

Abstract
The thyroid cancer is a rare oncological entity, representing no more than 1% of all human malignant neoplasms. Recently, it has been demonstrated a sharp increase in incidence of differentiated thyroid carcinoma, equally occurring in both sexes. So far, multiple genetic alterations have been identified in differentiated thyroid carcinoma, leading to investigate the clinical utility of genetic studies. In particular, molecular genetic approaches searching for gene mutations in the material collected by fine needle ago-biopsy may have a particular utility in small nodules and in those specimens with an indeterminate cytology. The expansion of knowledge about genetic mutations occurring in different thyroid tumors has characterized recent years, allowing the identification of a correlation between specific mutations and phenotypic characteristics of thyroid cancers, essential for their prognosis. This review will briefly report on the histological features and the new entity represented by thyroid microcarcinoma and will focus on both environmental and genetic aspects associated with the occurrence of thyroid cancer.
1. Introduction
The thyroid cancer (TC) is a rare oncological entity, representing no more than 1% of all human malignant neoplasms. However, it represents the most common malignant endocrine neoplasia whose incidence has progressively increased over the past two decades, according to the majority of Tumors Registries. In fact, the data collected at the Surveillance Epidemiology and End-Results Cancer Registries program (SEER) [1] indicate an annual average prevalence of thyroid carcinoma (TCa) of 6.6/100.000 (9.5 and 3.5/100.000 for women and men, resp.) with an annual increase >5% in the period 1975–2002.
In a very recent study in the United States (U.S.), a sharp increase in incidence of differentiated TC, equally occurring in both sexes and independently by the sizes of the lesions, has been reported. However, the detection at a preclinical stage, through neck ultrasounds (US), of a greater number of asymptomatic TCas and the progressive increase in both the prevalence and incidence of nodular thyroid disease cannot represent the only explanation for these increased incidence rates. Such findings are mainly due to an improved diagnostic histopathology with particular regard to the cytological diagnosis on fine needle ago-biopsy (FNAB). Consequently, other factors such as environmental influences and molecular alterations must be taken into account [2].
This review will briefly report on histological features and the new entity represented by thyroid microcarcinoma (TmCa) and will focus on both environmental and genetic aspects associated with the occurrence of TC.
2. Histological Features
TCs are divided into papillary carcinoma (PTC) (MIM #188550), follicular carcinoma (FTC) (MIM #188470), medullary thyroid carcinoma (MTC) (MIM #155240), anaplastic thyroid carcinoma (ATC), primary lymphoma of the thyroid (PLT), and primary sarcoma of the thyroid (PST).
The PTC accounts for 80% of all thyroid malignancies [3] whereas FTC, the second most common malignancy of TC, represents approximately 15% of cases [4]. The MTC represents 3% of thyroid malignancies [4]. ATC, approximately representing 2%, is the most aggressive form of TC, while PLT and PST are very rare (Figure 1).
Figure 1.
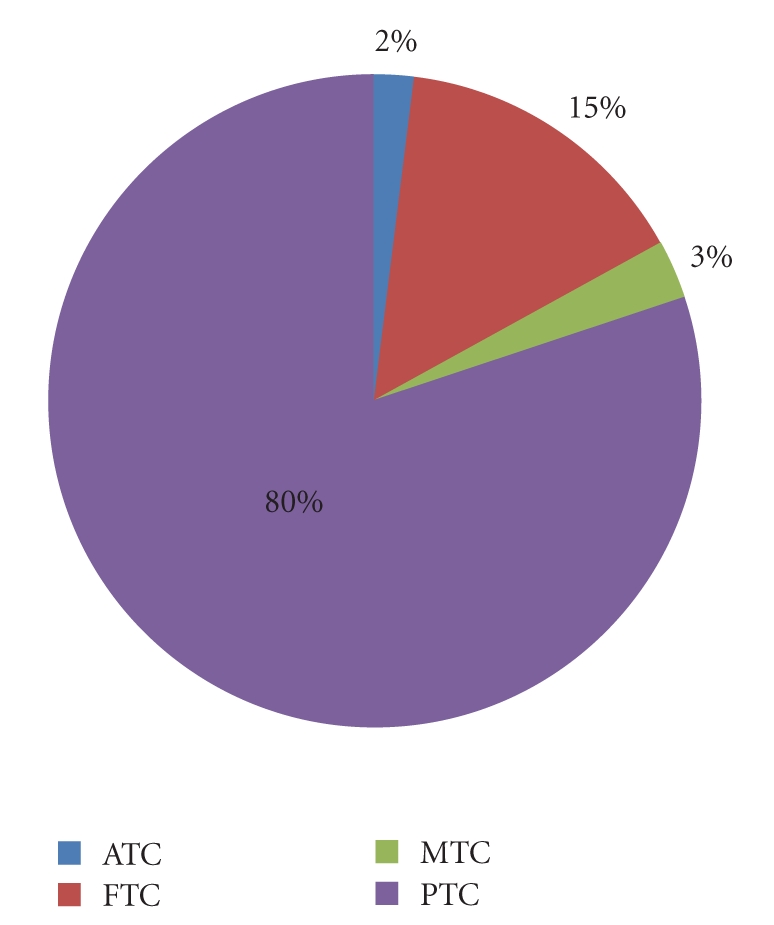
Frequency of thyroid neoplasms.
Through a careful revision of several published studies, a correlation between age of incidence and histological type can be established. In fact, PTC is more frequent in childhood and <50 years [5], FTC in patients <60 years [6], and the ATC 60–70 years [7] (Table 1).
Table 1.
Correlation between incidence and histological type of TCs.
| Histotype | Incidence |
|---|---|
| PTC | (i) Childhood |
| (ii) <50 years | |
| FTC | (iii) <60 years |
| ATC | (iv) 60–70 years |
3. Thyroid Microcarcinoma (TmCa): A “New Entity”
TmCa, diameter <1 cm, is an increasing pathological finding that could be regarded as a thyroid incidentaloma. In fact, as it happens for adrenal gland incidentaloma, TmCa is occasionally identified at US of the neck performed for other reasons. Most of TmCas are PTC with a sclerotic appearance of the nodule and similar prevalence in both sexes. TmCa is rare in children; whereas in autoptic series of adults, their frequency is similar in all of the age groups, indicating that in the absence of screening techniques enabling their identification TmCas occur in young adults and most cases regress or do not ever reach the clinical expression [8].
4. Mortality
Despite the significant increase in its incidence, mortality for TC has not increased in equal measure. It appears two times higher in female subjects with a rate of the annual mortality between 0.4–2.8 and 0.2–1.2/100.000, respectively, for women and men [1].
5. Risk Factors
The multifactor etiology of TC is the result of complex genetic and environmental factors interaction in individuals at risk. Epidemiological studies suggest the following main risk factors.
6. Gender and Age
TC is 2–4 times more frequent in women who generally exhibit a better prognosis than men in whom a higher malignant progression of nodules has been reported. It is rare in patients aged <16 years, presenting an annual incidence of 0.02–0.3/100.000, and extremely uncommon below the age of 10 years [9–11]. Its incidence increases with ageing and the average age at diagnosis is 45–50 years. However, the following issues have to be stressed: (1) although rare, the presence of TC in childhood accounts for a more advanced disease at diagnosis; and (2) in patients aged >60 years an increased risk for malignancy of thyroid nodules has been observed.
7. Ethnic Differences
A geographic and ethnic variability of TC incidence has been reported. In areas such as Iceland, Hawaii, the Philippines, Japan, and Israel its incidence is higher than in North America, Canada, and U.S.
In U.S., the TCa is more frequent in Caucasian descent subjects than Afro-American, Hispanic, Hawaiian, Chinese, or Japanese women, whose incidence is still twice as high in their countries of origin. All these findings suggest that such differences may be attributable to both environmental (e.g., dietary habits) and genetic factors [9, 12].
8. Environmental Factors
In this section we will consider the role of several parameters such as the (1) exposure to ionizing radiations, (2) age at the time of the exposure, (3) presence of a previous history of benign thyroid disease, (4) role of the dietary iodine intake, (5) role of the body mass index, and (6) role of hormonal factors.
8.1. Previous Exposure to Ionizing Radiation
The role of a previous exposure to ionizing radiation in thyroid carcinogenesis has been established since 1950 following the explosion of the atomic bomb in Japan.
Previous exposure to ionizing radiation for external irradiation of the neck increases the incidence of thyroid nodules, either benign or malignant, and palpable nodules are detected in 20%–30% of people exposed to radiation [13], as well it happens in pediatric patients undergoing radiation therapy for oncological and haematological malignancies such as lymphoma or leukemia [14, 15].
The minimum latency period between exposure and clinical evidence of thyroid disease has been reported to be at least 4-5 years, reaching the maximum peak 20 years from exposure to decrease thereafter. The risk increases from medium doses above 10 cGy, and for doses up to 1500 cGy a linear dose-cancer risk can be observed. For higher doses the risk decreases probably in relation to radio-induced cell necrosis.
8.2. Age at the Time of Irradiation
It represents the main risk factor and after 15–20 years there is no longer an increased risk. In children exposed to doses of 1 Gy, the excess risk for TCa is equal to 7.7 [16].
Several studies conducted after the Chernobyl nuclear disaster have shown an increased incidence of TCs in subjects that at that time were aged between 5 months and 10 years [17, 18]. The average age at diagnosis of TCa was 14 years with no substantial gender-related difference in the incidence.
According to the pathology findings, the most frequent histological features were represented by solid and follicular variants of PTC. At the time of diagnosis, the disease was in an advanced stage, already exhibiting lymph node and lung metastases, a more aggressive biological behaviour, and it resulted to be more frequently associated with autoimmune thyroiditis [17, 18].
8.3. Previous History of Benign Thyroid Disease
In subjects suffering from benign thyroid nodules and, to a lesser percentage, in subjects suffering from goiter, a higher frequency of TC has been demonstrated [19, 20]. Such a correlation suggests either the presence of common environmental causal factors or the lack of difference in the rate of malignancy of single or multiple nodules, not yet confirmed.
Several studies reported an increased incidence of malignant nodules (from 0.4% to 9.8%) in individuals with Basedow's disease (MIM #275000) [19, 20]. These studies also noted an increased risk for those subjects who had palpable nodules, and also evaluated through neck US or thyroid scintigraphy, compared with those with diffuse non-nodular goiter. Moreover, TCs that occurred in patients with Basedow's disease seem to have a more aggressive clinical behavior [21].
Although hyperthyroidism (toxic adenoma and toxic multinodular goiter) or Hashimoto's thyroiditis (MIM #140300) do not represent additional risk factors for TCa, affected subjects have a higher risk to develop a thyroid lymphoproliferative disease such as a thyroid lymphoma [22, 23].
8.4. Contribution of Iodine in the Food
In areas with iodine deficiency, a higher incidence of thyroid nodules and TCs has been observed. However, after correction for the greatest number of nodules, the percentage of TCa in thyroid nodules is similar to the one found in areas with normal intake of dietary iodine.
Different histotypes occur in accordance to the contribution of dietary iodine (Figure 2). In presence of a sufficient iodine intake, more than 80% of cancer consist of PTC; whereas in areas with iodine deficiency follicular and anaplastic figures are more frequently reported (approximately 2-3 times higher than observed in areas with adequate iodine intake) [11].
Figure 2.

Contribution of iodine in the food to the thyroid tumorigenesis.
8.5. Body Mass Index
Several case-control studies have shown an increased risk of TCa in patients with high body mass index (BMI). The risk would be increased by 5-fold in obese men and 2 times in obese women (>97 percentile), compared to the risk observed in patients with weight <3rd percentile. In women (especially in postmenopausal age) a weight gain >14% appears to positively correlate with the onset of TCa [24, 25].
8.6. Hormonal Factors
The male-to-female incidence ratio has been reporting to be different according to the period of life in which TC occurs. In women of childbearing age, this ratio is about 2–4 : 1 and is reduced to 1.5 : 1 in older prepuberal and menopause individuals [26, 27]. In pregnancy, the diagnosis of goiter or thyroid nodules is frequent and an increase in thyroid volume and nodules may occur. Consequently, it has been hypothesized as a role for hormonal factors or other factors related to pregnancy in the pathogenesis of TCa [28]. However, it is still unclear why a female predominance exists [29].
9. Genetic Factors
In order to explain the role of genetics in thyroid carcinogenesis, we will consider the (1) presence of a positive familial history for TC and associated diseases and (2) molecular genetic aspects including (a) fine mechanisms such as gene mutations, both at nuclear and mitochondrial level, (b) gross mechanisms represented by gene rearrangements and loss of heterozygosity (LOH), and (c) a brief paragraph concerning the more recent knowledge on the role of microRNAs.
9.1. Familial History and Associated Diseases
In this paragraph, only nonmedullary thyroid tumors will be considered. Familial MTCs will be treated in the paragraph of RET proto-oncogene.
In 3%–5% of patients with TC, a positive family history for thyroid tumors in first-degree relatives can be detected. Indeed, in the setting of familial nonmedullary TCa, (FNMTCa), PTC mainly occurs in more members of the pedigree, being inherited as a dominant autosomal trait with an incomplete penetrance. In the affected subjects, the aggressiveness of the tumor is higher than that observed in the general population (high frequency of multifocal form and an higher rate of relapse compared to patients with sporadic PTC), but this observation has not been confirmed in other series [30].
