

Abstract
Background
Myosin performs ATP free energy transduction into mechanical work in the motor domain of the myosin heavy chain (MHC). Energy transduction is the definitive systemic feature of the myosin motor performed by coordinating in a time ordered sequence: ATP hydrolysis at the active site, actin affinity modulation at the actin binding site, and the lever-arm rotation of the power stroke. These functions are carried out by several conserved sub-domains within the motor domain. Single nucleotide polymorphisms (SNPs) affect the MHC sequence of many isoforms expressed in striated muscle, smooth muscle, and non-muscle tissue. The purpose of this work is to provide a rationale for using SNPs as a functional genomics tool to investigate structurefunction relationships in myosin. In particular, to discover SNP distribution over the conserved sub-domains and surmise what it implies about sub-domain stability and criticality in the energy transduction mechanism.
Results
An automated routine identifying human nonsynonymous SNP amino acid missense substitutions for any MHC gene mined the NCBI SNP data base. The routine tested 22 MHC genes coding muscle and non-muscle isoforms and identified 89 missense mutation positions in the motor domain with 10 already implicated in heart disease and another 8 lacking sequence homology with a skeletal MHC isoform for which a crystallographic model is available. The remaining 71 SNP substitutions were found to be distributed over MHC with 22 falling outside identified functional sub-domains and 49 in or very near to myosin sub-domains assigned specific crucial functions in energy transduction. The latter includes the active site, the actin binding site, the rigid lever-arm, and regions facilitating their communication. Most MHC isoforms contained SNPs somewhere in the motor domain.
Conclusions
Several functional-crucial sub-domains are infiltrated by a large number of SNP substitution sites suggesting these domains are engineered by evolution to be too-robust to be disturbed by otherwise intrusive sequence changes. Two functional sub-domains are SNP-free or relatively SNP-deficient but contain many disease implicated mutants. These sub-domains are apparently highly sensitive to any missense substitution suggesting they have failed to evolve a robust sequence paradigm for performing their function.
Background
Single nucleotide polymorphisms (SNPs) are common single base DNA sequence variants that account for a sizable portion of the genetic variability between individuals. Some SNPs are common and have minor allele frequencies that approach 50%, while others are found much less frequently. There is some conceptual overlap between rare SNPs (with minor allele frequencies of less than 1%) and disease implicated mutations, but in common usage the term polymorphism is restricted to non-pathogenic sequence changes. Genome SNP patterns are fingerprints identifying subpopulations with common heritage that potentially instigate subpopulation specific traits exploitable for individualized treatment of disease. For the purposes of this discussion, it is implicit that the SNPs accumulate in the genome because they negligibly affect survival, however, we recognize that some SNPs may in fact have deleterious effects on the expression or function of protein products.
Another distinction that is germane to the present work is between synonymous and nonsynonymous SNPs. SNPs that lie within the coding region are synonymous if they do not change the amino acid specified by the codon and are nonsynonymous when they do. Even though synonymous SNPs will not change protein sequence they might still impact expression of a protein product by influencing the subcellular localization, stability or translational efficiency of mRNA [1,2]. The nonsynonymous SNP introduces a single amino acid change into the protein sequence. Assessing the impact of such protein sequence changes on protein function can be difficult. For most SNPs whose minor allele frequency is at least 5%, it is likely that their impact on genetic fitness is minor [3]. SNPs that clearly have harmful functional consequences only rarely become more common than this 5% level in the population as a whole. For less common SNPs, homozygous individuals that have two copies of the SNP are rare and deleterious consequences can often be masked by the presence of the more common functional allele. Another factor in deciding the significance of the nonsynonymous SNPs is genetic redundancy. The affected gene could be one copy among several present in the genome. These factors decide the prevalence of the affected protein in the tissues and may be more important for survival than how the SNP affects protein function. Furthermore, a SNP induced loss of functionality can sometimes be compensated by redundant functionality in the larger physiological system. An example is skeletal muscle where the relative composition of the myosin isoforms depends on environmental factors such as exercise [4-6] or zero gravity [7]. The muscle tissue may adapt to loss of function in the SNP affected isoform to assure survival. Finally, while SNPs accumulated in the data base are identified world wide without apparent prejudice, they are not necessarily a random sampling of the genome. SNP frequency in different gene regions could reflect uneven DNA sequencing accuracy and efficiency or other unknown biases inadvertently built into the data base. Our results are implications from the current data base that changes and grows daily.
Here we identify and discuss the myosin heavy chain nonsynonymous SNPs. We will include even scarce SNPs where homozygous individuals are rare. We do so in the spirit of being comprehensive, with the full understanding that neutrality of function cannot be assumed in these cases. Additionally, some SNPs discussed appear in the database, but have not been confirmed by other groups. Our purpose is to provide a rationale for using these reports to investigate structure-function relationships in myosin. Functional sub-domains within myosin have emerged as disturbance propagators essential to transduction. They are structural entities in which to identify SNP substitution sites and focus speculation on how SNPs influence myosin function.
The myosin heavy chain (MHC) consists of a globular head domain called subfragment 1 (S1) and C-terminal tail responsible for dimerization and cargo binding. In muscle, myosin dimers form (thick) filaments with interacting tails and head domains projecting outward from the filament to associate with complementary actin (thin) filaments in a muscle sarcomere [8]. Cellular myosins have several forms [9-12]. The cellular myosin V dimer is a processive motor utilizing the two heads cooperatively to move along actin filaments [13].
S1 contains functional sub-domains that retain their structure, but move relatively, during transduction [14]. ATPase transduction starts in the active site composed in part by the P-loop, Switch 1, and Switch 2 polypeptides (linked to a 7-stranded β-sheet separating the active site from the actin binding site) that sense the γ-phosphate position and coordination. ATP hydrolysis in the active site is not followed immediately by phosphate release because the Switch 1 R246 and Switch 2 E469 salt-bridge "back door" inhibits phosphate release until actin binds. With actin binding, the back door opens to release phosphate and the Switch 2 α-helix (or relay helix) transmits linear force originating from the active site to a converter domain converting linear force into torque to rotate the lever-arm. The lever-arm α-helix, rigidified by the bound myosin light chains (MLCs), amplifies displacement and impels myosin relative to actin [15,16]. A region between the switch 2 helix and the converter domain, the SH2/SH1 hinge, undergoes substantial conformation change during the transduction. The 7-stranded β-sheet, switch 2 helix, converter, lever-arm, and SH2/SH1 hinge are the sub-domains identified in the skeletal myosin crystal structure depicted in Figure 1[17].
Figure 1.
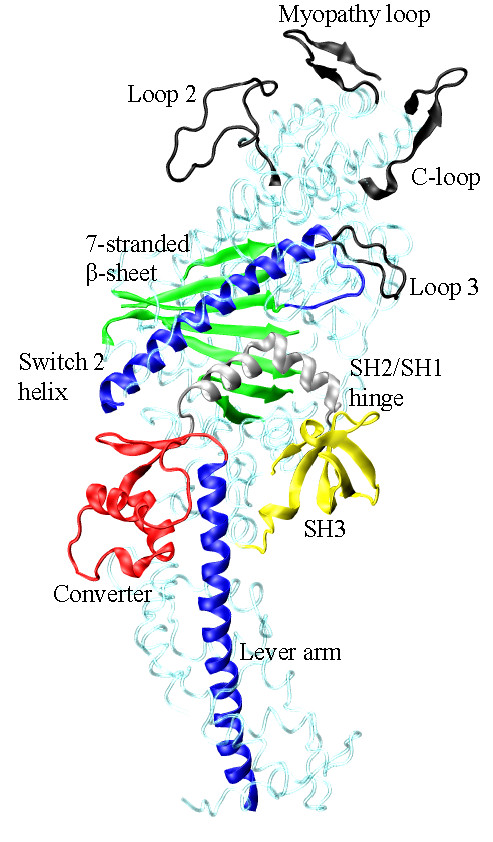
Several myosin peptides or domains identified with energy transduction of ATP hydrolysis free energy to the mechanical work of moving actin. Structured (Myopathy and Cloop, AA363-377 and 404-417) and unstructured (Loops 2 and 3, AA626-651 and 568-580) surface loops are actin binding peptides (black). The 7-stranded β-sheet (green, AA116-127, 170-180, 248-257, 265-271, 458-468, and 671-678) mediates ATP hydrolysis and actin binding affinity modulation. The Switch 2 helix (blue, AA469-509) transmits translational movement in the active site to the converter domain (red, AA716-772) where it is converted to the torque needed to rotate the lever-arm (blue, AA773-813). The SH2/SH1 hinge (silver, AA688-715) undergoes a large conformation change with lever-arm rotation. The function of SH3 (yellow, AA30-80) in energy transduction is not yet understood. Not depicted are actin-binding-peptides at two sites in the upper half of the molecule.
Multiple peptides within S1 (C-loop, Myopathy loop, and others) constitute the actin binding site. This site alternates between weak and strong actin binding affinity in coordination with the lever-arm swing. Phosphate release in muscle myosin ATPase (or the opening of the R246/E469 back door salt-bridge) is rate limiting due to its inhibition by a large entropic energy barrier in the absence of actin. Phosphate release initiates lever-arm swing hence the rate limiting step inhibits progress through the cycle until actin binds and a work producing lever-arm power stroke is possible. Coupling actin binding to reducing the energy barrier to phosphate release is an allosteric effect predicted for Myopathy or C-loop structured surface loops [18,19]. We showed the C-loop is the allosteric actin contact sensor mediating bidirectionally between the actin-binding and active sites [20]. The C-loop lowers the energy barrier to phosphate release by coupling structural transitions within the S1 that allow phosphate release without inducing strain in the 7-stranded β-sheet. Several actin binding myosin surface loops are identified in Figure 1.
In this paper we locate nonsynonymous SNPs within the functional sub-domains and evaluate their expected functional impact based on: i) how well the native residue is conserved among isoforms, ii) comparison of general physical characteristics (size, charge, hydrophobicity, etc.) between the native and substituted side chain, and iii) (when possible) the hypothetical role of the native residue in transduction and how this role could be affected by the substituted side chain. We find that SNPs infiltrate nearly every corner of the myosin head and that their significance may lie in where they are tolerated in contrast to where they are excluded.
Results
Twenty-two human myosin heavy chain genes MYH1-4, MYH6-15, MYH7B, MYOIE, MYO6, MYO7A and 7B, MYO9A and 9B, and MYO10 were searched for nonsynonymous SNPs as described in METHODS. We confined the search to SNPs in the S1 domain. Table 1 summarizes results with the SNP reference number locating it in the human genome and in the NCBI data base, the sequence position relative to the skeletal myosin 2× sequence, the MHC gene and myosin isoform it encodes, the position for the SNP within myosin domains, the average percentage of heterozygous genes (one allele normal the other SNP substituted) in the standard sub-populations, whether (Yes or No) there are SNP substituted homozygous genes (both alleles SNP substituted) in the standard sub-populations or observed in screened individuals, and the number of individuals screened. Missing data is denoted with a hyphen.
Table 1.
