

Abstract
Lynch syndrome is an inherited disease caused by a germline mutation in one of four DNA mismatch repair (MMR) genes. The clinical manifestations can be somewhat variable depending upon which gene is involved, and where the mutation occurs. Moreover, the approach to the diagnosis of Lynch syndrome is becoming more complex as more is learned about the disease, and one needs to understand how the DNA MMR proteins function, and what makes them malfunction, to have an optimal appreciation of how to interpret diagnostic studies such as microsatellite instability and immunohistochemistry of the DNA MMR proteins. Finally, an understanding of the role of the DNA MMR system in regulation of the cell cycle and the response to DNA damage helps illuminate the differences in natural history and response to chemotherapeutic agents seen in Lynch syndrome.
Keywords: Lynch syndrome, HNPCC, DNA mismatch repair, Microsatellite instability, MSH2, MLH1, MSH6, PMS2, Colorectal cancer, Familial cancer
Introduction
Lynch syndrome is the hereditary disease caused by a germline mutation in one of four different human DNA mismatch repair (MMR) genes. This disease is inherited in an autosomal dominant fashion, and gives rise to familial clusters of cancers of the colon, endometrium, and other sites; the tumors have some characteristic pathological features and have an unusually early age of onset. With one exception (the Muir-Torre syndrome), there is no premorbid phenotype for this disease. However the tumors are best recognized by virtue of the unique mutational signature, microsatellite instability (MSI), that is present in the tumor tissue [1].
This review will focus upon the basic biology of defective DNA MMR, and provide correlation between the underlying mechanism of the disease and the clinical spectrum observed in Lynch syndrome. The defective DNA MMR activity is thought to be responsible for the unique phenotypic abnormalities seen in these tumors, including the more rapid adenoma-to-carcinoma transition, and the differential response to chemotherapy seen in this disease. Also, depending upon which of the DNA MMR genes is mutated, a slightly different clinical spectrum of disease may occur.
The DNA MMR system
The DNA MMR system consists of several nuclear proteins that act in concert to detect and repair errors that occur during the S phase of DNA replication. The critical elements of the DNA MMR system most relevant to Lynch syndrome are the MutS homologue (MSH) family and MutL homologue (MLH) family of proteins. As illustrated in Fig. 1, the MSH proteins always work as heterodimers. MSH2 is an obligatory partner in the system, and it may dimerize with either of two other family members: MSH6 or MSH3 [2]. The MSH2–MSH6 (MutSα) heterodimer preferentially recognizes and binds single base pair mismatches, such as the G-T mismatch illustrated in Fig. 1A, or mononucleotide repeats as shown in Fig. 1B. Through an energy-requiring process, free MutSα interacts with DNA at the site of a mismatch, exchanges ADP for ATP, and forms a sliding clamp around the DNA [3]. Alternatively, the MSH2-MSH3 pair (called MutSβ) preferentially recognizes larger “loop-out” errors, which can occur at dinucleotide repeats or other repetitive sequences.
Fig. 1.

DNA MMR (A) Repair of a single nucleotide mismatch in S phase by MutSα. (MutL components not shown here) A. The polymerase has erroneously placed a G in the daughter strand across from a non-complementary T in the template, creating a mismatch. B. The heterodimer of hMSH2 and hMSH6 (MutSα), bound by ADP and in an open configuration, monitors newly synthesized DNA strand for mispairs. Upon encountering the G-T mispair, an exchange of ATP for ADP occurs, and MutSα switches to a closed, sliding clamp along the DNA. C. The sliding clamps can migrate in either direction from the mispair, and as this occurs, additional MutSα clamps may be recruited to the mismatch. The MutSα moving in the 5′ > 3′ direction will eventually encounter the PCNA-DNA polymerase complex, and according to one hypothesis, displace the enzymes involved in DNA synthesis. D. Exonuclease I (together with MutL homologue heterodimers) excises the newly synthesized daughter strand back to the site of the mismatch, removing the erroneous G. E. The error is corrected by resynthesis (from [43], adapted with permission from [44]). (B) Recognition of insertion/deletion errors at microsatellite sequences by MutSβ. Loop-out lesions are caused by “slippage” at repetitive sequences (such as An or [CA]n) during DNA replication, and are recognized by hMSH2 + hMSH3 (MutSβ). In this illustration, the slippage has created a short “loop out” on the nascent strand for the An sequence, which would lead to an insertion frame shift mutation after replication, whereas the slippage is shown on the template strand for the (CA)n repeat, which would lead to a deletion mutation after replication (from [43], adapted with permission from [44])
MutS heterodimers signal the site of mispairing, however additional proteins are required to complete the repair process. The MutL family of proteins consists of one obligatory partner, MLH1, which may dimerize with PMS2, PMS1, or MLH3 (not shown in Fig. 1). Little is known about the MutL family beyond the function of the MLH1-PMS2 heterodimer, which is called MutLα. This heterodimer has endonuclease activity [4], interacts with the MutS-DNA complex, and together with ExoI, PCNA, and other enzymes required for DNA synthesis, the newly synthesized strand containing the error is excised, followed by resynthesis [5]. Repair of replication errors is required for the transition from S phase through G2 to mitosis [6].
Microsatellite Instability (MSI)
The absence of MSH2 or MLH1 leads to complete loss of DNA MMR activity. As a result, DNA synthetic errors accumulate at an accelerated rate as the cells proliferate. Therefore, tumor DNA from cancers with deficient DNA MMR activity has the characteristic mutational signature called MSI. As illustrated in Fig. 2, one can select a panel of five microsatellite sequences, amplify them by PCR, and compare these with the amplicons obtained at the same sequences in that individual's normal tissues [7]. In most instances, the mutations at the microsatellite sequences result in a deletion of one or more of the mononucleotide or dinucleotide elements, resulting in a shorter sequence. This can be detected when the PCR products are resolved by size using HPLC. A significant degree of MSI is called MSI-high (MSI-H), and is defined by the presence of mutations at two or more of the 5 consensus microsatellite sequences. Most CRCs have no mutated microsatellite sequences, so they are called “microsatellite stable” (MSS). Occasional tumors have only 1/5 ofthe microsatellites sequences mutated, and these are referred to as MSI-low, or MSI-L. Biologically, MSI-H cancers behave differently than those that are MSS or MSI-L, and subsequently in this review, MSI means MSI-H.
