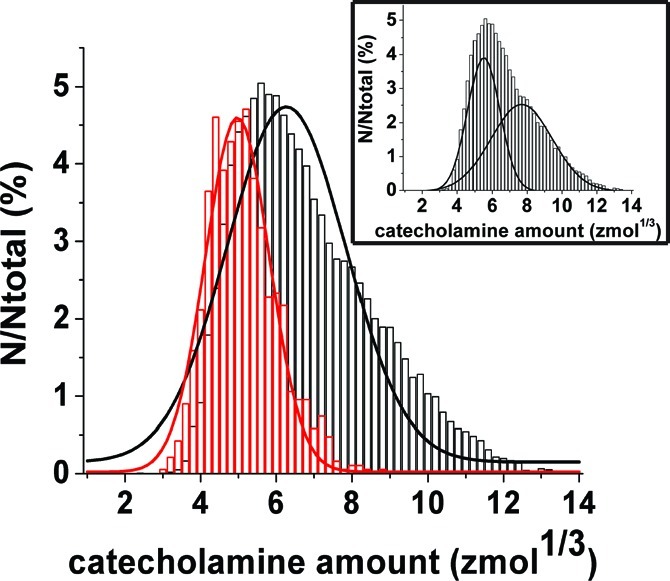
Normalized frequency histogram of the vesicular catecholamine amounts quantified from intact cells that underwent stimulated exocytosis (red) versus isolated vesicles (black) investigated on the microfluidic platform. Exocytosis at single cells was induced by application of elevated K+ solution. Data plotted as the cube root transform. Bin size = 0.2 zmol1/3. Fits were obtained from a Gaussian distribution of the data. Average amount of catecholamine measured from stimulated exocytosis of intact cells was 141 ± 2.67 zmol (n = 946 events), whereas that for isolated vesicles was 387 ± 1.87 zmol (n = 29 643 events). In the inset, two Gaussian distributions are fit to the data for isolated vesicles investigated with electrochemical cytometry. The mean of each distribution is greater than the Gaussian fit of the data from exocytosis experiments at single cells (4.97 zmol1/3 for single cell experiments versus 5.5 and 7.7 zmol1/3 for isolated vesicles). Correlation coefficient for fitting two distributions of these data is equal to 0.99 compared with 0.90 for fitting one distribution. Error is SEM.
This hypothesis that exocytosis does not result in complete expulsion of the transmitter in a vesicle is supported in the literature. Vesicular content measurements have been shown to vary with experimental changes in stimulation (16,30), the osmotic pressure differential between the vesicular and plasma membranes (31,32), vesicular pH manipulation (33), temperature gradients (34), and manipulation of second-messenger systems (35). It is possible that during exocytosis of large dense core vesicles, the event does not result in full distention of the fused vesicle and thus rapid closure takes place prior to full release. This is consistent with studies where evidence suggests that in PC12 cells 97% of exocytosis events are followed by rapid endocytosis (36). In addition, if one compares the data from patch amperometry to that from direct amperometry quantification of the former reveals 3−5 times the release of the latter (37). This is generally not addressed in the literature, but it is likely that the pressure applied during patch amperometry results in more complete distention, consistent with our data.
Vesicular Content Is Altered with Pharmacology at the Single Vesicle Level
The amount of catecholamine released was measured as a function of vesicle volume controlled by pharmacological manipulation. PC12 cells were treated with either L-DOPA (a synthetic precursor to dopamine) or reserpine (a potent inhibitor of the vesicular monoamine transporter (VMAT)), which are known to increase and decrease vesicular catecholamine levels, respectively (12,13). Measurements of the amount released were made at single cells and compared with isolated vesicles as described above. Representative individual current transients observed from both treated and untreated cells are shown in Figure 4A for each measurement. When cells are treated with reserpine the charge, Q, is markedly smaller than that for untreated cells; conversely, the charge increases with L-DOPA treatment. In addition, the charge observed from experiments performed on isolated vesicles is greater than that from stimulated release experiments within each of the treatments.
Figure 4.

Pharmacological treatment alters vesicular quantal size from both intact cells and individual vesicles isolated from cells. (A) Typical peak characteristics representing pharmacological manipulation of vesicular quantal size from both stimulated exocytosis at single PC12 cells and isolated vesicles. Data were collected from matched cell preparations. Cells were incubated with either 100 nM reserpine (a potent VMAT inhibitor) or 100 μM L-DOPA (a precursor in dopamine synthesis) for 90 min prior to stimulated exocytosis investigations and vesicle isolation. (B) Representative normalized frequency histograms describing the distributions of vesicular catecholamine amounts quantified from reserpine-treated (red), untreated (blue), and L-DOPA-treated (black) intact cells that underwent stimulated exocytosis. (C) Representative normalized frequency histograms of vesicular catecholamine amounts quantified from isolated vesicles from reserpine-treated (red), untreated (blue), and L-DOPA-treated (black) matched cell populations in panel B. Data plotted as cubed root amounts. Bin size = 0.2 zmol1/3. Fits were obtained from a Gaussian distribution of the data. (D) Cumulative analysis for the average number of molecules of catecholamine quantified per vesicle from stimulated exocytosis of intact cells (striped) versus individual isolated vesicles (white) under pharmacological manipulation. The number of events measured for stimulated exocytosis at single cells was 312, 946, and 1376 for reserpine-treated, untreated, and L-DOPA-treated cells, respectively. The number of events measured for the electrochemical cytometry of vesicles was 13 363, 29 643, and 22 270 for reserpine-treated, untreated, and L-DOPA-treated cells, respectively. Error in panel D is SEM.