Of course, genetic factors are responsible for some familial syndromes associated with high prevalence of nodular thyroid disease and TCa, such as familial polyposis of colon (FAP), Cowden's disease (CD), and Carney's complex (CNC).
In FAP (MIM #175100), the risk of developing multicentric PTC, in particular the cribriform variant [31], is about 100 times higher than the one observed in the general population [32]. Female subjects below 35 years are more frequently affected.
In CD (MIM #158350), an autosomal dominant disease with multiple hamartomas in different tissues, the risk of PTC, or FTC is higher than observed in the general population [33].
CNC (MIM #160980) is a multiple endocrine neoplasia syndrome inherited as an autosomal dominant trait. The disease is characterized by spotty skin pigmentation, cardiac and extracardiac myxomatosis, schwannomas, and endocrine tumors, like multiple hypersecretory adrenal nodules, growth hormone-secreting pituitary tumors, gonadal tumors, and thyroid neoplasms, either follicular or papillar form. CNC is caused by loss of function mutations in PRKAR1A gene, which encodes the type 1A regulatory subunit of protein kinase A, a modulator of intracellular signaling of PKA (PKA; cyclic AMP (cAMP)-dependent protein kinase) [34–36].
Sporadic thyroid tumors can rarely accommodate mutations, and in fact it is very rare that these tumors can accommodate somatic mutations of PRKAR1A [37, 38].
9.2. Molecular Genetic Aspects
In recent years, the molecular bases of thyroid carcinogenesis have been thoroughly investigating. The most frequent genetic alterations, detected by molecular biology studies over the past 20 years, are mainly represented by the activation of oncogenes such as BRAF, RAS, RET, and NTRK1 and the silencing of tumor suppressor genes such as PTEN, TP53.
9.2.1. Fine Mechanisms: Nuclear and Mitochondrial Gene Mutations
Nuclear Genes —
BRAF Gene —
BRAF gene (OMIM #115150) encodes a protein belonging to the family of serine-threonine kinases, activator of mitogen-activated protein kinase (MAPK) with a high affinity for MEK1 and MEK2, MAP kinase kinases, leading to their phosphorylation more efficiently than other RAF isoforms [39].
MAPKs respond to mitogenic extracellular stimuli and regulate gene expression, mitosis, differentiation, proliferation, and cell survival/apoptosis. MEK1 and MEK2 activate the serine/threonine specific protein kinases ERK1 and ERK2. Activated ERKs are pleiotropic effectors of cell physiology and play an important role in the control of gene expression involved in the cell division cycle, apoptosis, cell differentiation, and cell migration [40, 41].
BRAF and PTC —
BRAF mutations are the most common genetic alterations found in PTCs, being present in approximately 45% of these tumors [42–44] (Table 2). BRAF mutations are present in 40%–70% of PTCs with higher percentage of positivity in more aggressive variants such as “tall cell” dedifferentiated forms [45].
In particular, the genetic alteration with the higher prevalence in classical PTCs involves the nucleotide 1799 determining a valine-glutamate substitution at amino acid residue 600 (V600E) with consequent activation of BRAF kinase that results in a continuous phosphorylation of MEK and MAPK pathway effectors. Such a mutation is rare in FTC [43, 46, 47].
Two more rare activating mutations of BRAF have been also described in PTCs: (a) the K601E point mutation, small in-frame insertion or deletion surrounding codon 600 and determining a lysine-glutamic substitution, and (b) the AKAP9-BRAF rearrangement that is more common in those PTCs associated with a previous radiation exposure [48–51]. AKAP9 (A kinase (PRKA) anchor protein (yotiao) 9) gene encodes a member of the AKAP family, proteins which have the common function of binding to the regulatory subunit of protein kinase A (PKA) [52].
BRAF-PTC Genotype/Phenotype Correlation —
The characteristics of aggressiveness of PTCs, such as extrathyroidal extension, advanced presentation, presence of lymph node, or distant metastases have been associated in many studies with the presence of BRAF mutation [53–57], even if it has been proven to be an independent predictor of tumor recurrence, also at an early stage of disease [55, 58].
The BRAF mutation is thought to account for the impairment of the function of the sodium-iodine symporter (NIS) and other molecular pathways involved in the iodine metabolism of the follicular cell. In fact, BRAF mutation has been found to be associated either with a decreased iodine intake in some thyroid tumors or the failure of response to therapy in disease relapse [55, 59].
BRAF and ATC —
A BRAF mutation has been reported in 20% of ATCs, exhibiting also areas of well-differentiated PTC, and 15% of poorly differentiated TCs [53, 54, 60] (Table 2).
BRAF Mutant Animal Models —
The involvement of BRAF in thyroid tumorigenesis has been also suggested by studies on transgenic mice with thyroid-specific expression of BRAF V600E [61].
In fact, these mice developed a PTC with invasion of blood vessels, thyroid capsule, and perithyroid skeletal muscle. They are all features of aggressiveness, demonstrating a progression to poorly differentiated TC.
RAS Gene —
HRAS, KRAS, and NRAS genes are members of the RAS family (OMIM #109800) coding for a G-protein. When activated, RAS protein starts the intracellular signal transduction through the release of GTP and the activation of MAPK and PI3K/AKT pathways (see below). Therefore, an increase of the affinity for GTP and inactivation of the GTPase function are explained by the presence of point mutations in the RAS domains, especially in codons 12, 13, and 61, which determine a constantly active RAS mutant protein [62].
RAS and PTC —
Point mutations of RAS are found in 10%–20% of PTCs [63–65] (Table 2).
RAS and FTC/Follicular Adenomas —
RAS mutations have been also found in 40%–50% of FTCs and in 20%–40% of follicular adenomas, the latter with a prevalent microfollicular pattern of growth [66–70]. Often, the NRAS and HRAS mutations occur at codon 61. They have a low incidence in oncocytic tumor (designated as oncocytic if at least 75% of their constituent cells can be described as oncocytes) and, in fact, these mutations have been reported only in 0–4% of follicular adenomas and in 15%–25% of FTCs [68, 71, 72] (Table 2).
RAS and ATC —
Point mutations of RAS have been described in 18%–27% of poorly differentiated thyroid tumors and in 50%–60% of ATCs (Table 2). It is likely that mutant RAS facilitates a genomic instability predisposing to further genetic abnormalities as those of the TP53 gene and then the malignant progression. An example of this relation is a case of ATC that occurred into the context of a well-differentiated FTC where in both forms a RAS mutation has been found, whereas a TP53 mutation has been found only in the ATC [73].
Table 2.
Prevalence of major genetic alterations in all of the TCs but MTC.
| Genes | PTC | FTC | ATC |
|---|---|---|---|
| BRAF | 45% | RARE | 20% |
| RAS | 10%–20% | 40%–50% | 50%–60% |
| NTRK1 | <5% | — | — |
| PI3K/AKT and PTEN | — | 7% | 15% |
| TP53 | RARE | RARE | 60%–80% |
| CTNNB1 | RARE | RARE | 66% |
| RET/PTC | 25%–30% | — | — |
| PAX8/PPARγ | — | 35% | — |
RAS-PTC-FTC Genotype/Phenotype Correlation —
PTC —
The mutation of RAS in PTCs is most frequently associated with the follicular variant of PTC, then to well-capsulated tumors with low rates of nodal metastases [74, 75], although some studies have found a correlation between RAS mutation and PTC with a more aggressive behavior as the presence of distant metastases [76].
FTC —
In several studies, the association between RAS mutation and FTCs with a more aggressive behavior, presence of bone metastases, has been demonstrated. These findings support that RAS mutation can be associated with a less favorable prognosis of FTCs [70, 77–79].
In vitro studies showed that the mutant RAS protein could promote a chromosomal instability and then a consequent more aggressive behavior of the tumor [80, 81].
RET Gene —
The RET proto-oncogene (OMIM #164761) encodes a membrane receptor with a tyrosine kinase activity. This receptor has an extracellular domain, containing the signal peptide, the cadherin-like region and the cysteine-rich region, a single transmembrane domain, and an intra-cellular portion containing the tyrosine kinase domain [82]. The physiological ligands of RET belong to the glial-derived neurotrophic factors (GDNFs) family, composted by four members: neurturin, persephin, artemin, and GDNF, having a specific trophic effect on RET [83]. The formation of the ligand-coreceptor-receptor complex is responsible for both the activation of the kinase catalytic domain and the signal transduction which induces cells proliferation through a complex network of second messengers [84]. The tyrosine kinases are enzymes that stimulate other regulatory proteins through phosphorylation of their tyrosine residues and their subsequent activation stimulates the cell division [84].
As other members of this family, RET exhibits an oncogenic potential and plays a particularly important role in thyroid human cancers.
Activating chromosomal rearrangements of RET are involved in the tumorigenesis of some forms of PTC, and its activating point mutations account for both familial and sporadic MTC forms. In fact the 40% of PTCs are associated with somatic gene rearrangements [85].
The familial MTC is the most important clinical feature occurring within the Multiple Endocrine Neoplasia type 2 (MEN2) syndrome (OMIM #171400) [86].
MEN2 is an autosomal dominant disease described in hundreds of families throughout the World. Three distinct clinical variants of MEN2 have been reported: MEN2A, accounting for >80% of MEN2, MEN2B, and Familial Medullary Thyroid Carcinoma (FMTC). All variants of MEN2 show a high penetrance for MTC; in fact, 90% of MEN2 adult RET mutant gene carriers will eventually show evidence of MTC [87].
MEN2-associated RET germline mutations are mostly located in the cysteine-rich extracellular domain, particularly in MEN2A where they are present in 90% of cases [88–90], whereas in MEN 2B, RET germline mutation is predominantly at codon 918 in the intracellular tyrosine kinase domain of the protein (Table 3).
Interestingly, between 1% and 24% of individuals with MTC may have simple disease-causing germline mutations of RET gene [91–93] and for this reason many experts recommend DNA testing for RET in all patients with MTC [94].
Somatic mutations of the RET gene are present in 20%–80% of cases of sporadic MTCs [95, 96]. The vast majority of these mutations affect codon 918, although they have also been identified in a few other regions of the gene. Some of these somatic mutations have an heterogeneous distribution within the tumor or are detected only in a subset of metastatic nodules, thereby raising concerns that they may not be essential for carcinogenesis [95].
RET-MTC Genotype/Phenotype Correlation —
The reported strong correlation between genotype and clinical expression of MEN2-associated MTC have provided the opportunity to stratify three RET codon mutation categories of mutant carrier children [87] (Table 3).
Children with MEN2B and/or RET codon 883, 918, or 922 mutations are classified as having the highest risk from aggressive MTC and should be operated on within the first 6 months (Table 3).
Children with any RET codon 611, 618, 620, or 634 mutations are classified as intermediate level and should have thyroidectomy performed before the age of 5 years (Table 3).
Children with RET codon 609, 768, 790, 791, 804, and 891 mutations are classified as lower-risk level and may be operated on at a later stage (Table 3). For all groups, a more aggressive neck dissection should be performed if evidence of involved lymph nodes in the lateral neck [87] is found.
Patients with 791 mutation exhibit a lower penetrance concerning MTC o C-cell hyperplasia (CCH), suggesting that the neck surgery could be postponed up to the moment in which we assist to the increase of simulated calcitonin serum levels, underlying that some patients with this mutation will never undergo prophylactic surgery [97].
Similar findings have been obtained in patients with 649L (transmembrane domain) RET mutation. It has been reported that either patients carrying this mutation developed a mild MTC phenotype, thus delaying its diagnosis at an older age and confirming data already reported in literature [98, 99], or double RET mutants, 649L and 634, exhibited a more aggressive course, with the clinical phenotype dominated by the “more severe” mutation, C634 [96, 100, 101].
Prophylactic thyroidectomy in 649L mutant carriers should be correlated to the levels of stimulated calcitonin [101].
N T R K1 Gene —
NTRK1 (OMIM #155240) tyrosine kinase gene is located on chromosome 1q22 and encodes for the receptor for nerve growth factor (NGF) (Table 2). Its oncogenic activation occurs through a chromosomal rearrangement [102]. NTRK1 rearrangements are less frequent than reported for RET [103].
PI3K/AKT Pathway and PTEN Gene Mutations —
Protein RAS and fusion protein RET/PTC may activate the PI3K/AKT (phosphatidylinositol 3-kinase/Akt) signaling pathway through the loss of function of PTEN [104].
PI3K are a family of related intracellular signal transducer enzymes capable of phosphorylating the inositol ring of phosphatidylinositol. AKT protein family, whose members are also called protein kinases B (PKB), plays an important role in mammalian cellular signaling.
PTEN is a protein that, following activating mutations or amplifications of the genes encoding the effector proteins of PI3K/AKT pathway, inhibits PI3K signaling. Since the PI3K/AKT pathway is fundamental in regulating cell growth, proliferation, and survival, mutations of the PIK3CA gene (OMIM #114480), encoding the catalytic subunit of PI3K, have been searched in thyroid tumors and found in 6%–13% of FTCs and in 0%–6% of follicular adenomas [105–107].
As mentioned above, mutations of PTEN gene (OMIM #601728), involved in thyroid carcinogenesis, are responsible for CD which is characterized by the occurrence of hamartomas in multiple organs, benign thyroid lesions such as multinodular goiter and thyroid adenoma and exhibit an increased risk of thyroid cancer (especially FTC) and breast [33].