Fig. 2.

Microsatellite Instability Five mononucleotide repeat sequences (NR21, BAT26, NR24, NR27, and BAT 25) were amplified in a multiplex reaction and separated by HPLC. The upper lanes labeled “normal DNA” represent DNA from lymphocytes, and the lower lanes labeled “CRC tissue” represent DNA extracted from the cancer. In each instance shown here, the microsatellite sequence has undergone a deletion mutation, as indicated by the bold arrow. In this case, all five microsatellites were mutated, and the tumor was designated MSI-H
Target genes that mediate carcinogenesis in MSI tumors
It is thought that most of the mutations at the microsatellite sequences used to detect MSI in CRCs are not mechanistically responsible for the behavior of tumor cells. However, there are approximately 32 target genes in the human genome in which there is a mononucleotide repeat of seven or more elements (i.e., A10, G8, etc) in the coding sequence of the gene product [8]. The deletion of one of these elements leads to a frame shift, and loss of function of that gene. Longer repetitive elements are more prone to mutation than shorter ones.
The most commonly mutated gene in MSI-H tumors is the TGFβ1R2 gene, which contains an A10 sequence. This gene product is required for negative growth signaling through TGFβ-SMAD cascade. Most of the genes shown in Table 1 are either involved in the regulation of growth, the regulation of apoptosis, or are genes involved in DNA repair. Perhaps critical to the altered behavior of Lynch syndrome tumors, the BAX gene has a G8 sequence found in its coding region, and this sequence is mutated in approximately half of all tumors with MSI [9]. Curiously, each of the “minor” DNA MMR genes (i.e. MSH6, MSH3, MLH3, and PMS2) has a repetitive sequence of eight or more nucleotides in its coding sequence [10]. Furthermore, MSH2 has an encoded A7 sequence. Thus, once DNA MMR activity is reduced, other genes in the MMR family are prone to mutation at these repetitive sequences, leading to a progressive process in which the entire system can be inactivated [11]. Loss of DNA MMR activity greatly accelerates the rate of accumulation of mutations in genes responsible for restraining cell growth. This provides a reasonable hypothesis for the rapid growth of adenomas and transition to carcinoma seen in Lynch syndrome.
Table 1.
Mutational targets in CRCs with MSI [8]
Gene involved in apoptosis
Gene involved with growth control
Gene involved with DNA repair
Cell models used to study human DNA MMR
Several cell models are available to study the underlying biology of defective DNA MMR in vitro. The HCT116 colon cancer cell line has biallelic mutations in the MLH1 gene. The HEC59 and LoVo colon cell lines both have biallelic mutations of the MSH2 gene. The DLD1 colon cancer cell line has mutations of the MSH6 gene. Furthermore, cell lines have been developed from these by stable chromosome transfer, which permits one to study the biology of these cells after restoration of DNA MMR activity. The HCT116+chr3 cell line has undergone a stable transfer of chromosome of 3 into the cells, which corrects the defect of MLH1 [12]. HEC59+chr2 and LoVo+chr2 cells have both had stable transfer of chromosome 2, which corrects the MSH2 defect [13, 14]. The HEC59 cell line has mutations in MSH2 and MSH6, and both are corrected by the stable transfer of chromosome 2 into this cell line.
Most CRCs with MSI are not caused by Lynch syndrome. A more common mechanism for loss of DNA MMR activity in CRC is epigenetic inactivation of the MLH1 gene by methylation of the promoter. The cell lines SW48 and RKO both have this defect, and these cells can be rendered DNA MMR proficient by the use of demethylating agents in vitro, which leads to re-expression of MLH1 in 24–48 h [15].
The use of these in vitro models has permitted us to observe the protein–protein interactions in the DNA MMR family, and provides insight into the biological features of defective DNA MMR activity. Figure 3 is a series of PCR gels and Western blots in which the upper panel (RT-PCR) represents the reverse transcriptase-PCR analyses of gene expression, and the lower panels demonstrate protein expression in these cells [16]. The cell line SW480 is a control CRC cell line with intact DNA MMR activity. As shown in the second lane from the left, HCT116 cells have a weak MLH1 band in the upper (RT-PCR) panel because of biallelic mutations in MLH1. The lower panel demonstrates no expression of MLH1 protein, illustrating the pathological nature of the mutation. Importantly, in spite of the robust presence of mRNA for the PMS2 gene in the upper panel, there is no corresponding PMS2 protein in the lower panel. Thus, mutational inactivation of MLH1 leads to destabilization of the PMS2 protein, presumably because of rapid turnover in the absence of a stabilizing protein binding partner. As shown in the third lane, restoration of MLH1 by stable transfer chromosome 3 leads to an increase in the RT-PCR band for MLH1, and the appearance of both MLH1 and PMS2 proteins in the lower panels.
Fig. 3.
