

Abstract
HERG (KCNH2) and ether-à-go-go (eag) (KCNH1) are members of the same subfamily of voltage-gated K+ channels. In eag, voltage-dependent activation is significantly slowed by extracellular divalent cations. To exert this effect, ions bind to a site located between transmembrane segments S2 and S3 in the voltage sensor domain where they interact with acidic residues that are conserved only among members of the eag subfamily. In HERG channels, extracellular divalent ions significantly accelerate deactivation. To investigate the ionbinding site in HERG, acidic residues in S2 and S3 were neutralized singly or in pairs to alanine, and the functional effects of extracellular Mg2+ were characterized in Xenopus oocytes. To modulate deactivation kinetics in HERG, divalent cations interact with eag subfamily-specific acidic residues (D460 and D509) and also with an acidic residue in S2 (D456) that is widely conserved in the voltage-gated channel superfamily. In contrast, the analogous widely-conserved residue does not contribute to the ion-binding site that modulates activation kinetics in eag. We propose that structural differences between the ion-binding sites in the eag and HERG voltage sensors contribute to the differential regulation of activation and deactivation gating in these channels. A previously proposed model for S4 conformational changes during voltagedependent activation can account for the differential regulation of gating seen in eag and HERG.
Keywords: voltage-dependent, activation, deactivation, IKr, cardiac ion channel, magnesium, KCNH1, KCNH2, KCNE2
HERG (human ether-à-go-go related gene) (KCNH2) encodes two alternatively spliced voltage-gated K+ channel subunits (HERG1a and HERG1b) that co-assemble with MiRP1 (MinK-Related Protein 1) (KCNE2), a protein with a single transmembrane segment, to form the cardiac IKr channel.1,2 IKr channels play an essential role in repolarization of the cardiac action potential due to their unique gating kinetics. During action potential depolarization, IKr channels open and then very rapidly inactivate by a Ctype mechanism.3–7 Rapid inactivation limits the availability of IKr channels during the plateau phase of the action potential. As the membrane begins to repolarize, however, IKr channels rapidly recover from inactivation and reenter an open, conducting conformation. Channel closure (deactivation) from this state occurs slowly. As a result, IKr channels generate a resurgent K+ current that reinforces and accelerates repolarization of the cardiac action potential. These unique gating properties of cardiac IKr channels are an intrinsic feature of heterotetrameric channels composed of the HERG1a and HERG1b subunits, although coassembly with MiRP1 subunits may modify the kinetics and pharmacology of HERG channels to more closely resemble those of native cardiac channels.1,8
The essential role of HERG channels in the heart is underscored by the finding that mutations in HERG are responsible for Long QT Syndrome Type 2 (LQTS2).3,9,10 In LQTS, a defect in repolarization of the cardiac action potential results in a lengthened QT interval detected by electrocardiogram. Individuals with LQTS are at increased risk for ventricular arrhythmia and sudden death.11 Although congenital forms of LQTS are rare, acquired LQTS is a significant health problem and is typically due to drug interactions with HERG channels.12,13 In some cases, naturally occurring polymorphisms in HERG and MiRP1 may be responsible for increased drug sensitivity.14,15
The process and kinetics of deactivation in HERG channels are central to the physiological role of IKr channels. Interestingly, the rate of deactivation in HERG channels is modulated by relatively small changes in extracellular Mg2+ or Ca2+ in their physiological concentration ranges. These ions appear to act by modifying channel gating rather than by blocking the pore or changing its conduction properties.16–19 We have tested the hypothesis that extracellular divalent cations regulate gating kinetics by binding directly to the voltage sensor in HERG channels. This hypothesis is predicated on our previous work with the ether-à-go-go (eag) K+ channel. In eag, extracellular Mg2+ regulates the kinetics of channel activation by binding to two acidic residues located in transmembrane segments S2 and S3.20 Among voltage-gated K+ channels, these positions are occupied by acidic residues only in the eag subfamily, which explains why this form of ion regulation is uniquely found in this group. We now report that the HERG voltage sensor contains an ion-binding site that modulates gating kinetics. In HERG, ion binding affects the rates of both activation and deactivation, in contrast to eag, where effects on deactivation are not prominent.21,22 In HERG, the bound ion interacts with both eag specific and widely conserved acidic residues. This is in contrast to eag, where only eag subfamily-specific residues are implicated in ion binding.20 We propose that differences between the ion-binding sites explain why ion binding prominently modulates deactivation gating in HERG but not in eag. Despite these differences, the data indicate that the presence of an ion-binding site between S2 and S3 is a conserved feature of members of the eag subfamily of voltage-gated K+ channels.
EXPERIMENTAL PROCEDURES
Molecular biology
The wild-type HERG1a cDNA clone was kindly provided by Dr. Mark Keating (Harvard Medical School, Boston, MA). Mutations in HERG1a were generated by the QuikChange method (Stratagene, La Jolla, CA) or by polymerase chain reaction (PCR) using a four primer strategy.23 Mutated constructs were verified by Big Dye terminator sequencing (UCLA Sequencing Core Facility, Los Angeles, CA). Constructs in the pSP64 vector were linearized with EcoRI before in vitro transcription using the mMessage mMachine kit (Ambion, Austin, TX). RNA was injected into Xenopus oocytes for electrophysiological analysis.24
Electrophysiology
Ionic currents from wild-type and mutant HERG channels were recorded at room temperature (20–22°C) 2–4 days after RNA injection using a two electrode voltage clamp.24,25 Electrodes were filled with 3 M KCl and had resistances of 0.3–1.0 MΩ. Oocytes were bathed in a modified ND96 solution containing 96 mM NaCl, 2 mM KCl, 5 mM HEPES, pH 7.5. The concentrations of MgCl2 and CaCl2 were varied as indicated in each experiment. The bath solution was changed using a gravity-driven perfusion system that was governed by solenoid valves and controlled manually.
Voltage pulse protocols were generated and data were acquired using pClamp software and a TL1 Labmaster interface (Axon Instruments, Foster City, CA). Data were sampled at 2–40 kHz and filtered at one fifth the sampling frequency using an 8-pole Bessel filter (Frequency Devices, Haverhill, MA). Linear capacitative and leak currents were subtracted using a P/-4 protocol.26
HERG ionic currents were evoked by pulsing from a holding potential of −80 mV to voltages ranging from −100 mV to +80 mV or to +100 mV in 20 mV increments. Tail currents were recorded upon repolarization to −70 mV. The sampling rate was varied as needed to resolve deactivation kinetics. Tail currents were fitted with double or single exponential functions to derive time constants for deactivation (τdeact). Activation kinetics was characterized by an envelope of tails procedure.27 Values of τdeact and τact are provided as mean ± SEM. Statistical significance was assessed using a one-way ANOVA.
The probability of opening as a function of voltage was determined by measuring the amplitudes of isochronal tail currents evoked by repolarizing to −70 mV from various depolarized potentials. Tail current amplitudes were normalized to the maximum value and plotted versus pulse potential. The data were fitted with a single Boltzmann equation using Origin 7.0 (Origin Lab, Northampton, MA) to obtain slope and midpoint potential values.
RESULTS
Extracellular Mg2+ accelerates deactivation in wild-type HERG channels
HERG ionic currents were recorded in Xenopus oocytes in the presence or absence of 10 mM extracellular Mg2+ using a two-electrode voltage clamp (Fig. 1A). Currents were elicited by pulsing from a holding potential of −80 mV to voltages ranging from −100 to +80 mV. The membrane was then repolarized to −70 mV to record tail currents. Current amplitudes during the depolarizing pulses were low, as expected for the rapidly inactivating HERG channel.6 Upon repolarization and recovery from inactivation, however, large outward tail currents were recorded as the channel deactivated.
