

Abstract
Class I phosphoinositide 3-kinases (PI3Ks) are lipid kinases that regulate cell growth. One of these kinases, PI3Kα, is frequently mutated in diverse tumor types. The recently determined structure of PI3Kα reveals features that distinguish this enzyme from related lipid kinases. In addition, wild-type PI3Kγ differs from PI3Kα by a substitution identical to a PI3Kα oncogenic mutant (H1047R), explaining differences in the enzymatic activities of the normal α and γ enzymes. Comparison of the PI3K structures also identified structural features that could potentially be exploited for the design of isoform-specific inhibitors.
INTRODUCTION
Phosphatidylinositol 3,4,5-trisphosphate (PIP3) is a minor component of cellular membranes that can initiate signaling events that control the growth of normal as well as neoplastic cells. For example, PIP3 acts as a docking site for pleckstrin homology domain (PH)-containing proteins such as the AKT serine/threonine kinases (also known as protein kinase B, PKB) and for the 3-phosphoinositide-dependent protein kinase-1 (PDK1)1. Once associated with the membrane, AKTs are activated by phosphorylation at two sites and in turn phosphorylate numerous protein targets, including mTOR2, Tuberin3, GSK3β4, BAD5,6, MDM27,8, p21(WAF1/CIP1) 9,10, caspase 911, IKK12, and a subset of forkhead transcription factors13–15. The biological consequences of AKT activation are broad, and include regulation of cell proliferation, survival, and motility. The class I PI3Ks (PI3Kα, PI3Kβ, PI3Kδ and PI3Kγ ) are lipid kinases that phosphorylate phosphatidylinositol 4,5-bisphosphate (PI 4,5-P2) at the 3-position of the inositol ring, generating phosphatidylinositol 3,4,5-trisphosphate (PIP3) 1,16–18.
In addition to their importance in general signal transduction, PI3Ks play an important role in disease, particularly in cancers19–22. Recently, it has been shown that PIK3CA (which encodes p110α, the catalytic subunit of PI3Kα) was somatically mutated in diverse cancers, including those of the colon, rectum, breast, brain, liver and ovary 23–31. Deletion and truncation mutations in the regulatory subunit p85α of PI3K have also been found in human tumors, though less frequently than those in the catalytic subunit 32.
Because mutations in the catalytic domain (p110α) of PI3Kα that constitutively activate its kinase activity are common in cancers, many groups have targeted this enzyme for drug development 33. Most of the compounds characterized, however, are not specific and inhibit other PI3Ks as well as other kinases. Availability of the structure of the various PI3K enzymes could facilitate the development of more specific inhibitors. The structures of wild boar and human p110γ (PI3Kγ) are known 34–36, as is the structure of a complex between the N-terminal domain of p110α (residues 1 to 108) and the human p85α inter (i)SH2 domain (residues 431 to 600) 37. The structure of a complex between the full-length human p110α catalytic subunit and the domains of the p85α regulatory subunit critical for its binding have recently been reported38. The PI3Kα and PI3Kγ structures, coupled with information about the sequence and biochemistry of the PI3K enzymes, provide a wealth of information about conserved features as well as distinctive characteristics between members of this important family of enzymes.
The class I PI3K gene family
Two subclasses of class I PI3K enzymes have been described. Class IA enzymes are composed of catalytic subunits whose enzymatic activities are completely dependent on their binding to regulatory subunits. Human cells contain three genes (PIK3CA, PIK3CB, and PIK3CD) that encode the catalytic subunits of class IA PI3K enzymes (termed PI3Kα, PI3Kβ, and PI3Kδ, respectively). The major polypeptides produced by these three genes are p110α, p110β, and p110δ, respectively, collectively termed p110. p110α and p110β are expressed in most tissues, while p110δ is expressed primarily in leukocytes and in a small number of other cell types. The regulatory subunits of class IA enzymes are collectively referred to as p85 and are encoded by three genes in humans (PIK3R1, PIK3R2, and PIK3R3). PIK3R1 encodes p85α, p55α, and p50α as a result of alternative splicing, while PIK3R2 encodes only p85β and PIK3R3 encodes only p55γ. The p85α and p85β polypeptides are expressed in most cells, while the other isoforms are expressed in a more limited manner.