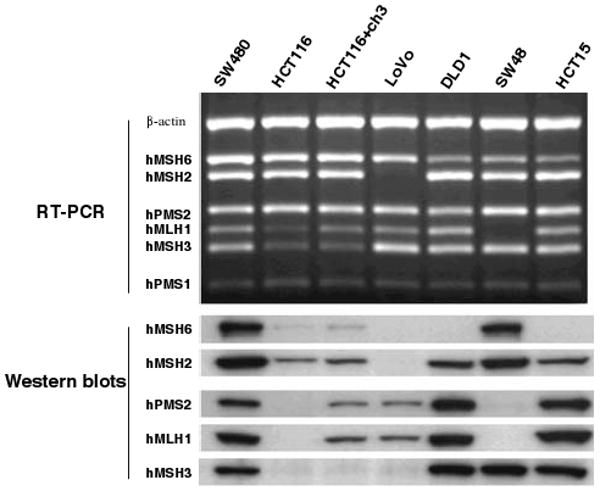
Analysis of mRNA and protein expression of the DNA MMR genes in CRC cell lines with mutations. The upper panel (labeled RT-PCR) with the dark background represents PCR gels. Each of the white bands is a PCR product. The lower panel (labeled Western blots) consists of dark protein bands. There are seven lanes which represent the results from the seven CRC cell lines, as indicated across the top. SW480 is MMR proficient, and is used as a positive control. mRNA is present for each of the MMR genes in the upper panels, and proteins are expressed as shown in each of the lower lanes. (No effective monoclonal antibody was available for PMS1, therefore there are no Western blots for this protein.) HCT116 cells are mutated and deficient for MLH1 and MSH3. HCT116+chr3 cells were derived from HCT116 by the stable transfer of chromosome 3 [12]. This cell line is MMR proficient, but is still MSH3 deficient. LoVo is an MSH2-deficient cell line. DLD1 is an MSH6-deficient cell line. SW48 is deficient in MMR activity because both alleles of MLH1 are methylated and silenced. HCT15 is MSH6-deficient. SW480 (first lane) expresses all six mRNAs for DNA MMR genes, and all five of the proteins, and serves as the normal control. HCT116 demonstrates the consequences of an inactivating mutation in MLH1. The missense mutation creates a weak RT-PCR band for MLH1 but completely removes the protein band for MLH1 and PMS2 (second lane). As an incidental consequence of MSI in this line, MSH3 has undergone a somatic mutation, which accounts for the weak protein band. Restoration of MLH1 by stable transfer of chromosome 3 in the HCT116+chr3 cell line results in a restoration of DNA MMR activity, and the appearance of protein bands for MLH1 and PMS2, as shown in the third lane. There is no change in the status of MSH3 in this cell line. LoVo has mutations that result in the total loss of the RT-PCR band for MSH2, and as a consequence, loss of the protein bands for MSH2, MSH6, and MSH3 (fourth lane). The DLD1 cell line has a mutation in the MSH6 gene. There is still an mRNA for MSH6, but the protein is abnormal, and no band is seen on the Western blot. SW48 has methylation-induced silencing of the MLH1 gene (lane 6). Consequently, there is no mRNA for this gene and no protein product for MLH1 or PMS2. Although not shown here, demethylation of the MLH1 promoter in vitro results in re-expression of MLH1, and restoration of the protein bands for MLH1 and PMS2. HCT15 was derived from DLD1, and has the same mutation in MSH6 (lane 7) [16]
The cell line LoVo has mutations in both alleles of MSH2, which leads to a complete loss of its mRNA, so there is no band in the upper RT-PCR panel. As shown in the lower panel, the loss of MSH2 protein leads to the loss of MSH2, MSH6, and MSH3 proteins, because the latter two require MSH2 for stabilization. The next lane illustrates DLD1, which has a missense mutation in the MSH6 gene. This mutation does not lead to loss of the mRNA; however there is complete loss of functional MSH6 protein. Because MSH6 is a “minor” MMR protein and plays no role in stabilizing other MutS proteins, its absence does not lead to the loss the other MutS proteins. MSH2 compensates for loss of MSH6 by binding to MSH3, as illustrated in the Western blots.
The cell line HCT15 (in the far right lane) is the parental line from which DLD1 was derived, and it shows the same molecular features as seen in DLD1. The cell line SW48 (in the 2nd lane from the right), has undergone methylation and silencing of the MLH1 gene. This leads to a total loss of the mRNA for MLH1, and a total loss of the proteins MLH1 and PMS2. Although not shown here, after demethylation of the MLH1 gene in vitro, both MLH1 and PMS2 are re-expressed.
Clinical implications of the biochemistry of human DNA MMR
Figure 3 demonstrated that MSH2 is required to stabilize both MSH6 and MSH3, but the reverse is not true. If MSH2 is absent, there is no DNA MMR activity. If either MSH6 or MSH3 is the only absent protein, some DNA MMR activity is preserved. Furthermore, whereas the absence of MSH2 will lead to the loss of all DNA MMR activity, isolated absence of MSH6 largely results in loss of single base mismatch repair. Thus, one may not see classical MSI in tumors associated with isolated MSH6 deficiency [17–20], there may not be dinucleotide repeat instability, and one could be misled by the consensus panel of microsatellite markers (which uses three dinucleotide repeats and two mononucleotide repeats) [7]. Some of these tumors occasionally have only MSI-L [20].
On the basis of the underlying biology of interactions between DNA MMR proteins, one can explain some of the variable phenotypic spectrum seen in Lynch syndrome. First, germline mutations in the two major DNA MMR genes, MSH2 and MLH1, result in the “classic” form of Lynch syndrome. In both types of families, one sees typical MSI-H and the full spectrum of tumors. Germline mutations in the MSH6 gene lead to an attenuated form of Lynch syndrome with later onset of the tumors [20]. This is presumably because MSH3 dimerizes with MSH2, which permits partial DNA MMR activity, and delays the appearance of tumors.
Missense mutations in DNA MMR genes may lead to alterations in the enzymatic activity of the DNA MMR protein, but the protein may still persist in the cell. Although not shown in Fig. 3, certain missense mutations in MLH1 or MSH6 can lead to the translation of a non-functioning protein that will be detected by immunological techniques. Consequently, one may detect these proteins using immunohistochemistry (IHC), although the expression may be weak or heterogeneous in the cancer tissue. Missense mutations are especially common in MSH6, whereas deletions of the gene or premature stop codons are more common with MSH2.