Vesicular catecholamine amounts were measured and plotted as normalized frequency histograms for the amount released by amperometry from intact cells (Figure 4B). This is compared with electrochemical cytometry measurements of the amount in vesicles isolated from the cell environment (Figure 4C). Results from the reserpine-treated cells (red), untreated cells (blue), and L-DOPA-treated cells (black) are shown for each measurement. A cumulative analysis of average vesicular catecholamine content for matched cell populations is presented in Figure 4D. The amount of catecholamine detected following L-DOPA treatment significantly increased versus control (one-way ANOVA, p < 0.001) whether release was observed by amperometry or the total vesicle amount was observed by electrochemical cytometry. Accordingly, the reserpine treatment significantly decreased the amount of catecholamine detected versus control for both release and vesicle content measurements (one-way ANOVA, p < 0.001 for the amount released from intact cells and p < 0.05 for the amount in vesicles isolated from the cell environment). In addition, the total vesicle content is significantly different than the amount detected in release experiments (one-way ANOVA, p < 0.001) for control, L-DOPA, and reserpine treatment. Vesicular catecholamine amounts can be directly linked to the number of molecules detected using Avogadro’s number. For stimulated exocytosis at single cells, the release of 72 000 ± 3100 molecules was detected for reserpine-treated cells, 85 000 ± 1600 molecules for untreated cells, and 117 000 ± 2700 molecules for L-DOPA-treated cells (n = 312, 946, and 1376 events; error is SEM). Vesicular amounts measured by electrochemical cytometry were 167 000 ± 1100 molecules following reserpine treatment, 220 000 ± 1100 molecules for untreated cells, and 320 000 ± 1600 molecules following L-DOPA treatment (n = 13 363, 29 643, and 22 270 events; error is SEM).
The average amount of catecholamine released following stimulated exocytosis at intact cells is significantly less than the total vesicular catecholamine amount. The fraction of average catecholamine amount released was 43% ± 2%, 39% ± 1%, and 37% ± 1% (error is SEM) for the reserpine-treated, untreated, and L-DOPA-treated cells, respectively, further indicating that the entire contents are not expelled from vesicles during the exocytosis processes. Moreover, the drug treatments yield similar results for both changes in release and total vesicle content. The average vesicular release of catecholamine relative to control cells was reduced by 15% ± 4% with reserpine treatment and was increased by 27% ± 4% for L-DOPA treatment. This trend was maintained in the analysis of isolated vesicles by electrochemical cytometry; a 24% ± 1% decrease in average vesicular catecholamine content for reserpine treatment and a 31% ± 1% increase with L-DOPA treatment was observed compared with control (error is SEM). Interestingly, the drug treatments demonstrated the utility of this novel cell-free method to serve as a simple and quantitative tool for performing high-throughput analyses to screen for direct physiological effects of pharmacological manipulation at the single vesicle level.
Size Measurements of Vesicles Validate the Electrochemical Cytometry Results
Our results comparing vesicular catecholamine content by electrochemical cytometry to electrochemical measurements of release consistently show that the amount released upon stimulated exocytosis at single cells is approximately 40% of that measured from isolated vesicles. To assign this fraction to the hypothesis that the vesicle does not release its entire contents during exocytosis, we must confirm several experimental parameters. These include (i) that the methods are capable of quantifying similar-sized vesicles, (ii) that the isolation procedures yield individual secretory vesicles for analysis, and (iii) that the isolated vesicles are representative of the vesicles sampled in single cell release experiments. The methods used are capable of quantifying similar-sized vesicles; our limits of detection (LOD) on the microfluidic-based platform are similar to those from release events in single cell experiments (LOD ≈ 5000 molecules for each method). Indeed, the smallest-sized vesicles measured at single cells are observed in isolated vesicle experiments, as indicated by the distributions in Figure 4B,C.
To confirm that the isolation procedures yield individual secretory vesicles for analysis, independent measurements of vesicle size from the treated and untreated PC12 cells investigated above were made using dynamic light scattering (DLS). As previously indicated, pharmacology can be used to alter vesicular catecholamine levels in this cell model, and these changes can be quantified directly from size measurements since vesicle volume is affected as a result of treatment (13,14). DLS is a noninvasive measurement that relates the diffusion velocity of a suspension of vesicles in the cell-free model to the average hydrodynamic diameter using the Stokes−Einstein equation (26). The average diameters for reserpine-treated, untreated, and L-DOPA-treated PC12 cell vesicles are listed in Table 1. DLS measurements were used to provide a qualitative analysis of both treated and untreated vesicle suspensions. These measurements indicate that the vesicle isolation procedures used in the cell-free model are valid since the mean diameter is in agreement with independent measurements of PC12 cell vesicular size obtained in this study (vide infra) and in previous reports (12,13). This was also verified by performing a Western Blot for synaptophysin, a known integral membrane protein present on neurosecretory vesicles (Figure S1, Supporting Information).
Table 1. Vesicle Size Measurements from Dynamic Light Scattering (DLS) and Transmission Electron Microscopy (TEM).
| treatment | vesicle diameter (nm) by DLSa | vesicle diameter (nm) by TEMb |
|---|---|---|
| reserpinec | 174 ± 47 | 141 ± 2 |
| untreatedd | 183 ± 24 | 153 ± 2 |
| L-DOPAe | 194 ± 39 | 186 ± 4 |
Error = SD.
Error = SEM.
For TEM data, 206 vesicles.
For TEM data, 217 vesicles.