The p110 subunits of class IA PI3Ks have five domains: an N-terminal domain called ABD (adaptor binding domain) that binds to the regulatory p85 family members, a Ras-binding domain (RBD), a C2 domain that has been proposed to bind to cellular membranes, a helical domain of unknown function, and a kinase catalytic domain. The p85 polypeptides also have five known domains: an N-terminal SH3 domain, a Rho-GAP domain, and two SH2 domains (the more N-terminal nSH2 and the C-terminal cSH2) separated by an iSH2 domain that is responsible for binding to the catalytic subunit. In the basal state, the p85 regulatory subunits bind to and inhibit the p110 catalytic subunits. Upon appropriate cellular stimuli, the nSH2 and cSH2 domains bind phosphorylated tyrosines in activated receptors and adaptor proteins, and this phosphotyrosine binding activates the p110 catalytic subunits. It is important to note that the phosphotyrosine binding does not release p85 from p110: the heterodimeric state persists after phosphopeptide binding.
The class IB PI3K consists of only one enzyme, PI3Kγ. It does not contain an N-terminal p85-binding motif and does not need to interact with a regulatory subunit in order to be enzymatically active. Instead, it appears to be activated by G protein-coupled receptors and regulated by heterotrimeric G proteins. The catalytic subunit of PI3Kγ, p110γ, is encoded by PIK3CG and is expressed in leukocytes and in a small number of other tissues. The p110γ polypeptide interacts with adaptor subunits called p101 and p84/87 39–41 that may help tether p110γ to the membrane and facilitate its interaction with G proteins. The p110γ polypeptide can also bind Ras in a fashion similar to the class IA catalytic subunits.
Structure of Class I PI3Ks
Mutation Sites in the ABD of p110α Make Different Contacts Than Other Class IA PI3Ks
While the sequences of p110β and p110δ are 55% identical, they are only 40% identical to p110α. In p110α/niSH238, the ABD and kinase domains interact with each other through close contacts between Arg38 and Arg88 from the ABD and Gln738, Asp743, and Asp746 from the kinase domain38. Based on this observation, the mutations at Arg38 and Arg88 observed in cancers were proposed to alter the catalytic activity of PI3Kα by affecting this ABD-kinase domain interaction38. Is this ABD-kinase domain interaction a common feature of all class IA PI3Ks? Surprisingly, although Arg38 and Arg88 are conserved in all the ABD of class IA PI3Ks, the residues of the kinase domain with which they interact are not: Gln738, Asp743, and Asp746 of p110α are replaced by Cys, Ala, and Glu in p110β and p110δ (Supplementary information S1 (figure)). These replacements highlight a critical difference among the intramolecular regulatory motifs in PI3Ks that distinguishes PI3Kα, the isoform that is frequently mutated in cancers, from the other class IA PI3Ks. It explains why mutations at these positions are more frequent in PI3Kα than in the other isoforms: analogous mutations in the other PI3K isoforms would likely have no effect on the kinase domain.
Comparison between the Structures of the Class IA PI3Kα and the Class IB PI3Kγ
RBD (Ras-binding domain)
Residues 255-267 within the RBD domain of p110γ are not ordered in the structure of the free enzyme but are ordered in the structure of the complex between p110γ and Ras 34. On the basis of this observation it was proposed that Ras binding results in ordering of this mobile loop 34. However, the corresponding region (residues 227–247) of p110α is ordered in the absence of Ras and is in a conformation different from that in the p110γ–Ras structure (Fig. 1A, 1B). In fact, part of this loop is locked in the ATP-binding site of the kinase domain of a neighboring molecule in the p110α niSH2 crystal (Fig. 1C) 38. This interaction may be a crystallization artifact but could represent a true physiological interaction. We hypothesize that in vitro such an interaction could lead to intermolecular inhibition of p110α: the RBD loop of one molecule competes with ATP binding to another molecule by occupying the binding site. Ras could then activate p110α by releasing the RBD loop from the ATP binding site, providing a mechanism of Ras activation that is different from the one operational in p110γ. Aruging against this hypothesis is the fact that gel filtration chromatography of the purified p110α/niSH2 complex carried out at a protein concentration of 0.5 mg/ml shows a single symmetrical peak corresponding to a molecular weight of 150 kD, consistent with a soluble monomer. This may indicate that this is a low affinity interaction that becomes relevant only when PI3Kα is associated with membrane.