For the reasons stated above, loss of MSH3 does not lead to total loss of DNA MMR activity. In spite of the fact that this gene is subject to somatic mutation in some MSI CRCs [21], no families with germline mutations in MSH3 have ever been reported, presumably because of the strong protective effects of the residual MutSα heterodimer (MSH2 + MSH6). In a similar fashion, germline mutations in the PMS2 gene lead to an attenuated form of the disease [22–24]. This is presumably because MLH1 can also heterodimerize with MLH3 or PMS1, providing sufficient activity to compensate for the absence of the MutLα heterodimer (MLH1 + PMS2), and retard the onset of disease. Finally, although there have been individuals with mutations in MLH3 or ExoI 1 who have had colon cancers, there are no large, documented families with Lynch syndrome attributable to these genes [25].
Genotype and phenotype in lynch syndrome
In the context of the biochemical basis of DNA MMR, it becomes understandable why one expects a “classic” Lynch syndrome phenotype when the germline mutation occurs in one of the two “major” DNA mismatch repair genes—MSH2 or MLH1. When there are mutations in either of these genes, one sees high degrees of penetrance and classic family histories. The CRCs in these families almost always show MSI, and the offending gene can often be found using IHC. Some experience with DNA MMR IHC is required to avoid making errors in the interpretation.
MSH2
When there are germline mutations in MSH2, one usually sees no expression of MSH2 and MSH6 proteins at IHC of CRCs. Most of the mutations in MSH2 create premature stop codons, altered splice sites, or are large deletions or rearrangements within the gene, which completely abrogate gene expression. Deletions of exons 1–6 are particularly common, and large deletions may comprise up to one third of all of germline mutations in MSH2 [26].
MLH1
Lynch syndrome of the MLH1 type is somewhat more complicated. When MLH1 is lost by a genetic deletion or a premature stop codon, one sees loss of both MLH1 and PMS2 at IHC. Most of the CRCs show classic MSI, with rare exceptions (e.g., MLH1 D132H) [27]. However, the frequency of missense mutations is somewhat higher with MLH1 than in MSH2. Some of these will lead to a loss of the enzymatic activity of MLH1, but expression of the MLH1 and PMS2 proteins at IHC can be preserved, leading the pathologist to a falsely “negative” reading (i.e. the protein appears normal). A common scenario is to find weak or ambiguous MLH1 staining, and absent PMS2 staining; this is most likely to represent a germline missense mutation in MLH1. Since MLH1 is expressed in lower abundance than MSH2 [28], the staining is weaker and the pathologist will require some experience in the interpretation of these slides. Furthermore, DNA MMR protein expression is down-regulated in response to oxidative stress [29] and hypoxia [30], which further can confound the IHC with this protein.
The use of IHC for PMS2 may be useful in confusing cases of MLH1-type Lynch syndrome. As mentioned, if there is ambiguity in the interpretation of MLH1 expression, the loss of PMS2 is a useful confirmatory finding. Also, some mutations in MLH1 (specifically, missense mutations) may leave residual MLH1 expression at IHC, in spite of a total loss of MMR function. In these instances (which are uncommon), one sees MSI, and PMS2 expression is absent from the tumor, leading one to the proper diagnosis. Missense mutations in MLH1 can be difficult to interpret, and are more likely to be pathogenic when they occur in the interactive domains between MLH1 and PMS2, between the MutS and MutL heterodimers, or with ExoI, as shown in Fig. 4 [31]. Ultimately, one must consult an accurate data base for each sequence variation to determine which ones are pathogenic mutations and which are non-pathogenic polymorphisms. The proper interpretation may require either data from a large pedigree, or the use of in vitro functional analyses of the variant proteins [32].
Fig. 4.
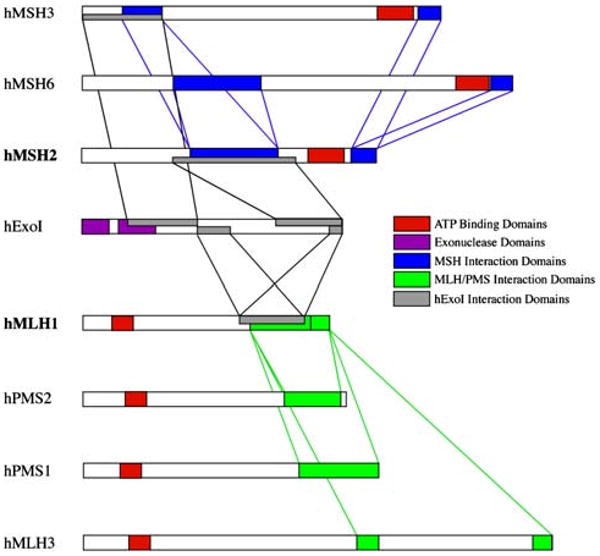
Interaction among the DNA MMR proteins. Each of the MutS homologues (MSH3, MSH6, and MSH2) interacts as a heterodimer with the MutL homologues (MLH1, PMS2, PMS1, and MLH3), acting as heterodimers, and the exonuclease, ExoI. Mutations that occur in the interactive domains may abrogate the ability of these proteins to interact and function in DNA MMR, but in some instances, the mutations may not lead to destabilization and loss of the protein product. The interactive domains among the MutS homologues, among the MutL homologues, and between one another are illustrated here. (taken from Boland and Fishel [31])
The other major complicating feature of MLH1 IHC is the fact that about 15% of all CRCs show MSI, and most of this is due to the acquired silencing of the MLH1 gene due to promoter methylation [33]. About 75–80% of the CRCs which show MSI (i.e., 10–12% of all CRCs) develop MSI due to this acquired defect, and the other 20–25% (i.e., 3–4% of all CRCs) are due to Lynch syndrome. The acquired silencing of MLH1 usually occurs in older patients, but occasionally can be found younger ones.
MSH6
Lynch syndrome of the MSH6 type produces an attenuated phenotype, because of the partial compensation provided by the MSH3 protein. The cancers in patients with germline mutations in MSH6 tend to occur later in their lives; therefore, the term “attenuation” is a reference to age of onset, but not the eventual prevalence of cancer. However, by age 70, 71% of women with MSH6-type Lynch syndrome will have developed endometrial cancer [20]. Furthermore, CRCs are as penetrant in this disease as in the classic forms, but tend to occur later in life (mean age ∼56 years) than in classic Lynch syndrome. Just as occurs with MLH1, some missense mutations in the MSH6 gene will increase the risk for cancer, but the tumors will still show staining of MSH6 protein at IHC.