PTEN and FTC —
Mutations of PTEN gene have been reported in about 7% of FTCs, whereas they have not been found in follicular adenomas [106, 107] (Table 2).
PTEN and ATC —
Point mutations of PIK3CA and PTEN genes have been reported in approximately 20% and 15% of ATCs cases, respectively [105, 106, 108] (Table 2).
T P53 Gene —
TP53 gene (OMIM #191170) encodes a protein that is essential to maintain the integrity of the genetic heritage, as it protects the body against genetic damage that induces cancer by stimulating the production of both proteins that inhibit proliferation and promote cell differentiation, either DNA repair or apoptosis.
Inactivating point mutations of TP53 make the encoded protein unable to enter the nucleus, so it cannot longer control the production of regulating proteins, and, therefore, the above mentioned events. This has been demonstrated by studies in which the recovery of the expression of TP53 in ATCs cultured cells would reduce the rate of proliferation, the reexpression of thyroid-specific genes (e.g., TPO, PAX-8), regaining the ability to respond to stimulation with thyroid hormone [109, 110].
Mutations of TP53 represent the most common genetic alterations of all types of human cancers.
In the thyroid these mutations are present in approximately 60%–80% of ATCs, in 30% of poorly differentiated tumors, and only rarely in FTCs and PTCs (Table 2), mostly involving the exons 5–8 of the gene [111–115].
C T N N B1 (β-Catenin) Gene —
CTNNB 1 gene (OMIM #116806) encodes β-catenin, a cytoplasmic protein which is an important intermediary in the wingless signaling pathway (WNT) [116, 117]. The Wnt signaling pathway consists of a complex network of proteins playing important roles in embryogenesis and cancer, and also involved in normal physiological processes in adult animals [118].
Point mutations at exon 3 of CTNNB1 gene have been found in 25% of poorly differentiated carcinomas and 66% of ATCs, respectively, but not in well-differentiated carcinoma [119, 120] (Table 2).
Table 3.
RET and MTC: Genotype-phenotype correlation.
| RET receptor protein | RET mutation | CMT progression | Level of the risk of progression |
|---|---|---|---|
| Exon 10 | Cys 609 | Partially slow 5–7 years | High |
| Cys 611 | |||
| Cys 618 | |||
| Cys 620 | |||
| Exon 11 | Cys 630 | Intermediated ~1 year | |
| Cys 634 | |||
|
| |||
| Transmembrane domain | RETS649L | Slow 6–22 years | Intermediated |
| Exon 13 | Glu 768 Asp | ||
| Leu 790 Phe | |||
| Tyr 791 Phe | |||
| Exon 14 | Val 804 Met/Le | ||
| Exon 15 | Ser 891 Ala | ||
|
| |||
| Exon 16 | Met 918 Thr | Fast <1 year | Very high |
Mitochondrial DNA: Gene Mutations —
Somatic point mutations and deletions of mitochondrial DNA have been found to be more frequent in adenomas and oncocytic carcinomas, whereas they are more rare in PTCs and FTCs [121].
Recently, in 15% of oncocytic tumors, but not in other types of TC, mutations in the GRIM-19 gene (OMIM 609435), encoding for a protein involved in the process of cell death and mitochondrial metabolism, have been identified, suggesting that the alteration of GRIM-19 gene may serve as a specific marker of such tumors [121, 122].
9.2.2. Gross Mechanisms: Gene Rearrangements and Loss of Heterozygosity
Gene/Chromosomal Rearrangements —
RET/PTC Genes —
In 40 % of PTCs, RET/PTC rearrangement has been described [85] (Table 2). The rearrangement involved the fusion between the portion 3′ of the gene for the receptor tyrosine kinase RET and the portion 5′ of heterologous genes [85]. RET/PTC1, RET/PTC3, and RET/PTC2 are the most frequent types of the rearrangement found in PTCs.
RET/PTC1 and RET/PTC3 are paracentric inversions because RET and H4 (OMIM #601985) or NCOA4 (ELE1) (OMIM 601984), the respective fusion partners, both reside on chromosome 10q where RET is located [123, 124].
Specifically, the abnormal fusion protein RET/PTC1(H4-RET) is a constitutively activated tyrosine kinase, whereas NCOA4 (Nuclear receptor coactivator 4) gene encodes an androgen receptor coactivator.
On the contrary, RET/PTC2 is due to an interchromosomal translocation between chromosome 10 and chromosome 17 [125].
Other types of RET/PTC rearrangements have been subsequently identified. Most of these rare types of RET/PTC have been found in 50%–80% of PTCs in patients with a history of previous environmental (such as the Chernobyl nuclear accident) or therapeutic exposure to ionizing radiation and in 40%–70% of PTCs of children and young adults [126–130].
Rearrangements such as RET/ELKS (OMIM #607127), t (10;12) (q11;p13), and RET/HOOK3 (OMIM *607825; Homo sapiens hook homolog 3) have also been seen in patients with sporadic PTC [131, 132].
In particular, ELKS gene localizes onto chromosome 12p13.3 and its amino acidic sequence is rich in glutamic acid (E), leucine (L), lysine (K), and serine (S).
All the fusion events leave the domain of the receptor tyrosine kinase RET intact and allow RET/PTC oncoprotein to bind SHC (OMIM #600560), involved in RAS regulation [133]; SHC (Src homology 2 domain containing) transforming protein 1, also known as SHC1, is a human gene [134].
In approximately 20% of sporadic PTCs RET/PTC rearrangements have been found [135, 136]. RET/PTC rearrangement is regarded as an early event in thyroid carcinogenesis and it is frequently found in papillary microcarcinoma [137]. Moreover, it has been also found in adenomas and other benign lesions of the thyroid. However, since it is present in most tumor cells, it is reasonable to consider it specific for PTCs [138–140].
P A X8/PPARγ Genes —
PAX8/PPARγ rearrangement, due to the fusion of the PAX8 (OMIM #167415) gene with the PPARy gene (OMIM #601487), results from t(2;3) (q13,p25) translocation [141]. PAX8 gene is a member of the paired box (PAX) family of transcription factors whereas P P A R γ gene encodes for nuclear receptor protein which functions as transcription factor regulating the expression of several genes.
PAX8/PPARγ rearrangement has been found in 35% FTCs, in 2%–10% of follicular adenomas, and at a lower percentage in the Hurtle's cell carcinoma [142–144] (Table 2) suggesting these injuries to be preinvasive (in situ) forms of FTCs.
Tumors expressing such a rearrangement occur at a young age and are small in size with a solid growth pattern or nests and vascular invasion. The mechanisms of cell transformation induced by PAX8/PPARγ are not fully known [143–145]. Immunohistochemical techniques allow us to detect that the rearrangement consists of an over-expression of the PPARγ protein although only an intense and diffuse nuclear staining correlates with the presence of the rearrangement [141, 146].
Several studies have been conducted on this rearrangement, and various mechanisms have been observed and assumed to have a possible role on (1) inhibition of the normal function of PPARγ by a dominant negative effect on the mutant protein PAX8/PPARγ on PPARγ wild type [141, 147], (2) activation of P P A R γ target genes in tumors expressing PAX8/PPARγ, (3) deregulation of PAX8 function, known to be crucial for the differentiation of thyroid cells, and (4) activation of genes not related either to the wild-type PPARγ or to the PAX8 pathways [148, 149].
LOH Studies —
Another genetic alteration, a gross alteration, reported in the TCs is LOH.
LOH represents the loss of the normal function of one allele of a gene in which the other allele was already inactivated at somatic or germline level.
LOH is detected on average in 6%–12% of follicular adenomas and in 30%–50% of FTCs.
The chromosomal regions most frequently involved are located on chromosomes 2p, 3p, 9q, 9p, 10q, 11p, 17p, and 15q [150–152].
LOH Genotype/Phenotype Correlation —
The frequency of LOH has been correlated with the aggressiveness of the tumor and the presence of relapse in patients with FTC. In fact, in the minimally invasive tumors LOH has been detected in 30% of cases, while its frequency was greater in 50% of more aggressive cases and in the presence of disease relapse [151, 153].
A study conducted on a small group of FTCs has suggested that allelic loss of the VHL gene on 3p26 may serve as important diagnostic and prognostic markers of FTCs being specific for malignancy, even though its clinical usefulness should be validated in a larger group of tumors [154–157].
LOH is more present in thyroid oncocytic tumors. The most affected regions resulted to be located on chromosomes 1q, 2p, 3q, 8q, 14q, and 18q [154, 156], but also the loci on chromosome 1q and 2p showed a significantly higher rate of LOH in oncocytic carcinomas than in adenomas, with a sensitivity of 100% and a specificity of 65% for the detection of malignant tumors [156].
9.2.3. MicroRNAs (miRNAs)
miRNAs are a class of small noncoding RNAs involved in a wide range of processes such as proliferation, development, apoptosis, metabolism, and response to stress. In studies conducted in different types of human tumors, it has been shown that miRNAs are abnormally expressed [158]. miRNAs profile expression analysis in human TCs, by microarray approach, detected an aberrant expression of several miRNAs in PTCs [159].
Overexpression of mir-221, -222, and-181b has been also demonstrated in transformed rat thyroid cell lines and in mouse models of thyroid carcinogenesis.
Functional studies, performed by blocking mir-221 function and by overexpressing mir-221 in human PTC-derived cell lines, have suggested a critical role of mir-221 overexpression in thyroid carcinogenesis. Taken together, these data have indicated the existence of an miRNA signature associated with PTCs, and suggested the miRNA deregulation as an important event in thyroid cell transformation [160].
Overexpression of mir-221 in PTC may drive gene expression patterns by directly and indirectly regulating numerous genes, including HOXB5 [161, 162]. HOXB5 gene encodes a nuclear protein with a homeobox DNA-binding domain and the encoded protein functions as a sequence-specific transcription factor that is involved in several tissue developments [163].
Molecular Therapy —
In oncology, understanding of the molecular mechanisms that control cell growth and survival has led to the development of new more selective and targeted pharmacological agents.
Compounds that have demonstrated a potential pre-clinical therapeutic application in TC are discussed below.
Inhibitors of BRAF Protein —
Since alterations of BRAF are widely represented in many solid tumors, such as malignant melanoma, it is clearly appreciated the considerable interest directed to the development of specific inhibitors of BRAF activity [45, 53]. Among these, the best known is the bi-aryl urea BAY 43-9006 (sorafenib) [164]. BAY 43-9006 is an inhibitor active against RAF multikinase and other proteinkinase (VEGFR-2 and PDGFR), which can effectively block the kinase activity of BRAF [46, 165]. Preclinical studies have demonstrated its ability to inhibit the BRAF signal and growth of all of the thyroid carcinoma cell lines expressing this oncogene [166]. BAY 43-9006 has been tested on several types of human carcinomas including TCa, and the preliminary results showed a minimal or partial response in some patients [167].
Currently, numerous other compounds are being studied. They include AAL881 and LBT613, which has not demonstrated in vitro an efficacy exceeding that of sorafenib [168].
Inhibitors of RET —
The somatic rearrangements of the RET proto-oncogene, in particular those resulting from rearrangements RET/PTC, form various oncoproteins that represent potential molecular targets for the development of specific inhibitors. Indeed, several studies have demonstrated their high frequency in PTCs and their ability to intervene early in the process of neoplastic transformation [169].
Several inhibitors of the enzyme activity of RET have been developed, some of natural origin such as herbimicine A, clavilactones, and other synthetics [170, 171]. Their mechanism of action interferes with the ATP-binding site at the catalytic domain of RET molecule. The most effective compounds belong to the class of indolocarbazoles (CEP-701, CEP 751) [172], pyrazolopyrimidine (PP1, PP2) [173, 174], quinazoline (ZD6474) [175, 176], and indolinones (RPI-1) [177–179].
These compounds, in addition to directly inhibit the activity of RET, can also act on other downstream kinases involved by activated RET as the inhibition of Fak from PP2 [174].
ZD6474 is a tyrosinkinase inhibitor, belonging to the family of quinazoline, blocking effectively RET and the type 2 receptor of VEGF [175, 180]. In an in vitro study ZD6474 also stops the growth of human PTC cell lines expressing RET/PTC1 and prevents the growth of fibroblasts in mice expressing RET/PTC3 [181, 182].
As mentioned above, BAY 43-9006 binds to RAF-1, BRAF, and other receptor tyrosine kinases responsible for neoangiogenesis and tumor progression (VEGFR-2, VEGFR-3, FLT-3, PDGFR-B, and KIT). Moreover, BAY 43-9006 also seems to be able to inhibit the activity of RET and the tumoral growth [183].
Multikinase inhibitor, SU12248 (sunitinib), has proven to be effective in inhibiting the RET/PTC kinase signal in experimental models and it has been tested in a phase II study on differentiated TC, refractory to radiometabolic therapy and not surgically resectable [184].
EEE 788 (cetuximab) has been shown to have antiangiogenetic effects by blocking EGF-R and VEGF-R in cell lines of TC [185].