Fig. 1.
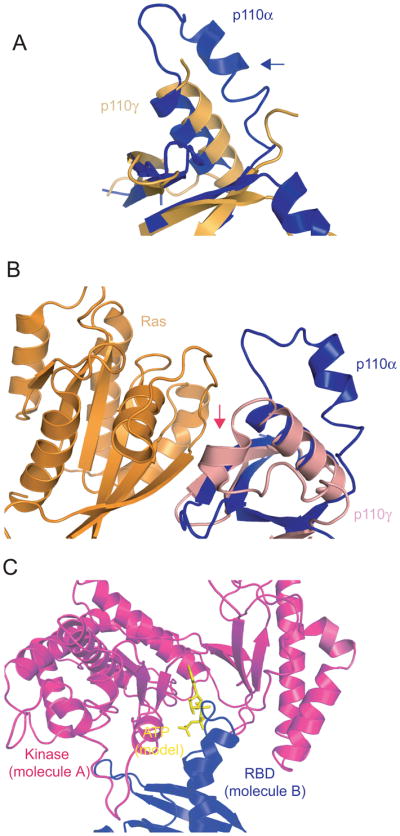
Comparison of the RBDs (Ras-binding domains) of p110α and p110γ
A. Comparison of the RBDs of p110α (blue) and free p110γ (gold). The blue arrow shows the ordering of residues 227–247 in p110α.
B. Comparison of the RBDs of p110α and Ras/p110γ complex. The red arrow indicates the position of residues 255–267 in p110γ that become ordered by the binding of Ras.
C. Interaction between p110α molecules in the crystal structure. The RBD (blue, molecule B) is locked in the ATP binding pocket of the kinase domain from a neighboring molecule (magenta, molecule A)
If the interaction between the RBD and the kinase domain observed in the crystal is physiological, how is the formation of the infinite chain of molecules found in the crystal prevented? A possible solution to this conundrum is provided by the observation that parts of p85α, including cSH2, are omitted in our construct. Were these parts present, they could clash with a third molecule, allowing only two molecules to interact with each other and precluding concatamerization.
Based on the comparison of the structures of the free p110γ and Ras/p110γ complex, Pacold et al. identified an allosteric effect induced by the binding of Ras to p110γ34. In particular, they suggested that the C2 and kinase domains “spread apart” in the Ras-bound structure. In the p110α/niSH2 structure, the C2 domain appears to be even farther apart from the kinase domain than in the Ras-bound p110γ, suggesting that p85 binding to p110α has an effect similar to, or even greater than, that induced by the Ras binding to p110γ, but in p110α this effect does not enhance the catalytic activity 42.
C2 domain
The interaction of p110α with its regulatory subunit p85α involves close contacts between the C2 domain of p110α and the iSH2 domain of p85α. One of the C2 domain residues responsible for this interaction, Asn345, which is mutated to lysine in some cancers, is located on a loop (CBR1, residues 342–355). There is no sequence similarity in the CBR1 loops of p110α and p110γ, even though the region preceding the loop contains the conserved sequence R(I/V)KI (Fig. 2). It is not known whether the C2 domain of the class IB p110γ also contacts its regulatory subunit p101, but substantial differences exist between the conformations of the CBR1 loops of p110α and p110γ: when the structures of p110γ and p110α/niSH2 are aligned, the Cα carbons of some residues in the CBR1 loop of p110γ (residues 370–379) and that of p110α are >7 Å apart. In fact, the conformation of the CBR1 loop of p110γ is incompatible with binding to p85 because it would clash with the iSH2 (Fig. 2). This explains why the catalytic subunits p110γ uses an entirely different regulatory protein (p101). Conversely, we predict that the C2 domains of the other class IA catalytic subunits, p110β and p110δ, will be found to interact with the iSH2 domains of p85 using the same contacts as p110α (See Box 1).