PMS2
Lynch syndrome caused by germline mutations in PMS2 is the most challenging form of the disease. These mutations lead to an attenuated phenotype with weaker family histories and older ages of onset. However, there is some suggestion that germline mutations in PMS2 may be as common as those in MSH2 [23]. The tumors in PMS2-type Lynch syndrome will show MSI, and isolated loss of PMS2 staining at IHC. The diagnostic challenge is to distinguish this disease from certain missense mutations in MLH1 which lead to destabilization and isolated of the PMS2 protein at IHC [31]. The genetic diagnosis of this disease remains a great challenge because of the large number of pseudogenes which confound diagnostic DNA sequencing [34]. Unlike genetic testing for MSH2, MLH1, and MSH6, there are no commercially available tests for PMS2 at the time of this writing.
Clinical uses of MSI in CRC
Approximately 15% of all CRCs show MSI. There are three clinical uses of this information in CRC patients. The principal use of MSI testing is to find Lynch syndrome, as described above. Secondly, survival is significantly better in young patients with MSI (Fig. 5). This is not because these tumors are detected at earlier stages. Improved survival is found at all stages of disease. Importantly, patients with Stage I or II CRCs have five year survivals exceeding 90% [35]. The third clinical implication of MSI in CRC is the fact that these tumors do not have the same response to the administration of chemotherapy as do MSS (or MSI-L) tumors.
Fig. 5.
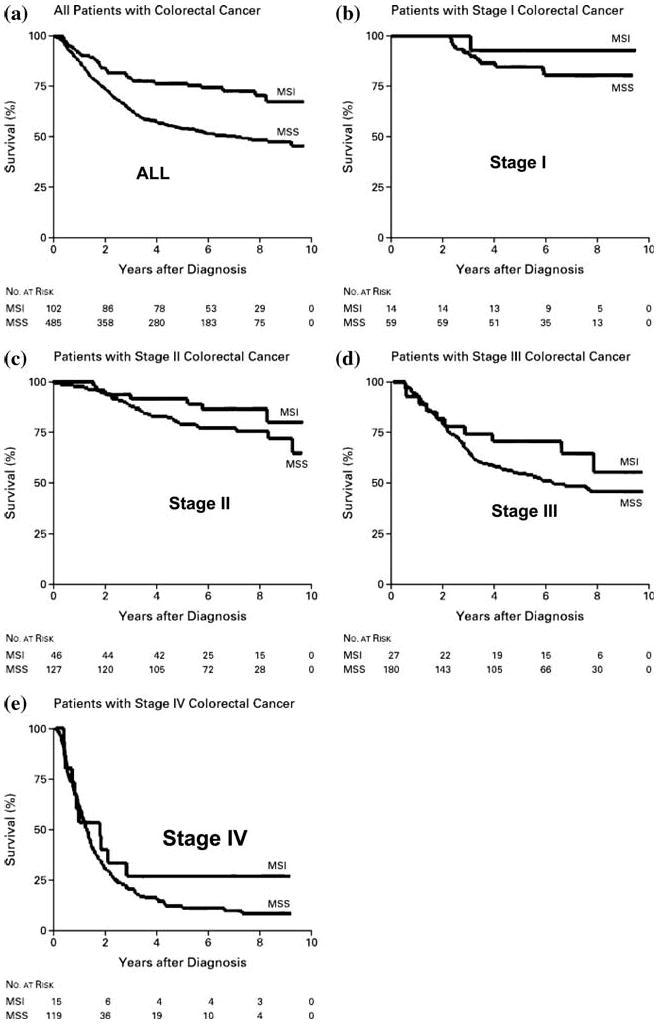
Improved survival in young patients who have CRC with MSI. The ten year survival is significantly better for patients whose CRC tumors are MSI compared with those MSS. As illustrated, this is true for tumors detected at each of the stages. In each instance, survival in the top curve is from patients under age 50 with CRC showing MSI [40]
Our group demonstrated over a decade ago that CRC cell lines with defective DNA MMR are relatively resistant to the cytotoxic effects of several drugs, including alkylating agents and 5-fluorouracil [6, 36, 37]. In fact, DNA MMR-deficient tumors are broadly resistant to chemotherapeutic agents that damage DNA [38]. More importantly, one can restore sensitivity to these DNA-damaging agents by restoring DNA MMR activity, either through stable chromosome transfer, or by re-expression of the MLH1 gene after exposure to demethylating agents [15]. As shown in Fig. 6, one sees a significant difference in cloning efficiency for DNA MMR-deficient cell lines after exposure to either the alkylating agent MNNG [6] or 5-FU [39]. This was also observed using clonagenic assays after demethylating the MLH1 gene with 5-azacytidine [15]. This led to the suggestion that patients with MSI CRCs would see less benefit from treatment with 5-FU compared against patients with MSS CRCs.
Fig. 6.