Figure 1.

Extracellular Mg2+ accelerates deactivation of wild-type HERG channels. (A) Wild-type HERG currents were recorded in the absence (left) or presence (middle) of 10 mM extracellular Mg2+. From a holding potential of −80 mV, 2 s test pulses ranging from −100 mV to +80 mV were applied in 20 mV increments prior to repolarization to −70 mV for 8 sec. Representative current traces are shown. Right, Tail currents evoked upon return from +60 mV to −70 mV in the absence (solid trace) or presence (dotted trace) of 10 mM Mg2+ have been scaled and superimposed to compare the time course of deactivation. Tail currents were fitted with two exponential components (dashed lines). The bath solution contained 96 mM NaCl, 2 mM KCl, 0.5 mM CaCl2, 5 mM HEPES, pH 7.5. The pulse protocol is shown below the current traces. (B) Activation kinetics of wild-type HERG channels were measured using an envelope of tails protocol. Upper, Representative current traces were evoked at +40 mV by test pulses of different durations. Tail currents were recorded upon repolarization to −80 mV. Arrows indicate the peak tail currents. The pulse protocol is shown below the current traces. Lower, Normalized peak tail current amplitudes obtained in the presence (□) or absence (■) of 10 mM Mg2+ have been plotted versus pulse duration. Data are shown as mean ± SEM, n = 5. Each data set was fitted with a single exponential component to obtain values for τact in the presence (dashed curve) or absence (solid curve) of Mg2+. (C) Itail,0 Mg/Itail,max,0 Mg (■) and Itail,10 Mg/Itail,max,10 Mg (□) are plotted versus V. (D) Left, Values of the deactivation time constants τfast (□) and τslow (■) obtained upon return from +40 mV to −70 mV have been plotted versus Mg2+ concentration. The horizontal bar indicates the physiological range of Mg2+ concentration. Data are shown as mean ± SEM, n = 7. In this and subsequent figures, if error bars are not visible, they are smaller than the size of the symbol. Right, The difference between the fitted τfast or τslow values (Δτfast (□) and Δτslow (■)) obtained in the absence and presence of Mg2+ were normalized and plotted versus Mg2+ concentration. In this figure, Δτfast = τfast 0 Mg − τfast + Mg and Δτslow = τslow 0 Mg − τslow + Mg. The half maximal effective concentration for Mg2+ was estimated as described in the text.
Extracellular Mg2+ significantly accelerated tail currents (Fig. 1A). The kinetics of deactivation at −70 mV were quantified by fitting two exponential components to the tail current records, providing values for two time constants, τfast and τslow (Table 1). In the presence of 10 mM Mg2+, the values of τfast and τslow at −70 mV were reduced by approximately 2-fold and 3-fold, respectively (Table 1). Mg2+ also accelerated deactivation about 2-fold when HERG was co-expressed with MiRP1 (data not shown).
Table 1.
Deactivation time constants for HERG and mutant constructs in the absence and presence of 10 mM Mg2+
| 0 mM Mg2+ | 10 mM Mg2+ | n | Vtest | |
|---|---|---|---|---|
| WT (slow)** | 2567 ± 129 | 789 ± 82 | 11 | +40 mV |
| WT (fast)** | 480 ± 25 | 219 ± 21 | 11 | +40 mV |
| D456A (slow) | 201 ± 21 | 165 ± 10 | 6 | +40 mV |
| D456A (fast)** | 52 ± 2 | 39 ± 1 | 6 | +40 mV |
| D460A* | 99 ± 5 | 76 ± 3 | 8 | +40 mV |
| D466A* | 111 ± 5 | 84 ± 7 | 5 | +40 mV |
| D509A* | 47 ± 1 | 42 ± 1 | 8 | +40 mV |
| D456A/D460A (slow) | 163 ± 7 | 166 ± 9 | 8 | +60 mV |
| D456A/D460A (fast) | 24 ± 1 | 26 ± 2 | 8 | +60 mV |
| D456A/D509A | 63 ± 3 | 59 ± 3 | 7 | +60 mV |
| D460A/D509A* | 50 ± 2 | 44 ± 1 | 5 | +60 mV |
| D466A/D509A** | 76 ± 2 | 53 ± 2 | 11 | +60 mV |
Values for the deactivation time constants (in ms, mean ± SEM) were derived from single or double exponential fits to the tail currents (see Figs. 1A and 3B). The number of experiments, n, and the test pulse, Vtest, are indicated.
and ** denote significant differences in the presence and absence of 10 mM Mg2+ p < 0.001 and p < 0.05, respectively, using one-way ANOVA.
Extracellular Mg2+ slows activation in wild-type HERG channels
In eag, Mg2+ slows channel opening with little effect on channel closing.21,22,28,29 In HERG channels, any effect of Mg2+ on activation kinetics is masked by the fast rate of inactivation. To investigate whether ion binding affects activation kinetics in HERG channels, we used an envelope of tails procedure (Fig. 1B).27 To activate the channel, the membrane was stepped from −80 mV to +40 mV for pulses of increasing duration in the presence or absence of Mg2+. Peak tail current amplitudes were measured upon repolarization to −80 mV. The time course of ionic current activation was then inferred from the increase in tail current amplitude as a function of pulse duration (Fig. 1B). At +40 mV, 10 mM Mg2+ slowed activation by approximately 2-fold (Table 2).
Table 2.
Activation time constants for HERG and mutant constructs in the absence and presence of 10 mM
| 0 mM Mg2+ | 10 mM Mg2+ | n | Vtest | |
|---|---|---|---|---|
| WT** | 56 ± 5 | 136 ± 7 | 5 | +40 mV |
| D456A | 137 ± 14 | 166 ± 27 | 5 | +40 mV |
| D460A | 63 ± 5 | 69 ± 5 | 5 | +60 mV |
| D466A | 72 ± 2 | 86 ± 7 | 5 | +40 mV |
| D509A | 51 ± 6 | 62 ± 3 | 4 | +60 mV |
| D456A/D460A | 330 ± 32 | 422 ± 41 | 5 | +70 mV |
| D456A/D509A | 129 ± 15 | 154 ± 13 | 3 | +70 mV |
| D460A/D509A* | 146 ± 8 | 192 ± 15 | 5 | +70 mV |
| D466A/D509A | 194 ± 13 | 203 ± 16 | 6 | +70 mV |
Values for the activation time constants (in ms, mean ± SEM) were derived from single exponential fits to the currents evoked by an envelope of tails protocol (see Figs. 1B and 5A). The number of experiments, n, and the test pulse, Vtest, are indicated.
and ** denote significant differences in the presence and absence of 10 mM Mg2+ p < 0.001 and p < 0.05, respectively, using a one-way ANOVA.
Our results indicate that extracellular Mg2+ modulates both activation and deactivation gating in HERG channels. Together, these effects reduce the maximum conductance measured in the presence of Mg2+ by about 20% (Fig. 1C).