For TEM data, 122 vesicles.
Transmission electron microscopy (TEM) is a well-established quantitative method for measuring subcellular domains within the construct of the cell environment and, therefore, was employed for size analysis of vesicles. Representative electron micrographs for vesicle analyses in single cells are pictured in Figure 5A and measurements that quantify these data are listed in Table 1. To investigate whether vesicles from similar pools were being sampled in the single cell release and isolated vesicle experiments, we developed a set of criteria for two types of vesicles and compared their sizes as determined by TEM. The first group included vesicles close to or already primed onto the plasma membrane (<65 nm, less than the approximate average radius of the vesicles measured). These vesicles are considered to be in the readily releasable pool and presumably the vesicles measured during stimulated release at single cells. We then defined a second group (>65 nm from the membrane), designated as the reserve pool. In other secretory cell models, the reserve pool is thought to contain larger, more mature vesicles (34). It is important to note that the procedures employed for vesicle isolation sample from the entire cell, which should yield components from both the readily releasable and reserve pools.
Figure 5.

Electron microscopy investigation of PC12 cell vesicular size and volume. (A) Representative TEM images from treated and untreated PC12 cells. Scale bar = 400 nm. (B) Plot of average vesicle size versus distance from the plasma membrane determined by TEM for untreated PC12 cells. Error is SEM. Values marked ns are not statistically different, p > 0.05, Mann−Whitney U-test. (C) Plot describing average volume of dense core vesicles and their constituents upon reserpine (white) treatment, in untreated (black) cells, and upon L-DOPA (striped) treatment measured by TEM. Values marked with ∗∗∗ in C are statistically different from untreated cells, with p < 0.0001, Mann−Whitney U-test. (D) Correlation between the amount of catecholamine detected in intact vesicles (cytometry; black boxes) and released from vesicles (amperometry; red circles) versus the volume in the vesicle halo (from TEM measurements). Data points for each of the measurements going from left to right are reserpine-treated, untreated, and L-DOPA-treated cells. Amperometric measured amounts of catecholamine are plotted against TEM measurements of halo volume. The slope is the concentration of catecholamine in the halo and the intercept is the amount of catecholamine in the dense core. Values listed in Table 2. Linear regression results are available in Methods.
The average diameter of vesicles in the readily releasable pool as measured by TEM was 148 ± 3 nm (n = 68; error is SEM). Notably, vesicles in the reserve pool, which are further from the plasma membrane, possessed an average diameter of 153 ± 2 nm (n = 147; error is SEM). This difference is not statistically significant (Mann−Whitney U-test, p = 0.30). The plot in Figure 5B illustrates this by showing that vesicles in these cells are the same size whether they are primed for exocytosis close to the plasma membrane or far from the membrane (measurements recorded for vesicles up to 3 μm from the membrane). This validates that the isolated vesicles are representative of the vesicles sampled in single cell release experiments and that measurements quantified from isolated vesicles are due to excess catecholamine retained during exocytotic release at single cells. In fact, it is likely that even less than 40% is released due to possible losses during the vesicle isolation procedure. Indeed, if catecholamine were to escape the vesicle after cell fractionation, this would cause an underestimation of the amount in each vesicle.
Quantification of Intravesicular Catecholamine Stores: Halo vs the Dense Core
Once it was determined that the vesicle does not release its entire contents during exocytosis, we examined where the excess catecholamine remains after full fusion. TEM was used to map the size characteristics of catecholamine stores at the subvesicle level. The TEM images in Figure 5A reveal the prime components of the large dense core vesicles in this neurosecretory cell model, namely, a dark granule composed of a semicrystalline matrix of acidic proteins and semisoluble catecholamine (referred to as the dense core) encased within a membrane containing solubilized catecholamine that surrounds the dense core (referred to as the halo). The volumes of each intravesicular domain were calculated from TEM size measurements (Figure 5C). The data reveal that various constituents of the vesicle are altered upon pharmacological manipulation of the catecholamine stores as previously reported for this cell line (12), also demonstrating the existence of the halo as a genuine vesicular feature. A significant decrease in vesicle volume is observed when catecholamine levels are depleted with reserpine treatment, and conversely, a significant increase is observed with L-DOPA treatment (Mann−Whitney U-test, p < 0.001). The increase in halo volume with L-DOPA treatment (Mann−Whitney U-test, p < 0.001) resulted in a much more pronounced effect than dense core size, which was not statistically different from control (Mann−Whitney U-test, p > 0.05). This phenomenon was conserved with reserpine treatment where a statistically significant amount of catecholamine was depleted from the halo compared with control (Mann−Whitney U-test, p < 0.005).