AMG706 is another multikinase inhibitor with anticancer and antiangiogenetic effects, directed selectively against VEGF receptors (VEGFR1, VEGFR2, VEGFR3), PDGF, Kit, and RET [186]. In a recent phase 1 clinical study on a small group of patients with different histotypes of TC, AMG706 seems to be well tolerated and potentially reduces either the volume of the tumor or cancer biochemical markers [187].
Practical Applications of Molecular Genetics to the Clinical Approach for TCs —
In recent years, multiple genetic alterations have been identified in differentiated TC, leading to test the clinical utility of genetic studies. In particular, molecular genetic approaches searching for gene mutations in the material collected by FNAB may have a particular utility in small nodules and in those specimen with an indeterminate cytology.
Considering that FNA presents a false-negative rate of 1% to 11%, a false-positive rate of 1% to 8%, a sensitivity of 65% to 98%, and a specificity of 72% to 100%, its limitations are related to the skill of the operator, the expertise of the cytologist, and the difficulty in distinguishing some benign cellular adenomas from their malignant counterparts [188].
BRAF Mutations —
The detection of somatic BRAF mutations provided the diagnosis of PTC in 15%–30% of cases with doubtful cytology [189, 190]. The search for somatic BRAF mutations, performed on specimen obtained by FNAB, not only may allow a preoperative diagnosis, but also it is easy to be performed on small amounts of DNA and not particularly expensive since it is mainly restricted to a single mutation [191–193]. Importantly, detection of BRAF V600E mutation can be successfully achieved by various molecular techniques using DNA isolated from fresh or fixed FNAB samples. Four different detection methods revealed a comparable and high sensitivity of the detection in archival FNAB smears [194].
RAS Mutations —
The diagnostic value of searching for somatic RAS mutations is still controversial because it is not a specific indicator of malignancy, being also present in benign follicular adenomas. However, RAS mutations arise frequently in FTC and follicular variant of PTC, both histotypes difficult to diagnose at cytology performed by FNAB. Considering the key role played by mutant RAS, both in the progression of MTC and undifferentiating cancer, it has been recommended the surgical removal of RAS-positive adenomas in order to avoid the potential transformation into malignant forms [195].
In a prospective study aimed to analyze the role of the search for different mutations in improving the preoperative diagnosis of thyroid nodules by FNAB, the identification of RAS mutations has been found effective in ameliorating the diagnostic accuracy and allowing the diagnosis of malignant tumors in many samples with a previous negative or inadequate cytological diagnosis [196].
RET/PTC Genes Rearrangements —
The RET/PTC rearrangements may be sought for diagnostic purposes in the samples obtained by cytology performed with FNAB for a better definition of the preoperative diagnosis of thyroid nodules, especially in samples with indeterminate cytology or having an insufficient quantity of cells for a diagnostic purpose [140, 190, 197, 198].
Searching for BRAF Mutations and RET/PTC Rearrangements: A Comparison among the Specificities —
When compared to the search for BRAF mutations, the molecular approach for RET/PTC rearrangements detection needs a more deep investigation since it requires the extraction of RNA and the search for at least two rearrangements of RET (RET/PTC1 and RET/PTC3).
The specificity is lower because, unlike the BRAF mutations present only in PTCs, RET/PTC rearrangements may also be found in benign thyroid diseases (sporadic follicular adenomas, benign thyroid nodules, and Hashimoto's thyroiditis) [137, 139, 199–201].
P A X8/P P A R γ Genes Rearrangements —
The analysis for the PAX8/PPARy rearrangement may be useful in diagnosis, even if it has to be taken into account that this genetic alteration is not unique to the FTC, but it can also be found in follicular adenoma and PTC.
The identification of the PAX8/PPARy rearrangement needs sophisticated and expensive methods such as RT-PCR (reverse transcription-polymerase chain reaction), which is a highly sensitive technique for mRNA detection and quantitation [202], FISH (fluorescence in situ hybridization) [203], or immunohistochemistry.
It has been suggested that the detection of an intense and diffuse immunoreactivity of P P A R γ in tumor cells may justify the analysis of new sections of the tumor capsule and a more accurate appraisal of the suspicious areas in the search of capsular and/or vascular invasion [204].
10. Conclusions
TC is one of the most important malignant tumors of the endocrine system. Its incidence is increasing over the years, approximately 1% of all the new diagnoses of cancer. Its etiology appears to be multifactorial, being due to the interaction between environmental factors, among which the most important are exposure to radiation and the lack of iodine in the diet, and genetic factors. The expansion of knowledge about genetic mutations occurring in different thyroid tumors has characterized recent years, allowing the identification of a correlation between specific mutations and phenotypic characteristics of thyroid cancers, essential for their prognosis. First example is represented by BRAF mutation that appears to be an indicator of an aggressive behavior of PTC. Studies of this mutation were later extended to the development of new targeted therapies for TC such as the ones represented by inhibitors of RET- and BRAF-dependent tyrosine kinase activity, as also other molecular targets, currently under development or already in stages of clinical trial. The results of these trials should provide us with the therapeutic efficacy of these treatments and their potential use, whether developed as monotherapy or as associations of multiple drugs, especially in the treatment of aggressive thyroid tumors such as medullary carcinoma, the poorly differentiated cancer, and anaplastic carcinoma.
The application of genetic research of particular mutations to the diagnosis has allowed to improve the cytological diagnosis in those samples with FNA cytology indeterminate and/or atypical. The identification of BRAF mutation is particularly promising since the simple realization and the high specificity of the analysis for the determination of malignancy.
However, despite the analysis of all of the several known mutations, up to now the molecular test alone is not sufficient to detect all cases of malignancy. In fact, in various percentages in both the PTC and FTC and especially in oncocytic carcinomas, these mutations do not reach a high degree of specificity.
Acknowledgment
This work was supported by F.I.R.M.O. Fondazione Raffaella Becagli (to MLB).
References
- 1.Edwards BK, Brown ML, Wingo PA, et al. Annual report to the nation on the status of cancer, 1975–2002, featuring population-based trends in cancer treatment. Journal of the National Cancer Institute. 2005;97(19):1407–1427. doi: 10.1093/jnci/dji289. [DOI] [PubMed] [Google Scholar]
- 2.Chen AY, Jemal A, Ward EM. Increasing incidence of differentiated thyroid cancer in the United States, 1988–2005. Cancer. 2009;115(16):3801–3807. doi: 10.1002/cncr.24416. [DOI] [PubMed] [Google Scholar]
- 3.Cooper DS, Doherty GM, Haugen BR, et al. Management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid. 2006;16(2):109–141. doi: 10.1089/thy.2006.16.109. [DOI] [PubMed] [Google Scholar]
- 4.De Lellis RA, Lloyd RV, Heitz PU, Eng C, editors. World Health Organization International Classification of Tumors. Pathology and Genetics of Tumors of Endocrine Organs. Lyon, France: IARC Press; 2004. [Google Scholar]
- 5.Hay ID. Papillary thyroid carcinoma. Endocrinology and Metabolism Clinics of North America. 1990;19(3):545–576. [PubMed] [Google Scholar]
- 6.Cooper DS, Schneyer CR. Follicular and Hurthle cell carcinoma of the thyroid. Endocrinology and Metabolism Clinics of North America. 1990;19(3):577–591. [PubMed] [Google Scholar]
- 7.Chiacchio S, Lorenzoni A, Boni G, Rubello D, Elisei R, Mariani G. Anaplastic thyroid cancer: prevalence, diagnosis and treatment. Minerva Endocrinologica. 2008;33(4):341–357. [PubMed] [Google Scholar]
- 8.Harach HR, Franssila KO, Wasenius V-M. Occult papillary carcinoma of the thyroid. A “normal” finding in Finland. A systematic autopsy study. Cancer. 1985;56(3):531–538. doi: 10.1002/1097-0142(19850801)56:3<531::aid-cncr2820560321>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 9.Franceschi S, Boyle P, Maisonneuve P, et al. The epidemiology of thyroid carcinoma. Critical Reviews in Oncogenesis. 1993;4(1):25–52. [PubMed] [Google Scholar]
- 10.Landis SH, Murray T, Bolden S, Wingo PA. Cancer statistics. CA: A Cancer Journal for Clinicians. 1998;48(6):p. 329. doi: 10.3322/canjclin.48.1.6. [DOI] [PubMed] [Google Scholar]
- 11.Belfiore A, La Rosa GL, La Porta GA, et al. Cancer risk in patients with cold thyroid nodules: relevance of iodine intake, sex, age, and multinodularity. American Journal of Medicine. 1992;93(4):363–369. doi: 10.1016/0002-9343(92)90164-7. [DOI] [PubMed] [Google Scholar]
- 12.Parkin DM, Whelan SL, Ferlay J, Powell J, Teppo L. Cancer Incidence in Five Continents. Vol. 8. Lyon, France: IARC Press; 2003. (IARC Scientific Publication no. 155). [Google Scholar]
- 13.Hanson GA, Komorowski RA, Cerletty JM, Wilson SD. Thyroid gland morphology in young adults: normal subjects versus those with prior low-dose neck irradiation in childhood. Surgery. 1983;94(6):984–988. [PubMed] [Google Scholar]
- 14.Pui C-H, Cheng C, Leung W, et al. Extended follow-up of long-term survivors of childhood acute lymphoblastic leukemia. The New England Journal of Medicine. 2003;349(7):640–649. doi: 10.1056/NEJMoa035091. [DOI] [PubMed] [Google Scholar]
- 15.Hancock SL, Cox RS, McDougall IR. Thyroid diseases after treatment of Hodgkin’s disease. The New England Journal of Medicine. 1991;325(9):599–605. doi: 10.1056/NEJM199108293250902. [DOI] [PubMed] [Google Scholar]
- 16.Ron E, Lubin JH, Shore RE, et al. Thyroid cancer after exposure to external radiation: a pooled analysis of seven studies. Radiation Research. 1995;141(3):259–277. [PubMed] [Google Scholar]
- 17.Nikiforov Y, Gnepp DR. Pediatric thyroid cancer after the Chernobyl disaster: pathomorphologic study of 84 cases (1991-1992) from the Republic of Belarus. Cancer. 1994;74(2):748–766. doi: 10.1002/1097-0142(19940715)74:2<748::aid-cncr2820740231>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 18.Leenhardt L, Aurengo A. Post-Chernobyl thyroid carcinoma in children. Best Practice and Research: Clinical Endocrinology and Metabolism. 2000;14(4):667–677. doi: 10.1053/beem.2000.0109. [DOI] [PubMed] [Google Scholar]
- 19.Farbota LM, Calandra DB, Lawrence AM, Paloyan E. Thyroid carcinoma in Graves’ disease. Surgery. 1985;98(6):1148–1153. [PubMed] [Google Scholar]
- 20.Pacini F, Elisei R, Di Coscio GC, et al. Thyroid carcinoma in thyrotoxic patients treated by surgery. Journal of Endocrinological Investigation. 1988;11(2):107–112. doi: 10.1007/BF03350115. [DOI] [PubMed] [Google Scholar]
- 21.Pellegriti G, Belfiore A, Giuffrida D, Lupo L, Vigneri R. Outcome of differentiated thyroid cancer in Graves’ patients. Journal of Clinical Endocrinology and Metabolism. 1998;83(8):2805–2809. doi: 10.1210/jcem.83.8.4997. [DOI] [PubMed] [Google Scholar]