Myosin heavy chain SNPsa
| SNP (rs number) | Skeletal 2× sequence | Skeletal 2× residue if different | Gene | Isoform | Positionb | <Hetero>c (Homo-zygotes?) e | Countd |
|---|---|---|---|---|---|---|---|
| 7-stranded β-sheet (Figure 2) | |||||||
| 61745307 | I88M | MYH13 | super-fast | near β1 | - | - | |
| 442275 | E89Q | MYH6 | α-cardiac | near β1 | 0.5(N) | 525 | |
| 61739660 | H98Q | MYH1 | 2× | near β1 | - | - | |
| 2754166 | D108E | E108 | MYH7 | β-cardiac | near β1 | 0(N) | 525 |
| 7737765 | H170Y | R170 | MYO10 | myosin X | β 4 | - | - |
| 61745058 | D171E | E171 | MYH7B | β-cardiac | β 4 | - | - |
| 61745059 | N172D | MYH7B | β-cardiac | β 4 | - | - | |
| 45516091 | R240W | MYH7 | β-cardiac | SW1 | 0(Y) | 0f | |
| 28934610 | R246H | MYO7A | myosin VIIA | SW1 | - | - | |
| 3218713 | R252Q (disease implicated) | MYH7 | β-cardiac | β 6 | 0(Y) | 525 | |
| 55645295 | I253V | MYH14 | iso1/iso2 | β 6 | - | - | |
| 28936390 | E257V | T257 | MYO6 | myosin VI | β6 | - | - |
| 34416201 | T258K | MYH13 | super-fast | β 6/β 7 | 2.5(N) | 39 | |
| 4299484 | R445Q | MYH15 | unknown | near β7 | 18.0(?) | 1178 | |
| 61731179 | N447K | MYH6 | α-cardiac | near β7 | - | - | |
| 61745057 | L463P | MYH7B | β-cardiac | β 5 | - | - | |
| 4981473 | E469Q | MYH7 | β-cardiac | SW2 | 0(N) | 525 | |
| 28932773 | R675Q (disease implicated) | MYH8 | perinatal | β 3 | - | - | |
| 11539755 | E681K | MYOIE | myosin 1e | near β 3 | - | - | |
| Switch 2 helix (Figure 4) | |||||||
| 61734198 | Q478H | MYH11 | sm1A/sm2B | SW2 hx | - | - | |
| 1064307 | Y483H | F483 | MYO9B | myosin IXB | SW2 hx | - | - |
| 28934903 | N485I | MYO7A | myosin VIIA | SW2 hx | - | - | |
| 61745301 | H494D | MYH13 | super-fast | SW2 hx | - | - | |
| 9868484 | H495Y | MYH15 | unknown | SW2 hx | 38.3(Y) | 1121 | |
| 1052031 | F497L | MYO7A | myosin VIIA | SW2 hx | - | - | |
| 3218715 | E502K (disease implicated) | MYH7 | β-cardiac | SW2 hx | 0(Y) | 525 | |
| 3729813 | K505N | MYH7 | β-cardiac | SW2 hx | 0(N) | 525 | |
| SH2/SH1 hinge, converter, & lever-arm (Figure 3) | |||||||
| 73974725 | M688V | MYH1 | 2× | SH2/SH1 hn | - | - | |
| 34515627 | V694E | L694 | MYO7A | myosin VIIA | SH2/SH1 hn | - | - |
| 41298143 | R695H | H695 | MYO7A | myosin VIIA | SH2/SH1 hn | - | - |
| 2190729 | G701R | MYH13 | super-fast | swivel | 2.8(Y) | 384 | |
| 28940307 | R707S | MYH14 | iso1/iso2 | SH2/SH1 hn | - | - | |
| 3181426 | R710S | MYH7 | β-cardiac | SH2/SH1 hn | - | - | |
| 35641839 | V720I (disease implicated) | MYO7A | myosin VIIA | Converter | - | - | |
| 1136661 | Y723C(disease imp)K723 | MYO10 | myosin X | Converter | - | - | |
| 3746442 | P735S (disease implicated) | MYH7B | β-cardiac | Converter | g | 525 | |
| 26740 | R743Q | K743 | MYO10 | myosin X | Converter | 13.5(?) | 1122 |
| 11847151 | L785M | MYH6 | α-cardiac | Lever-Arm | 0(N) | 525 | |
| 36090425 | D787N | Q787 | MYO7A | myosin VIIA | Lever-Arm | - | - |
| 3218716 | A801T (disease implicated) | MYH7 | β-cardiac | LA & ELC IQ | 0(Y) | 690 | |
| SH3 (Figure 5) | |||||||
| 17092199 | P31T | MYH7B | β-cardiac | SH3 | discordant(?)h | 1013 | |
| 28711516 | G57R | MYH6 | α-cardiac | SH3 | -(Y) | - | |
| 17707947 | V60I | MYO10 | myosin X | SH3 | 12.9(?) | 1092 | |
| 41309316 | K68N | A68 | MYH7B | β-cardiac | SH3 | - | - |
| Actin binding (Figure 6) | |||||||
| 45629132 | R370H | K370 | MYO7A | myosin VIIA | C-loop | - | - |
| 35222064 | Q371P | MYH13 | super-fast | C-loop | 2.5(N) | 39 | |
| 2515926 | P378Q | MYH6 | α-cardiac | C-loop | - | - | |
| 3218714 | R406W (disease implicated) | MYH7 | β-cardiac | Myopathy | 0(Y) | 525 | |
| 45522831 | R408C | K408 | MYH7B | β-cardiac | Myopathy | - | - |
| 35349985 | F534L | MYH4 | 2b | actin bind | 2.8(N) | 39 | |
| 59922029 | R570K | K570 | MYO10 | myosin X | Loop 3 | - | - |
| 61735348 | V572A/D | A572 | MYH3 | embryonic | Loop 3 | - | - |
| 28565077 | A621V | MYH15 | unknown | near Loop 2 | - | - | |
| 61745053 | T631N | E631 | MYH7B | β-cardiac | Loop 2 | - | - |
| 34693726 | A637V | G637 | MYH8 | perinatal | Loop 2 | 9.2(N) | 310 |
| 2276282 | E643K | K643 | MYO7A | myosin VIIA | Loop 2 | - | - |
| 61743282 | F655S | MYO7B | myosin VIIB | actin bind | - | - | |
| N-term, U50, and L50 | |||||||
| 3729993 | D4A | MYH7 | β-cardiac | N-term | 0(Y) | 525 | |
| 34042358 | D4E | MYH13 | super-fast | N-term | 3.2(?) | 62 | |
| 45511396 | R18C | MYH7 | β-cardiac | N-term | 0.4(Y) | 0f | |
| 61730792 | R24P/L | MYH1 | 2× | N-term | - | - | |
| 41312286 | T25M | I25 | MYH7B | β-cardiac | N-term | - | - |
| 590722 | P(<32)Ti | - | MYH14 | iso1/iso2 | undefined | - | - |
| 2404991 | G(<48)Si | - | MYO7B | myosin VIIB | undefined | - | - |
| 1052030 | L(<58)Si | - | MYO7A | myosin VIIA | undefined | - | - |
| 35218876 | R(<64)Q | - | MYO9B | myosin IXB | undefined | 2.5(?) | 39 |
| 10518970 | P(<82)Li | - | MYO9A | myosin IXA | undefined | 4.3(?) | 1224 |
| 17855105 | R(<82)Ki | - | MYO9A | myosin IXA | undefined | - | - |
| 2929516 | T(<82)Ii | - | MYO9A | myosin IXA | undefined | discordant(?)h | 1122 |
| 34773557 | M142I | A142 | MYH14 | iso1/iso2 | N-term | 2.8(?) | 70 |
| 6174305 | R145G | G145 | MYO10 | myosin X | N-term | - | - |
| 41298131 | I148T | R148 | MYO7A | myosin VIIA | N-term | - | - |
| 58359270 | L199F | I199 | MYO9B | myosin IXB | N-term | - | - |
| 28936391 | H287R | MYO6 | myosin VI | U50 | - | - | |
| 6870170 | E300D | L300 | MYO10 | myosin X | U50 | 1.3(?) | 908 |
| 35512085 | T307P | MYH4 | 2b | U50 | - | - | |
| 34498817 | P320A | E320 | MYH14 | iso1/iso2 | U50 | 5.5(?) | 309 |
| 35315400 | S323C | V323 | MYH14 | iso1/iso2 | U50 | 2.5(?) | 78 |
| 34124921 | I326T | MYH8 | perinatal | U50 | 1.2(N) | 39 | |
| 35984286 | Q329R (disease implicated) | MYH4 | 2b | U50 | 2.5(N) | 39 | |
| 34846075 | V335I | T335 | MYO10 | myosin X | U50 | 2.5(?) | 39 |
| 41298135 | R336H | D336 | MYO7A | myosin VIIA | U50 | - | - |
| 34419805 | T345A | MYH8 | perinatal | U50 | 2.8(N) | 35 | |
| 61756677 | V350D/A | MYH2 | 2a | U50 | - | - | |
| 11750538 | R350W | V350 | MYO10 | myosin X | U50 | 50(?) | 1264 |
| 1724577 | E389D | Y389 | MYH12 | myosin V | U50 | 17.2(Y) | 1262 |
| 61732664 | Y(389-390)Hj | MYO6 | myosin VI | U50 | - | - | |
| 61742021 | I514T (disease implicated) | MYO7B | myosin VIIB | L50 | - | - | |
| 12949680 | A594T | MYH4 | 2b | L50 | 7.4(N) | 39 | |
a Abbreviations SW1 or SW2 denote Switch 1 or 2, hx is helix, sb is salt-bridge, hn is hinge, and hyphen (-) is
no data submitted.
b Position names are defined in the MHC sequence as follows:
1. SH3 like β-barrel: AA30-80
2. 7-stranded β-sheet: β1 AA116-120; β2 121-127; β3 671-678; β4 170-180; β5 458-468; β6 248-257; β7 265-271
3. Loop 1 AA200-220
4. Upper 50 k (U50) AA220-468
5. C-loop AA363-377
6. Myopathy loop AA404-417
7. Switch 2 helix AA469-509
8. Lower 50 k (L50) AA469-620
9. Actin binding peptides: hydrophobic helical peptides AA529-560 and 652-661, Loop 2, Loop 3,
C--loop, Myopathy loop
10. Loop 3 actin binding site: AA568-580
11. Strut loop: AA600-604
12. Loop 2 AA626-651
13. SH2/SH1 hinge AA688-715
14. Converter domain: AA716-772
15. Lever-arm AA773-813
cAverage percentage of heterozygous genes (one allele normal, the other SNP substituted) in the standard subpopulations.
d Individuals sequenced.
e Are there SNP substituted homozygous genes (both alleles SNP substituted) in the standard sub-populations or
observed in screened individuals?
f Unexplained inconsistency.
g All individuals sequenced are homozygous in the SNP replacement over 4 population groups.
h Discordant genotype implies conflicting reports.
i Sequence segment not aligned in BLAST.
j Sequence segment SLEYCAE in myosin VI inserts between AA389 and 390 in MYH1. This is not Insert 1 or
2 mentioned in the text.
Most MHC isoforms contribute SNPs including the fast skeletal muscle isoforms 2×, 2a, 2b, embryonic, and perinatal; α- and β-cardiac myosins; smooth muscle variants sm1A and sm2B; myosin V; super-fast myosin; two isoforms of MYH14; MYH15; and other unconventional myosins including IE, VI, VIIA, VIIB, IXA, IXB, and X. Myosin 2b (MYH4) is a fast skeletal myosin isoform apparently expressed in a skeletal jaw closing (masseter) muscle [21]. The embryonic and perinatal isoforms (MYH3 and 8) are developmentally regulated [22,23]. The embryonic isoform also expresses in regenerating muscle and in extraocular muscles (EOM) [24]. A mutation in perinatal myosin is implicated in a rare genetic disease causing distal arthrogryposis syndrome [25]. The α-cardiac isoform (MYH6) is predominantly expressed in the atrium [26]. The β-cardiac isoform (MYH7) is predominantly expressed in the heart ventriculum and in slow or type 1 skeletal muscle fibers. Numerous heart disease implicated mutations occur in the β-cardiac MHC [27]. Another myosin isoform (MYH7B/MYH14) on chromosome 20 is called a β-cardiac myosin with 85% homology with the β-cardiac myosin coded by MYH7.