Fig. 2.

Comparison between the C2 domains of p110α and p110γ
The C2 domains from p110α (cyan) and p110γ (pink) are aligned. The incompatibility between CBR1 of p110γ and iSH2 binding is indicated by red arrow (see text)
Box 1. A New Structural Feature that is Common to all Class IA PI3Ks.
In p110α/niSH2, Asn345 of C2 is within hydrogen bonding distance of Asn564 and Asp560 of the iSH2 coiled-coil of the p85 regulatory subunit. Although the overall sequence identity among the C2 domains of the class IA PI3Ks is relatively low (~36%), Asn345 in p110α is conserved in p110β (Asn344) and p110δ (Asn334) (Supplementary information S1 (Figure)). In addition, Asn564 and Asp560 in p85α are conserved in the regulatory subunits of class IA PI3Ks (p85β and p55γ). Thus, this hydrogen bond appears to mediate a conserved interaction between the catalytic and the regulatory subunits in all class IA PI3Ks.
Helical domain
Two mutations, E542K and E545K, in the helical domain of p110α occur with high frequency in cancers,. While E545 is conserved in all class IA PI3Ks, the corresponding residue is Ala in p110γ (Supplementary information S1 (Figure)). Both E542 and E545 occur at the interface between the helical domain of p110α and the nSH2 domain of p85α 37,38. Furthermore, this contact is in a region of nSH2 that also makes contact with the kinase domain of p110α. These mutations were therefore hypothesized to alter the contact between the helical domain and nSH2 in such a way that the presence of nSH2 no longer inhibits the kinase activity. This interpretation is supported by the recent experiments of Carson et al. 43. They showed that helical domain mutations increase the activity of PI3Kα by a factor of 2–4 but that the activity is not further increased by tyrosine phosphorylated peptides that normally activate the wild-type enzyme.
Kinase domain (residues 697–1068 of p110α and 726–1102 of p110γ)
The kinase domains of p110α and p110γ represent the most conserved residues in the two proteins. It is therefore striking that the positions of two equivalent helices in these domains, helix α K12 in p110α (residues 1032–1048) and helix kα11 in p110γ (residues 1064–1078), constitute one of the most divergent features of the two structures (root mean square distance [r.m.s.d.] 3.2 Å; Fig. 3A). Furthermore, the residues following the αK12 helix are disordered in the p110α/niSH2 structure, while the equivalent residues (1081–1090) form a short helix at the end of the p110γ structure (Fig. 3A). These differences may be of mechanistic importance. The αK12 helix is spatially close to the activation loop (residues 933–957) of p110α, which determines the substrate specificity 44,45 and possibly the activation status of PI3Ks. In addition, the nSH2 domain of p85α, which was shown to inhibit the activity of PI3Kα 42, was tentatively placed in a region close to αK12 of p110α on the basis of weak electron density. We hypothesize that the position of αK12 in p110α, which is influenced by the nSH2 domain and possibly by other factors, regulates enzyme activity through its effect on the activation loop.
Fig. 3.

Comparison between the Kinase domains of p110α and p110γ
A. The two equivalent helices, αK12 of p110α and kα11 of p110γ, are shown in magenta and orange, respectively. The positions of the helices in each of the structures are shown by the arrows.
B. The interaction between His1047 and Leu956 in p110α is shown.
C. The interaction between Arg1076 and Lys1000 in p110γ is shown. Superposition of the p110α shows the different position of αK12 helix compared with kα11 helix of p110γ.