Physiological concentrations of Mg2+ modulate HERG gating
To investigate the concentration dependence of Mg2+ modulation in HERG, deactivation kinetics were compared at Mg2+ concentrations up to 20 mM (Fig. 1D, left). Concentrations as low as 0.5 mM significantly accelerated deactivation; 10 mM Mg2+ gave a near maximal effect. To estimate the apparent affinity of HERG for Mg2+, changes in τfast and τslow in the absence and presence of Mg2+ were normalized (Δτfast/Δτmax and Δτslow/Δτmax) and plotted versus Mg2+ concentration (Fig. 1D, right). The data were fitted with the equation Δτ/Δτmax =[Mg2+]/([Mg2+] + K0.5), providing estimates for the half maximal effective concentrations for Δτfast and Δτslow of 2.2 and 1.8 mM respectively. Given experimental variability, these values do not differ significantly (Fig. 1D). The steepest change in deactivation kinetics occurred between 0 and 3 mM. Since the normal concentration of Mg2+ in serum ranges from 0.8 to 1.2 mM, Mg2+ modulation of deactivation in HERG channels is likely to be important physiologically.
Extracellular Ca2+ modulates HERG gating
In eag, the ion-binding site between S2 and S3 can accommodate different divalent cations, including Mg2+, Mn2+ and Ni2+, all of which modulate activation gating.20,30,31 To investigate whether multiple ions modulate HERG gating, we measured activation and deactivation kinetics in the presence and nominal absence of extracellular Ca2+ (Fig. 2). Like Mg2+, Ca2+ accelerated deactivation and slowed activation (Fig. 2).
Figure 2.
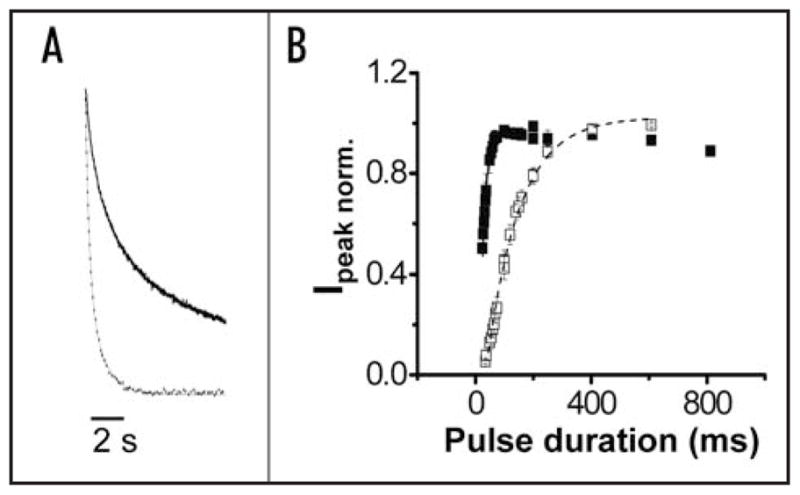
Extracellular Ca2+ modulates gating kinetics of wild-type HERG channels. (A) Wild-type HERG tail currents evoked upon return from +40 mV to −70 mV in the absence (solid trace) or presence (dotted trace) of 10 mM Ca2+ have been scaled and superimposed to compare the time course of deactivation. Tail currents were fitted with two exponential components (thin dashed lines). The bath solution contained 96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 5 mM HEPES, pH 7.5. Values of τfast and τslow in the absence of Ca2+ were: 609 ± 58 ms and 3371 ± 402 ms, respectively (n = 4). Values of τfast and τslow in the presence of Ca2+ were: 206 ± 10 ms and 729 ± 55 ms, respectively (n = 4). (B) Using the envelope of tails procedure described in Figure 1B, normalized peak tail current amplitudes were obtained in the presence (□) or absence (■) of 10 mM Ca2+ and plotted versus pulse duration. Data are shown as mean ± SEM, n = 4. Each data set was fitted with a single exponential component to obtain values for τact in the presence (dashed curve) or absence (solid curve) of Ca2+. Values of τact in the absence and presence of Ca2+ were 20 ± 4 ms and 121 ± 11 ms, respectively.
Mg2+ binds between S2 and S3 to modulate deactivation and activation kinetics
The HERG protein contains a number of acidic residues in transmembrane segments S2 and S3 (Fig. 3A). Three of these (D456 and D466 in S2, D501 in S3) correspond to acidic residues that are widely conserved among members of the voltage-gated ion channel superfamily.32 Two others (D460 in S2 and D509 in S3) correspond to acidic residues that are conserved only among members of the eag subfamily of voltage-gated K+ channels (Fig. 3A).33,34 In eag, these subfamily-specific acidic residues contribute to a divalent ion-binding site that modulates activation gating.20,30,31,35
Figure 3.
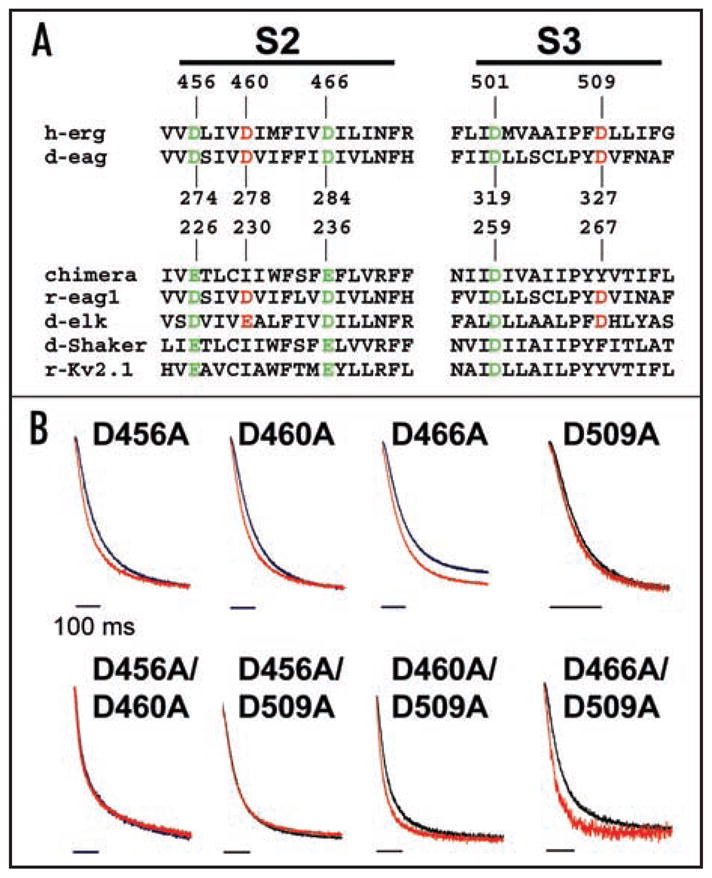
Mg2+ modulates deactivation kinetics by binding between S2 and S3. (A) Sequences of S2 and S3 segments from voltage-gated K+ channels have been aligned. Acidic residues conserved throughout the voltage-gated channel superfamily are shown in green; acidic residues conserved only in the eag subfamily of K+ channels are shown in red. Sequences from eag family members, representative members of the Kv1 and Kv2 families, and a chimeric channel in which the Kv2.1 voltage sensor paddle has been inserted into Kv1.2 are included.31,33,34,42 Members of the eag subfamily contain acidic amino acids in S2 and S3 that are not present in other voltage-gated K+ channels.30 Numbering is shown for HERG, d-eag and the chimeric channel sequences. Abbreviations used are: h, human; d, Drosophila; r, rat; erg, eag-related gene; elk, eag-like K+ channel gene. (B) Tail currents evoked upon return to −70 mV from +40 mV for single mutants or +60 mV for double mutants in the absence (black trace) or presence (red trace) of 10 mM Mg2+ have been scaled and superimposed to compare the time course of deactivation. Tail currents were fitted with single or double exponential components (thin dashed lines). Bars: 100 ms. The bath solution contained 96 mM NaCl, 2 mM KCl, 0.5 mM CaCl2, 5 mM HEPES, pH 7.5.