The MYH11 gene encodes the 4 smooth muscle myosin isoforms that differ near the C-terminus (isoforms 1 and 2) or at Loop 1 where a seven amino acid peptide is omitted (isoform A) or inserted (isoform B) [28]. The Loop 1 insert correlates with tissue localization [29] and enzyme kinetics [30,31]. Nucleotide-induced fluorescence intensity increase in the highly conserved W511 occurs in all but one myosin isoform implying it results from common origin within the conserved motor core. The smA isoform has nucleotide-induced tryptophan fluorescence enhancement like other myosin isoforms but we showed with site-directed mutagenesis that the contributing tryptophan is probably not W511. These results suggested that smA and smB myosin isoforms have different influence propagating pathways emanating from the active site through the switch 2 helix containing W511 to the converter domain [32].
Processive myosin V (MYH12) translates over actin filaments using a hand-over-hand mechanism [33-35]. The super-fast isoform (MYH13) expresses in EOMs performing diverse functions including eye movement [24]. The EOMs are more complex than limb muscles with 9 or more different MHCs expressed in adult muscle including the fast skeletal, β-cardiac, and embryonic MHC. The super-fast myosin supports higher velocity and lower tension contractions [36]. The MYH14 gene codes for 2 myosin isoforms (iso1/iso2 in Table 1) expressed in muscle and non muscle tissue. The MYH14 gene is implicated in hearing disorders [37]. SNP variants in MYH15 are associated with elevated risk for heart disease [38].
Myosin IE (MYOIE) is a cellular myosin with an extended C-loop thought to interact with tropomyosin in tropomyosin-containing actin [39,40]. Myosin VI is a reversedirection motor with core structure resembling forward-direction motors except for two inserts (1 & 2). Insert 1, near the active site and Switch 1, is probably partially responsible for the slow ADP release (A·M^·D→A·M in Scheme 1, METHODS)[41]. Insert 2 modifies the converter domain to re-direct lever-arm swing for reverse-directed motility [41,42]. Mutations in myosin VII Aassociate with Us her syndrome that in its most severe phenotype is a deafness-blindness disorder [43,44]. Myosin IXB is a single headed processive motor that is unique because its rate limiting step is not ADP release but ATP hydrolysis (M*·T→M**·D·P in Scheme 1) suggesting it remains actin bound even in a weak actin binding state [45]. Slow dissociation from actin is attributed to tethering by a unique Loop 2 insertion. Myosin X is a membrane associated motor involved in filopodia motility [46] that has novel monomeric and dimeric conformations at physiological protein concentrations [11]. Calmodulin or calmodulin like protein (CLP) are the lever-arm bound myosin light chains in myosin X [47]. Myosin X is believed to be processive consistent with its function in the cell.
SNP implications for motor function
Three SNPs, R246H, E469Q (Figure 2) and G701R (Figure 3), are remarkable because of the functional significance of the residues they modify. R246 and E469 form the Switch 1/Switch 2 salt-bridge at the γ-phosphate in bound ATP preventing product release following hydrolysis in the active site of S1. Site-directed mutagenesis of Dictyostelium discoideum (Dicty) myosin produced R246E, E469R, and the double mutant R246E/E469R reversing the polarity of the salt-bridge [48]. Only the double mutant remained functional but with altered ATPase kinetics. Similar experiments using expressed smooth muscle myosin suggested R246 is significant for back door closure after ATP binding to the active site and that E469 is important for ATP hydrolysis by its positioning of a water molecule for nucleophilic attack on the γ-phosphate [49]. The E469Q SNP substitution does not necessarily prohibit the latter mechanism although E469A/R mutants eliminated the phosphate burst and actin-activated ATPase. The R246H SNP substitution alters size but not charge of the side chain while R246A/E mutants likewise eliminated the phosphate burst and actin-activated ATPase. The loss of the phosphate burst and actin-activated ATPase indicates the phosphate release step is no longer rate limiting undermining the efficient energy transduction in muscle because the ATPase cycle disengages from the work producing interaction with actin. The SNPs suggests it would be interesting to test the R246H and E469Q substitutions in an in vitro system. R246H occurs in myosin VIIA where ATPase is rate limited by ADP release rather than phosphate release possibly explaining its tolerance to substitution. E469Q occurs in β-cardiac myosin where ATPase is rate limited by phosphate release. R246H and E469Q occur with unknown or apparently low probability for expression, respectively.
Figure 2.
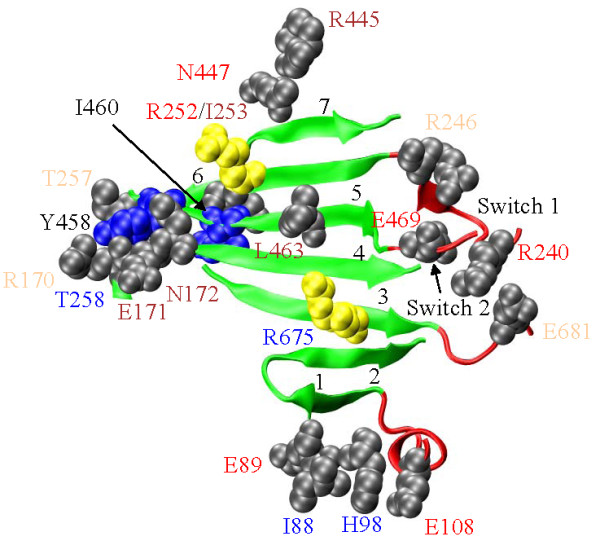
The 7-stranded β-sheet (green) and the adjacent structures (red). SNP substitution sites in the vicinity are depicted in grey or yellow with space filling models of the unsubstituted side chains. The yellow residues are also implicated in disease. SNP residue annotation color coding corresponds to: cardiac myosin II (red), skeletal myosin II (blue), non-muscle myosin II (brown), and unconventional myosin (tan). Residues annotated in black are not SNPs. The 7 strands are numbered at the tip indicating the sequence number increasing direction. R675 on β3 (AA671-678); R170, E171, and N172 on β4 (AA170-180); L463 on β5 (AA458-468); and R252, I253, and T257 on β6 (AA248-257) modify the structure. E681 is three residues past the end of β3, E469 is on Switch 2 just two residues past the end of β5, R240 and R246 are on Switch 1 just before the start of β6, and T258 lies between β6 and β7 (AA265-271). Residues I88, E89, H98, E108, R445, and N447 are distant in sequence but spatially associated with the 7-stranded β-sheet structure. E89 is the most distant at ~7.5 angstroms. Blue side chains for residues Y458 and I460 are not SNPs but are proposed to contribute substantially to the energy barrier determining the rate limiting step to myosin ATPase product release in the absence of actin.
Figure 3.
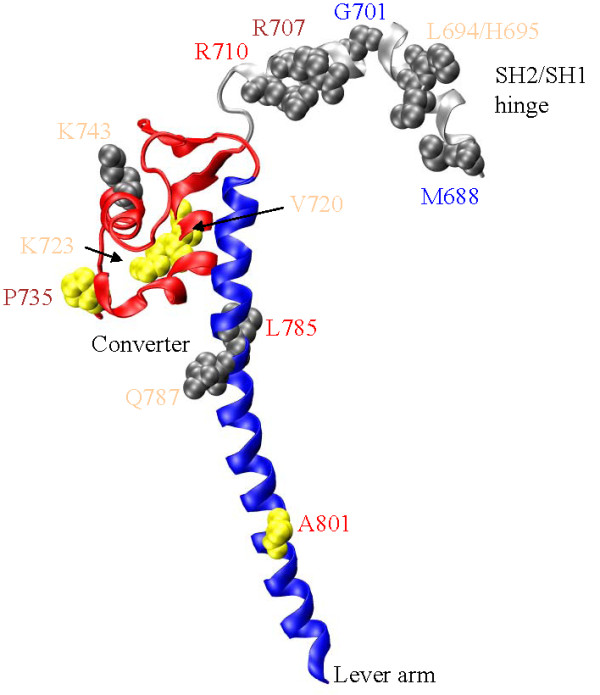
The SH2/SH1 hinge (silver, AA688-715), converter (red, AA716-772), and lever-arm (blue, AA773-813). SNP residue annotation color coding corresponds to: cardiac myosin II (red), skeletal myosin II (blue), non-muscle myosin II (brown), and unconventional myosin (tan). Thirteen SNP substitution sites at M688, L694, H695, G701, R707, R710, V720, K723, P735, K743, L785, Q787, and A801 are depicted in gray or yellow with space filling models of the unsubstituted side chains. The yellow residues are also implicated in disease. R710 is adjacent to the reactive thiol residue (SH1 at C709) on the SH2/SH1 hinge. L785, Q787, and A801 are on the lever-arm and within the ELC binding region. The converter domain receives impulses from the Switch 2 helix that it converts into the torque needed rotate the lever-arm while the SH2/SH1 hinge changes conformation.
G701R in super-fast myosin replaces the glycine swivel with a residue containing a bulky side chain inhibiting free rotation in the Ramachandran angles that is characteristic only to glycine [50]. The highly conserved G701 pivot was found to be essential for motor activity in skeletal [51] and Dicty [52] myosins. S1 crystal structures depicting myosin conformation in M** [53] and M* [17] lever-arm orientation states indicate G701 swivels 40-50 deg about two axes with the lever-arm swing [54]. Unlike nearby swivel-candidate residues, G705 and G712, the G701 maintains conformations in the pre- and post-powerstroke states that only glycine can readily accommodate. If the G701R substituted protein truly functions in healthy individuals, it suggests the super-fast myosin has a substantially modified transduction mechanism to accomplish its high velocity contraction. The G701R substitution occurs with an average heterozygous population in percent (AHP) of 2.8% and with homozygous individuals identified.
Figure 2 shows the 7-stranded β-sheet separating the active from the actin binding sites and numerous residue positions with SNP substitutions. Substitutions H170Y (R170 in MYH1), D171E (E171 in MYH1), and N172D modify β4; L463P modifies β5; while I253V and E257V (T257 in MYH1) modify β6 within the β-sheet structure. SNP substitution R675Q modifying β3 is implicated distal arthrogryposis syndrome [25] and in heart disease together with SNP substitution R252Q modifying β6 [27]. The R/Q substitution replaces a large +charged side chain with a polar, neutral, and smaller side chain where total surface area changes by ~52 Å2. I/V, D/E, H/Y and N/D substitutions are conservative of charge, size, and polarity. E/V substitution is not conservative of charge, size, or polarity. The L/P substitution could be the most perturbative due to proline's backbone restrictive geometry inducing strain in the β-sheet. Distortions in this β-sheet accompany events associated with ATP hydrolysis and product release since the P-loop, Switch 1, and Switch 2 are closely linked with the β4, β6, and β5 strands, respectively [55]. Furthermore, crystal structures of myosin V without bound nucleotide models the actin-attached rigor conformation and suggests strong binding with actin closes a cleft within the 50 kDa domain of S1 [56] causing distortions in the β1, β2, and β3 strands [55]. Thus many aspects of energy transduction impact the 7-stranded β-sheet and in particular, strands 3-6 containing the SNP substitutions. Our MCMD simulation suggests distortion of the β-sheet in the β5 and β7 strands provides a substantial part of the entropy dominated energy barrier to product release in the absence of actin [19]. In particular, β5 residues Y458 and I460 (Figure 2) have solvent accessible surface areas (ASA) that undergo dramatic increases with barrier transition contributing significantly to the entropy dominated free energy barrier. Simulation suggests residues on β5 and β7 should more likely perturb the contraction mechanism implying the SNP substitution L463P modifying β5 may again have special significance. These SNPs occur in a variety of myosin genes and isoforms but most (including L463P) are relatively new entries that have not undergone further verification to test their presence in sub-population data bases.