During the exocytotic full fusion processes, it is thought that crystalline catecholamine present in the dense core granule interacts with extracellular fluid, swells, and is solubilized as it is extruded through the fusion pore concomitantly with the halo contents (17). Our results show a ∼60% decrease in electrochemically measured catecholamine amounts between release at single cells and total content from the vesicles in the cell-free model. To account for this discrepancy, we hypothesize that catecholamine constituents intercalated in both the halo and dense core are retained postfusion. This was experimentally addressed by correlating the electrochemical measurements from each of the methods with volume data elucidated from TEM intravesicular size measurements as a function of pharmacological manipulation of catecholamine content. The result is shown in Figure 5D, where catecholamine amount data from release at single cells (red) and isolated vesicles (black) is plotted versus the volume of the halo, acquired from TEM data of treated and untreated PC12 cells. Linear regression analyses for the model systems investigated provide information about the average amount of catecholamine detected from the dense core (the intercept) and the concentration of catecholamine in the halo (the slope). The differences in the slopes of the linear regression for each method (Table 2) are not statistically significant (Student’s t test, p = 0.15). However, the intercept of these data reveals that single cells expel an average of 50% of catecholamine associated with the dense core during exocytosis (Student’s t test, p < 0.05). The notion of vesicular catecholamine stores being retained postfusion is supported in the literature for partial release via processes such as “kiss and run” (14,38) exocytosis and a “flickering fusion pore” (39). During these mechanisms, the vesicle is thought to momentarily fuse with the cell membrane forming a small pore through which transmitter can escape (39,40). The pore then closes prior to full distention of vesicular contents. In single cell experiments, the amperometric signals for exocytosis are generally thought to represent complete vesicle distension. However, the electrochemical cytometry data reported here demonstrate incomplete expulsion of vesicular catecholamine. To explain this and the retention of the catecholamine in both the dense core and halo, we propose that the average dense core vesicle does not fully open during exocytosis, a process in between kiss and run and full membrane distension.
Table 2. Determining Intravesicular Catecholamine Stores of PC12 Cell Vesicles Using Amperometric and TEM Measurementsa.
| amt detected (zmol) | amt in dense core (zmol) | [halo] (mM) | |
|---|---|---|---|
| stimulated exocytosis at single cells | 141 ± 3 | 75 ± 10 | 39 ± 7 |
| isolated vesicles | 387 ± 2 | 153 ± 70 | 125 ± 35 |
The average amounts of catecholamine were measured directly using amperometric detection of stimulated release at single cells and electrochemical cytometry of isolated vesicles. Error is SEM. Amount in the dense core is the intercept from Fig. 5D (error is SD). The concentration in the halo is the slope from Fig. 5D (error is SD).
Conclusions
We have demonstrated that individual vesicles isolated from the cell environment can be separated and probed to quantify transmitter content, a process we call electrochemical cytometry. These amounts can be directly altered with pharmacological treatment and variances due to the treatments can be resolved at the single vesicle level in a high-throughput manner. By correlating data from single cell release experiments with our cell-free model, we have demonstrated that vesicles release only 40% of their total catecholamine load during exocytosis, a premise that contradicts classical assumptions of complete quantal release. Furthermore, we have shown that independent measurements of vesicular and subvesicular size both validate our novel method for monitoring transmitter content and also contribute information regarding in which intravesicular domain excess catecholamine is stored. The implications of these results to neuroscience are large; namely, vesicular neurotransmitter and hormone release is not necessarily all-or-none. Therefore, exocytotic release might be regulated within a single event, which could have implications in neural disease states. Moreover, controlling the fraction of release from vesicles is a potential pharmaceutical target.
Methods
Reagents
Sodium chloride (NaCl), potassium chloride (KCl), magnesium chloride (MgCl2), glucose, N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), calcium chloride (CaCl2), N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid (TES), sodium hydroxide (NaOH), sodium dodecyl sulfate (SDS), reserpine, 3,4-dihydroxy-l-phenyl-alanine (L-DOPA), hydrofluoric acid (aq 48%), and the Synaptic Vesicles Isolation Kit were obtained from Sigma Aldrich (St. Louis, MO). All chemicals were used as received. Isotonic physiological saline used in single cell amperometry experiments was prepared with 150 mM NaCl, 5 mM KCl, 1.2 mM MgCl2, 5 mM glucose, 10 mM HEPES, and 2 mM CaCl2. For individual vesicle measurements on the microfluidic-based platform, the electrophoretic separation buffer consisted of 50 mM TES with 2% 1-propanol. The lysis buffer was 50 mM TES with 5% (w/v) SDS. All buffers were adjusted to pH 7.4 using NaOH and filtered through 0.2 μm pore size filters (Nalgene, Rochester, NY).
Cell Culture
Rat pheochromocytoma cells (PC12) were purchased from the American Type Culture Collection (Manassas, VA) and maintained as previously described (41). For stimulated exocytosis experiments at single cells, PC12 cells were grown on poly(d-lysine)-coated culture dishes (Becton Dickinson, Bedford, MA) in supplemented RPMI-1640 medium. For isolated vesicle experiments, cells were cultured on 75 cm3 poly(d-lysine)-coated flasks. Cells were maintained in a 7% CO2 atmosphere at 37 °C and used when confluency was reached.
Vesicle Isolation
A Synaptic Vesicle Isolation Kit (Sigma-Aldrich) was used to extract vesicles from the cells with a modified procedure provided by the manufacturer. All buffer constituents and isolation procedures are listed as proprietary knowledge of Sigma Aldrich. Modifications were made to the protocol to ensure a sufficient amount of cells to suit the recommended procedures, which have been tested on rat and rabbit brain tissue. To recreate a tissue-like model from this immortalized cell line, confluent cells from three 75 cm3 flasks were forcefully released from the poly(d-lysine)-coated surface and the three suspensions were combined into one fraction. This fraction was then centrifuged at 1500 × g for 5 min to pellet the suspension. The supernatant containing growth media was then removed and discarded. According to the manufacturer-recommended protocol, cells were subjected to lysis in the presence of 10 mL of hypoosmotic buffer. This fraction was then centrifuged at 20 000 × g at 4 °C for 25 min. This allowed for removal of other organelles present in the cell, as they pelleted to the bottom of the centrifuge tube during this treatment. The supernatant, containing vesicles, was then recovered and subjected to ultracentrifugation at 70 000 × g at 4 °C for 1 h. Vesicles were recovered as a pellet in the bottom of the centrifuge tube. The supernatant was discarded, and 2 mL of vesicle storage buffer was added to the pellet, resulting a crude suspension of vesicles for analysis. A Western Blot was performed to confirm successful vesicle isolation by targeting synaptophysin, a known integral membrane protein present on neuroendocrine vesicles (Figure S1, Supporting Information).