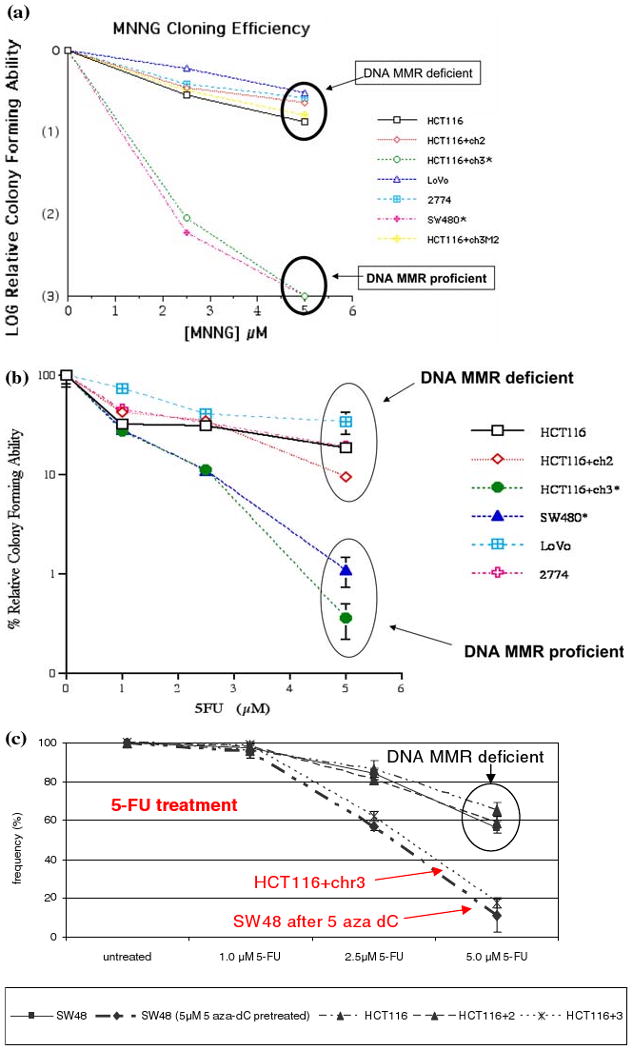
In vitro evidence for resistance to the alkylating agent MNNG by CRC cell lines with deficient DNA MMR activity. (A) The cell lines HCT116, HCT116+ch3M2, LoVo, and 2774 are all DNA MMR deficient, and have intact colony forming ability in the presence of the mutagenic alkylating agent MNNG. The CRC cell lines SW480 and HCT116+ch3 are DNA MMR proficient, and cannot form colonies after exposure to 5 μM MNNG [6]. (B) Similarly, after exposure to 5 μM 5-FU, which is a pharmacologically relevant dose of this drug, the DNA MMR deficient cell lines all tolerated treatment, where as the DNA MMR proficient cell line (HCT116+ch3 and SW480) were relatively unable to form colonies. Note that the y-axis is a log scale [39]. (C) The cell line SW48 is DNA MMR-deficient because of methylation-induced silencing of the MLH1 gene. However, after treatment with the demethylating agent 5 aza-dC, the MLH1 protein is re-expressed, and the cell in rendered MMR proficient. Using a clonagenic assay, the DNA MMR deficient cell lines (HCT116, HCT116+ch2, and SW48) are all resistant to the cytotoxic effects of 5-FU, whereas the MMR proficient cell line HCT116+chr3 and SW48 after demethylation both show sensitivity to 5-FU [15]
Clinical confirmation of this has come from several groups, but the concept has not been free of controversy. Ribic et al. demonstrated in a retrospective study of patients who had been prospectively randomized into 5-FU-containing chemotherapeutic regimens that those with MSI CRCs failed to benefit from 5-FU. Moreover, there was a 2-fold, but not statistically significant, increase in mortality in these patients (Fig. 7) [40]. Similar findings have been reported by three other groups [37, 41, 42]. Thus, it is reasonable to exercise caution regarding chemotherapy in Lynch syndrome patients with Stage III, or especially Stage II, CRCs. It is possible that drugs will found in the future that will be selectively beneficial in this disease.
Fig. 7.
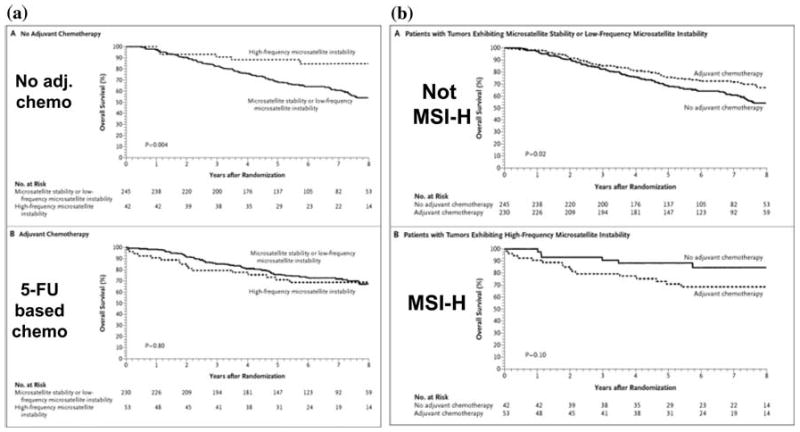
Clinical outcomes in CRC patients treated with 5-FU adjuvant chemotherapy depends upon the DNA MMR status. (A) The upper panel shows survival in patients with Stage II or Stage III CRC not given adjuvant chemotherapy. The dotted line on the top cohort (those whose tumors showed MSI) survived significantly longer than patients whose CRCs did not show MSI. In the lower panel, the use of 5-FU based chemotherapy results in an improvement in patients whose tumor are MSS, and the two survival curves converge [40]. (B) Survival was examined in patients with CRC, and analyzed based upon MSI status. The upper panel shows the impact of 5-FU based chemotherapy in patients with no MSI. The dotted line shows that patients treated with adjuvant chemotherapy had a significant improvement in survival. However, in the lower panel, patients had tumors with MSI, and the addition of 5-FU-based chemotherapy did not improve survival, and may have resulted in a slight reduction in overall survival (difference is not significant, P = 0.10). This indicates that 5-FU based chemotherapy does not provide a survival advantage in patients with MSI CRCs, and helps in the selection of patients who might benefit best from this intervention [40]
Summary
In summary, the biochemistry of the DNA MMR system has been studied in a number of CRC cell lines, which has helped confirmed the predictions initially made in E. coli and in yeast. The human DNA MMR system is more complicated than that in lower organisms, in that all of the human DNA MMR proteins require heterodimerization to function, and selective losses of individual proteins has differential impact on the MMR system. This complexity in DNA MMR results in clinical heterogeneity among different families with Lynch syndrome. The role of DNA MMR in regulating the cell cycle and signaling apoptosis in response to DNA damage is of particular clinical importance, since these patients require additional consideration when planning possible cancer chemotherapy.