Figure 2 also shows Switch 1 and the R240W and R246H SNP sites adjacent to the 7-stranded β-sheet, E681K adjacent to β3, Switch 2 and the E469Q SNP site adjacent to β5, T258K appearing between the β6 (AA248-257) and β7 (AA265-271) strands, and D108E (E108 in MYH1) upstream from β1 and adjacent to the 7-stranded β-sheet. Additionally, I88M, E89Q, and H98Q are within a few angstroms of β1, while R445Q and N447K are within a few angstroms of β7. The R240W substitution provides an opportunity to insert a harmless spectroscopic probe (i.e., W240) into the Switch 1 peptide to sense directly the back-door dynamics and induced strain in the 7-stranded β-sheet. The D108E SNP reiterates the natural cardiac/skeletal myosin isoform sequence substitution. I88M, E89Q, and H98Q connect the 7-stranded β-sheet to SH3. N447K appears within a cluster of heart disease linked mutations [27]. R445Q has an AHP of 18% and comes from MYH15. The next largest AHP SNP at 2.5% from around the 7-stranded β-sheet is T258K in super-fast myosin. Seventeen SNP substitutions (excluding disease linked R252Q and R675Q) in, or proximal to, the 58 residue 7-stranded β-sheet is unexpected given the significance attributed to the region.
Figure 4 shows the Switch 2 helix underlying the 7-stranded β-sheet where β5 and the Switch 2 helix are linked by Switch 2, and sites for the eight SNP substitutions, Q478H, Y483H (F483 in MYH1), N485I, H494D, H495Y, F497L, and K505N, modifying the Switch 2 helix. SNP substitution E502K is implicated in heart disease [27]. The Switch 2 α-helix transmits linear force originating from the active site to the converter domain. Helix rigidity could be essential to force transmission. Q478 is a highly conserved residue at the beginning of the helix where it could maximally affect force transmission efficiency by reducing rigidity. The Q478H substitution tests this hypothesis because the Q/H substitution is helix destabilizing [57] suggesting helix stability may not be crucial. Other SNP substitutions, N485I, H495Y, F497L and E502K, are helix stabilizing while Y483H, H494D and K505N are destabilizing. K505N is at the C-terminal end of the helix where destabilization would be least detrimental. H495 is a potentially sensitive point where the helix develops a kink in the pre-power stroke M** conformation [53]. The H495Y substitution from MYH15 has an AHP of 38% and homozygous individuals were identified. A H495W mutation might sense Switch 2 helix movement by tryptophan fluorescence changes and represents a desirable signal donor in the transduction mechanism. Highly conserved residues on the tip of the switch 2 helix, Y504 and I509, are thought to mediate interaction with the converter domain but are not affected by known SNPs.
Figure 4.
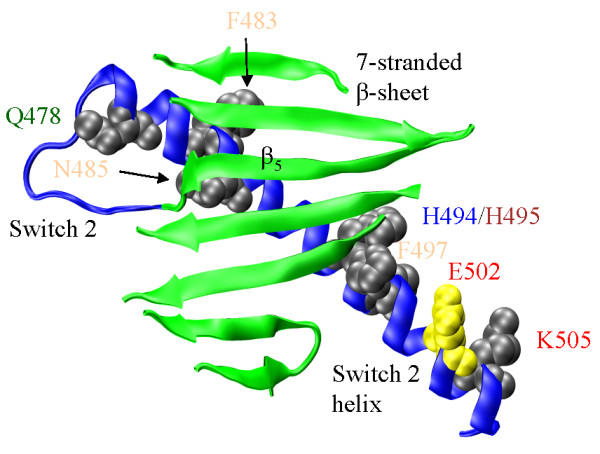
The 7-stranded β-sheet (green), Switch 2, and the Switch 2 helix (blue). SNP residue annotation color coding corresponds to: cardiac myosin II (red), skeletal myosin II (blue), smooth muscle myosin II (green), non-muscle myosin II (brown), and unconventional myosin (tan). Eight SNP substitution sites at Q478, F483, N485, H494, H495, F497, E502, and K505 on the Switch 2 helix are depicted in gray or yellow with space filling models of the unsubstituted side chains. The yellow residue is also implicated in disease. Movement at Switch 2 propagates to the Switch 2 helix and to the converter domain (not shown) via the tip of the helix near K505.
Figure 3 shows the SNP substitutions sites for the SH2/SH1 hinge [M688V, V694E (L694 in MYH1), R695H (H695 in MYH1), G701R, R707S, R710S], converter [R743Q (K743 in MYH1)], and lever-arm [L785M and D787N (Q787 in MYH1)]. SNP substitutions V720I, Y723C, and P735S in the converter, and A801T in the lever-arm, are implicated in heart disease [27]. The converter domain position P735 is implicated is heart disease by a P735L substitution in β-cardiac MHC [27]. The P735S SNP replacement is homozygous in the 4 subpopulation groups tested suggesting the reference sequence is incorrect.
The converter domain has been implicated as the element undergoing elastic distortion in the myosin accompanying force development prior to mechanical work production [58,59]. Site directed mutagenesis in the SH2/SH1 hinge--converter interface, and in the converter domain proper, provided clues for how it performs the conversion of linear translation in the Switch 2 helix to the rotation of the lever-arm [60]. Mutations F713A or F768A completely eliminated or sharply inhibited the ability of smooth muscle myosin to function mechanically. Standard kinetics experiments looking at the ATP binding, hydrolysis, product release, and the fluorescence from the ATP sensitive tryptophan (W511) indicate active site kinetics and transmission of force through the Switch 2 helix are fully intact in the mutants. The findings suggest that the mutations perturb only the converter function. Other experiments, specifically fluorescence resonance energy transfer (FRET), actin sliding assay, and single myosin force development show that both mutants are compromised mechanically. The F713A mutant causes a catastrophic mechanical failure due to the loss of the hydrophobic contact between the SH2/SH1 hinge and converter. The F768A mutant looses the rigidity of the Switch 2 helix/converter contact at F768 making a motor that can rotate its lever-arm but with less force. The SH2/SH1 hinge has a high density of SNP substitutions with 6 occurring in a 28 residue peptide. SNP sites at R707 and R710 are highly conserved among myosin isoforms but do not appear to enter into the crucial interaction with the converter domain. M688 is not conserved among the myosin isoforms.
The sole converter domain SNP substitution not implicated in disease is R743Q that is also common with an AHP of 13.5%. The SNP occurs in myosin X that has a low converter domain sequence homology with myosin 2×. Myosin VI, VII, and X have a unique structure at the head-tail junction wherein a monomeric single α-helix (SAH) competes with the dimeric coiled-coil [9-11]. The SAH domain might function as an elastic element undermining converter domain significance in this isoform. Hypothetically, the converter domain is sensitive to mutation because it usually needs to perform the two functions of the linear-to-rotary motion converter and elastic element.
The L785M, D787N (Q787 in MYH1), and A801T SNP substitutions modify the leverarm. Mutation in this region could interfere with essential light chain (ELC) binding. The loss of light chains, either regulatory (RLC) or ELC, affects myosin morphology and its motor function probably by lowering the rigidity of the lever-arm domain [61]. Either substitution could also directly perturb the lever-arm structure and rigidity also leading to loss of function. None of these possibilities may be happening due to the lever-arm SNP substitutions because their consequences would be severe. Aside from G701R and R743Q, SNPs shown in Figure 3 affect a small or unknown percentage of the population.
Figure 5 shows the SH3 domain (AA30-80) and the sites for the P31T, G57R, V60I, and K68N (A68 in MYH1) substitutions. The SH3 domain is a β-barrel located close to the myosin active site but with uncertain function. The SH3 domain undergoes significant conformational change during ATP hydrolysis including at the G57 site where Ramachandran angle changes consistent with a glycine residue will be inhibited by steric clash with the arginine side chain. Smooth muscle myosin contains tryptophan residues in SH3 and its vicinity that sense conformational change in the N-terminal domain with nucleotide binding to the active site [62]. N-terminus truncated constructs of the Dicty S1 showed SH3 played a role is structural stabilization of S1 and in communication among functional domain within the myosin head [63].
Figure 5.
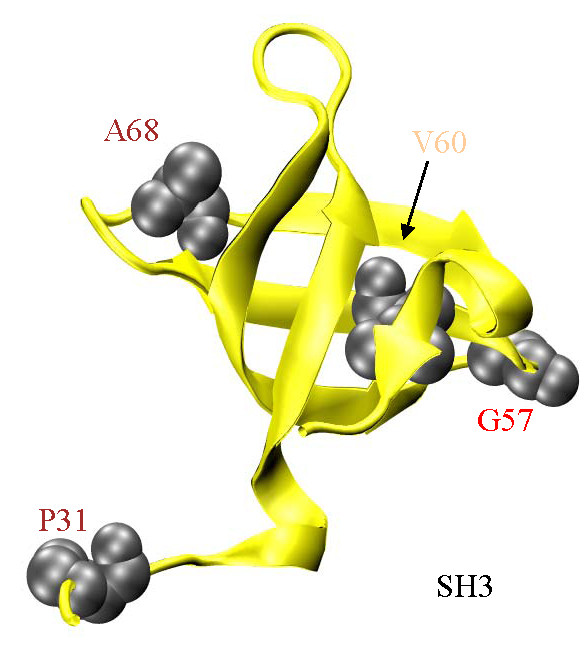
The SH3 domain (yellow, AA30-80) and 4 SNP substitution sites at P31, G57, V60, and A68 are depicted in gray with space filling models of the unsubstituted side chains. SNP residue annotation color coding corresponds to: cardiac myosin II (red), non-muscle myosin II (brown), and unconventional myosin (tan). The function of SH3 is unknown but it is implicated in transmission of influence from ELC to the active site and in supporting the binding of the ELC N-terminus to actin. Several SNP substituted residues adjacent to the 7-stranded β-sheet (Figure 2) are also adjacent to SH3 including I88, E89, H98, and E108.
The essential light chain (ELC) appears to couple SH3 and lever-arm sub-domains in S1. LC1 and LC3 are ELC isoforms expressed in skeletal muscle with the S1 heavy chain binding LC1/RLC or LC3/RLC pairs in equal amounts. LC1 differs from LC3 by a 40 residue extension at the N-terminus. Cardiac myosin has only LC1. The flexible LC1 N-terminus is not resolved in S1 crystal structures and is not shown in Figure 1. Electron cryomicroscopy reconstructions suggest SH3 and the N-terminus of LC1 interact in skeletal muscle myosin [64]. There it is suggested that the SH3/LC1 interaction could facilitate the (N-term)LC1/(N-term)actin interaction known to affect the overall actomyosin interaction in skeletal and cardiac myosins [65,66] and that SH3 could contain an LC1/active-site path of influence known to modulate myosin ATPase [67]. The P31T substitution in MYH14 and V60I in myosin X are common.
SNP implications for actomyosin
Protein-protein contacts in actomyosin were identified by experimental structural studies combined with simulated docking of the myosin and actin crystal structures [68-72], by detecting changes in actin binding strength, actin-activated myosin ATPase, and in vitro motility caused by the mutation of small peptide segments or individual residues in myosin [73-81]. Primary hydrophobic actin contacts are helical segments, AA529-560 and AA652-661, while the unstructured surface Loop 2 (AA626-651) maintains ionic interactions with the actin N-terminus. Secondary sites are an unstructured surface loop, Loop 3 (AA568-580), and the structured Myopathy loop (AA404-417) [68] also on the S1 surface. Figure 6 shows the actin binding myosin surface loops including Loops 2, 3, Myopathy and C-loop. Also shown are actin binding site SNP substitutions F534L and F655S that fall within a primary hydrophobic actin contact.