Electrochemical Recordings for Stimulated Exocytosis at Single Cells
Carbon-fiber disk microelectrodes (5 μm diameter) were constructed as described previously (42) and back-filled with 3 M KCl. Electrode tips were polished at a 45° angle on a diamond dust-embedded micropipet beveling wheel (model BV-10; Sutter Instrument, Novato, CA). Amperometric recordings were collected as described previously (13). Briefly, electrodes were held at +0.65 V vs a Ag/AgCl reference electrode (World Precision Instruments, Inc., Sarasota, FL) using a commercially available patch-clamp instrument (Axopatch 200B; MDS Analytical Technologies, Sunnyvale, CA) configured as described previously (32). The output was filtered at 2 kHz using a four-pole low-pass Bessel filter and digitized at 5 kHz. Data were displayed in real time and stored in the computer with no further filtering. Exocytosis was measured from 40 s intervals of measured current transients evoked with a 5 s, 20 psi pulse (Picospritzer II; General Valve, Fairfield, NJ) of physiological saline containing elevated potassium (100 mM KC1). All experiments were performed at 37 ± 1 °C. Exocytotic spikes were identified and the areas (fC) were determined using a multipass algorithm described previously (21). Mini Analysis (Synaptosoft Inc., Fort Lee, NJ) was used for analysis of the resultant peaks to determine area. Signals were designated as exocytotic events if their amplitude values were five times the rms noise of the baseline compared with a 1 s portion of stable baseline recorded before the first stimulation. All peaks identified by the program were inspected visually, and complex peaks were excluded manually from the data sets.
Microfluidic Device Fabrication for the Separation, Lysis, and Electrochemical Detection of Individual Vesicles
A hybrid capillary−microfluidic device was developed as previously described to investigate catecholamine amounts from individual vesicles isolated from the cell environment (26). Briefly, microfluidic channels were fabricated using standard photo- and soft-lithography methods. A master mold was developed by spin-coating 125 μm of SU-8 100 negative photoresist (MicroChem Corp., Newton, MA) on a 3-in. silicon wafer (Silicon Quest International, Inc., Santa Clara, CA). A photolithographic mask containing imprinted device features was made from a high-resolution laser photoplot (CAD/Art Services, Inc., Bandon, OR). The mask was placed over the wafer, which was then exposed to ultraviolet light, and developed according to the resist manufacturer protocol.
Soft lithography was carried out using a Sylgard 184 silicone elastomer kit (Dow Corning Corp., Midland, MI). A 10:1 ratio of poly(dimethylsiloxane) (PDMS) prepolymer base to curing agent was cast onto the master mold and cured at 70 °C for 2 h. The PDMS layer was then peeled from the master, revealing an impression of microfluidic channels. The center channel served to secure the separation capillary in the finished device. The other two channels, each 200 μm wide, are set at a 30° angle to the center channel and used to direct lysis buffer to the capillary outlet in a sheath-flow format. The three channels converge into a 2 mm wide channel where the electrode is placed for detection of catecholamine quantified from individual lysed vesicles. A buffer reservoir for capillary electrophoresis was cut into a 2 mm layer of PDMS and plasma-bonded to a glass microslide (Corning Inc., Corning, NY). The layer containing imprinted microfluidic channels was then plasma-bonded onto the reservoir layer to form a three-layer device (100 W, 1 min.).
Separation and Detection of Isolated Vesicles
Fused-silica capillaries (45 cm in length, 15 μm i.d./150 μm o.d., Polymicro Technologies, Phoenix, AZ) were prepared for electrochemical detection by removing 2 mm of the polyimide coating with a flame and subsequently etching the exposed fused silica by purging He (250 psi) for 15 min in a HF bath. This resulted in an etch with a frustum geometry measuring approximately 40 μm wide at the base, which serves to both ease placement of the electrode at the capillary outlet and decouple the applied separation voltage from the electrochemical cell (43).
Electrokinetic injections of vesicles were performed for 5 s at 111 V/cm, and separations were carried out at 333 V/cm using a high-voltage supply (Spellman, Hauppauge, NY). Capillaries were conditioned before each separation to prevent nonspecific binding of the vesicle membrane to the fused silica by rinsing at 333 V/cm with 1 M NaOH for 2 min, Ultratrol Dynamic Pre-Coat-HN (Target Discovery, Palo Alto, CA) for 5 min, and separation buffer for 10 min. A syringe pump (KD Scientific, Holliston, MA) was used to control volumetric flow of lysis buffer via 1 mL plastic syringes continuously through the microchannels at a rate of 2 μL/min (0.05 cm/s).
Amperometric electrochemical detection was carried out with a two-electrode format. A 5 μm-diameter carbon fiber was sealed in a glass capillary and cut to a length of approximately 500 μm from the glass seal to fabricate a cylindrical microelectrode as previously described (44). The electrode was held at 0.90 V versus a silver wire quasi-reference electrode (Ag QRE, 0.25 mm diameter, Alfa Aesar, Ward Hill, MA), and the carbon-fiber electrode was positioned at the outlet of the capillary using an x,y,z-micromanipulator (Newport, Irvine, CA). Previous measurements and modeling indicated that this system provided a coulometric efficiency of 87% for catecholamine in vesicles; however, with less filtering, we have attained greater than 95% coulometric efficiency.