To test the hypothesis that acidic residues in S2 and/or S3 contribute to an ion-binding site that modulates HERG gating, the five aspartate residues in S2 and S3 were mutated singly and in pairs to alanine. D501A in S3 eliminated functional expression and was not studied further. Other mutant constructs produced functional channels in Xenopus oocytes. Gating properties of the mutant channels were qualitatively similar to those of wild-type HERG, with small currents during depolarizing pulses and large tail currents evoked upon repolarization (data not shown). However, all of the mutant constructs deactivated more rapidly than wild-type HERG (Table 1). To determine whether the mutations reduced the effect of Mg2+ on deactivation kinetics, ionic currents were recorded in the presence or absence of 10 mM Mg2+ and tail currents were elicited upon repolarization to −70 mV. The prominent modulation of deactivation kinetics evident in HERG wild-type channels was dramatically reduced in several of the mutant constructs (Fig. 3B).
Unlike wild-type HERG, some of the mutant channels had deactivation kinetics that were well-fitted by a single exponential component. Tail currents were fitted with one or two exponential components, as needed, to quantify deactivation kinetics in the presence and absence of Mg2+ (Fig. 4 and Table 1). In the single mutant constructs, the sensitivity of deactivation kinetics to Mg2+ was reduced to varying degrees, but the residual modulation was statistically significant (Table 1). For D456A, however, the effect of Mg2+ was significant only for the faster of two components of deactivation. Combination of D456A with D460A or D509A abolished Mg2+ modulation of deactivation kinetics. In contrast, D460A + D509A and D466A + D509A retained a small but statistically significant sensitivity to Mg2+. It is noteworthy that neutralization of both eag subfamily-specific acidic residues, D460 in S2 and D509 in S3, was not sufficient to eliminate Mg2+ modulation of deactivation kinetics. Rather, neutralization of either eag subfamily-specific acidic residue in combination with D456A was required. D456 in S2 corresponds to an acidic residue widely conserved among voltage-gated ion channels. In contrast, in eag, the analogous residue, D274, does not contribute to the ion-binding site.20,30 Our results are consistent with the conclusion that HERG channels contain an ion-binding site between S2 and S3 that modulates deactivation kinetics.
Figure 4.
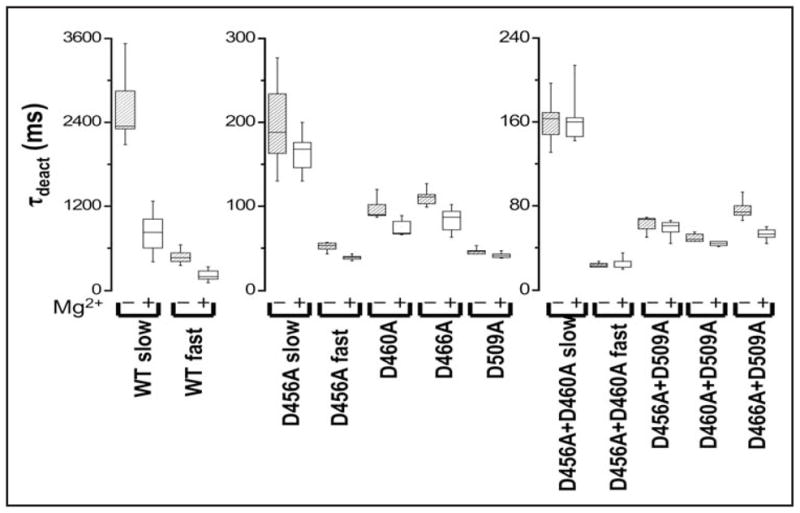
Mutating acidic residues in S2 and S3 reduces Mg2+ sensitivity of deactivation kinetics. Box plots of τdeact values obtained for wild-type and mutant channels in the absence or presence of 10 mM Mg2+ are shown. Tail currents were evoked upon repolarizing from +40 mV (+60 mV for double mutants) to −70 mV and fitted with single or double exponential functions. Values of τdeact for HERG wild-type and mutant channels are provided in Table 1.
We also investigated whether mutating acidic residues in S2 and S3 affected Mg2+ modulation of activation kinetics (Fig. 5). Single and double mutant constructs, including D466A located near the cytoplasmic end of S2, dramatically reduced or abolished the slowing of activation kinetics mediated by extracellular Mg2+ (Fig. 5 and Table 2).
Figure 5.
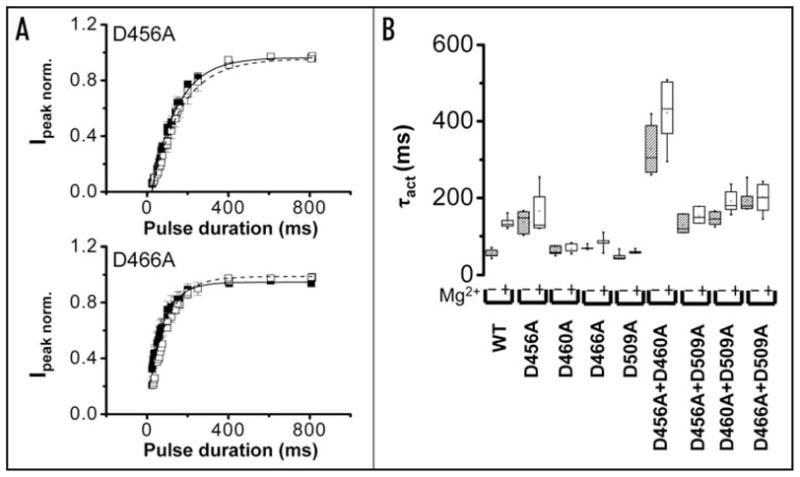
Mutating acidic residues in S2 or S3 reduces Mg2+ sensitivity of activation kinetics. (A) Using the envelope of tails procedure described in Figure 1B, normalized peak tail current amplitudes were obtained for D456A (upper) and D466A (lower) channels in the presence (□) or absence (■) of 10 mM Mg2+ and plotted versus pulse duration. Each data set was fitted with a single exponential component to obtain values for τact in the presence (dashed curve) or absence (solid curve) of Mg2+. Data are shown as mean ± SEM, n = 5. The bath solution contained 96 mM NaCl, 2 mM KCl, 0.5 mM CaCl2, 5 mM HEPES, pH 7.5. (B) Box plots of τact values obtained for wild-type and mutant channels in the absence or presence of 10 mM Mg2+ are shown. The voltage was stepped to +40 mV (+60 mV for D460A, D509A, +70 mV for double mutants) followed by repolarization to −70 mV. Values of τact for HERG wild-type and mutant channels are provided in Table 2.