Figure 6.
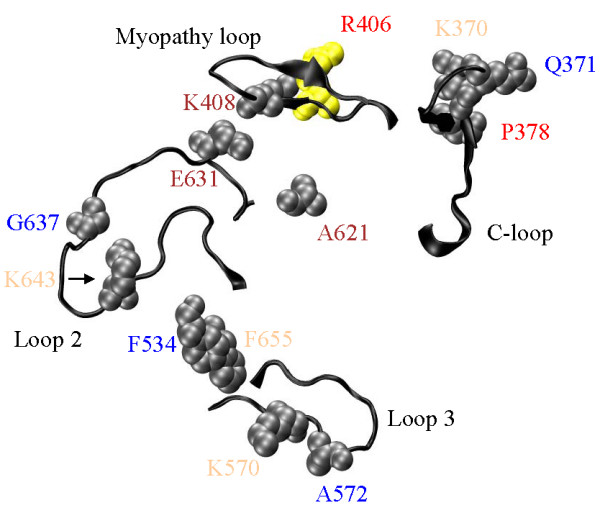
Elements of the actin binding site on S1. Structured surface loops (Myopathy and Cloop, AA404-417 and 363-377), unstructured surface loops (Loops 2 and 3, AA626-651 and 568-580), and F534 and F655 in the primary hydrophobic actin contacts (AA529-560 and 652-661) are actin binding peptides. SNP residue annotation color coding corresponds to: cardiac myosin II (red), skeletal myosin II (blue), non-muscle myosin II (brown), and unconventional myosin (tan). Thirteen SNP substitution sites K370, Q371 and P378 (C-loop); R406 and K408 (Myopathy loop); F534 and F655 (primary hydrophobic contact); K570 and A572 (Loop 3), and, A621, E631, G637, and K643 (Loop 2) are depicted in gray or yellow with space filling models of the unsubstituted side chains. The yellow residue is also implicated in disease.
Experimental work comparing tertiary structure of skeletal and β-cardiac MHC identified the C-loop (AA363-377), Figures 1 and 6, having influence on active site coupling to actin and possibly a direct interaction with actin [82]. Limited proteolysis of skeletal S1 cleaves the heavy chain at Loop 1 (AA200-220) and Loop 2 producing 25, 50, and 20 kDa molecular mass fragments [83]. These loops are involved in regulation of substrate release (Loop 1) and in actin binding and regulation of actin-activated ATPase (Loop 2) [75,78,84-86]. Limited proteolysis of βS1 cleaves the heavy chain at equivalent points and at the C-loop within the 50 kDa fragment [82]. The C-loop cleavage dramatically affects βS1 Mg++ATPase suggesting it participates in energy transduction [82]. Actin binding protected Loop 2 from proteolysis in skeletal S1 indicating Loop 2 involvement in actin binding [87,88]. Actin binding to βS1 fails to inhibit Loop 2 cleavage [82,89] but does inhibit C-loop cleavage [82]. These observations highlight differences in skeletal S1 and βS1 conformation and identify the C-loop as a possible actin binding site. The C-loop was proposed as a site of actin binding in skeletal S1 [71,72], in myosin V [90], and in myosin IE where it is called Loop 4 [39]. We constructed single site mutations in the smooth muscle myosin C-loop and chimeric proteins containing C-loop sequences from βS1 and skeletal S1 to investigate how sequence perturbs C-loop function [20,91]. Based on this work we proposed that the C-loop is an allosteric actin contact sensor initiating actin-activation of the myosin ATPase.
The C-loop SNP substitutions, R370H (K370 in MYH1), Q371P, and P378Q, occur in the myosin VIIA, super-fast myosin and α-cardiac myosin, respectively. In super-fast myosin, the Q/P substitution can be expected to perturb and rigidify the C-loop structure with the proline. We proposed structural rigidity in the C-loop is probably necessary for its effectiveness [19] but overall structure also plays a role and impacts myosin kinetics [20]. Each of these substitutions is expected to affect myosin kinetics related to its interaction with actin, e.g. actin-activated ATPase and motility. Heterozygous Q371P alleles are significantly abundant although no homozygous individuals were detected. Abundance of the other SNPs is untested.
Loop 2 and near Loop 2 SNP substitutions are A621V, T631N (E631 in MYH1), A637V (G637 in MYH1), and E643K (K643 in MYH1). A/V substitution replaces the apolar alanine with the larger apolar valine, T/N exchanges two neutral polar groups, and E/K reverses charge. The effect of Loop 2 on myosin ATPase and myosin interaction with actin has been thoroughly studied with site directed mutagenesis. In Dicty myosin II, Loop 2 tolerates significant sequence changes without affecting function. Function seems to depend on charged or apolar residue distribution but not on specific structural features because the loop is flexible. A/V and T/N substitutions are conservative and unlikely to impact function. The A637V substitution is from perinatal myosin and abundant in this isoform although no homozygous individuals were detected. Abundance of the other Loop 2 SNPs is untested.
The Myopathy Loop is a structured surface loop containing an unusual clustering of heart disease implicated mutations including R406Q/W/L, V407M, V409M, and G410V [27]. It was shown to be an actin binding site [69,92] and modulator of actin activated ATPase [81,93]. At R406, the smooth muscle heavy meromyosin (HMM) model for β-cardiac myosin has largely reiterated the clinical phenotype [94]. The G410V mutant shows modest changes in actomyosin affinity possibly indicating a generally degraded fit between myosin and actin like the R406 mutants [20]. The R406W mutant appears in the SNP data base in apparently healthy individuals. R408C (K408 in MYH1) has not been implicated in disease. None of the Myopathy loop SNPs have significant abundance although homozygous individuals carrying R406W were identified.
The Loop 3 SNP substitutions, R570K (K570 in MYH1) and V572A/D (A572 in MYH1), and the hydrophobic actin binding site residue substitutions, F534L and F655S, are conservative except for F655S. Among the myosin isoforms, all of these residues are variable. The heterozygous allele for F534L in skeletal myosin 2b has significant abundance (2.8%) but no homozygous individuals were identified.
Other myosin S1 SNPs
SNPs appearing in Table 1 but not discussed in the context of Figures 2, 3, 4, 5, and 6 locate to the N-terminus (AA1-219), the upper 50 k (AA220-468) and lower 50 k (AA469-620). The upper 50 k and lower 50 k domains (U50 and L50) designate peptides within the 50 k of S1 on either side of the cleft that closes on strong binding to actin. N-term SNP substitutions include D4A in MYH7 and D4E in MYH13, R18C, R24P/L, T25M (I25 in MYH1), M142I (A142 in MYH1), R145G (G145 in MYH1), I148T (R148 in MYH1), and L199F (I199 in MYH1). The U50 SNP substitutions include H287R, E300D (L300 in MYH1), T307P, P320A (E320 in MYH1), S323C (V323 in MYH1), I326T, V335I (T335 in MYH1), R336H (D335 in MYH1), T345A, V350D/A from myosin 2b and R350W (V350 in MYH1) from myosin X. L50 SNP substitutions are I514T and A594T. SNP substitutions Q329R and I514T are disease implicated. SNPs in the "other myosin S1" category do not fall within, or are not spatially proximal to, presently recognized S1 functional elements directly participating in transduction. The average heterozygous SNP allele frequency for this set ranges from 0 to 17.2% (17.2% for E389D in myosin V with homozygous individuals identified) with most heterozygous substitutions having significant abundance and some untested.
Identifying sites for probing motor dynamics
Figure 7 shows the amino acid sequence of myosin 2× vs AHP for the SNPs in Table 1. Myosin regions or domains identified with specific functions in energy transduction are identified by the broken red line and with a vertical label. Many SNPs have unknown or zero AHP that are both plotted as zero. The remaining substitution sites have a significant-to-large AHP and are identified with a horizontal label. Healthy individuals with homozygous SNP substituted alleles are implied by the significant heterozygous populations. Figure 7A, specifying SNPs in the AA1-400 portion of the MHC show numerous substitutions at the N-terminus and in the AA280-360 peptide segments. These regions are not directly related to core function because they do not fall into the identified functional subdomains. The C-loop and β6-β7 junction are functional elements that are the exceptions in this region because they are SNP modified. Figure 7B, specifying SNPs in the AA401-813 portion of the MHC, shows a somewhat different picture with all but one significant SNP substitution modifying a functional sub-domain. The high AHP sites are candidates for modification by mutagenesis to introduce probes that monitor motor dynamical structure without altering native behavior. Most crucial regions are covered where in introduction of Trp or Cys would facilitate direct (Trp) or indirect, through a specific modification by an extrinsic probe (Cys), observation of myosin dynamics. Remarkably, the G701R substitution in the SH2/SH1 hinge falls into this category.
Figure 7.
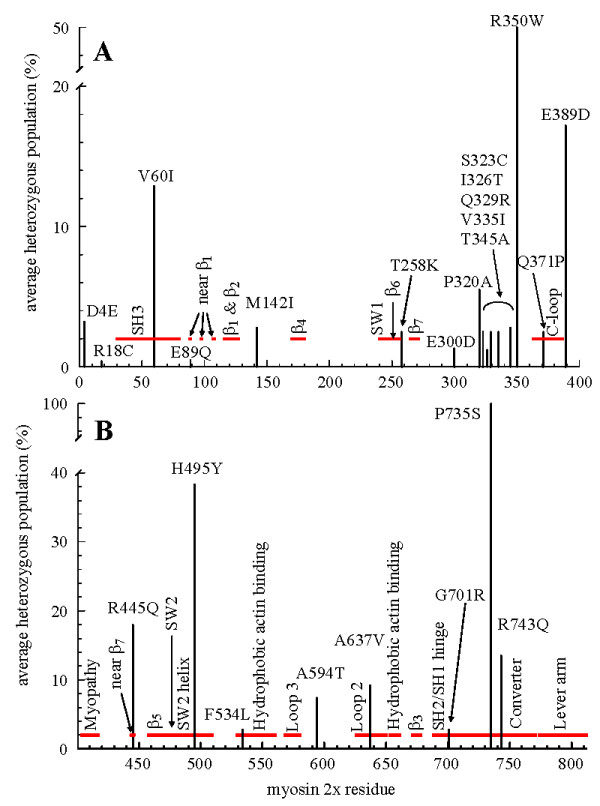
Function-neutral candidate SNP missense substitutions in myosin heavy chain. The x-axis is the myosin 2× (MYH1) sequence from 1-400 in Panel A and 401-812 in Panel B. The y-axis indicates AHP in percent. Substitutions with AHP ≥ 0.5% are denoted by a vertical bar and by the residue substitution and sequence number in a horizontal label. The thick broken horizontal red line indicates myosin functional domain positions described in the text. Domain names are denoted with vertical labels. Converter domain (Panel B) SNP substitution P735S has an AHP showing 100% substitution in the 4 subpopulations tested suggesting the reference sequence is incorrect.
Discussion
SNPs account for about 75% of the number of sequence differences between individuals in a population [95]. They have significant utility in gene mapping studies and there are an increasing number of SNPs that have been shown to have a significant influence on an individual's susceptibility to disease and response to various drug therapies. In some well-studied genes and gene families, patterns of SNPs within the coding sequences have also provided some insight into protein function. Perhaps the best known example of this is the SNP (rs334) that leads to sickle cell anemia. This Glu6Val change in the protein sequence has an allele frequency of about 25% in Yoruban populations and creates well-known negative and presumed positive consequences for the individuals that carry this sequence variant. Myosin SNPs could likewise create negative and positive consequences for certain populations with specific functional requirements that should be explored experimentally. Nonsynonymous SNPs that have truly neutral functional consequences also provide important information about protein function, because they highlight regions of protein structure that are tolerant of structural variation. We discuss three scenarios for interpreting SNP data denoted, the function-neutral, the too-robust, and the too-sensitive.