Current was measured using a Keithly model 427 (15 Hz bandpass) current amplifier (Cleveland, OH) and digitized at 500 Hz with a National Instruments PCI-6221 DAQ card using LabView 8.0 software (National Instruments, Austin, TX) written in-house. OriginLab 8.0 (Northampton, MA) was used to generate electropherograms and Mini Analysis (Synaptosoft Inc., Fort Lee, NJ) was used for analysis of the resultant peaks to determine area (fC). Events were quantified if the signal was greater than five times the rms baseline noise.
Western Blot of Isolated Vesicles
Vesicles isolated from the cell environment were investigated for the presence of synaptophysin, a known integral membrane protein present in synaptic and neuroendocrine vesicles. A 50 μL suspension of vesicles was combined with 50 μL of lysis solution containing 5% (w/v) SDS in 50 mM TES buffer. The mixture was vortexed vigorously for 1 min. Aliquots of this solution containing ∼20 μg of protein were loaded into four lanes of a 12% SDS−PAGE gel. Separations were carried by applying 200 V for 2 h to the gel. A SeeBlue Plus2 prestained standard protein kit (Invitrogen, Carlsbad, CA) was used to visualize the separation. Following this, the gel was transferred onto nitrocellulose paper, followed by blocking and incubation with the primary antibody, monoclonal anti-synaptophysin (2 h incubation at room temperature; 1:1000 dilution of antibody). Monoclonal anti-synaptophysin antibody was provided in the Synaptic Vesicle Isolation Kit (Sigma Aldrich)). The secondary antibody, HRP-conjugated antimouse IgG, was used for chemiluminescent detection of the targeted protein (ProteoQwest Chemiluminescent Western Blotting Kit, Sigma Aldrich). Results revealed a prominent band at 38 kDa, confirming the presence of synaptophysin in the vesicle lysate (Supporting Information).
Dynamic Light Scattering of Isolated Vesicles
A Zetasizer Nano S (Malvern Instruments, Worcestershire, UK) was used to acquire light scattering data. Measurements were collected using a 4 mW HeNe laser operated at 633 nm. Vesicle suspensions were diluted 10-fold in buffer. Data were collected in size mode at 25 °C and fit using instrument software.
Transmission Electron Microscopy of Single Cells
PC12 cells were rinsed with RPMI-1640 medium without serum and detached from the flasks. Single cell suspensions were transferred to Microfuge tubes and pelleted at 100 × g for 10 min. Cell pellets were fixed with an ice-cold fixative containing 2% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4, at room temperature for 1 h and then incubated overnight at 4 °C. The cells were postfixed in 1% OsO4 for 1 h and dehydrated by serial treatment in solutions of graded ethanol and embedded in Eponite 12. The areas of interest were selected under a dissecting microscope, and 80-nm-thick sections were produced in an ultramicrotome (Reichart Microscopy, Depew, NY). Sections were contrast-enhanced with uranyl acetate and lead citrate and examined with a JEOL JEM 1200 EXII transmission electron microscope (JEOL, Peabody, MA) at 80 kV. Quantitative analysis of vesicle structures was performed using Image J 1.37v (Wayne Rasband, NIH, Bethesda, MD). Transmission electron microscopy images were imported into this software, and the limiting membrane of each vesicle as well as the perimeter of its dense core were traced. Once each object was inscribed, Image J determined its diameter (the average distance of the major and minor axis on the initial trace). Only vesicles in which a dense core could be clearly identified were measured. Vesicle diameter and dense core diameter are measured and converted to volume from the equation of a sphere (V = 4/3πr3). Halo volume is the difference between the volumes calculated from the vesicle and dense core. Measured values were adjusted to account for thickness of the sample tissue slice (80 nm) as previously reported (45). For the correlation between the amount of catecholamine detected (from amperometry measurements) and the volume in the halo (from TEM measurements) for measured release at single cells and isolated vesicles in Figure 4D, linear regression of release experiments on single cells yielded the equation y = 38.97x + 74.76 (R2 = 0.985) and that for isolated vesicle experiments yielded y = 124.7x + 153.2 (R2 = 0.925).
Acknowledgments
The authors thank Dr. Richard Cyr and Dr. Bernhard Luscher of Penn State’s Department of Biology for use of the ultracentrifuge and Western blot supplies, respectively, Dr. Sujin Yun from Penn State’s Department of Chemistry for performing a Western Blot on the isolated vesicles, and Dr. Gang Ning from Penn State’s Veterinary and Biomedical Sciences Department for TEM assistance.