Contributor Information
C. Richard Boland, Email: rickbo@baylorhealth.edu, Department of Internal Medicine and Sammons Cancer Center, Baylor University Medical Center (250 Hoblitzelle), 3500 Gaston Avenue, Dallas, TX 75246, USA.
Minoru Koi, Email: minourk@baylorhealth.edu, Department of Internal Medicine and Sammons Cancer Center, Baylor University Medical Center (250 Hoblitzelle), 3500 Gaston Avenue, Dallas, TX 75246, USA.
Dong K. Chang, Email: dkchang@skku.edu, Sungkyunkwan University School of Medicine, Samsung Medical Center, Seoul, Korea.
John M. Carethers, Email: jcarethers@ucsd.edu, Department of Medicine, University of California, San Diego, CA, USA.
References
- 1.Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348(10):919–932. doi: 10.1056/NEJMra012242. [DOI] [PubMed] [Google Scholar]
- 2.Acharya S, Wilson T, Gradia S, et al. hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc Natl Acad Sci USA. 1996;93(24):13629–13634. doi: 10.1073/pnas.93.24.13629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gradia S, Subramanian D, Wilson T, et al. hMSH2-hMSH6 forms a hydrolysis-independent sliding clamp on mismatched DNA. Mol Cell. 1999;3(2):255–261. doi: 10.1016/s1097-2765(00)80316-0. [DOI] [PubMed] [Google Scholar]
- 4.Kadyrov FA, Dzantiev L, Constantin N, Modrich P. Endonucleolytic function of MutLalpha in human mismatch repair. Cell. 2006;126(2):297–308. doi: 10.1016/j.cell.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 5.Heinen CD, Schmutte C, Fishel R. DNA repair and tumorigenesis: lessons from hereditary cancer syndromes. Cancer Biol Ther. 2002;1(5):477–485. doi: 10.4161/cbt.1.5.160. [DOI] [PubMed] [Google Scholar]
- 6.Carethers JM, Hawn MT, Chauhan DP, et al. Competency in mismatch repair prohibits clonal expansion of cancer cells treated with N-methyl-N′-nitro-N-nitrosoguanidine. J Clin Invest. 1996;98(1):199–206. doi: 10.1172/JCI118767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58(22):5248–5257. [PubMed] [Google Scholar]
- 8.Duval A, Hamelin R. Mutations at coding repeat sequences in mismatch repair-deficient human cancers: toward a new concept of target genes for instability. Cancer Res. 2002;62(9):2447–2454. [PubMed] [Google Scholar]
- 9.Rampino N, Yamamoto H, Ionov Y, et al. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science. 1997;275(5302):967–969. doi: 10.1126/science.275.5302.967. [DOI] [PubMed] [Google Scholar]
- 10.Chang DK, Metzgar D, Wills C, Boland CR. Microsatellites in the eukaryotic DNA mismatch repair genes as modulators of evolutionary mutation rate. Genome Res. 2001;11(7):1145–1146. doi: 10.1101/gr.186301. [DOI] [PubMed] [Google Scholar]
- 11.Perucho M. Microsatellite instability: the mutator that mutates the other mutator. Nat Med. 1996;2(6):630–631. doi: 10.1038/nm0696-630. [DOI] [PubMed] [Google Scholar]
- 12.Koi M, Umar A, Chauhan DP, et al. Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N′-nitro-N-nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. Cancer Res. 1994;54(16):4308–4312. [PubMed] [Google Scholar]
- 13.Umar A, Koi M, Risinger JI, et al. Correction of hypermutability, N-methyl-N′-nitro-N-nitrosoguanidine resistance, and defective DNA mismatch repair by introducing chromosome 2 into human tumor cells with mutations in MSH2 and MSH6. Cancer Res. 1997;57(18):3949–3955. [PubMed] [Google Scholar]
- 14.Watanabe Y, Haugen-Strano A, Umar A, et al. Complementation of an hMSH2 defect in human colorectal carcinoma cells by human chromosome 2 transfer. Mol Carcinog. 2000;29(1):37–49. [PubMed] [Google Scholar]
- 15.Arnold CN, Goel A, Boland CR. Role of hMLH1 promoter hypermethylation in drug resistance to 5-fluorouracil in colorectal cancer cell lines. Int J Cancer. 2003;106(1):66–73. doi: 10.1002/ijc.11176. [DOI] [PubMed] [Google Scholar]
- 16.Chang DK, Ricciardiello L, Goel A, Chang CL, Boland CR. Steady-state regulation of the human DNA mismatch repair system. J Biol Chem. 2000;275(24):18424–18431. doi: 10.1074/jbc.M001140200. [DOI] [PubMed] [Google Scholar]
- 17.Kolodner RD, Tytell JD, Schmeits JL, et al. Germ-line msh6 mutations in colorectal cancer families. Cancer Res. 1999;59(20):5068–5074. [PubMed] [Google Scholar]
- 18.Miyaki M, Konishi M, Tanaka K, et al. Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Genet. 1997;17(3):271–272. doi: 10.1038/ng1197-271. [DOI] [PubMed] [Google Scholar]
- 19.Kariola R, Hampel H, Frankel WL, Raevaara TE, de la Chapelle A, Nystrom-Lahti M. MSH6 missense mutations are often associated with no or low cancer susceptibility. Br J Cancer. 2004;91(7):1287–1292. doi: 10.1038/sj.bjc.6602129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hendriks YM, Wagner A, Morreau H, et al. Cancer risk in hereditary nonpolyposis colorectal cancer due to MSH6 mutations: impact on counseling and surveillance. Gastroenterology. 2004;127(1):17–25. doi: 10.1053/j.gastro.2004.03.068. [DOI] [PubMed] [Google Scholar]