- 22.Segal K, Ben-Bassat M, Avraham A, Har-El G, Sidi J. Hashimoto’s thyroiditis and carcinoma of the thyroid gland. International Surgery. 1985;70(3):205–209. [PubMed] [Google Scholar]
- 23.Holm LE, Blomgren H, Lowhagen T. Cancer risks in patients with chronic lymphocytic thyroiditis. The New England Journal of Medicine. 1985;312(10):601–604. doi: 10.1056/NEJM198503073121001. [DOI] [PubMed] [Google Scholar]
- 24.Dal Maso L, Vecchia CL, Franceschi S, et al. A pooled analysis of thyroid cancer studies. V. Anthropometric factors. Cancer Causes and Control. 2000;11(2):137–144. doi: 10.1023/a:1008938520101. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki T, Matsuo K, Hasegawa Y, et al. Anthropometric factors at age 20 years and risk of thyroid cancer. Cancer Causes and Control. 2008;19(10):1233–1242. doi: 10.1007/s10552-008-9194-x. [DOI] [PubMed] [Google Scholar]
- 26.Negri E, Dal Maso L, Ron E, et al. A pooled analysis of case-control studies of thyroid cancer. II. Menstrual and reproductive factors. Cancer Causes and Control. 1999;10(2):143–155. doi: 10.1023/a:1008880429862. [DOI] [PubMed] [Google Scholar]
- 27.Franceschi S, Preston-Martin S, Dal Maso L, et al. A pooled analysis of case-control studies of thyroid cancer. IV. Benign thyroid diseases. Cancer Causes and Control. 1999;10(6):583–595. doi: 10.1023/a:1008907227706. [DOI] [PubMed] [Google Scholar]
- 28.Goodman MT, Kolonel LN, Wilkens LR. The association of body size, reproductive factors and thyroid cancer. British Journal of Cancer. 1992;66(6):1180–1184. doi: 10.1038/bjc.1992.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ron E, Lunenfeld B, Menczer J, et al. Cancer incidence in a cohort of infertile women. American Journal of Epidemiology. 1987;125(5):789–790. doi: 10.1093/oxfordjournals.aje.a114595. [DOI] [PubMed] [Google Scholar]
- 30.Malchoff CD, Malchoff DM. Familial papillary thyroid carcinoma. Cancer Treatment and Research. 2004;122:381–387. doi: 10.1007/1-4020-8107-3_22. [DOI] [PubMed] [Google Scholar]
- 31.Li Volsi VA, Albores-Saavedra J, Asa SL, et al. Papillary carcinoma. In: DeLellis RA, Lloyd RV, Heitz PU, Eng C, editors. Tumors of Endocrine Organs. 2004. pp. 57–66. (World Health Organization Classification of Tumors). [Google Scholar]
- 32.Harach HR, Williams GT, Williams ED. Familial adenomatous polyposis associated thyroid carcinoma: a distinct type of follicular cell neoplasm. Histopathology. 1994;25(6):549–561. doi: 10.1111/j.1365-2559.1994.tb01374.x. [DOI] [PubMed] [Google Scholar]
- 33.Pilarski R. Cowden syndrome: a critical review of the clinical literature. Journal of Genetic Counseling. 2009;18(1):13–27. doi: 10.1007/s10897-008-9187-7. [DOI] [PubMed] [Google Scholar]
- 34.Kirschner LS, Sandrini F, Monbo J, Lin J-P, Carney JA, Stratakis CA. Genetic heterogeneity and spectrum of mutations of the PRKAR1A gene in patients with the Carney complex. Human Molecular Genetics. 2000;9(20):3037–3046. doi: 10.1093/hmg/9.20.3037. [DOI] [PubMed] [Google Scholar]
- 35.Casey M, Vaughan CJ, He J, et al. Mutations in the protein kinase A R1α regulatory subunit cause familial cardiac myxomas and Carney complex. Journal of Clinical Investigation. 2000;106(5):R31–R38. doi: 10.1172/JCI10841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bertherat J, Horvath A, Groussin L, et al. Mutations in regulatory subunit type 1A of cyclic adenosine 5′-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. Journal of Clinical Endocrinology and Metabolism. 2009;94(6):2085–2091. doi: 10.1210/jc.2008-2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sandrini F, Matyakhina L, Sarlis NJ, et al. Regulatory subunit type I-α of protein kinase A (PRKARIA): a tumor-suppressor gene for sporadic thyroid cancer. Genes Chromosomes and Cancer. 2002;35(2):182–192. doi: 10.1002/gcc.10112. [DOI] [PubMed] [Google Scholar]
- 38.Kirschner LS, Kusewitt DF, Matyakhina L, et al. A mouse model for the carney complex tumor syndrome develops neoplasia in cyclic AMP-responsive tissues. Cancer Research. 2005;65(11):4506–4514. doi: 10.1158/0008-5472.CAN-05-0580. [DOI] [PubMed] [Google Scholar]
- 39.Peyssonnaux C, Eychene A. The Raf/MEK/ERK pathway: new concepts of activation. Biology of the Cell. 2001;93(1-2):53–62. doi: 10.1016/s0248-4900(01)01125-x. [DOI] [PubMed] [Google Scholar]
- 40.Dérijard B, Raingeaud J, Barrett T, et al. Independent human MAP kinase signal transduction pathways defined by MEK and MKK isoforms. Science. 1995;267(5198):682–685. doi: 10.1126/science.7839144. [DOI] [PubMed] [Google Scholar]
- 41.Pearson G, Robinson F, Beers Gibson T, et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocrine Reviews. 2001;22(2):153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 42.Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Research. 2003;63(7):1454–1457. [PubMed] [Google Scholar]
- 43.Xing M. BRAF mutation in thyroid cancer. Endocrine-Related Cancer. 2005;12(2):245–262. doi: 10.1677/erc.1.0978. [DOI] [PubMed] [Google Scholar]
- 44.Cohen Y, Xing M, Mambo E, et al. BRAF mutation in papillary thyroid carcinoma. Journal of the National Cancer Institute. 2003;95(8):625–627. doi: 10.1093/jnci/95.8.625. [DOI] [PubMed] [Google Scholar]
- 45.Karasarides M, Chiloeches A, Hayward R, et al. B-RAF is a therapeutic target in melanoma. Oncogene. 2004;23(37):6292–6298. doi: 10.1038/sj.onc.1207785. [DOI] [PubMed] [Google Scholar]
- 46.Wan PTC, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116(6):855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 47.Adeniran AJ, Zhu Z, Gandhi M, et al. Correlation between genetic alterations and microscopic features, clinical manifestations, and prognostic characteristics of thyroid papillary carcinomas. American Journal of Surgical Pathology. 2006;30(2):216–222. doi: 10.1097/01.pas.0000176432.73455.1b. [DOI] [PubMed] [Google Scholar]
- 48.Trovisco V, Vieira de Castro I, Soares P, et al. BRAF mutations are associated with some histological types of papillary thyroid carcinoma. Journal of Pathology. 2004;202(2):247–251. doi: 10.1002/path.1511. [DOI] [PubMed] [Google Scholar]
- 49.Carta C, Moretti S, Passeri L, et al. Genotyping of an Italian papillary thyroid carcinoma cohort revealed high prevalence of BRAF mutations, absence of RAS mutations and allowed the detection of a new mutation of BRAF oncoprotein (BRAFV599Ins) Clinical Endocrinology. 2006;64(1):105–109. doi: 10.1111/j.1365-2265.2005.02401.x. [DOI] [PubMed] [Google Scholar]
- 50.Hou P, Liu D, Xing M. Functional characterization of the T1799-1801del and A1799-1816ins BRAF mutations in papillary thyroid cancer. Cell Cycle. 2007;6(3):377–379. doi: 10.4161/cc.6.3.3818. [DOI] [PubMed] [Google Scholar]
- 51.Ciampi R, Knauf JA, Kerler R, et al. Oncogenic AKAP9-BRAF fusion is a novel mechanism of MAPK pathway activation in thyroid cancer. Journal of Clinical Investigation. 2005;115(1):94–101. doi: 10.1172/JCI23237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. http://www.ncbi.nlm.nih.gov/sites/entrez?Db=gene&Cmd=ShowDetailView&TermToSearch=10142.
- 53.Nikiforova MN, Kimura ET, Gandhi M, et al. BRAF mutations in thyroid tumors are restricted to papillary carcinomas and anaplastic or poorly differentiated carcinomas arising from papillary carcinomas. Journal of Clinical Endocrinology and Metabolism. 2003;88(11):5399–5404. doi: 10.1210/jc.2003-030838. [DOI] [PubMed] [Google Scholar]
- 54.Namba H, Nakashima M, Hayashi T, et al. Clinical implication of hot spot BRAF mutation, V599E, in papillary thyroid cancers. Journal of Clinical Endocrinology and Metabolism. 2003;88(9):4393–4397. doi: 10.1210/jc.2003-030305. [DOI] [PubMed] [Google Scholar]
- 55.Xing M, Westra WH, Tufano RP, et al. BRAF mutation predicts a poorer clinical prognosis for papillary thyroid cancer. Journal of Clinical Endocrinology and Metabolism. 2005;90(12):6373–6379. doi: 10.1210/jc.2005-0987. [DOI] [PubMed] [Google Scholar]
- 56.Fugazzola L, Mannavola D, Cirello V, et al. BRAF mutations in an Italian cohort of thyroid cancers. Clinical Endocrinology. 2004;61(2):239–243. doi: 10.1111/j.1365-2265.2004.02089.x. [DOI] [PubMed] [Google Scholar]
- 57.Trovisco V, Soares P, Preto A, et al. Type and prevalence of BRAF mutations are closely associated with papillary thyroid carcinoma histotype and patients’ age but not with tumour aggressiveness. Virchows Archiv. 2005;446(6):589–595. doi: 10.1007/s00428-005-1236-0. [DOI] [PubMed] [Google Scholar]
- 58.Kim TY, Kim WB, Rhee YS, et al. The BRAF mutation is useful for prediction of clinical recurrence in low-risk patients with conventional papillary thyroid carcinoma. Clinical Endocrinology. 2006;65(3):364–368. doi: 10.1111/j.1365-2265.2006.02605.x. [DOI] [PubMed] [Google Scholar]
- 59.Riesco-Eizaguirre G, Gutiérrez-Martínez P, García-Cabezas MA, Nistal M, Santisteban P. The oncogene BRAFV600E is associated with a high risk of recurrence and less differentiated papillary thyroid carcinoma due to the impairment of Na+/I− targeting to the membrane. Endocrine-Related Cancer. 2006;13(1):257–269. doi: 10.1677/erc.1.01119. [DOI] [PubMed] [Google Scholar]
- 60.Begum S, Rosenbaum E, Henrique R, Cohen Y, Sidransky D, Westra WH. BRAF mutations in anaplastic thyroid carcinoma: implications for tumor origin, diagnosis and treatment. Modern Pathology. 2004;17(11):1359–1363. doi: 10.1038/modpathol.3800198. [DOI] [PubMed] [Google Scholar]
- 61.Knauf JA, Ma X, Smith EP, et al. Targeted expression of BRAFV600E in thyroid cells of transgenic mice results in papillary thyroid cancers that undergo dedifferentiation. Cancer Research. 2005;65(10):4238–4245. doi: 10.1158/0008-5472.CAN-05-0047. [DOI] [PubMed] [Google Scholar]
- 62.Liu R-T, Hou C-Y, You H-L, et al. Selective occurrence of RAS mutations in benign and malignant thyroid follicular neoplasms in Taiwan. Thyroid. 2004;14(8):616–621. doi: 10.1089/1050725041692882. [DOI] [PubMed] [Google Scholar]
- 63.Namba H, Rubin SA, Fagin JA. Point mutations of RAS oncogenes are an early event in thyroid tumorigenesis. Molecular Endocrinology. 1990;4(10):1474–1479. doi: 10.1210/mend-4-10-1474. [DOI] [PubMed] [Google Scholar]
- 64.Ezzat S, Zheng L, Kolenda J, Safarian A, Freeman JL, Asa SL. Prevalence of activating RAS mutations in morphologically characterized thyroid nodules. Thyroid. 1996;6(5):409–416. doi: 10.1089/thy.1996.6.409. [DOI] [PubMed] [Google Scholar]
- 65.Vasko VV, Gaudart J, Allasia C, et al. Thyroid follicular adenomas may display features of follicular carcinoma and follicular variant of papillary carcinoma. European Journal of Endocrinology. 2004;151(6):779–786. doi: 10.1530/eje.0.1510779. [DOI] [PubMed] [Google Scholar]
- 66.Lemoine NR, Mayall ES, Wyllie FS, et al. High frequency of RAS oncogene activation in all stages of human thyroid tumorigenesis. Oncogene. 1989;4(2):159–164. [PubMed] [Google Scholar]
- 67.Suarez HG, du Villard JA, Severino M, et al. Presence of mutations in all three RAS genes in human thyroid tumors. Oncogene. 1990;5(4):565–570. [PubMed] [Google Scholar]
- 68.Esapa CT, Johnson SJ, Kendall-Taylor P, Lennard TWJ, Harris PE. Prevalence of RAS mutations in thyroid neoplasia. Clinical Endocrinology. 1999;50(4):529–535. doi: 10.1046/j.1365-2265.1999.00704.x. [DOI] [PubMed] [Google Scholar]
- 69.Motoi N, Sakamoto A, Yamochi T, Horiuchi H, Motoi T, Machinami R. Role of RAS mutation in the progression of thyroid carcinoma of follicular epithelial origin. Pathology Research and Practice. 2000;196(1):1–7. doi: 10.1016/S0344-0338(00)80015-1. [DOI] [PubMed] [Google Scholar]