Supporting Information Available
Western Blot for synaptophysin, which was used to confirm the vesicle isolation procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by the NIH grant 5R01EB00352-10 and by a grant from the Swedish Science Foundation (VR). A.G.E. is supported by a Marie Curie Chair from the European Union’s sixth Framework. This publication was supported by The Pennsylvania State University Materials Research Institute Nano Fabrication Network and the National Science Foundation Cooperative Agreement No. 0335765, National Nanotechnology Infrastructure Network, with Cornell University.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Valtorta F.; Fesce R.; Grohovaz F.; Haimann C.; Hurlbut W. P.; Iezzi N.; Tarelli F. T.; Villa A.; Ceccarelli B. (1990) Neurotransmitter release and synaptic vesicle recycling. Neuroscience 35, 477–489. [DOI] [PubMed] [Google Scholar]
- Edwards R. H. (2007) The neurotransmitter cycle and quantal size. Neuron 55, 835–858. [DOI] [PubMed] [Google Scholar]
- Sudhof T. C. (2004) The synaptic vesicle cycle. Annu. Rev. Neurosci. 27, 509–547. [DOI] [PubMed] [Google Scholar]
- Rahamimoff R.; Fernandez J. M. (1997) Pre- and postfusion regulation of transmitter release. Neuron 18, 17–27. [DOI] [PubMed] [Google Scholar]
- Wightman R. M.; Jankowski J. A.; Kennedy R. T.; Kawagoe K. T.; Schroeder T. J.; Leszczyszyn D. J.; Near J. A.; Diliberto E. J.; Viveros O. H. (1991) Temporally resolved catecholamine spikes correspond to single vesicle release from individual chromaffin cells. Proc. Natl. Acad. Sci. U.S.A. 88, 10754–10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amatore C.; Arbault S.; Bonifas I.; Guille M.; Lemaitre F.; Verchier Y. (2007) Relationship between amperometric pre-spike feet and secretion granule composition in chromaffin cells: An overview. Biophys. Chem. 129, 181–189. [DOI] [PubMed] [Google Scholar]
- Pothos E. N. (2002) Regulation of dopamine quantal size in midbrain and hippocampal neurons. Behav. Brain Res. 130, 203–207. [DOI] [PubMed] [Google Scholar]
- Alvarez de Toledo G.; Fernandezchacon R.; Fernandez J. M. (1993) Release of secretory products during transient vesicle fusion. Nature 363, 554–558. [DOI] [PubMed] [Google Scholar]
- Hochstetler S. E.; Puopolo M.; Gustincich S.; Raviola E.; Wightman R. M. (2000) Real-time amperometric measurements of zeptomole quantities of dopamine released from neurons. Anal. Chem. 72, 489–496. [DOI] [PubMed] [Google Scholar]
- Gubernator N. G.; Zhang H.; Staal R. G. W.; Mosharov E. V.; Pereira D. B.; Yue M.; Balsanek V.; Vadola P. A.; Mukherjee B.; Edwards R. H.; Sulzer D.; Sames D. (2009) Fluorescent false neurotransmitters visualize dopamine release from individual presynaptic terminals. Science 324, 1441–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pothos E. N.; Davila V.; Sulzer D. (1998) Presynaptic recording of quanta from midbrain dopamine neurons and modulation of the quantal size. J. Neurosci. 18, 4106–4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colliver T. L.; Pyott S. J.; Achalabun M.; Ewing A. G. (2000) VMAT-mediated changes in quantal size and vesicular volume. J. Neurosci. 20, 5276–5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sombers L. A.; Hanchar H. J.; Colliver T. L.; Wittenberg N.; Cans A.; Arbault S.; Amatore C.; Ewing A. G. (2004) The effects of vesicular volume on secretion through the fusion pore in exocytotic release from PC12 cells. J. Neurosci. 24, 303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D.; Edwards R. (2000) Vesicles: Equal in neurotransmitter concentration but not in volume. Neuron 28, 5–7. [DOI] [PubMed] [Google Scholar]
- Gong L. W.; Hafez I.; Alvarez de Toledo G.; Lindau M. (2003) Secretory vesicles membrane area is regulated in tandem with quantal size in chromaffin cells. J. Neurosci. 23, 7917–7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pothos E. N.; Mosharov E.; Liu K. P.; Setlik W.; Haburcak M.; Baldini G.; Gershon M. D.; Tamir H.; Sulzer D. (2002) Stimulation-dependent regulation of the pH, volume and quantal size of bovine and rodent secretory vesicles. J. Physiol. 542, 453–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerberg J.; Curran M.; Cohen F. S.; Brodwick M. (1987) Simultaneous electrical and optical measurements show that membrane-fusion precedes secretory granule swelling during exocytosis of beige mouse mast-cells. Proc. Natl. Acad. Sci. U.S.A. 84, 1585–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciobanu L.; Rubakhin S. S.; Stuart J. N.; Fuller R. R.; Webb A. G.; Sweedler J. V. (2004) Characterization of the physicochemical parameters of dense core atrial gland and lucent red hemiduct vesicles in Aplysia californica. Anal. Chem. 76, 2331–2335. [DOI] [PubMed] [Google Scholar]
- Ge S. C.; White J. G.; Haynes C. L. (2009) Quantal release of serotonin from platelets. Anal. Chem. 81, 2935–2943. [DOI] [PubMed] [Google Scholar]
- Albillos A.; Dernick G.; Horstmann H.; Almers W.; Alvarez de Toledo G.; Lindau M. (1997) The exocytotic event in chromaffin cells revealed by patch amperometry. Nature 389, 509–512. [DOI] [PubMed] [Google Scholar]
- Mosharov E. V.; Sulzer D. (2005) Analysis of exocytotic events recorded by amperometry. Nat. Methods 2, 651–658. [DOI] [PubMed] [Google Scholar]