SNPs reside in healthy people, otherwise they are deleted by natural selection, hence it might be hypothesized that the amino acid substitution is function-neutral. In this case, SNPs identify residues or components of the motor not directly related to core function making them candidate sites for adventitious modification. An example is the R240W substitution that places a spectroscopic probe in the 7-stranded β-sheet at the interface of the active and actin-binding sites. It is thought that coordination of ATP hydrolysis with actin binding and release is mediated by this β-sheet and an optical probe of its structure would be valuable. R240 is not highly conserved among species but infrequently substituted by aromatic residues. Its negligible AHP raises uncertainty about potential use although homozygous individuals were reported. G701 is the pivot for the lever-arm swing and thought to be necessary for myosin function. The G701R SNP substitution has 2.8% AHP but it is difficult to understand how this substitution could be function-neutral in the swinging lever-arm picture for force production. Other unlikely function-neutral candidates are the R246H and E469Q substitutions with unknown and insignificant AHP, respectively. R246 and E469 form the SW1/SW2 salt-bridge regulating phosphate release in ATPase in the rate limiting step for muscle myosins. R246H originates from the cellular myosin VII isoform where the native rate limiting ADP release minimizes R246 significance. E469Q originates from β-cardiac myosin using the phosphate release mechanism. Under the function-neutral hypothesis, the collection of SNPs at R246, E469 and the vicinity of the 7-stranded β-sheet would imply a diminished role for this structure in energy transduction.
Figure 7 identifies the most likely candidates for the function-neutral SNPs because they have significant to large AHP implying the presence of healthy individuals carrying homozygous substituted alleles. Here SNPs cluster in a few regions of the myosin head not identified with core functionality but also cover most function-crucial regions in the S1. SNPs in Figure 7 may be most useful as leads for the introduction of probes that monitor motor dynamical structure without altering native behavior.
An alternative to the function-neutral hypothesis is that SNP substitutions modify functional structures too-robust to be disturbed by otherwise intrusive sequence changes. Excluding for a moment the E469Q and G701R special cases, several structures in S1 fulfill expectations implied by this hypothesis. The 7-stranded β-sheet and vicinity collected 17 SNP substitution sites (excluding known disease related sites, see Table 1) over 14 of the 22 myosin genes where SNPs were identified. The too-robust hypothesis implies that the energy transduction mechanism contained in the 7-stranded β-sheet is too well designed by evolution to be compromised by any of the SNP substitutions. Actin binding by S1, modified by SNPs in the main hydrophobic binding site (F534L and F655S), in Loop 3 (R570K and V572A/D), Loop 2 (A621V, T631N, A637V, and E643K), the Myopathy loop (R408C, not including the disease implicated R406W), and the C-loop (R370H, Q371P and P378Q), is another potential too-robust site. The switch 2 helix, charged with propagating linear motion from the active site to the converter domain, suffers seven SNP substitutions and functions while the SH2/SH1 hinge and lever-arm successfully manage impacts of six and two SNP substitutions, respectively. Returning to E469Q and G701R, the functional impact of E469Q (7-stranded β-sheet) has not been evaluated in vitro, however, other substitutions at this site were disruptive to muscle myosin function except when the polarity of the R246/E469 salt-bridge was reversed. Then the mutant S1 functioned near normally. The E469Q substitution seems likely to disrupt the interaction with R246 thereby altering specific rates in the ATPase cycle but potentially within boundaries that do not overly impact function. The G701R substitution is difficult to rationalize even in the too-robust hypothesis. The functional impact of G701R has likewise not been evaluated in vitro but a glycine swivel is disrupted by the presence of any side chain with a β-carbon. How super-fast myosin could function with G701R needs clarification.
The too-robust hypothesis is often the implicit working hypothesis for those utilizing site directed mutagenesis to study protein mechanisms. It may sometimes be too optimistic an assumption for interpreting SNPs. The SNP substitution may affect protein function compensated by redundant functionality outside the affected protein in the larger physiological system. Different SNPs will have different applicable hypotheses and the challenge is to identify the correct one to apply.
We anticipated SNP clustering in a few regions of the myosin head identifying the expendable components of the system unrelated to core functionality. This occurred to some extent within the narrow context of the significant AHP SNPs. Residues 1-400 in MHC show significant AHP SNP substitutions at the N-terminus and in the AA280-360 peptide segments that do not fall into the identified functional sub-domains. However, residues 401-812 show a different picture with all but one significant AHP SNP substitution modifying functional sub-domains. Overviewing the total set of substitutions, the S1 SNPs are distributed spatially over the protein more uniformly than expected. Figure 8 shows the annotated S1 from Figure 1 with the SNP substitution sites identified by the solid atom rendering in yellow. Most SNPs locate within, or to the vicinity of, domains intimately connected to the myosin core functionality. They do not seem to select any one structural feature for a diminished role in the core functionality except possibly the 7-stranded β-sheet that attracted so many SNP substitutions.
Figure 8.
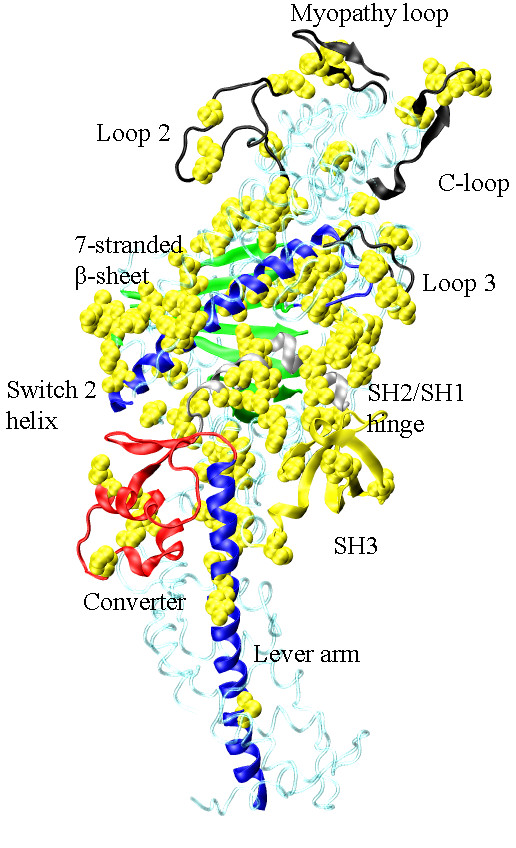
The S1 crystal structure depicted in Figure 1 and with the heavy chain SNP substitution sites indicated with space filling models of the unsubstituted side chains. SNP substitutions locate throughout the myosin S1 heavy chain.
The Myopathy loop and the converter domain nearly escaped modification by SNPs not already disease related. They each have one SNP substitution not known to be disease implicated. As suggested by its name, known mutations in the Myopathy loop accompany heart disease [27]. SNPs do not appear in the Myopathy loop because most substitutions cause serious functional deficit. The same appears to be true for the converter where there are 10 or more heart disease linked mutations. These structures are too-sensitive to substitution to be a target for SNPs. The large AHP for the SNP substitution R743Q in the converter domain (Figure 7) appears to have identified a unique site in the domain that can be safely modified. At least for myosin, regions or domains excluded from SNP substitution may be the most precisely defined. These high sensitivity regions are anti-targets for SNPs because they are not functionally robust (i.e. too-sensitive) rather than because they are the only domains necessary for function.
Conclusions
Single nucleotide polymorphisms of the myosin heavy chain were mined from the NCBI data base using computer programs written in perl that identified human nonsynonymous SNP amino acid missense substitutions for any MHC gene. Twenty-two MHC genes were searched including muscle and non-muscle myosin isoforms. Excluding disease implicated residues and sites without homology with skeletal myosin 2×, the 71 SNPs identified were distributed spatially homogeneously over MHC with 22 falling outside specific functional domains. The remaining 49 SNPs were spatially related to the 7-stranded β-sheet mediating active site and actin binding site communication (17 SNPs), the actin binding peptides (12 SNPs), switch 2 helix (7 SNPs), SH2/SH1 hinge (6 SNPs), SH3 (4 SNPs), lever-arm (2 SNPs), and converter (1 SNP). Two functional elements were almost devoid of SNPs, the converter domain and the Myopathy loop. These elements are burdened with an unusually high number of disease implicated mutations. The SNP substitution sites in MHC suggest they can infiltrate domains that are engineered by evolution to be "too-robust" to be disturbed by otherwise intrusive sequence changes, and, "function-neutral" sites where substitution is immaterial. Disease implicated mutations and the absence of SNP substitutions map to regions "too-sensitive" to be modified because they have not evolved a robust sequence paradigm for performing their function.
Methods
Automated SNP retrieval
SNP data records were retrieved using a computer program written in perl. Perl was chosen because of its text-parsing and internet capabilities; and because it is widely available. The basic distribution of perl is available for use with various operating systems, including Microsoft Windows, linux/unix, and Macintosh. The first program (get_snps_flt.pl) uses NCBI eUtils to search for missense SNPs in the human myosin genes. The results are saved locally as flat text files then parsed into tables using a second perl program (make_table.pl). The get_snps_flt.pl and make_table.pl programs are listed in the additional file 1 where there are instructions for their use.
The NCBI SNP database stores individual SNP submissions and assigns them unique identifiers that are prefixed with ss (submitter sequence). These individual records are eventually assigned to groups so that all SNPs that occur at a particular position in a gene are in the same cluster. Each cluster has a unique numeric identifier that is prefixed with rs (reference sequence). Table 2 shows a representative tabular output from make_table.pl. In every cluster, the reference (or "normal") allele is compared to the SNP substituted allele to determine the missense mutation in the expressible protein. Up to three SNPs may be reported in a cluster, corresponding to the three possible non-reference bases. The residue sequence in Table 2 is the sequence of the gene product, or myosin isoform, in which the SNP is detected. Sequence alignment with BLAST positioned the SNP substitutions on the myosin crystal structure.
Table 2.
SNPs for smooth muscle myosin MYH11
| ID of SNP Cluster | Gene | Function | Base Change | Amino Acid Change | Peptide Sequence#a |
|---|---|---|---|---|---|
| rs61734198b | MYH11 | missense | G -->C | Q -->H | 473 |
| MYH11 | missense | G -->T | Q -->H | 473 | |
| MYH11 | missense | G -->C | Q -->H | 480 | |
| MYH11 | missense | G -->T | Q -->H | 480 | |
| rs16967494 | MYH11 | missense | G -->A | A -->T | 1234 |
| MYH11 | missense | G -->A | A -->T | 1241 | |
| rs16967494 | MYH11 | missense | G -->A | A -->T | 1234 |
| MYH11 | missense | G -->A | A -->T | 1241 | |
| rs35176378 | MYH11 | missense | A -->G | M -->V | 1508 |
| MYH11 | missense | A -->G | M -->V | 1515 | |
| rs35035518 | MYH11 | missense | C -->T | S -->L | 883 |
| MYH11 | missense | C -->T | S -->L | 890 | |
| rs34321232 | MYH11 | missense | A -->C | K -->Q | 1621 |
| MYH11 | missense | A -->C | K -->Q | 1628 | |
| rs34263860 | MYH11 | missense | G -->A | A -->T | 1104 |
| MYH11 | missense | G -->A | A -->T | 1111 | |
| rs16967510 | MYH11 | missense | T -->C | V -->A | 1289 |
| MYH11 | missense | T -->C | V -->A | 1296 | |
| rs16967494 | MYH11 | missense | G -->A | A -->T | 1234 |
| MYH11 | missense | G -->A | A -->T | 1241 | |
| rs12149651 | MYH11 | missense | C -->A | L -->M | 1053 |
| MYH11 | missense | C -->A | L -->M | 1060 | |
| rs7196804 | MYH11 | missense | G -->A | V -->M | 1310 |
| MYH11 | missense | G -->A | V -->M | 1317 | |
| rs1801902 | MYH11 | missense | A -->G | T -->A | 864 |
| MYH11 | missense | A -->G | T -->A | 871 |
aSNPs corresponding to amino acid residues in the tail portion of myosin appear in the output for the automated SNP retrieval program but lie outside the motor domain of myosin and are not discussed in the text.
bSNP rs61734198 is an example of a two base change (G→C and G→T) SNP where His substitutes for Gln in both cases. The MYH11 gene product includes splicing variants with different sequences hence the same SNP has two sequence numbers in the variants that are in this case sm1A (AA473) and sm2B (AA480).