Mg2+ shifts Po-V curve in wild-type and mutant channels
Extracellular divalent ions have been reported to shift the voltage dependence of HERG activation in the depolarized direction.18,19,36,37 The size of the shift varies among different reports, perhaps because different ionic conditions were used.18,19,36,37 We found that extracellular Mg2+ shifted the voltage dependence of activation in the depolarized direction in wild-type channels (Fig. 6 and Table 3). To determine whether mutating acidic residues in S2 and S3 reduced or eliminated the effect of Mg2+ on the steady state properties of activation, we characterized the probability of opening as a function of voltage (Po-V) in mutant channels in the presence and absence of Mg2+. With the exception of D466A, the single and double mutations shifted the voltage dependence of HERG activation to more depolarized potentials in the absence of Mg2+. An additional positive shift was seen when 10 mM Mg2+ was added to the bath solution (Fig. 6 and Table 3). Thus, the effect of Mg2+ on the voltage dependence of activation was not primarily due to ion binding between S2 and S3. It is likely that Mg2+ exerted a non-specific surface charge effect on the voltage sensor in both wild-type and mutant channels.38 In our experiments, the bath solution contained 0.5 mM Ca2+ with or without 10 mM Mg2+. Adding Ca2+ improved the quality of our electrophysiological recordings and served to partially offset surface charge effects due to added Mg2+. However, in all likelihood Ca2+ binds to the HERG voltage sensor, accounting for its effects on opening and closing kinetics (see Fig. 2). The use of 0.5 mM Ca2+ was therefore a compromise, reducing the effect of Ca2+ on gating kinetics in Mg2+-free recordings, while at the same time stabilizing the membrane and providing some surface charge screening. Not surprisingly, 0.5 mM Ca2+ was insufficient to screen entirely the non-specific surface charge effects of 10 mM Mg2+. We therefore focused on the kinetic effects of Mg2+ to identify the ion-binding site in the HERG voltage sensor domain.
Figure 6.
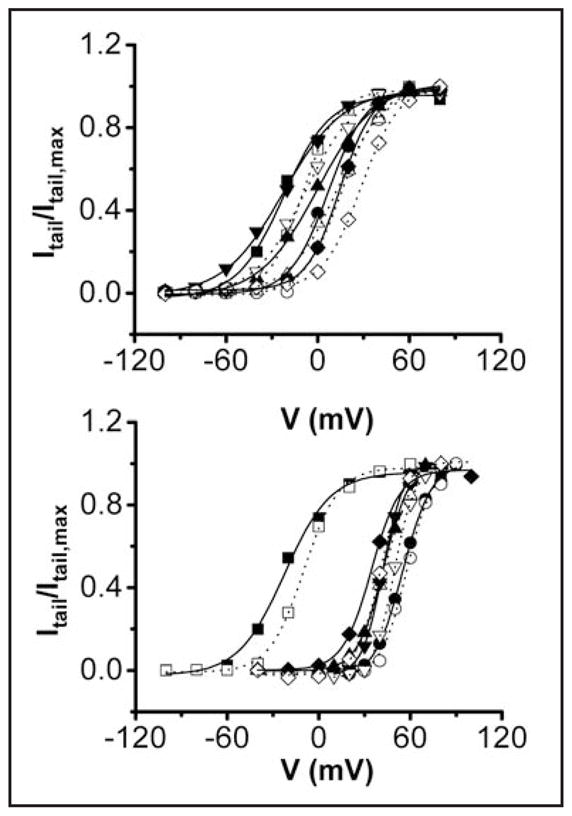
Mg2+ shifts the voltage dependence of activation in both wild-type and mutant channels. Upper: Isochronal tail current amplitudes in the presence or absence of Mg2+ were measured at −70 mV and normalized to the maximum tail current amplitude obtained in the absence of Mg2+. Itail/Itail,max has been plotted versus voltage for wild type (■, □); D456A (●,❍); D460A (▲,Δ); D466A (▼;,∇); and D509A (◇,◆) channels in the absence (filled symbols) or presence (open symbols) of 10 mM Mg2+. Bottom: Itail/Itail,max has been plotted versus voltage for wild type (■,□); D456A + D460A (●,❍); D456A + D509A (▲,Δ); D460A + D509A (▼;,∇); and D466A + D509A (◇,◆) channels in the absence (filled symbols) or presence (open symbols) of 10 mM Mg2+. The bath solution contained 96 mM NaCl, 2 mM KCl, 0.5 mM CaCl2, 5 mM HEPES, pH 7.5. After repolarization from various test potentials to −70 mV, isochronal tail current amplitudes were measured and normalized to the maximum amplitude obtained in the experiment. Data points represent the average of 5–6 experiments. Error bars are omitted for clarity. Each data set was fitted with a single Boltzmann function (solid curves, 0 mM Mg2+; dotted curves, 10 mM Mg2+) to obtain values for the midpoint potential (V½) and slope factor (see Table 3 legend). V½ values are provided in Table 3.
Table 3.
Activation parameters for HERG and mutant constructs in the absence and presence of 10 mM Mg2+
| 0 mM Mg2+ | 10 mM Mg2+ | n | ΔV½ | |
|---|---|---|---|---|
| WT | −22 ± 3 | −10 ± 2 | 5 | 12 mV |
| D456A | 7 ± 1 | 15 ± 2 | 6 | 8 mV |
| D460A | −4 ± 1 | 12 ± 1 | 6 | 16 mV |
| D466A | −29 ± 2 | −10 ± 4 | 5 | 19 mV |
| D509A | 14 ± 1 | 27 ± 2 | 6 | 13 mV |
| D456A/D460A | 55 ± 2 | 59 ± 1 | 6 | 4 mV |
| D456A/D509A | 39 ± 2 | 44 ± 2 | 5 | 5 mV |
| D460A/D509A | 42 ± 1 | 50 ± 1 | 5 | 8 mV |
| D466A/D509A | 34 ± 1 | 41 ± 2 | 6 | 7 mV |
Isochronal tail current amplitudes in the presence or absence of Mg2+ were measured at −70 mV and normalized to the maximum tail current amplitude obtained in the absence of Mg2+. Values for the midpoint potential (V1/2) (in mV, mean ± SEM) were derived from plots of Itail/Itail,max versus V fitted with a Boltzmann equation of the form: Itail/Itail,max=1/(1+exp[(V1/2 − V)/A]). The number of experiments, n, and the shift of V1/2, ΔV1/2, induced by the presence of 10 mM Mg2+ are provided.
DISCUSSION
Modulation of HERG gating by extracellular divalent cations may help counteract detrimental cardiac effects of hypomagnesemia and hypocalcemia
Modulation of HERG gating by Mg2+ and Ca2+ is likely to be physiologically important because a major target of ion regulation is the slow deactivation process, which is a critical factor in repolarization of the cardiac action potential.39,40 Furthermore, significant changes in deactivation kinetics result from small changes in extracellular Mg2+ and Ca2+ in and near the normal concentration range. Hypomagnesemia and hypocalcemia increase the excitability of the heart and contribute to cardiac arrhythmia.41 However, such changes would also slow the rate of HERG deactivation, leading to additional outward K+ current that would be expected to counteract the hyperexcitability. Therefore, ionic modulation of deactivation gating in HERG may serve as a partially protective mechanism in the heart.