- 70.Basolo F, Pisaturo F, Pollina LE, et al. N-RAS mutation in poorly differentiated thyroid carcinomas: correlation with bone metastases and inverse correlation to thyroglobulin expression. Thyroid. 2000;10(1):19–23. doi: 10.1089/thy.2000.10.19. [DOI] [PubMed] [Google Scholar]
- 71.Schark C, Fulton N, Jacoby RF, Westbrook CA, Straus FH, II, Kaplan EL. N-RAS 61 oncogene mutations in Hurthle cell tumors. Surgery. 1990;108(6):994–1000. [PubMed] [Google Scholar]
- 72.Tallini G, Hsueh A, Liu S, Garcia-Rostan G, Speicher MR, Ward DC. Frequent chromosomal DNA unbalance in thyroid oncocytic (Hurthle cell) neoplasms detected by comparative genomic hybridization. Laboratory Investigation. 1999;79(5):547–555. [PubMed] [Google Scholar]
- 73.Asakawa H, Kobayashi T. Multistep carcinogenesis in anaplastic thyroid carcinoma: a case report. Pathology. 2002;34(1):94–97. doi: 10.1080/00313020120105732. [DOI] [PubMed] [Google Scholar]
- 74.Adeniran AJ, Zhu Z, Gandhi M, et al. Correlation between genetic alterations and microscopic features, clinical manifestations, and prognostic characteristics of thyroid papillary carcinomas. American Journal of Surgical Pathology. 2006;30(2):216–222. doi: 10.1097/01.pas.0000176432.73455.1b. [DOI] [PubMed] [Google Scholar]
- 75.Zhu Z, Gandhi M, Nikiforova MN, Fischer AH, Nikiforov YE. Molecular profile and clinical-pathologic features of the follicular variant of papillary thyroid carcinoma: an unusually high prevalence of RAS mutations. American Journal of Clinical Pathology. 2003;120(1):71–77. doi: 10.1309/ND8D-9LAJ-TRCT-G6QD. [DOI] [PubMed] [Google Scholar]
- 76.Hara H, Fulton N, Yashiro T, et al. N-RAS mutation: an independent prognostic factor for aggressiveness of papillary thyroid carcinoma. Surgery. 1994;116(6):1010–1016. [PubMed] [Google Scholar]
- 77.Garcia-Rostan G, Zhao H, Camp RL, et al. RAS mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer. Journal of Clinical Oncology. 2003;21(17):3226–3235. doi: 10.1200/JCO.2003.10.130. [DOI] [PubMed] [Google Scholar]
- 78.Karga H, Lee J-K, Vickery AL, Jr., Thor A, Gaz RD, Jameson JL. RAS oncogene mutations in benign and malignant thyroid neoplasms. Journal of Clinical Endocrinology and Metabolism. 1991;73(4):832–836. doi: 10.1210/jcem-73-4-832. [DOI] [PubMed] [Google Scholar]
- 79.Manenti G, Pilotti S, Re FC, Della Porta G, Pierotti MA. Selective activation of RAS oncogenes in follicular and undifferentiated thyroid carcinomas. European Journal of Cancer A. 1994;30(7):987–993. doi: 10.1016/0959-8049(94)90130-9. [DOI] [PubMed] [Google Scholar]
- 80.Fagin JA. Minireview: branded from the start-distinct oncogenic initiating events may determine tumor fate in the thyroid. Molecular Endocrinology. 2002;16(5):903–911. doi: 10.1210/mend.16.5.0838. [DOI] [PubMed] [Google Scholar]
- 81.Saavedra HI, Knauf JA, Shirokawa JM, et al. The RAS oncogene induces genomic instability in thyroid PCCL3 cells via the MAPK pathway. Oncogene. 2000;19(34):3948–3954. doi: 10.1038/sj.onc.1203723. [DOI] [PubMed] [Google Scholar]
- 82.Takahashi M, Buma Y, Iwamoto T, Inaguma Y, Ikeda H, Hiai H. Cloning and expression of the ret proto-oncogene encoding a tyrosine kinase with two potential transmembrane domains. Oncogene. 1988;3(5):571–578. [PubMed] [Google Scholar]
- 83.Robertson K, Mason I. The GDNF-KET signalling partnership. Trends in Genetics. 1997;13(1):1–3. doi: 10.1016/s0168-9525(96)30113-3. [DOI] [PubMed] [Google Scholar]
- 84.Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80(2):179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 85.Tallini G, Santoro M, Helie M, et al. RET/PTC oncogene activation defines a subset of papillary thyroid carcinomas lacking evidence of progression to poorly differentiated or undifferentiated tumor phenotypes. Clinical Cancer Research. 1998;4(2):287–294. [PubMed] [Google Scholar]
- 86.Gagel RF, Marx SJ. Multiple endocrine neoplasia. In: Larsen PR, Kronenberg H, Melmed S, Polonsky KS, editors. Book Multiple Endocrine Neoplasia. 10th edition. Philadelphia, Pa, USA: W. B. Saunders; 2003. pp. 1762–1771. [Google Scholar]
- 87.Brandi ML, Gagel RF, Angeli A, et al. Consensus: guidelines for diagnosis and therapy of MEN type 1 and type 2. Journal of Clinical Endocrinology and Metabolism. 2001;86(12):5658–5671. doi: 10.1210/jcem.86.12.8070. [DOI] [PubMed] [Google Scholar]
- 88.Mulligan LM, Marsh DJ, Robinson BG, et al. Genotype-phenotype correlation in multiple endocrine neoplasia type 2: report of the International RET Mutation Consortium. Journal of Internal Medicine. 1995;238(4):343–346. doi: 10.1111/j.1365-2796.1995.tb01208.x. [DOI] [PubMed] [Google Scholar]
- 89.Santoro M, Melillo RM, Carlomagno F, et al. Molecular biology of the MEN2 gene. Journal of Internal Medicine. 1998;243(6):505–508. doi: 10.1046/j.1365-2796.1998.00330.x. [DOI] [PubMed] [Google Scholar]
- 90.Hansford JR, Mulligan LM. Multiple endocrine neoplasia type 2 and RET: from neoplasia to neurogenesis. Journal of Medical Genetics. 2000;37(11):817–827. doi: 10.1136/jmg.37.11.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Eng C, Mulligan LM, Smith DP, et al. Low frequency of germline mutations in the RET protooncogene in patients with apparently sporadic medullary thyroid carcinoma. Clinical Endocrinology. 1995;43(1):123–127. doi: 10.1111/j.1365-2265.1995.tb01903.x. [DOI] [PubMed] [Google Scholar]
- 92.Decker RA, Peacock ML, Borst MJ, et al. Progress in genetic screening of multiple endocrine neoplasia type 2A: is calcitonin testing obsolete? Surgery. 1995;118(2):257–264. doi: 10.1016/s0039-6060(05)80332-0. [DOI] [PubMed] [Google Scholar]
- 93.Kitamura Y, Goodfellow PJ, Shimizu K, et al. Novel germline RET proto-oncogene mutations associated with medullary thyroid carcinoma (MTC): mutation analysis in Japanese patients with MTC. Oncogene. 1997;14(25):3103–3106. doi: 10.1038/sj.onc.1201102. [DOI] [PubMed] [Google Scholar]
- 94.Lips CJM. Clinical management of the multiple endocrine neoplasia syndromes: results of a computerized opinion poll at the Sixth International Workshop on Multiple Endocrine Neoplasia and von Hippel-Lindau Disease. Journal of Internal Medicine. 1998;243(6):589–594. doi: 10.1046/j.1365-2796.1998.00375.x. [DOI] [PubMed] [Google Scholar]
- 95.Eng C, Mulligan LM, Healey CS, et al. Heterogeneous mutation of the RET proto-oncogene in subpopulations of medullary thyroid carcinoma. Cancer Research. 1996;56(9):2167–2170. [PubMed] [Google Scholar]
- 96.Alberti L, Carniti C, Miranda C, Roccato E, Pierotti MA. RET and NTRK1 proto-oncogenes in human diseases. Journal of Cellular Physiology. 2003;195(2):168–186. doi: 10.1002/jcp.10252. [DOI] [PubMed] [Google Scholar]
- 97.Frank-Raue K, Machens A, Scheuba C, Niederle B, Dralle H, Raue F. Difference in development of medullary thyroid carcinoma among carriers of RET mutations in codons 790 and 791. Clinical Endocrinology. 2008;69(2):259–263. doi: 10.1111/j.1365-2265.2008.03215.x. [DOI] [PubMed] [Google Scholar]
- 98.Wiench M, Wygoda Z, Gubala E, et al. Estimation of risk of inherited medullary thyroid carcinoma in apparent sporadic patients. Journal of Clinical Oncology. 2001;19(5):1374–1380. doi: 10.1200/JCO.2001.19.5.1374. [DOI] [PubMed] [Google Scholar]
- 99.Vierhapper H, Bieglmayer C, Heinze G, Baumgartner-Parzer S. Frequency of RET proto-oncogene mutations in patients with normal and with moderately elevated pentagastrin-stimulated serum concentrations of calcitonin. Thyroid. 2004;14(8):580–583. doi: 10.1089/1050725041692990. [DOI] [PubMed] [Google Scholar]
- 100.Machens A, Niccoli-Sire P, Hoegel J, et al. Early malignant progression of hereditary medullary thyroid cancer. The New England Journal of Medicine. 2003;349(16):1517–1525. doi: 10.1056/NEJMoa012915. [DOI] [PubMed] [Google Scholar]
- 101.Colombo-Benkmann M, Li Z, Riemann B, et al. Characterization of the RET protooncogene transmembrane domain mutation S649L associated with nonaggressive medullary thyroid carcinoma. European Journal of Endocrinology. 2008;158(6):811–816. doi: 10.1530/EJE-07-0817. [DOI] [PubMed] [Google Scholar]
- 102.Greco A, Roccato E, Pierotti MA. TRK oncogenes in papillary thyroid carcinoma. Cancer Treatment and Research. 2004;122:207–219. doi: 10.1007/1-4020-8107-3_12. [DOI] [PubMed] [Google Scholar]
- 103.Bongarzone I, Vigneri P, Mariani L, Collini P, Pilotti S, Pierotti MA. RET/NTRK1 rearrangements in thyroid gland tumors of the papillary carcinoma family: correlation with clinicopathological features. Clinical Cancer Research. 1998;4(1):223–228. [PubMed] [Google Scholar]
- 104.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(8):4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.García-Rostán G, Costa AM, Pereira-Castro I, et al. Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Research. 2005;65(22):10199–10207. doi: 10.1158/0008-5472.CAN-04-4259. [DOI] [PubMed] [Google Scholar]
- 106.Hou P, Liu D, Shan Y, et al. Genetic alterations and their relationship in the phosphatidylinositol 3-kinase/Akt pathway in thyroid cancer. Clinical Cancer Research. 2007;13(4):1161–1170. doi: 10.1158/1078-0432.CCR-06-1125. [DOI] [PubMed] [Google Scholar]
- 107.Wang Y, Hou P, Yu H, et al. High prevalence and mutual exclusivity of genetic alterations in the phosphatidylinositol-3-kinase/Akt pathway in thyroid tumors. Journal of Clinical Endocrinology and Metabolism. 2007;92(6):2387–2390. doi: 10.1210/jc.2006-2019. [DOI] [PubMed] [Google Scholar]
- 108.Dahia PLM, Marsh DJ, Zheng Z, et al. Somatic deletions and mutations in the Cowden disease gene, PTEN, in sporadic thyroid tumors. Cancer Research. 1997;57(21):4710–4713. [PubMed] [Google Scholar]
- 109.Moretti F, Farsetti A, Soddu S, et al. p53 re-expression inhibits proliferation and restores differentiation of human thyroid anaplastic carcinoma cells. Oncogene. 1997;14(6):729–740. doi: 10.1038/sj.onc.1200887. [DOI] [PubMed] [Google Scholar]
- 110.Fagin JA, Tang S-H, Zeki K, Di Lauro R, Fusco A, Gonsky R. Reexpression of thyroid peroxidase in a derivative of an undifferentiated thyroid carcinoma cell line by introduction of wild-type p53. Cancer Research. 1996;56(4):765–771. [PubMed] [Google Scholar]
- 111.Fagin JA, Matsuo K, Karmakar A, Chen DL, Tang S-H, Koeffler HP. High prevalence of mutations of the p53 gene in poorly differentiated human thyroid carcinomas. Journal of Clinical Investigation. 1993;91(1):179–184. doi: 10.1172/JCI116168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Donghi R, Longoni A, Pilotti S, Michieli P, Della Porta G, Pierotti MA. Gene p53 mutations are restricted to poorly differentiated and undifferentiated carcinomas of the thyroid gland. Journal of Clinical Investigation. 1993;91(4):1753–1760. doi: 10.1172/JCI116385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dobashi Y, Sugimura H, Sakamoto A, et al. Stepwise participation of p53 gene mutation during dedifferentiation of human thyroid carcinomas. Diagnostic Molecular Pathology. 1994;3(1):9–14. doi: 10.1097/00019606-199403010-00003. [DOI] [PubMed] [Google Scholar]
- 114.Ho Y-S, Tseng S-C, Chin T-Y, Hsieh L-L, Lin J-D. p53 gene mutation in thyroid carcinoma. Cancer Letters. 1996;103(1):57–63. doi: 10.1016/0304-3835(96)04196-1. [DOI] [PubMed] [Google Scholar]
- 115.Takeuchi Y, Daa T, Kashima K, Yokoyama S, Nakayama I, Noguchi S. Mutations of p53 in thyroid carcinoma with an insular component. Thyroid. 1999;9(4):377–381. doi: 10.1089/thy.1999.9.377. [DOI] [PubMed] [Google Scholar]
- 116.Kraus C, Liehr T, Hülsken J, et al. Localization of the human β-catenin gene (CTNNB1) to 3p21: a region implicated in tumor development. Genomics. 1994;23(1):272–274. doi: 10.1006/geno.1994.1493. [DOI] [PubMed] [Google Scholar]