D. Conformations of the activation and catalytic loops of p110α and p110γ.
The His1047Arg oncogenic mutant of p110α
His1047Arg, in helix αK12 of p110α, is one of the two most frequently observed oncogenic mutations in p110α. Interestingly, the residue corresponding to 1047 of p110α is Arg1076 in p110γ. In the p110α/niSH2 complex, His1047 is within hydrogen bonding distance of the main-chain carbonyl of Leu956 (Fig. 3B), which corresponds to Leu987 of p110γ and is therefore conserved between α and γ. In the p110γ structure, however, no interaction between Arg1076 and Leu987 is observed. Instead, Arg1076 is within hydrogen bonding distance of the main-chain carbonyl of Lys1000 (Fig. 3C). This hydrogen bonding shift results in the movement of helix kα11 away from the activation loop in p110γ The change places the C-terminal end of the activation loop in p110γ in a conformation that is more open than that in p110α (Fig. 3D). The oncogenic His1047Arg mutation in p110α could lead to the formation of a new hydrogen bond involving the Arg residue, resulting in the movement of αK12 to a position similar to that of kα11 in p110γ and allowing easier access of substrates to the catalytic site. It is known that nSH2 inhibits the activity of p110α42 and that the His1047Arg mutation increases p110α activity 29,46,47. These observations suggest that the positions of αK12 and the activation loop observed in the p110α/niSH2 structure correspond to those of the inhibited state and that the positions of kα11 and the activation loop in the p110γ structure correspond to the activated state. Accordingly, p110γ can be regarded as a naturally occurring His1047Arg mutant.
Insights into Inhibitor Selectivity
As noted above, the ATP-binding pocket in the p110α/niSH2 crystal is occupied by a loop of the RBD from a neighboring molecule. As most PI3K inhibitors interact with the ATP-binding pocket, it is not possible to obtain inhibitor-bound crystals in the same crystal form. Nevertheless, structures of several inhibitors bound to p110γ have been reported 35,48, and comparisons between the active site conformations of p110α/niSH2 and inhibitor-bound p110γ can provide insights into the basis for selectivity.
The residues that line the ATP-binding pockets of p110α and p110γ are highly conserved and have similar three-dimensional structures, suggesting that inhibitors bind to these two PI3K isoforms in a similar manner (Supplementary information S2 (Figure A)). For example, wortmannin forms a covalent bond with Lys833 and makes hydrogen bonds with Asp964, Ile963, Val882, and Ser806 in p110γ 35. All five of these residues are conserved in p110α and show little deviation of Cα carbons (0.6~1.8Å) when the two structures are aligned (Supplementary information S2 (Figure A). Only slight movements of the active site loops would be required for p110α to bind wortmannin in a mode identical to that of p110γ. However, the loop between residues 771 and 779 (IMSSAKRPL) in p110α adopts a different conformation than the corresponding loop in p110γ (VMASKKKPL, residues 803–811). This difference in loop conformations is not the result of a change of p110γ induced by inhibitor binding, as the free p110γ 35 adopts a loop conformation similar to the inhibitor-bound p110γ rather than that of p110α/niSH2 (Supplementary information S2 (Figure C)).
One inhibitor that shows selectivity for the various PI3K enzymes is the quinazolinone purine PIK-39 48. The structure of PIK-39 bound to p110γ, determined by x-ray diffraction 48, shows that the quinazolinone moiety of PIK-39 extends perpendicular to the plane in which the aromatic moieties of most PI3K inhibitors reside, causing a conformational change of the side chain of Met804. Modeling of PIK-39 in the binding site of p110α places the Cα and Cβ of Met772 in p110α (Met772 of p110α corresponds to Met804 in p110γ) at only 2.5 and 1.1 Å away from the tip of the quinazolinone of PIK-39. This proximity is the result of the different conformation of the loop containing residues 771 to 779 in p110α noted above. It would therefore be difficult for p110α to accommodate PIK-39, even with a conformational change of the Met772 side chain (Supplementary information S2 (Figure D)). This observation suggests that the different conformations of the loops containing residues 771 to 779 could be exploited for the design of isoform-specific inhibitors. Indeed, the conformation of Met804 in PI3Kγ has been proposed as a possible target of isoform-specific inhibitors48.
Summary and Conclusions
The availability of the structure of the p110α/niSH2 complex allows a detailed comparison of class IA PI3Ks as well as comparisons between class IA and class IB PI3Ks, revealing features that distinguish the two subclasses of enzymes and providing mechanistic insights into the regulation of class I PI3K activities. Importantly, a critical helix in the kinase domain appears to regulate the catalytic activity in both physiological and pathological conditions.