Our experiments were conducted using the HERG1a splice variant, whereas cardiac channels contain HERG1a and 1b subunits.2 HERG1a and 1b are identical except at the amino terminus.8 Both subunits contain the ion-binding site residues in the voltage sensor, consistent with the idea that divalent ions would accelerate deactivation in native cardiac channels. It is worth noting that the deactivation kinetics of heterotetrameric HERG1a/HERG1b and homotetrameric HERG1a channels differ.8 Therefore deactivation kinetics in HERG1a/HERG1b channels measured in the presence and absence of extracellular divalent ions would be expected to deviate from the values reported in this study.8
Mg2+ interacts with D456, D460 and D509 to modulate deactivation gating in HERG
The prominent modulation of activation and deactivation in HERG channels indicates that extracellular divalent cations are able to bind to voltage sensors in the resting conformation to affect the rate of opening and in the activated conformation to affect the rate of closing. Our data support the conclusion that Mg2+ interacts with D456 and D460 in S2 and D509 in S3 to accelerate channel closing. This conclusion is compatible with the results of Fernandez et al.37 They found that the non-physiological ion Cd2+ binds to a site formed by the same three residues and thereby shifts the voltage-dependence of activation. Fernandez and co-workers measured shifts in activation caused by 0.5 mM Cd2+ in the constant presence of 1 mM Ca2+ and 2 mM Mg2+. The presence of excess Ca2+ and Mg2+ presumably reduced non-specific surface charge effects caused by Cd2+ binding to negatively charged lipid headgroups or acidic amino acids located in extracellular loops,38 making it feasible to measure shifts in activation caused by Cd2+ binding to the voltage sensor site. In contrast, much of the Mg2+-induced shift in the voltage dependence of activation measured in our experiments is likely to result from a non-specific surface charge effect rather than specific-binding to the HERG voltage sensor.
Mg2+ binding to the resting conformation may depend on the negative electrostatic potential of the cleft between S2 and S3
Our data suggest that Mg2+ also binds between S2 and S3 to slow channel opening. Interestingly, the mutations had differential effects on Mg2+ modulation of activation and deactivation gating. Whereas only double mutant combinations containing D456A abolished Mg2+ modulation of closing rate, each single mutant eliminated modulation of activation kinetics, including D456A and D466A, located near the extracellular and intracellular ends of S2, respectively. These results suggest that divalent ions affect HERG activation and deactivation kinetics by different mechanisms. On the basis of cysteine modification experiments, Tseng and colleagues have proposed that HERG contains a long, externally-facing aqueous cleft that extends to D466 in S2 in the resting conformation. 19 Our results suggest that binding of divalent cations to the resting voltage sensor depends more on the negative electrostatic potential of this cleft than on interactions with specific acidic residues. Reduction of the negative potential by neutralization of even one acidic residue appears to be sufficient to eliminate ion binding to the resting conformation. In contrast, our work on eag did not reveal a similar mechanism in that channel, likely due to subtle structural differences between the clefts in eag and HERG.
Differential regulation of activation and deactivation by ion binding in eag and HERG
Divalent cation modulation of deactivation in HERG and activation in eag are mechanistically related because both phenomena involve ion binding to a specific site that includes the eag subfamily-specific acidic residues in S2 and S3. In HERG, the widely conserved acidic residue D456 is also involved in ion binding, whereas the analogous residue in eag is not.20 We propose that differences in the eag and HERG binding sites contribute to the differential regulation of opening and closing kinetics in these channels.
High resolution structural information is not available for eag or HERG. However, the structures of voltage-gated K+ channels are likely to be highly conserved. To compare the eag and HERG binding sites, we generated tentative models based on the X-ray structure of a chimeric K+ channel in which the voltage sensor paddle of Kv2.1 has been inserted into Kv1.2 (Fig. 7).42 Although the sequences of HERG and eag diverge from that of the chimeric channel, S2 and S3 can be aligned unambiguously due to the presence of highly conserved residues (Fig. 3A).
Figure 7.

Speculative models for the ion-binding sites in eag and HERG. Models for (A) eag and (B) HERG were based on the X-ray structure of a chimeric K+ channel in an activated conformation.42 S1 (blue, corresponding to residues P161-E183 in the chimeric channel), S2 (pale green, residues D220-C244), S3 (pink, residues N253-S275) and S4 (cyan, residues V288-S307) are shown in ribbon form. The side chains of I230 in S2 and Y267 in S3 have been changed to aspartate. Additionally, E226 has been changed to aspartate in the HERG model. A Mg2+ ion (green sphere) is shown equidistant from the γ carbons of the acidic residues that contribute to each binding site. Accession number for the structure of the chimeric channel in the protein data base is 2R9R. Models were generated using Win-coot v.0.3.3 and PyMOL.
To model the binding sites, residues I230 in S2 and Y267 in S3 in the chimeric channel sequence were changed to aspartate residues (Figs. 3A and 7A).42 For the HERG model, the conserved S2 residue E226 was also changed to an aspartate residue (Figs. 3A and 7B). Using numbers corresponding to the chimeric sequence, the eag binding site comprises D230 and D267, whereas the HERG binding site comprises D226, D230 and D267 (Fig. 7A and B). For simplicity, we assumed that Mg2+ binds equidistant from the γ-carbon atoms of the aspartate residues that contribute to the binding site.
In the eag model, Mg2+ is located 3.8 Å from the γ-carbons of D230 and D267 in excellent agreement with equivalent distances in divalent cation binding sites of known structure (Fig. 7A).43–46 Mg2+ binds closer to the extracellular side of the membrane in HERG than in eag due to the participation of D226 in the binding site (Fig. 7B). In the HERG model, the distance between a centrally-located Mg2+ ion and the γ carbons of D226, D230 and D267 is 8.2 Å, placing the carbonyl oxygens too far away for optimal coordination. This suggests that the structures of HERG and the chimeric channel differ in the vicinity of the binding site or that ion binding in HERG draws the coordinating residues closer together.
Previous work suggests a mechanistic explanation for the differential modulation of gating in HERG and eag. In K+ channels, the first four arginine residues in S4, called R1-R4, carry the bulk of the gating charge across the transmembrane electric field during voltage-dependent gating.47–49 Based on data obtained with Shaker and eag channels, we proposed a model for voltage sensor conformational changes that occur late during the activation process.30,50 In this model, R3 and R4 move sequentially into the extracellular cleft where they interact with the widely conserved acidic residue in S2 (Fig. 3A).30,50 (This residue, corresponding to D456 in HERG, D274 in eag and E226 in the chimeric channel, will be referred to as D/E1 to simplify the following discussion.) In our model, D/E1 in S2 and R4 in S4 interact at short range in the activated confirmation. 30,50 This electrostatic interaction has since been confirmed by the X-ray structures of Kv1.2 and the chimeric channel in the activated state.42,51,52 Significantly, the D/E1-R4 interaction is stable in molecular dynamics simulations using the Kv1.2 structure embedded in a lipid bilayer.53
Ion binding in eag slows the rate determining step in activation, during which R3 moves into proximity with D/E1 to generate an intermediate closed state.22,30,35 In eag, D/E1 does not contribute to the binding site, indicating that bound ions are located below the position of D/E1 (see Fig. 7).20,30 We propose that bound ions reduce the negative electrostatic potential near the bottom of the cleft, slowing the rate determining transition for activation in which R3 enters the cleft to move towards D/E1.
In contrast, ion binding in HERG accelerates deactivation. Channel closing involves S4 movement, consistent with the observation that ionic tail currents and off gating currents have very similar kinetics in a variety of voltage-gated channels, including Shaker and eag.54,55 As the channel closes, S4 moves inward, disrupting the electrostatic interaction between D/E1 and R4. Because D/E1 contributes to the binding site, the electrostatic interaction between R4 and D/E1 would be weakened in the presence of a divalent ion, destabilizing the activated conformation and accelerating deactivation.