- Martin A. R. (1966) Quantal nature of synaptic transmission. Physiol. Rev. 46, 51–66. [Google Scholar]
- Finnegan J. M.; Pihel K.; Cahill P. S.; Huang L.; Zerby S. E.; Ewing A. G.; Kennedy R. T.; Wightman R. M. (1996) Vesicular quantal size measured by amperometry at chromaffin, mast, pheochromocytoma, and pancreatic beta-cells. J. Neurochem. 66, 1914–1923. [DOI] [PubMed] [Google Scholar]
- Monck J. R.; Fernandez J. M. (1992) The exocytotic fusion pore. J. Cell Biol. 119, 1395–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerink R. H. S.; Ewing A. G. (2008) The PC12 cell as model for neurosecretion. Acta Physiol. 192, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omiatek D. M.; Santillo M. F.; Heien M. L.; Ewing A. G. (2009) Hybrid capillary−microfluidic device for the separation, lysis, and electrochemical detection of vesicles. Anal. Chem. 81, 2294–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D.; Pothos E. N. (2000) Regulation of quantal size by presynaptic mechanisms. Rev. Neurosci. 11, 159–212. [DOI] [PubMed] [Google Scholar]
- Fuller K. M.; Arriaga E. A. (2003) Analysis of individual acidic organelles by capillary electrophoresis with laser-induced fluorescence detection facilitated by the endocytosis of fluorescently labeled microspheres. Anal. Chem. 75, 2123–2130. [DOI] [PubMed] [Google Scholar]
- Chiu D. T.; Lillard S. J.; Scheller R. H.; Zare R. N.; Rodriguez-Cruz S. E.; Williams E. R.; Orwar O.; Sandberg M.; Lundqvist J. A. (1998) Probing single secretory vesicles with capillary electrophoresis. Science 279, 1190–1193. [DOI] [PubMed] [Google Scholar]
- Elhamdani A.; Palfrey H. C.; Artalejo C. R. (2001) Quantal size is dependent on stimulation frequency and calcium entry in calf chromaffin cells. Neuron 31, 819–830. [DOI] [PubMed] [Google Scholar]
- Sombers L. A.; Maxson M. M.; Ewing A. G. (2005) Loaded dopamine is preferentially stored in the halo portion of PC12 cell dense core vesicles. J. Neurochem. 93, 1122–1131. [DOI] [PubMed] [Google Scholar]
- Borges R.; Travis E. R.; Hochstetler S. E.; Wightman R. M. (1997) Effects of external osmotic pressure on vesicular secretion from bovine adrenal medullary cells. J. Biol. Chem. 272, 8325–8331. [DOI] [PubMed] [Google Scholar]
- Camacho M.; Machado J. D.; Montesinos M. S.; Criado M.; Borges R. (2006) Intragranular pH rapidly modulates exocytosis in adrenal chromaffin cells. J. Neurochem. 96, 324–334. [DOI] [PubMed] [Google Scholar]
- Haynes C. L.; Siff L. N.; Wightman R. M. (2007) Temperature-dependent differences between readily releasable and reserve pool vesicles in chromaffin cells. Biochim. Biophys. Acta 1773, 728–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado J. D.; Morales A.; Gomez J. F.; Borges R. (2001) cAMP modulates exocytotic kinetics and increases quantal size in chromaffin cells. Mol. Pharmacol. 60, 514–520. [PubMed] [Google Scholar]
- Cans A. S.; Hook F.; Shupliakov O.; Ewing A. G.; Eriksson P. S.; Brodin L.; Orwar O. (2001) Measurement of the dynamics of exocytosis and vesicle retrieval at cell populations using a quartz crystal microbalance. Anal. Chem. 73, 5805–5811. [DOI] [PubMed] [Google Scholar]
- Montesinos M. S.; Machado D.; Camacho M.; Diaz J.; Morales Y. G.; de la Rosa D. A.; Carmona E.; Castaneyra A.; Viveros O. H.; O'Connor D. T.; Mahata S. K.; Borges R. (2008) The crucial role of chromogranins in storage and exocytosis revealed using chromaffin cells from chromogranin a null mouse. J. Neurosci. 28, 3350–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Li Y.; Tsien R. W. (2009) The dynamic control of kiss-and-run and vesicular reuse probed with single nanoparticles. Science 323, 1448–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal R. G.; Mosharov E. V.; Sulzer D. (2004) Dopamine neurons release transmitter via a flickering fusion pore. Nat. Neurosci. 7, 341–346. [DOI] [PubMed] [Google Scholar]
- Ales E.; Tabares L.; Poyato J. M.; Valero V.; Lindau M.; Alvarez de Toledo G. (1999) High calcium concentrations shift the mode of exocytosis to the kiss-and-run mechanism. Nat. Cell Biol. 1, 40–44. [DOI] [PubMed] [Google Scholar]
- Kozminski K. D.; Gutman D. A.; Davila V.; Sulzer D.; Ewing A. G. (1998) Voltammetric and pharmacological characterization of dopamine release from single exocytotic events at rat pheochromocytoma (PC12) cells. Anal. Chem. 70, 3123–3130. [DOI] [PubMed] [Google Scholar]
- Robinson D. L.; Venton B. J.; Heien M. L. A. V.; Wightman R. M. (2003) Detecting subsecond dopamine release with fast-scan cyclic voltammetry in vivo. Clin. Chem. 49, 1763–1773. [DOI] [PubMed] [Google Scholar]
- Powell P. R.; Woods L. A.; Ewing A. G. (2005) Characterization of etched electrochemical detection for electrophoresis in micron inner diameter capillaries. J. Sep. Sci. 28, 2540–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawagoe K. T.; Zimmerman J. B.; Wightman R. M. (1993) Principles of voltammetry and microelectrode surface-states. J. Neurosci. Methods 48, 225–240. [DOI] [PubMed] [Google Scholar]
- Parsons T. D.; Coorssen J. R.; Horstmann H.; Almers W. (1995) Docked granules, the exocytic burst, and the need for ATP hydrolysis in endocrine cells. Neuron 15, 1085–1096. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.