- 117.Van Hengel J, Nollet F, Berx G, van Roy N, Speleman F, van Roy F. Assignment of the human β-catenin gene (CTNNB1) to 3p22 → p21.3 by fluorescence in situ hybridization. Cytogenetics and Cell Genetics. 1995;70(1-2):68–70. doi: 10.1159/000133994. [DOI] [PubMed] [Google Scholar]
- 118.Lie D-C, Colamarino SA, Song H-J, et al. Wnt signalling regulates adult hippocampal neurogenesis. Nature. 2005;437(7063):1370–1375. doi: 10.1038/nature04108. [DOI] [PubMed] [Google Scholar]
- 119.Garcia-Rostan G, Camp RL, Herrero A, Carcangiu ML, Rimm DL, Tallini G. β-catenin dysregulation in thyroid neoplasms: down-regulation, aberrant nuclear expression, and CTNNB1 exon 3 mutations are markers for aggressive tumor phenotypes and poor prognosis. American Journal of Pathology. 2001;158(3):987–996. doi: 10.1016/s0002-9440(10)64045-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Miyake N, Maeta H, Horie S, et al. Absence of mutations in the β-catenin and adenomatous polyposis coli genes in papillary and follicular thyroid carcinomas. Pathology International. 2001;51(9):680–685. doi: 10.1046/j.1440-1827.2001.01269.x. [DOI] [PubMed] [Google Scholar]
- 121.Máximo V, Soares P, Lima J, Cameselle-Teijeiro J, Sobrinho-Simões M. Mitochondrial DNA somatic mutations (point mutations and large deletions) and mitochondrial DNA variants in human thyroid pathology: a study with emphasis on Hurthle cell tumors. American Journal of Pathology. 2002;160(5):1857–1865. doi: 10.1016/S0002-9440(10)61132-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Máximo V, Botelho T, Capela J, et al. Somatic and germline mutation in GRIM-19, a dual function gene involved in mitochondrial metabolism and cell death, is linked to mitochondrion-rich (Hurthle cell) tumours of the thyroid. British Journal of Cancer. 2005;92(10):1892–1898. doi: 10.1038/sj.bjc.6602547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Grieco M, Santoro M, Berlingieri MT, et al. PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell. 1990;60(4):557–563. doi: 10.1016/0092-8674(90)90659-3. [DOI] [PubMed] [Google Scholar]
- 124.Santoro M, Dathan NA, Berlingieri MT, et al. Molecular characterization of RET/PTC3; a novel rearranged version of the RETproto-oncogene in a human thyroid papillary carcinoma. Oncogene. 1994;9(2):509–516. [PubMed] [Google Scholar]
- 125.Bongarzone I, Monzini N, Borrello MG, et al. Molecular characterization of a thyroid tumor-specific transforming sequence formed by the fusion of ret tyrosine kinase and the regulatory subunit RIα of cyclic AMP-dependent protein kinase A. Molecular and Cellular Biology. 1993;13(1):358–366. doi: 10.1128/mcb.13.1.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fugazzola L, Pierotti MA, Vigano E, Pacini F, Vorontsova TV, Bongarzone I. Molecular and biochemical analysis of RET/PTC4, a novel oncogenic rearrangement between RET and ELE1 genes, in a post-Chernobyl papillary thyroid cancer. Oncogene. 1996;13(5):1093–1097. [PubMed] [Google Scholar]
- 127.Nikiforov YE, Rowland JM, Bove KE, Monforte-Munoz H, Fagin JA. Distinct pattern of ret oncogene rearrangements in morphological variants of radiation-induced and sporadic thyroid papillary carcinomas in children. Cancer Research. 1997;57(9):1690–1694. [PubMed] [Google Scholar]
- 128.Soares P, Fonseca E, Wynford-Thomas D, Sobrinho-Simões M. Sporadic ret-rearranged papillary carcinoma of the thyroid: a subset of slow growing, less aggressive thyroid neoplasms? Journal of Pathology. 1998;185(1):71–78. doi: 10.1002/(SICI)1096-9896(199805)185:1<71::AID-PATH42>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 129.Rabes HM, Demidchik EP, Sidorow JD, et al. Pattern of radiation-induced RET and NTRK1 rearrangements in 191 post-chernobyl papillary thyroid carcinomas: biological, phenotypic, and clinical implications. Clinical Cancer Research. 2000;6(3):1093–1103. [PubMed] [Google Scholar]
- 130.Fenton CL, Lukes Y, Nicholson D, Dinauer CA, Francis GL, Tuttle RM. The ret/PTC mutations are common in sporadic papillary thyroid carcinoma of children and young adults. Journal of Clinical Endocrinology and Metabolism. 2000;85(3):1170–1175. doi: 10.1210/jcem.85.3.6472. [DOI] [PubMed] [Google Scholar]
- 131.Nakata T, Kitamura Y, Shimizu K, et al. Fusion of a novel gene, ELKS, to RET due to translocation t(10;12)(q11;p13) in a papillary thyroid carcinoma. Genes Chromosomes and Cancer. 1999;25(2):97–103. doi: 10.1002/(sici)1098-2264(199906)25:2<97::aid-gcc4>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 132.Ciampi R, Giordano TJ, Wikenheiser-Brokamp K, Koenig RJ, Nikiforov YE. HOOK3-RET: a novel type of RET/PTC rearrangement in papillary thyroid carcinoma. Endocrine-Related Cancer. 2007;14(2):445–452. doi: 10.1677/ERC-07-0039. [DOI] [PubMed] [Google Scholar]
- 133.Knauf JA, Kuroda H, Basu S, Fagin JA. RET/PTC-induced dedifferentiation of thyroid cells is mediated through Y1062 signaling through SHC-RAS-MAP kinase. Oncogene. 2003;22(28):4406–4412. doi: 10.1038/sj.onc.1206602. [DOI] [PubMed] [Google Scholar]
- 134.Ravichandran KS. Signaling via Shc family adapter proteins. Oncogene. 2001;20(44):6322–6330. doi: 10.1038/sj.onc.1204776. [DOI] [PubMed] [Google Scholar]
- 135.Tallini G, Asa SL. RET oncogene activation in papillary thyroid carcinoma. Advances in Anatomic Pathology. 2001;8(6):345–354. doi: 10.1097/00125480-200111000-00005. [DOI] [PubMed] [Google Scholar]
- 136.Nikiforov YE. RET/PTC rearrangement in thyroid tumors. Endocrine Pathology. 2002;13(1):3–16. doi: 10.1385/ep:13:1:03. [DOI] [PubMed] [Google Scholar]
- 137.Viglietto G, Chiappetta G, Martinez-Tello FJ, et al. RET/PTC oncogene activation is an early event in thyroid carcinogenesis. Oncogene. 1995;11(6):1207–1210. [PubMed] [Google Scholar]
- 138.Sheils OM, O’Eary JJ, Uhlmann V, Luttich K, Sweeney EC. ret/PTC-1 activation in Hashimoto thyroiditis. International Journal of Surgical Pathology. 2000;8(3):185–189. doi: 10.1177/106689690000800305. [DOI] [PubMed] [Google Scholar]
- 139.Elisei R, Romei C, Vorontsova T, et al. RET/PTC rearrangements in thyroid nodules: studies in irradiated and not irradiated, malignant and benign thyroid lesions in children and adults. Journal of Clinical Endocrinology and Metabolism. 2001;86(7):3211–3216. doi: 10.1210/jcem.86.7.7678. [DOI] [PubMed] [Google Scholar]
- 140.Cheung CC, Carydis B, Ezzat S, Bedard YC, Asa SL. Analysis of ret/PTC gene rearrangements refines the fine needle aspiration diagnosis of thyroid cancer. Journal of Clinical Endocrinology and Metabolism. 2001;86(5):2187–2190. doi: 10.1210/jcem.86.5.7504. [DOI] [PubMed] [Google Scholar]
- 141.Kroll TG, Sarraf P, Pecciarini L, et al. PAX8-PPARγ1 fusion in oncogene human thyroid carcinoma. Science. 2000;289(5483):1357–1360. doi: 10.1126/science.289.5483.1357. [DOI] [PubMed] [Google Scholar]
- 142.French CA, Alexander EK, Cibas ES, et al. Genetic and biological subgroups of low-stage follicular thyroid cancer. American Journal of Pathology. 2003;162(4):1053–1060. doi: 10.1016/S0002-9440(10)63902-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nikiforova MN, Lynch RA, Biddinger PW, et al. RAS point mutations and PAX8-PPARγ rearrangement in thyroid tumors: evidence for distinct molecular pathways in thyroid follicular carcinoma. Journal of Clinical Endocrinology and Metabolism. 2003;88(5):2318–2326. doi: 10.1210/jc.2002-021907. [DOI] [PubMed] [Google Scholar]
- 144.Dwight T, Thoppe SR, Foukakis T, et al. Involvement of the PAX8/peroxisome proliferator-activated receptor γ rearrangement in follicular thyroid tumors. Journal of Clinical Endocrinology and Metabolism. 2003;88(9):4440–4445. doi: 10.1210/jc.2002-021690. [DOI] [PubMed] [Google Scholar]
- 145.Marques AR, Espadinha C, Catarino AL, et al. Expression of PAX8-PPARγ1 rearrangements in both follicular thyroid carcinomas and adenomas. Journal of Clinical Endocrinology and Metabolism. 2002;87(8):3947–3952. doi: 10.1210/jcem.87.8.8756. [DOI] [PubMed] [Google Scholar]
- 146.Nikiforova MN, Biddinger PW, Caudill CM, Kroll TG, Nikiforov YE. PAX8-PPARγ rearrangement in thyroid tumors: RT-PCR and immunohistochemical analyses. American Journal of Surgical Pathology. 2002;26(8):1016–1023. doi: 10.1097/00000478-200208000-00006. [DOI] [PubMed] [Google Scholar]
- 147.Gregory Powell J, Wang X, Allard BL, et al. The PAX8/PPARγ fusion oncoprotein transforms immortalized human thyrocytes through a mechanism probably involving wild-type PPARγ inhibition. Oncogene. 2004;23(20):3634–3641. doi: 10.1038/sj.onc.1207399. [DOI] [PubMed] [Google Scholar]
- 148.Giordano TJ, Au AYM, Kuick R, et al. Delineation, functional validation, and bioinformatic evaluation of gene expression in thyroid follicular carcinomas with the PAX8-PPARG translocation. Clinical Cancer Research. 2006;12(7):1983–1993. doi: 10.1158/1078-0432.CCR-05-2039. [DOI] [PubMed] [Google Scholar]
- 149.Reddi HV, McIver B, Grebe SKG, Eberhardt NL. Minireview: the paired box-8/peroxisome proliferator-activated receptor-γ oncogene in thyroid tumorigenesis. Endocrinology. 2007;148(3):932–935. doi: 10.1210/en.2006-0926. [DOI] [PubMed] [Google Scholar]
- 150.Ward LS, Brenta G, Medvedovic M, Fagin JA. Studies of allelic loss in thyroid tumors reveal major differences in chromosomal instability between papillary and follicular carcinomas. Journal of Clinical Endocrinology and Metabolism. 1998;83(2):525–530. doi: 10.1210/jcem.83.2.4550. [DOI] [PubMed] [Google Scholar]
- 151.Hunt JL, Livolsi VA, Baloch ZW, et al. A novel microdissection and genotyping of follicular-derived thyroid tumors to predict aggressiveness. Human Pathology. 2003;34(4):375–380. doi: 10.1053/hupa.2003.61. [DOI] [PubMed] [Google Scholar]
- 152.Rodrigues-Serpa A, Catarino A, Soares J. Loss of heterozygosity in follicular and papillary thyroid carcinomas. Cancer Genetics and Cytogenetics. 2003;141(1):26–31. doi: 10.1016/s0165-4608(02)00686-6. [DOI] [PubMed] [Google Scholar]
- 153.Hunt JL, Yim JH, Carty SE. Fractional allelic loss of tumor suppressor genes identifies malignancy and predicts clinical outcome in follicular thyroid tumors. Thyroid. 2006;16(7):643–649. doi: 10.1089/thy.2006.16.643. [DOI] [PubMed] [Google Scholar]
- 154.Zedenius J, Wallin G, Svensson A, et al. Allelotyping of follicular thyroid tumors. Human Genetics. 1995;96(1):27–32. doi: 10.1007/BF00214182. [DOI] [PubMed] [Google Scholar]
- 155.Tung WS, Shevlin DW, Kaleem Z, Tribune DJ, Wells SA, Jr., Goodfellow PJ. Allelotype of follicular thyroid carcinomas reveals genetic instability consistent with frequent nondisjunctional chromosomal loss. Genes Chromosomes and Cancer. 1997;19(1):43–51. [PubMed] [Google Scholar]
- 156.Segev DL, Saji M, Phillips GS, et al. Polymerase chain reaction-based microsatellite polymorphism analysis of follicular and Hurthle cell neoplasms of the thyroid. Journal of Clinical Endocrinology and Metabolism. 1998;83(6):2036–2042. doi: 10.1210/jcem.83.6.4882. [DOI] [PubMed] [Google Scholar]
- 157.Hunt JL, Yim JH, Tometsko M, et al. Loss of heterozygosity of the VHL gene identifies malignancy and predicts death in follicular thyroid tumors. Surgery. 2003;134(6):1043–1048. doi: 10.1016/j.surg.2003.07.023. [DOI] [PubMed] [Google Scholar]
- 158.Israel A, Sharan R, Ruppin E, Galun E. Increased MicroRNA activity in human cancers. PLoS One. 2009;4(6, article e6045) doi: 10.1371/journal.pone.0006045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Jazdzewski K, Liyanarachchi S, Swierniak M, et al. Polymorphic mature microRNAs from passenger strand of pre-miR-146a contribute to thyroid cancer. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(5):1502–1505. doi: 10.1073/pnas.0812591106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pallante P, Visone R, Ferracin M, et al. MicroRNA deregulation in human thyroid papillary carcinomas. Endocrine-Related Cancer. 2006;13(2):497–508. doi: 10.1677/erc.1.01209. [DOI] [PubMed] [Google Scholar]