In conclusion, we propose that the location of the bound ion in the cleft plays a major role in determining whether divalent cations affect primarily the rate-determining step for opening, as in eag, or the closing transition that blocks ion conduction through the pore, as in HERG. This difference between the channels is intriguing given the essential role of HERG deactivation in heart function.
Acknowledgments
We are grateful to Allan Mock for excellent technical assistance. We thank Drs. Jeff Abramson and Vincent Chaptal for assistance with Figure 7, and Drs. William Silverman, Muriel Lainé, John Bannister and members of the Papazian laboratory for helpful discussions. This work was supported by grants from the NIH (GM43459) and the Laubisch Fund for Cardiovascular Research at UCLA to Diane M. Papazian.
ABBREVIATIONS
- eag
ether-à-go-go
- HERG
human ether-à-go-go related gene
- LQTS
long QT syndrome
- MiRP1
MinK-related protein 1
- PCR
polymerase chain reaction
References
- 1.Abbott GW, Sesti F, Splawsi I, Buck ME, Lehmann MH, Timothy KW, Keating MT, Goldstein SAN. MiRP1 forms IKr with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–87. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- 2.Jones EM, Roti Roti EC, Wang J, Delfosse SA, Robertson GA. Cardiac Ikr channels minimally comprise hERG 1a and 1b subunits. J Biol Chem. 2004;279:44690–4. doi: 10.1074/jbc.M408344200. [DOI] [PubMed] [Google Scholar]
- 3.Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–9. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- 4.Schönherr R, Heinemann SH. Molecular determinants for activation and inactivation of HERG, a human inward rectifier potassium channel. J Physiol. 1996;493:635–42. doi: 10.1113/jphysiol.1996.sp021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith PL, Baukrowitz T, Yellen G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature. 1996;379:833–6. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- 6.Spector PS, Curran ME, Zou A, Keating MT, Sanguinetti MC. Fast inactivation causes rectification of the IKr channel. J Gen Physiol. 1996;107:611–9. doi: 10.1085/jgp.107.5.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang S, Morales MJ, Liu S, Strauss HC, Rasmusson RL. Time, voltage and ionic concentration dependence of rectification of h-erg expressed in Xenopus oocytes. FEBS Lett. 1996;389:167–73. doi: 10.1016/0014-5793(96)00570-4. [DOI] [PubMed] [Google Scholar]
- 8.London B, Trudeau MC, Newton KP, Beyer AK, Copeland NG, Gilbert DJ, Jenkins NA, Satler CA, Robertson GA. Two isoforms of the mouse ether-à-go-go-related gene coassemble to form channels with properties similar to the rapidly activating component of the cardiac delayed rectifier K+ current. Circ Res. 1997;81:870–8. doi: 10.1161/01.res.81.5.870. [DOI] [PubMed] [Google Scholar]
- 9.Curran ME, Splawski I, Timothy KW, Vincent GW, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- 10.Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- 11.Dumaine R, Antzelevitch C. Molecular mechanisms underlying the long QT syndrome. Curr Opin Cardiol. 2002;17:36–42. doi: 10.1097/00001573-200201000-00006. [DOI] [PubMed] [Google Scholar]
- 12.Walker BD, Krahn AD, Klein GJ, Skanes AC, Wang J, Hegele RA, Yee R. Congenital and acquired long QT syndromes. Can J Cardiol. 2003;19:76–87. [PubMed] [Google Scholar]
- 13.Sanguinetti MC, Mitcheson JS. Predicting drug-hERG channel interactions that cause acquired long QT syndrome. Trends Pharmacol Sci. 2005;26:119–24. doi: 10.1016/j.tips.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Sesti F, Abbott GW, Wei J, Murray KT, Saksena S, Schwartz PJ, Priori SG, Roden DM, George AG, Jr, Goldstein SAN. A common polymorphism associated with antibiotic-induced cardiac arrhythmia. Proc Natl Acad Sci USA. 2000;97:10613–8. doi: 10.1073/pnas.180223197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paavonen KJ, Chapman H, Laitinen PJ, Fodstad H, Piippo K, Swan H, Toivenon L, Viitasalo M, Kontula K, Pasternak M. Functional characterization of the common amino acid 897 polymorphism of the cardiac potassium channel KCNH2 (HERG) Cardiovasc Res. 2003;59:603–11. doi: 10.1016/s0008-6363(03)00458-9. [DOI] [PubMed] [Google Scholar]
- 16.Johnson JP, Mullins FM, Bennett PB. Human ether-à-go-go-related gene K+ channel gating probed with extracellular Ca2+: Evidence for two distinct voltage sensors. J Gen Physiol. 1999;113:565–80. doi: 10.1085/jgp.113.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Po SS, Wang DW, Yang IC, Johnson JP, Jr, Nie L, Bennett PB. Modulation of HERG potassium channels by extracellular magnesium and quinidine. J Cardiovasc Pharmacol. 1999;33:181–5. doi: 10.1097/00005344-199902000-00002. [DOI] [PubMed] [Google Scholar]
- 18.Johnson JP, Balser JR, Bennett PB. A novel extracellular calcium sensing mechanism in voltage-gated potassium ion channels. J Neurosci. 2001;21:4143–53. doi: 10.1523/JNEUROSCI.21-12-04143.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu J, Zhang M, Jiang M, Tseng G-N. Negative charges in the transmembrane domains of HERG K channel are involved in the activation- and deactivation-gating processes. J Gen Physiol. 2003;121:599–614. doi: 10.1085/jgp.200308788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silverman WR, Tang C-Y, Mock AF, Huh K-B, Papazian DM. Mg2+ modulates voltage-dependent activation in ether-à-go-go potassium channels by binding between transmembrane segments S2 and S3. J Gen Physiol. 2000;116:663–77. doi: 10.1085/jgp.116.5.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang C-Y, Bezanilla F, Papazian DM. Extracellular Mg2+ modulates slow gating transitions and the opening of Drosophila ether-à-go-go potassium channels. J Gen Physiol. 2000;115:319–37. doi: 10.1085/jgp.115.3.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bannister JPA, Tang C-Y, Chanda B, Bezanilla F, Papazian DM. Evidence for a second open state in eag channels that may correspond to an inactivated but conducting conformation. Biophys J. 2006;90:116–3. [Google Scholar]
- 23.Landt O, Grunert HP, Hahn U. A general method for rapid site-directed mutagenesis using the polymerase chain reaction. Gene. 1990;96:125–8. doi: 10.1016/0378-1119(90)90351-q. [DOI] [PubMed] [Google Scholar]
- 24.Timpe LC, Schwarz TL, Tempel BL, Papazian DM, Jan YN, Jan LY. Expression of functional potassium channels from Shaker cDNA in Xenopus oocytes. Nature. 1988;333:143–5. doi: 10.1038/331143a0. [DOI] [PubMed] [Google Scholar]
- 25.Papazian DM, Timpe LC, Jan YN, Jan LY. Alteration of voltage-dependence of Shaker potassium channel by mutations in the S4 sequence. Nature. 1991;349:305–10. doi: 10.1038/349305a0. [DOI] [PubMed] [Google Scholar]