Because PI3Kα is an enzyme that is frequently activated by mutations in cancers, it is regarded as a promising target for anticancer therapeutics. One current challenge is the development of isoform-specific inhibitors that would presumably minimize adverse effects. Despite the high degree of conservation of the enzyme active site, the structures of p110α/niSH2 and p110γ show significant differences in the conformation of a loop that explains the selectivity of at least one class of inhibitor. This structural feature may provide a basis for the design and optimization of isoform-specific PI3K inhibitors in the future.
Supplementary Material
References
- 1.Vanhaesebroeck B, Alessi DR. The PI3K-PDK1 connection: more than just a road to PKB. Biochem J. 2000;346(Pt 3):561–76. [PMC free article] [PubMed] [Google Scholar]
- 2.Nave BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J. 1999;344(Pt 2):427–31. [PMC free article] [PubMed] [Google Scholar]
- 3.Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151–62. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- 4.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–9. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 5.Datta SR, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 6.del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–9. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 7.Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci U S A. 2001;98:11598–603. doi: 10.1073/pnas.181181198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou BP, et al. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol. 2001;3:973–82. doi: 10.1038/ncb1101-973. [DOI] [PubMed] [Google Scholar]
- 9.Lawlor MA, Rotwein P. Insulin-like growth factor-mediated muscle cell survival: central roles for Akt and cyclin-dependent kinase inhibitor p21. Mol Cell Biol. 2000;20:8983–95. doi: 10.1128/mcb.20.23.8983-8995.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rossig L, et al. Akt-dependent phosphorylation of p21(Cip1) regulates PCNA binding and proliferation of endothelial cells. Mol Cell Biol. 2001;21:5644–57. doi: 10.1128/MCB.21.16.5644-5657.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cardone MH, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–21. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 12.Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- 13.Brunet A, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–68. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 14.Guo S, et al. Phosphorylation of serine 256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J Biol Chem. 1999;274:17184–92. doi: 10.1074/jbc.274.24.17184. [DOI] [PubMed] [Google Scholar]
- 15.Rena G, Guo S, Cichy SC, Unterman TG, Cohen P. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J Biol Chem. 1999;274:17179–83. doi: 10.1074/jbc.274.24.17179. [DOI] [PubMed] [Google Scholar]
- 16.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–19. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 17.Katso R, et al. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001;17:615–75. doi: 10.1146/annurev.cellbio.17.1.615. [DOI] [PubMed] [Google Scholar]
- 18.Vogt PK, Bader AG, Kang S. Phosphoinositide 3-kinase: from viral oncoprotein to drug target. Virology. 2006;344:131–8. doi: 10.1016/j.virol.2005.09.027. [DOI] [PubMed] [Google Scholar]
- 19.Chang HW, et al. Transformation of chicken cells by the gene encoding the catalytic subunit of PI 3-kinase. Science. 1997;276:1848–50. doi: 10.1126/science.276.5320.1848. [DOI] [PubMed] [Google Scholar]
- 20.Li J, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–7. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 21.Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol. 2004;22:2954–63. doi: 10.1200/JCO.2004.02.141. [DOI] [PubMed] [Google Scholar]
- 22.Steck PA, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–62. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 23.Bachman KE, et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther. 2004;3:772–5. doi: 10.4161/cbt.3.8.994. [DOI] [PubMed] [Google Scholar]
- 24.Broderick DK, et al. Mutations of PIK3CA in anaplastic oligodendrogliomas, high-grade astrocytomas, and medulloblastomas. Cancer Res. 2004;64:5048–50. doi: 10.1158/0008-5472.CAN-04-1170. [DOI] [PubMed] [Google Scholar]