Monte Carlo Molecular Dynamics (MCMD) simulation of myosin dynamics
The dominant kinetic pathway for the actomyosin ATPase cycle is summarized by solid arrows in Scheme 1.
Scheme 1.
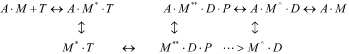
M, M*, M**, and M^ are intermediates corresponding to distinct myosin conformations, A is actin, T is ATP, D is ADP, and P is inorganic phosphate. M* and M** weakly bind actin while M and M^ strongly bind actin. The broken arrow (···>) in the bottom row indicates the rate limiting product release step in the absence of actin. Work production occurs when M** ·D·P weakly binds actin (vertical transition), releases product P with the opening of the active site "back door" (R246/E469 salt-bridge), and forms the strong actin bond A·M^·D. The lever-arm rotates to impel actin ending in the low free energy rigor state A.M. Cross-bridge repriming begins with ATP binding to A.M initiating dissociation from actin, ATP hydrolysis, and reversal of the lever-arm power stroke rotation. The power stroke, envisioned by comparison of the M** and M* crystal structures, rotates the lever-arm through ~70 degrees [17,53]. We performed a dynamics simulation for myosin joining the known M** and M* transient intermediate structures in the Scheme 1pathway using a non-equilibrium Monte Carlo molecular dynamics (MCMD) simulation of the M** → M* conformation trajectory [19].
The lever-arm swing conformation change in myosin occurs on the millisecond time domain and involves many atoms. We developed the new dynamics simulation strategy because system complexity excludes use of standard molecular dynamics methods. All conformation trajectories confirm that during ATPase, an entropy dominated structural transition in the actin binding domain of S1 is a free energy barrier ensuring that product release is rate limiting in the absence of actin and prior to the lever-arm swing in agreement with the conventional myosin mechanism. MCMD simulation of the power stroke indicated that two complementary peptides, designated U50a and U50b (AA145-361 and AA362-462 peptides), mainly in the 50 kDa actin binding domain encompass the free energy barrier to product release and the anti-barrier, respectively. Simulation shows that coupling the U50a and U50b conformation transitions removes the free energy barrier to product release. The C-loop is a structured surface loop linking U50a and U50b whose structure would be perturbed with actin binding. These circumstances suggest that perturbation of the C-loop with actin binding couples U50a and U50b transitions causing product release and power stroke initiation.
Homology Modeling
Homology modeling uses a crystal structure template to constrain a target sequence of unknown structure. The template is the chicken skeletal myosin S1 (2mys, [17]) and the target sequence is the human fast skeletal MHC isoform 2× (MYH1) and essential light chain 3 (ELC3). Homology modeling was done with Modeller 9.4 [96]. All structures shown depict S1 from myosin 2×.
Accurate model building depends on target and template sequence similarity and alignment. Template and target sequences for the MHC and ELC are practically identical (>91% sequence identity) suggesting Modeller produces a reliable structure [96]. We did alignment using the NCBI BLAST 2 sequences protocol http://www.ncbi.nlm.nih.gov/blast/bl2seq/wblast2.cgi. Unstructured surface loops on myosin were not resolved in the crystal structures but were added using the appropriate Modeller routine. Homology model accuracy was evaluated using the Discrete Optimized Protein Energy (DOPE) score as suggested in the Modeller tutorial. The DOPE score is proportional to potential energy per residue, smoothed over a 15 residue window, and normalized by the number of restraints acting on each residue. It indicates problem regions in the homology model when profiles are compared from target and template sequences. The DOPE profile showed no unusual problem regions over the entire peptide.
Visualizations of myosin structures were created in Visual Molecular Dynamics (VMD) [97] then rendered in POV-Ray http://www.povray.org/ and output as bitmap files.
Abbreviations
(AA): amino acid; (AHP): average heterozygous population; (ASA): accessible surface area; (βS1): β-ventricular myosin subfragment 1; (Dicty): Dictyostelium discoideum; (DOPE): discrete optimized protein energy; (ELC): myosin essential light chain; (EOM): extraocular muscle; (L50): lower 50 k domain of S1 containing AA469-620; (LC1): long ELC isoform; (LC3)short ELC isoform; (MHC): myosin heavy chain; (MLC): myosin light chain; (NCBI): National Center for Biotechnology Information; (RLC): myosin regulatory light chain; (S1): myosin subfragment 1; (SAH): single α-helix; (SNP): single nucleotide polymorphism; (U50): upper 50 k domain of S1 containing AA220-468; (U50a): U50 sub-domain (AA145-361): containing free energy barrier to phosphate release in myosin ATPase; (U50b): U50 sub-domain (AA362-462): containing free energy anti-barrier to phosphate release in myosin ATPase
Authors' contributions
TPB performed the SNP searches, aligned protein sequences, and drafted the manuscript. KLN wrote the perl software and adapted it for use on the project. EDW and KA provided conceptual guidance and helped draft the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Instructions for retrieving SNPs. Two perl programs provide automated retrieval and organization of SNP data from the NCBI database. Additional file 1 contains the perl programs and instructions for their use.
Contributor Information
Thomas P Burghardt, Email: burghardt@mayo.edu.
Kevin L Neff, Email: neff.kevin@mayo.edu.
Eric D Wieben, Email: wieben.eric@mayo.edu.
Katalin Ajtai, Email: ajtai.katalin@mayo.edu.
Acknowledgements
This work was supported by NIH-NIAMS grant R01AR049277 and by the Mayo Clinic Rochester. We thank Dr Alan R. Penheiter at the Mayo Molecular Medicine Program for guiding us at the start of this project.
References
- Jarjanazi H, Savas S, Pabalan N, Ozcelik H. Biological implications of SNPs in signal peptide domains of human proteins. Proteins Structure, Function and Bioinformatics. 2008;70:394–403. doi: 10.1002/prot.21548. [DOI] [PubMed] [Google Scholar]
- Knöll R, Kostin S, Klede S, Savvatis K, Klinge L, Stehle I, Gunkel S, Kötter S, Babicz K, Sohns M, Miocic S, Didié M, Knöll G, Zimmermann WH, Thelen P, Bickeböller H, Maier L, Schaper W, Schaper J, Kraft T, Tschöpe C, Linke WA, Chien KR. A Common MLP (Muscle LIM Protein) Variant Is Associated With Cardiomyopathy. Circ Res. 2010;107:1–10. doi: 10.1161/CIRCRESAHA.109.206243. [DOI] [PubMed] [Google Scholar]
- Boyko AR, Williamson SH, Indap AR, Degenhardt JD, Hernandez RD, Lohmueller KE, Adams MD, Schmidt S, Sninsky JJ, Sunyaev SR, White TJ, Nielsen R, Clark AG, Bustamante CD. Assessing the Evolutionary Impact of Amino Acid Mutations in the Human Genome. PLoS Genet. 2008;4:e1000083. doi: 10.1371/journal.pgen.1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen JL, Gruschy-Knudsen T, Sandri C, Larsson L, Schiaffino S. Bed rest increases the amount of mismatched fibers in human skeletal muscle. J Appl Physiol. 1999;86:455–460. doi: 10.1152/jappl.1999.86.2.455. [DOI] [PubMed] [Google Scholar]
- Harridge SDR. Plasticity of human skeletal muscle: gene expression to in vivo function. Exp Physiol. 2007;92:783–797. doi: 10.1113/expphysiol.2006.036525. [DOI] [PubMed] [Google Scholar]
- Williamson DL, Godard MP, Porter DA, Costill DL, Trappe SW. Progressive resistance training reduces myosin heavy chain coexpression in single muscle fibers from older men. J Appl Physiol. 2000;88:627–633. doi: 10.1152/jappl.2000.88.2.627. [DOI] [PubMed] [Google Scholar]
- Zhou M-Y, Klitgaard H, Saltin B, Roy RR, Edgerton VR, Gollnick PD. Myosin heavy chain isoforms of human muscle after short term spaceflight. J Appl Physiol. 1995;78:1740–1744. doi: 10.1152/jappl.1995.78.5.1740. [DOI] [PubMed] [Google Scholar]
- Huxley HE. The mechanism of muscular contraction. Science. 1969;164:1356–1366. doi: 10.1126/science.164.3886.1356. [DOI] [PubMed] [Google Scholar]
- Phichith D, Travaglia M, Yang Z, Liu X, Zong AB, Safer D, Sweeney HL. Cargo binding induces dimerization of myosin VI. Proc Natl Acad Sci USA. 2009;106:17320–17324. doi: 10.1073/pnas.0909748106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peckham M, Knight PJ. When a predicted coiled coil is really a single α-helix, in myosins and other proteins. Soft Matter. 2009;5:2493–2503. [Google Scholar]
- Knight PJ, Thirumurugan K, Xu Y, Wang F, Kalverda AP, Stafford WF III, Sellers JR, Peckham M. The Predicted Coiled-coil Domain of Myosin 10 Forms a Novel Elongated Domain That Lengthens the Head. J Biol Chem. 2005;280:34702–34708. doi: 10.1074/jbc.M504887200. [DOI] [PubMed] [Google Scholar]
- Sinard JH, Stafford WF, Pollard TD. The Mechanism of Assembly of Acanthamoeba Myosin-II Minifilaments: Minifilaments Assemble by Three Successive Dimerization Steps. J Cell Biol. 1989;109:1537–1547. doi: 10.1083/jcb.109.4.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta AD, Rock RS, Rief M, Spudich JA, Mooseker MS, Cheney RE. Myosin-V is a processive actin-based motor. Nature. 1999;400:590–593. doi: 10.1038/23072. [DOI] [PubMed] [Google Scholar]
- Onishi H, Morales MF. A closer look at energy transduction in muscle. Proc Natl Acad Sci USA. 2007;104:12714–12719. doi: 10.1073/pnas.0705525104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houdusse A, Cohen C. Structure of the regulatory domain of scallop myosin at 2 A resolution: implications for regulation. Structure. 1996;4:21–32. doi: 10.1016/S0969-2126(96)00006-8. [DOI] [PubMed] [Google Scholar]
- Highsmith S. Lever arm model of force generation by actin-myosin-ATP. Biochemistry. 1999;38:9792–9797. doi: 10.1021/bi9907633. [DOI] [PubMed] [Google Scholar]