- 21.Akiyama Y, Tsubouchi N, Yuasa Y. Frequent somatic mutations of hMSH3 with reference to microsatellite instability in hereditary nonpolyposis colorectal cancers. Biochem Biophys Res Commun. 1997;236(2):248–252. doi: 10.1006/bbrc.1997.6942. [DOI] [PubMed] [Google Scholar]
- 22.de Jong AE, van Puijenbroek M, Hendriks Y, et al. Microsatellite instability, immunohistochemistry, and additional PMS2 staining in suspected hereditary nonpolyposis colorectal cancer. Clin Cancer Res. 2004;10(3):972–980. doi: 10.1158/1078-0432.ccr-0956-3. [DOI] [PubMed] [Google Scholar]
- 23.Truninger K, Menigatti M, Luz J, et al. Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer. Gastroenterology. 2005;128(5):1160–1171. doi: 10.1053/j.gastro.2005.01.056. [DOI] [PubMed] [Google Scholar]
- 24.Hendriks YM, Jagmohan-Changur S, van der Klift HM, et al. Heterozygous mutations in PMS2 cause hereditary nonpolyposis colorectal carcinoma (Lynch syndrome) Gastroenterology. 2006;130(2):312–322. doi: 10.1053/j.gastro.2005.10.052. [DOI] [PubMed] [Google Scholar]
- 25.Hienonen T, Laiho P, Salovaara R, et al. Little evidence for involvement of MLH3 in colorectal cancer predisposition. Int J Cancer. 2003;106(2):292–296. doi: 10.1002/ijc.11218. [DOI] [PubMed] [Google Scholar]
- 26.Wijnen J, van der Klift H, Vasen H, et al. MSH2 genomic deletions are a frequent cause of HNPCC. Nat Genet. 1998;20(4):326–328. doi: 10.1038/3795. [DOI] [PubMed] [Google Scholar]
- 27.Lipkin SM, Rozek LS, Rennert G, et al. The MLH1 D132H variant is associated with susceptibility to sporadic colorectal cancer. Nat Genet. 2004;36(7):694–699. doi: 10.1038/ng1374. [DOI] [PubMed] [Google Scholar]
- 28.Chang DK, Ricciardiello L, Goel A, Chang CL, Boland CR. Steady-state regulation of the human DNA mismatch repair system. J Biol Chem. 2000;275(37):29178. [PubMed] [Google Scholar]
- 29.Chang CL, Marra G, Chauhan DP, et al. Oxidative stress inactivates the human DNA mismatch repair system. Am J Physiol Cell Physiol. 2002;283(1):C148–C154. doi: 10.1152/ajpcell.00422.2001. [DOI] [PubMed] [Google Scholar]
- 30.Koshiji M, To KK, Hammer S, et al. HIF-1alpha induces genetic instability by transcriptionally downregulating MutSalpha expression. Mol Cell. 2005;17(6):793–803. doi: 10.1016/j.molcel.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 31.Boland CR, Fishel R. Lynch syndrome: form, function, proteins, and basketball. Gastroenterology. 2005;129(2):751–755. doi: 10.1016/j.gastro.2005.05.067. [DOI] [PubMed] [Google Scholar]
- 32.Raevaara TE, Korhonen MK, Lohi H, et al. Functional significance and clinical phenotype of nontruncating mismatch repair variants of MLH1. Gastroenterology. 2005;129(2):537–549. doi: 10.1016/j.gastro.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 33.Kane MF, Loda M, Gaida GM, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57(5):808–811. [PubMed] [Google Scholar]
- 34.Chadwick RB, Meek JE, Prior TW, Peltomaki P, de la Chapelle A. Polymorphisms in a pseudogene highly homologous to PMS2. Hum Mutat. 2000;16(6):530. doi: 10.1002/1098-1004(200012)16:6<530::AID-HUMU15>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 35.Gryfe R, Kim H, Hsieh ET, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med. 2000;342(2):69–77. doi: 10.1056/NEJM200001133420201. [DOI] [PubMed] [Google Scholar]
- 36.Hawn MT, Umar A, Carethers JM, et al. Evidence for a connection between the mismatch repair system and the G2 cell cycle checkpoint. Cancer Res. 1995;55(17):3721–3725. [PubMed] [Google Scholar]
- 37.Carethers JM, Smith EJ, Behling CA, et al. Use of 5-fluorouracil and survival in patients with microsatellite-unstable colorectal cancer. Gastroenterology. 2004;126(2):394–401. doi: 10.1053/j.gastro.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 38.Aebi S, Kurdi-Haidar B, Gordon R, et al. Loss of DNA mismatch repair in acquired resistance to cisplatin. Cancer Res. 1996;56(13):3087–3090. [PubMed] [Google Scholar]
- 39.Carethers JM, Chauhan DP, Fink D, et al. Mismatch repair proficiency and in vitro response to 5-fluorouracil. Gastroenterology. 1999;117(1):123–131. doi: 10.1016/s0016-5085(99)70558-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349(3):247–257. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benatti P, Gafa R, Barana D, et al. Microsatellite instability and colorectal cancer prognosis. Clin Cancer Res. 2005;11(23):8332–8340. doi: 10.1158/1078-0432.CCR-05-1030. [DOI] [PubMed] [Google Scholar]
- 42.Jover R, Zapater P, Castells A, et al. Mismatch repair status in the prediction of benefit from adjuvant fluorouracil chemotherapy in colorectal cancer. Gut. 2006;55(6):848–855. doi: 10.1136/gut.2005.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jascur T, Boland CR. Structure and function of the components of the human DNA mismatch repair system. Int J Cancer. 2006;119(9):2030–2035. doi: 10.1002/ijc.22023. [DOI] [PubMed] [Google Scholar]
- 44.Fishel R. The selection for mismatch repair defects in hereditary nonpolyposis colorectal cancer: revising the mutator hypothesis. Cancer Res. 2001;61(20):7369–7374. [PubMed] [Google Scholar]