- 25.Campbell IG, et al. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 2004;64:7678–81. doi: 10.1158/0008-5472.CAN-04-2933. [DOI] [PubMed] [Google Scholar]
- 26.Lee JW, et al. PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene. 2005;24:1477–80. doi: 10.1038/sj.onc.1208304. [DOI] [PubMed] [Google Scholar]
- 27.Levine DA, et al. Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin Cancer Res. 2005;11:2875–8. doi: 10.1158/1078-0432.CCR-04-2142. [DOI] [PubMed] [Google Scholar]
- 28.Saal LH, et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2005;65:2554–9. doi: 10.1158/0008-5472-CAN-04-3913. [DOI] [PubMed] [Google Scholar]
- 29.Samuels Y, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 30.Vogt PK, Kang S, Elsliger MA, Gymnopoulos M. Cancer-specific mutations in phosphatidylinositol 3-kinase. Trends Biochem Sci. 2007;32:342–9. doi: 10.1016/j.tibs.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Helland A, Holm R, Kristensen GB, Borresen-Dale AL. PIK3CA mutations in advanced ovarian carcinomas. Hum Mutat. 2005;25:322. doi: 10.1002/humu.9316. [DOI] [PubMed] [Google Scholar]
- 32.Philp AJ, et al. The phosphatidylinositol 3′-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001;61:7426–9. [PubMed] [Google Scholar]
- 33.Knight ZA, Shokat KM. Chemically targeting the PI3K family. Biochem Soc Trans. 2007;35:245–9. doi: 10.1042/BST0350245. [DOI] [PubMed] [Google Scholar]
- 34.Pacold ME, et al. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell. 2000;103:931–43. doi: 10.1016/s0092-8674(00)00196-3. [DOI] [PubMed] [Google Scholar]
- 35.Walker EH, et al. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol Cell. 2000;6:909–19. doi: 10.1016/s1097-2765(05)00089-4. [DOI] [PubMed] [Google Scholar]
- 36.Walker EH, Perisic O, Ried C, Stephens L, Williams RL. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature. 1999;402:313–20. doi: 10.1038/46319. [DOI] [PubMed] [Google Scholar]
- 37.Miled N, et al. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science. 2007;317:239–42. doi: 10.1126/science.1135394. [DOI] [PubMed] [Google Scholar]
- 38.Huang CH, et al. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007;318:1744–8. doi: 10.1126/science.1150799. [DOI] [PubMed] [Google Scholar]
- 39.Stephens LR, et al. The G beta gamma sensitivity of a PI3K is dependent upon a tightly associated adaptor, p101. Cell. 1997;89:105–14. doi: 10.1016/s0092-8674(00)80187-7. [DOI] [PubMed] [Google Scholar]
- 40.Suire S, et al. p84, a new Gbetagamma-activated regulatory subunit of the type IB phosphoinositide 3-kinase p110gamma. Curr Biol. 2005;15:566–70. doi: 10.1016/j.cub.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 41.Voigt P, Brock C, Nurnberg B, Schaefer M. Assigning functional domains within the p101 regulatory subunit of phosphoinositide 3-kinase gamma. J Biol Chem. 2005;280:5121–7. doi: 10.1074/jbc.M413104200. [DOI] [PubMed] [Google Scholar]
- 42.Yu J, Wjasow C, Backer JM. Regulation of the p85/p110alpha phosphatidylinositol 3′-kinase. Distinct roles for the n-terminal and c-terminal SH2 domains. J Biol Chem. 1998;273:30199–203. doi: 10.1074/jbc.273.46.30199. [DOI] [PubMed] [Google Scholar]
- 43.Carson JD, et al. Effects of oncogenic p110alpha subunit mutations on the lipid kinase activity of phosphoinositide 3-kinase. Biochem J. 2008;409:519–24. doi: 10.1042/BJ20070681. [DOI] [PubMed] [Google Scholar]
- 44.Bondeva T, et al. Bifurcation of lipid and protein kinase signals of PI3Kgamma to the protein kinases PKB and MAPK. Science. 1998;282:293–6. doi: 10.1126/science.282.5387.293. [DOI] [PubMed] [Google Scholar]
- 45.Pirola L, et al. Activation loop sequences confer substrate specificity to phosphoinositide 3-kinase alpha (PI3Kalpha ). Functions of lipid kinase-deficient PI3Kalpha in signaling. J Biol Chem. 2001;276:21544–54. doi: 10.1074/jbc.M011330200. [DOI] [PubMed] [Google Scholar]
- 46.Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A. 2005;102:802–7. doi: 10.1073/pnas.0408864102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ikenoue T. Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res. 2005;65:4562–4567. doi: 10.1158/0008-5472.CAN-04-4114. [DOI] [PubMed] [Google Scholar]
- 48.Knight ZA, et al. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell. 2006;125:733–47. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.