- 26.Bezanilla F, Armstrong CM. Inactivation of the sodium channel. I. Sodium current experiments. J Gen Physiol. 1977;70:549–66. doi: 10.1085/jgp.70.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Viloria CG, Barros F, Giraldez T, Gomez-Varela D, de la Pena P. Differential effects of amino-terminal distal and proximal domains in the regulation of human erg K+ channel gating. Biophys J. 2000;79:231–46. doi: 10.1016/S0006-3495(00)76286-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Terlau H, Ludwig J, Steffan R, Pongs O, Stühmer W, Heinemann SH. Extracellular Mg2+ regulates activation of rat eag potassium channel. Pflügers Arch. 1996;432:301–12. doi: 10.1007/s004240050137. [DOI] [PubMed] [Google Scholar]
- 29.Schönherr R, Mannuzzu LM, Isacoff EY, Heinemann SH. Conformational switch between slow and fast gating modes: allosteric regulation of voltage sensor mobility in the EAG K+ channel. Neuron. 2002;35:935–49. doi: 10.1016/s0896-6273(02)00869-3. [DOI] [PubMed] [Google Scholar]
- 30.Silverman WR, Roux B, Papazian DM. Structural basis of two-stage voltage-dependent activation in K+ channels. Proc Natl Acad Sci USA. 2003;100:2935–40. doi: 10.1073/pnas.0636603100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Silverman WR, Bannister JPA, Papazian DM. Binding site in eag voltage sensor accommodates a variety of ions and is accessible in closed channel. Biophys J. 2004;87:3110–21. doi: 10.1529/biophysj.104.044602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chandy KG, Gutman GA. Voltage-gated potassium channel genes. In: North RA, editor. Ligand- and Voltage-Gated Ion Channels. Boca Raton, FL: CRC Press; 1995. pp. 1–71. [Google Scholar]
- 33.Warmke JW, Ganetzky B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc Natl Acad Sci USA. 1994;91:3438–42. doi: 10.1073/pnas.91.8.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ganetzky B, Robertson GA, Wilson GF, Trudeau MC, Titus SA. The eag family of K+ channels in Drosophila and mammals. Annu NY Acad Sci. 1999;868:356–69. doi: 10.1111/j.1749-6632.1999.tb11297.x. [DOI] [PubMed] [Google Scholar]
- 35.Bannister JPA, Chanda B, Bezanilla F, Papazian DM. Optical detection of rate-determining ion-modulated conformational changes of the ether-à-go-go K+ channel voltage sensor. Proc Natl Acad Sci USA. 2005;102:18718–23. doi: 10.1073/pnas.0505766102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanchez-Chapula JA, Sanguinetti MC. Altered gating of HERG potassium channels by cobalt and lanthanum. Pflugers Arch. 2000;440:264–74. doi: 10.1007/s004240000263. [DOI] [PubMed] [Google Scholar]
- 37.Fernandez D, Ghanta A, Kinard KI, Sanguinetti MC. Molecular mapping of a site for Cd2+-induced modification of human ether-à-go-go-related gene (hERG) channel activation. J Physiol. 2005;567:737–55. doi: 10.1113/jphysiol.2005.089094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elinder F, Arhem P. Metal ion effects on ion channel gating. Q Rev Biophys. 2003;36:373–427. doi: 10.1017/s0033583504003932. [DOI] [PubMed] [Google Scholar]
- 39.Hancox JC, Levi AJ, Witchel HJ. Time course and voltage dependence of expressed HERG current compared with native “rapid” delayed rectifier K current during the cardiac ventricular action potential. Pflugers Arch. 1998;436:843–53. doi: 10.1007/s004240050713. [DOI] [PubMed] [Google Scholar]
- 40.Tseng G-N. IKr: The hERG channel. J Mol Cell Cardiol. 2001;33:835–49. doi: 10.1006/jmcc.2000.1317. [DOI] [PubMed] [Google Scholar]
- 41.Janeira LF. Torsades de pointes and long QT syndromes. Am Fam Physician. 1995;52:1447–53. [PubMed] [Google Scholar]
- 42.Long SB, Tao X, Campbell EB, MacKinnon R. Atomic structure of a voltage-dependent K+ in a lipid membrane-like environment. Nature. 2007;450:376–82. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- 43.Andersson M, Malmendal A, Linse S, Iversson I, Forsen S, Svensson LA. Structural basis for the negative allostery between Ca2+ and Mg2+ binding in the intracellular Ca2+ receptor calbindin D9k. Protein Sci. 1997;6:1139–47. doi: 10.1002/pro.5560060602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Courcot B, Firley D, Fraisse B, Becker P, Gillet JM, Pattison P, Chernyshov D, Sghaier M, Zouhiri F, Desmaële D, d’Angelo J, Bonhomme F, Geiger S, Ghermani NE. Crystal and electronic structures of magnesium(II), copper(II) and mixed magnesium(II)-copper(II) complexes of the quinoline half of styrylquinoline-type HIV-1 integrase inhibitors. J Phys Chem B. 2007;111:6042–50. doi: 10.1021/jp0687551. [DOI] [PubMed] [Google Scholar]
- 45.Goldgur Y, Cohen GH, Fugiwara T, Yoshinaga T, Fuishita T, Sugimoto H, Endo T, Murai H, Davies DR. Structure of the HIV-1 integrase catalytic domain complexed with an inhibitor: a platform for antiviral drug design. Proc Natl Acad Sci USA. 1999;96:13040–3. doi: 10.1073/pnas.96.23.13040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Larsen TM, Benning MM, Wesenberg GE, Rayment I, Reed GH. Ligand-induced domain movement in pyruvate kinase: structure of the enzyme from rabbit muscle with Mg2+, K+ and L-phospholactate at 2.7 Å resolution. Arch Biochem Biophys. 1997;345:199–206. doi: 10.1006/abbi.1997.0257. [DOI] [PubMed] [Google Scholar]
- 47.Aggarwal SK, MacKinnon R. Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron. 1996;16:1169–77. doi: 10.1016/s0896-6273(00)80143-9. [DOI] [PubMed] [Google Scholar]
- 48.Schoppa NE, McCormack K, Tanouye MA, Sigworth FJ. The size of gating charge in wild-type and mutant Shaker potassium channels. Science. 1992;255:1712–5. doi: 10.1126/science.1553560. [DOI] [PubMed] [Google Scholar]
- 49.Seoh SA, Sigg D, Papazian DM, Bezanilla F. Voltage-sensing residues in the S2 and S4 segments of the Shaker K+ channel. Neuron. 1996;16:1159–67. doi: 10.1016/s0896-6273(00)80142-7. [DOI] [PubMed] [Google Scholar]
- 50.Tiwari-Woodruff SK, Lin MA, Schulteis CT, Papazian DM. Voltage-dependent structural interactions in the Shaker K+ channel. J Gen Physiol. 2000;115:123–38. doi: 10.1085/jgp.115.2.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Long SB, Campbell EB, MacKinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 52.Long SB, Campbell EB, MacKinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 2005;309:903–8. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- 53.Jogini V, Roux B. Dynamics of the Kv1.2 voltage-gated K+ channel in a membrane environment. Biophys J. 2007;93:3070–82. doi: 10.1529/biophysj.107.112540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bezanilla F, Perozo E, Papazian DM, Stefani E. Molecular basis of gating charge immobilization in Shaker potassium channels. Science. 1991;254:679–83. doi: 10.1126/science.1948047. [DOI] [PubMed] [Google Scholar]
- 55.Tang CY, Bezanilla F, Papazian DM. Extracellular Mg2+ modulates slow gating transitions and the opening of Drosophila ether-à-go-go potassium channels. J Gen Physiol. 2000;115:319–38. doi: 10.1085/jgp.115.3.319. [DOI] [PMC free article] [PubMed] [Google Scholar]