- 161.Kim HJ, Kim YH, Lee DS, Chung J-K, Kim S. In vivo imaging of functional targeting of miR-221 in papillary thyroid carcinoma. Journal of Nuclear Medicine. 2008;49(10):1686–1693. doi: 10.2967/jnumed.108.052894. [DOI] [PubMed] [Google Scholar]
- 162.Nikiforova MN, Chiosea SI, Nikiforov YE. MicroRNA expression profiles in thyroid tumors. Endocrine Pathology. 2009;20(2):85–91. doi: 10.1007/s12022-009-9069-z. [DOI] [PubMed] [Google Scholar]
- 163.Galang CK, Hauser CA. Cooperative DNA binding of the highly conserved human Hox 2.1 homeodomain gene product. The New Biologist. 1992;4(5):558–568. [PubMed] [Google Scholar]
- 164.Lyons JF, Wilhelm S, Hibner B, Bollag G. Discovery of a novel Raf kinase inhibitor. Endocrine-Related Cancer. 2001;8(3):219–225. doi: 10.1677/erc.0.0080219. [DOI] [PubMed] [Google Scholar]
- 165.Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Research. 2004;64(19):7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 166.Wan PTC, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116(6):855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 167.Baudin E, Schlumberger M. New therapeutic approaches for metastatic thyroid carcinoma. Lancet Oncology. 2007;8(2):148–156. doi: 10.1016/S1470-2045(07)70034-7. [DOI] [PubMed] [Google Scholar]
- 168.Ouyang B, Knauf JA, Smith EP, et al. Inhibitors of Raf kinase activity block growth of thyroid cancer cells with RET/PTC or BRAF mutations in vitro and in vivo. Clinical Cancer Research. 2006;12(6):1785–1793. doi: 10.1158/1078-0432.CCR-05-1729. [DOI] [PubMed] [Google Scholar]
- 169.Kodama Y, Asai N, Kawai K, et al. The RET proto-oncogene: a molecular therapeutic target in thyroid cancer. Cancer Science. 2005;96(3):143–148. doi: 10.1111/j.1349-7006.2005.00023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Taniguchi M, Uehara Y, Matsuyama M, Takahashi M. Inhibition of ret tyrosine kinase activity by herbimycin A. Biochemical and Biophysical Research Communications. 1993;195(1):208–214. doi: 10.1006/bbrc.1993.2031. [DOI] [PubMed] [Google Scholar]
- 171.Cassinelli G, Lanzi C, Pensa T, et al. Clavilactones, a novel class of tyrosine kinase inhibitors of fungal origin. Biochemical Pharmacology. 2000;59(12):1539–1547. doi: 10.1016/s0006-2952(00)00278-1. [DOI] [PubMed] [Google Scholar]
- 172.Strock CJ, Park J-I, Rosen M, et al. CEP-701 and CEP-751 inhibit constitutively activated RET tyrosine kinase activity and block medullary thyroid carcinoma cell growth. Cancer Research. 2003;63(17):5559–5563. [PubMed] [Google Scholar]
- 173.Carlomagno F, Vitagliano D, Guida T, et al. The kinase inhibitor PP1 blocks tumorigenesis induced by RET oncogenes. Cancer Research. 2002;62(4):1077–1082. [PubMed] [Google Scholar]
- 174.Carlomagno F, Vitagliano D, Guida T, et al. Efficient inhibition of RET/papillary thyroid carcinoma oncogenic kinases by 4-amino-5-(4-chloro-phenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2) Journal of Clinical Endocrinology and Metabolism. 2003;88(4):1897–1902. doi: 10.1210/jc.2002-021278. [DOI] [PubMed] [Google Scholar]
- 175.Carlomagno F, Vitagliano D, Guida T, et al. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Research. 2002;62(24):7284–7290. [PubMed] [Google Scholar]
- 176.Vidal M, Wells S, Ryan A, Cagan R. ZD6474 suppresses oncogenic RET isoforms in a Drosophila model for type 2 multiple endocrine neoplasia syndromes and papillary thyroid carcinoma. Cancer Research. 2005;65(9):3538–3541. doi: 10.1158/0008-5472.CAN-04-4561. [DOI] [PubMed] [Google Scholar]
- 177.Lanzi C, Cassinelli G, Pensa T, et al. Inhibition of transforming activity of the Ret/Ptc1 oncoprotein by a 2-indolinone derivative. International Journal of Cancer. 2000;85(3):384–390. [PubMed] [Google Scholar]
- 178.Lanzi C, Cassinelli G, Cuccuru G, et al. Inactivation of Ret/Ptc1 oncoprotein and inhibition of papillary thyroid carcinoma cell proliferation by indolinone RPI-1. Cellular and Molecular Life Sciences. 2003;60(7):1449–1459. doi: 10.1007/s00018-003-2381-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Cuccuru G, Lanzi C, Cassinelli G, et al. Cellular effects and antitumor activity of RET inhibitor RPI-1 on MEN2A-associated medullary thyroid carcinoma. Journal of the National Cancer Institute. 2004;96(13):1006–1014. doi: 10.1093/jnci/djh184. [DOI] [PubMed] [Google Scholar]
- 180.Wedge SR, Ogilvie DJ, Dukes M, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Research. 2002;62(16):4645–4655. [PubMed] [Google Scholar]
- 181.Carlomagno F, Vitagliano D, Guida T, et al. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Research. 2002;62(24):7284–7290. [PubMed] [Google Scholar]
- 182.Santoro M, Carlomagno F. Drug insight: small-molecule inhibitors of protein kinases in the treatment of thyroid cancer. Nature Clinical Practice Endocrinology and Metabolism. 2006;2(1):42–52. doi: 10.1038/ncpendmet0073. [DOI] [PubMed] [Google Scholar]
- 183.Carlomagno F, Anaganti S, Guida T, et al. BAY 43-9006 inhibition of oncogenic RET mutants. Journal of the National Cancer Institute. 2006;98(5):326–334. doi: 10.1093/jnci/djj069. [DOI] [PubMed] [Google Scholar]
- 184.Kim DW, Jo YS, Jung HS, et al. An orally administered multitarget tyrosine kinase inhibitor, SU11248, is a novel potent inhibitor of thyroid oncogenic RET/papillary thyroid cancer kinases. Journal of Clinical Endocrinology and Metabolism. 2006;91(10):4070–4076. doi: 10.1210/jc.2005-2845. [DOI] [PubMed] [Google Scholar]
- 185.Hoffmann S, Burchert A, Wunderlich A, et al. Differential effects of cetuximab and AEE 788 on epidermal growth factor receptor (EGF-R) and vascular endothelial growth factor receptor (VEGF-R) in thyroid cancer cell lines. Endocrine. 2007;31(2):105–113. doi: 10.1007/s12020-007-0008-9. [DOI] [PubMed] [Google Scholar]
- 186.Polverino A, Coxon A, Starnes C, et al. AMG 706, an oral, multikinase inhibitor that selectively targets vascular endothelial growth factor, platelet-derived growth factor, and kit receptors, potently inhibits angiogenesis and induces regression in tumor xenografts. Cancer Research. 2006;66(17):8715–8721. doi: 10.1158/0008-5472.CAN-05-4665. [DOI] [PubMed] [Google Scholar]
- 187.Boughton D, Rosen L, Van Vugt A, et al. Safety and antitumor activity of AMG 706 in patients with thyroid cancer: a subset analysis from a phase 1 dose-finding study. Journal of Clinical Oncology. 2006;24(18, supplement):p. 3030. [Google Scholar]
- 188.Gharib H, Goellner JR. Fine-needle aspiration biopsy of the thyroid: an appraisal. Annals of Internal Medicine. 1993;118(4):282–289. doi: 10.7326/0003-4819-118-4-199302150-00007. [DOI] [PubMed] [Google Scholar]
- 189.Cohen Y, Rosenbaum E, Clark DP, et al. Mutational analysis of BRAF in fine needle aspiration biopsies of the thyroid: a potential application for the preoperative assessment of thyroid nodules. Clinical Cancer Research. 2004;10(8):2761–2765. doi: 10.1158/1078-0432.ccr-03-0273. [DOI] [PubMed] [Google Scholar]
- 190.Salvatore G, Giannini R, Faviana P, et al. Analysis of BRAF point mutation and RET/PTC rearrangement refines the fine-needle aspiration diagnosis of papillary thyroid carcinoma. Journal of Clinical Endocrinology and Metabolism. 2004;89(10):5175–5180. doi: 10.1210/jc.2003-032221. [DOI] [PubMed] [Google Scholar]
- 191.Xing M, Tufano RP, Tufaro AP, et al. Detection of BRAF mutation on fine needle aspiration biopsy specimens: a new diagnostic tool for papillary thyroid cancer. Journal of Clinical Endocrinology and Metabolism. 2004;89(6):2867–2872. doi: 10.1210/jc.2003-032050. [DOI] [PubMed] [Google Scholar]
- 192.Chung K-W, Yang SK, Lee GK, et al. Detection of BRAFV600E mutation on fine needle aspiration specimens of thyroid nodule refines cyto-pathology diagnosis, especially in BRAFV600E mutation-prevalent area. Clinical Endocrinology. 2006;65(5):660–666. doi: 10.1111/j.1365-2265.2006.02646.x. [DOI] [PubMed] [Google Scholar]
- 193.Kumagai A, Namba H, Akanov Z, et al. Clinical implications of pre-operative rapid BRAF analysis for papillary thyroid cancer. Endocrine Journal. 2007;54(3):399–405. doi: 10.1507/endocrj.k06-194. [DOI] [PubMed] [Google Scholar]
- 194.Jin L, Sebo TJ, Nakamura N, et al. BRAF mutation analysis in fine needle aspiration (FNA) cytology of the thyroid. Diagnostic Molecular Pathology. 2006;15(3):136–143. doi: 10.1097/01.pdm.0000213461.53021.84. [DOI] [PubMed] [Google Scholar]
- 195.Vitagliano D, Portella G, Troncone G, et al. Thyroid targeting of the N-RAS(Gln61Lys) oncogene in transgenic mice results in follicular tumors that progress to poorly differentiated carcinomas. Oncogene. 2006;25(39):5467–5474. doi: 10.1038/sj.onc.1209527. [DOI] [PubMed] [Google Scholar]
- 196.Nikiforova MN, Zhao Wen Z, Robinson-Smith T, et al. Molecular testing of thyroid FNA samples: feasibility and significance for preoperative diagnosis of thyroid tumors (abstract) Modern Pathology. 2004;17(supplement 1):p. 77A. [Google Scholar]
- 197.Domingues R, Mendoça E, Sobrinho L, Bugalho MJ. Searching for RET/PTC rearrangements and BRAF V599E mutation in thyroid aspirates might contribute to establish a preoperative diagnosis of papillary thyroid carcinoma. Cytopathology. 2005;16(1):27–31. doi: 10.1111/j.1365-2303.2004.00223.x. [DOI] [PubMed] [Google Scholar]
- 198.Nikiforova MN, E Nikiforov Y. Molecular genetics of thyroid cancer: implications for diagnosis, treatment and prognosis. Expert Review of Molecular Diagnostics. 2008;8(1):83–95. doi: 10.1586/14737159.8.1.83. [DOI] [PubMed] [Google Scholar]
- 199.Bounacer A, Wicker R, Caillou B, et al. High prevalence of activating ret proto-oncogene rearrangements, in thyroid tumors from patients who had received external radiation. Oncogene. 1997;15(11):1263–1273. doi: 10.1038/sj.onc.1200206. [DOI] [PubMed] [Google Scholar]
- 200.Elisei R, Romei C, Vorontsova T, et al. RET/PTC rearrangements in thyroid nodules: studies in irradiated and not irradiated, malignant and benign thyroid lesions in children and adults. Journal of Clinical Endocrinology and Metabolism. 2001;86(7):3211–3216. doi: 10.1210/jcem.86.7.7678. [DOI] [PubMed] [Google Scholar]
- 201.Nikiforova MN, Caudill CM, Biddinger P, Nikiforov YE. Prevalence of RET/PTC rearrangements in Hashimoto’s thyroiditis and papillary thyroid carcinomas. International Journal of Surgical Pathology. 2002;10(1):15–22. doi: 10.1177/106689690201000104. [DOI] [PubMed] [Google Scholar]
- 202.Nikiforova MN, Biddinger PW, Caudill CM, Kroll TG, Nikiforov YE. PAX8-PPARγ rearrangement in thyroid tumors: RT-PCR and immunohistochemical analyses. American Journal of Surgical Pathology. 2002;26(8):1016–1023. doi: 10.1097/00000478-200208000-00006. [DOI] [PubMed] [Google Scholar]
- 203.Castro P, Rebocho AP, Soares RJ, et al. PAX8-PPAR γ rearrangement is frequently detected in the follicular variant of papillary thyroid carcinoma. Journal of Clinical Endocrinology and Metabolism. 2006;91(1):213–220. doi: 10.1210/jc.2005-1336. [DOI] [PubMed] [Google Scholar]
- 204.Gustafson KS, LiVolsi VA, Furth EE, Pasha TL, Putt ME, Baloch ZW. Peroxisome proliferator-activated receptor γ expression in follicular-patterned thyroid lesions. Caveats for the use of immunohistochemical studies. American Journal of Clinical Pathology. 2003;120(2):175–181. doi: 10.1309/2E6Q-GJRG-GETV-T8K9. [DOI] [PubMed] [Google Scholar]