- Rayment I, Rypniewski WR, Schmidt-Base K, Smith R, Tomchick DR, Benning MM, Winkelmann DA, Wesenberg G, Holden HM. Three-dimensional structure of myosin subfragment-1: A molecular motor. Science. 1993;261:50–58. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- Kirshenbaum K, Young M, Highsmith S. Predicting allosteric switches in myosins. Protein Sci. 1999;8:1806–1815. doi: 10.1110/ps.8.9.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burghardt TP, Hu JY, Ajtai K. Myosin dynamics on the millisecond time scale. Biophys Chem. 2007;131:15–28. doi: 10.1016/j.bpc.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajtai K, Halstead MF, Nyitrai M, Penheiter AR, Zheng Y, Burghardt TP. The Myosin CLoop Is an Allosteric Actin Contact Sensor in Actomyosin. Biochemistry. 2009;48:5263–5275. doi: 10.1021/bi900584q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton MJ, Brandon CA, Morris TJ, Braun TW, Yaw KM, Sciote JJ. Abundant expression of myosin heavy-chain IIB RNA in a subset of human masseter muscle fibres. Archives of Oral Biology. 2001;46:1039–1050. doi: 10.1016/S0003-9969(01)00066-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsch-Mizrachi I, Travis M, Blau H, Leinwand LA. Expression and DNA sequence analysis of a human embryonic skeletal muscle myosin heavy chain gene. Nucleic Acid Research. 1989;17:6167–6179. doi: 10.1093/nar/17.15.6167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feghali R, Leinwand LA. Molecular genetic characterization of a developmentally regulated human perinatal myosin heavy chain. The Journal of Cell Biology. 1989;108:1791–1797. doi: 10.1083/jcb.108.5.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZB, Rossmanith GH, Hoh JFY. Cross-Bridge Kinetics of Rabbit Single Extraocular and Limb Muscle Fibers. Invest Ophthalmol Vis Sci. 2000;41:3770–3774. [PubMed] [Google Scholar]
- Toydemir RM, Chen H, Proud VK, Martin R, van Bokhoven H, Hamel BC, Tuerlings JH, Stratakis CA, Jorde LB, Bamshad MJ. Trismus-Pseudocamptodactyly Syndrome Is Caused by Recurrent Mutation of MYH8. Am J Med Gen. 2006;140:2387–2393. doi: 10.1002/ajmg.a.31495. [DOI] [PubMed] [Google Scholar]
- Yamauchi-Takihara K, Sole MJ, Liew JC, Ing D, Liew C-C. Characterization of human cardiac myosin heavy chain genes. Proc Natl Acad Sci USA. 1989;86:3504–3508. doi: 10.1073/pnas.86.10.3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Driest SL, Jaeger MA, Ommen SR, Will ML, Gersh BJ, Tajik AJ, Ackerman MJ. Comprehensive analysis of the beta-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy. Journal of the American College of Cardiology. 2004;44:602–610. doi: 10.1016/j.jacc.2004.04.039. [DOI] [PubMed] [Google Scholar]
- Babu GJ, Warshaw DM, Perisamy M. Smooth muscle myosin heavy chain isoforms and their role in muscle physiology. Microscopy Research and Techniques. 2000;50:532–540. doi: 10.1002/1097-0029(20000915)50:6<532::AID-JEMT10>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Léguillette R, Gil FR, Zitouni N. (+)Insert smooth muscle myosin heavy chain (SM-B) isoform expression in human tissues. Am J Physiol. 2005;289:C1277–C1285. doi: 10.1152/ajpcell.00244.2004. [DOI] [PubMed] [Google Scholar]
- Sweeney HL, Rosenfeld SS, Brown F, Faust L, Smith J, Xing J, Stein LA, Sellers JR. Kinetic tuning of myosin via a flexible loop adjacent to the nucleotide binding pocket. J Biol Chem. 1998;273:6262–6270. doi: 10.1074/jbc.273.11.6262. [DOI] [PubMed] [Google Scholar]
- Lauzon A-M, Tyska MJ, Rovner AS, Freyzon Y, Warshaw DM, Trybus KM. A 7-amino-acid insert in the heavy chain nucleotide binding loop alters the kinetics of smooth muscle myosin in the laser trap. Journal of Muscle Research and Cell Motility. 1998;19:825–837. doi: 10.1023/A:1005489501357. [DOI] [PubMed] [Google Scholar]
- Halstead MF, Ajtai K, Penheiter AR, Spencer JD, Zheng Y, Morrison EA, Burghardt TP. An unusual transduction pathway in human tonic smooth muscle myosin. Biophys J. 2007;93:3555–3566. doi: 10.1529/biophysj.106.100818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker ML, Burgess SA, Sellers JR, Wang F, Hammer JA, Trinick J, Knight PJ. Twoheaded binding of a processive myosin to F-actin. Nature. 2000;405:807. doi: 10.1038/35015598. [DOI] [PubMed] [Google Scholar]
- Sakamoto T, Amitani I, Yokota E, Ando T. Direct observation of processive movement by individual myosin V molecules. Biochem Biophys Res Commun. 2000;272:586–590. doi: 10.1006/bbrc.2000.2819. [DOI] [PubMed] [Google Scholar]
- Komori Y, Iwane AH, Yanagida T. Myosin-V makes two brownian 90° rotations per 36-nm step. Nat Struct Mol Biol. 2007;14:968–973. doi: 10.1038/nsmb1298. [DOI] [PubMed] [Google Scholar]
- Schachat F, Briggs MM. Phylogenetic implications of the superfast myosin in extraocular muscles. J Exp Biol. 2002;205:2189–2201. doi: 10.1242/jeb.205.15.2189. [DOI] [PubMed] [Google Scholar]
- Yang T, Pfister M, Blin N, Zenner HP, Pusch CC, Smith RJ. Genetic heterogeneity of deafness phenotypes linked to DFNA4. Am J Med Gen A. 2005;139:9–12. doi: 10.1002/ajmg.a.30989. [DOI] [PubMed] [Google Scholar]
- Bare LAP, Morrison ACP, Rowland CMM, Shiffman DP, Luke MMM, Iakoubova OAM, Kane JPM, Malloy MJM, Ellis SGM, Pankow JSP, Willerson JTM, Devlin JJP, Boerwinkle EP. Five common gene variants identify elevated genetic risk for coronary heart disease. Genetics in Medicine. 2007;9:682–689. doi: 10.1097/GIM.0b013e318156fb62. [DOI] [PubMed] [Google Scholar]
- Kollmar M, Durrwang U, Kliche W, Manstein DJ, Kull FJ. Crystal structure of the motor domain of a class-l myosin. EMBO Journal. 2002;21:2517–2525. doi: 10.1093/emboj/21.11.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieto-Trivedi A, Dash S, Coluccio LM. Myosin surface loop 4 modulates inhibition of actomyosin 1b ATPase activity by tropomyosin. Biochemistry. 2007;46:2779–2786. doi: 10.1021/bi602439f. [DOI] [PubMed] [Google Scholar]
- Menetrey J, Bahloul A, Wells AL, Yengo CM, Morris CA, Sweeney HL, Houdusse A. The structure of the myosin VI motor reveals the mechanism of directionality reversal. Nature. 2005;435:779–785. doi: 10.1038/nature03592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Li A, Chen LQ, Houdusse A, Selvin PR, Sweeney HL. The unique insert at the end of the myosin VI motor is the sole determinant of directionality. Proc Natl Acad Sci USA. 2007;104:778–783. doi: 10.1073/pnas.0610066104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Gibbs D, Lillo C, Azarian SM, Legacki E, Zhang XM, Yang XJ, Williams DS. Lentiviral gene replacement therapy of retinas in a mouse model for Usher syndrome type 1B. Gene Ther. 2007;14:584–594. doi: 10.1038/sj.gt.3302897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson SG, Cideciyan AV, Aleman TS, Sumaroka A, Roman AJ, Gardner LM, Prosser HM, Mishra M, Bech-Hansen NT, Herrera W, Schwartz SB, Liu XZ, Kimberling WJ, Steel KP, Williams DS. Usher syndromes due to MYO7A, PCDH15, USH2A or GPR98 mutations share retinal disease mechanism. Hum Mol Genet. 2008;17:2405–2415. doi: 10.1093/hmg/ddn140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambara T, Ikebe M. A Unique ATP Hydrolysis Mechanism of Single-headed Processive Myosin, Myosin IX. J Biol Chem. 2006;281:4949–4957. doi: 10.1074/jbc.M509141200. [DOI] [PubMed] [Google Scholar]
- Berg JS, Cheney RE. Myosin-X is an unconventional myosin that undergoes intrafilopodial motility. Nature Cell Biology. 2002;4:246–250. doi: 10.1038/ncb762. [DOI] [PubMed] [Google Scholar]
- Bennett RD, Mauer AS, Strehler EE. Calmodulin-like Protein Increases Filopodiadependent Cell Motility via Up-regulation of Myosin-10. J Biol Chem. 2007;282:3205–3212. doi: 10.1074/jbc.M607174200. [DOI] [PubMed] [Google Scholar]
- Furch M, Fujita-Becker S, Geeves MA, Holmes KC, Manstein DJ. Role of the saltbridge between switch-1 and switch-2 of Dictyostelium myosin. J Mol Biol. 1999;290:797–809. doi: 10.1006/jmbi.1999.2921. [DOI] [PubMed] [Google Scholar]
- Onishi H, Ohki T, Mochizuki N, Morales MF. Early stages of energy transduction by myosin: Roles of Arg in Switch l, of Glu in Switch II, and of the salt-bridge between them. Proc Natl Acad Sci USA. 2002;99:15339–15344. doi: 10.1073/pnas.242604099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran GN, Venkatachalam CM, Krimm S. Stereochemical criteria for polypeptide and protein chain conformations III. Helical and hydrogen-bonded polypeptide chains. Biophys J. 1966;6:849–872. doi: 10.1016/S0006-3495(66)86699-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinose F, Wang SX, Kidambi US, Moncman CL, Winkelmann DA. Glycine 699 is pivotal for the motor activity of skeletal muscle myosin. J Cell Biol. 1996;134:895–909. doi: 10.1083/jcb.134.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson B, Ruppel KM, Wu Y, Spudich JA. Cold-sensitive mutants G680V and G691C of Dictyostelium Myosin II confer dramatically different biochemical defects. J Biol Chem. 1997;272:27612–27617. doi: 10.1074/jbc.272.44.27612. [DOI] [PubMed] [Google Scholar]
- Dominguez R, Freyzon Y, Trybus KM, Cohen C. Crystal structure of a vertebrate smooth muscle myosin motor domain and its complex with the essential light chain: visualization of the pre-power stroke state. Cell. 1998;94:559–571. doi: 10.1016/S0092-8674(00)81598-6. [DOI] [PubMed] [Google Scholar]
- Geeves MA, Holmes KC. Structural mechanism of muscle contraction. Annu Rev Biochem. 1999;68:687–728. doi: 10.1146/annurev.biochem.68.1.687. [DOI] [PubMed] [Google Scholar]
- Geeves MA, Fedorov R, Manstein DJ. Molecular mechanism of actomyosin-based motility. Cellular and Molecular Life Sciences. 2005;62:1462–1477. doi: 10.1007/s00018-005-5015-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coureux P-D, Sweeney HL, Houdusse A. Three myosin V structures delineate essential features of chemo-mechanical transduction. EMBO J. 2004;23:4527–4537. doi: 10.1038/sj.emboj.7600458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace CN, Scholtz JM. A Helix Propensity Scale Based on Experimental Studies of Peptides and Proteins. Biophys J. 1998;75:422–427. doi: 10.1016/S0006-3495(98)77529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler J, Winkler G, Schulte I, Scholz T, McKenna W, Brenner B, Kraft T. Mutation of the myosin converter domain alters cross-bridge elasticity. Proc Natl Acad Sci USA. 2002;99:3557–3562. doi: 10.1073/pnas.062415899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebohm B, Martinmehr F, Köhler J, Francino A, Navarro-Lopez F, Perrot A, Özcelik C, McKenna WJ, Brenner B, Kraft T. Cardiomyopathy mutations reveal variable region of myosin converter as a major element of cross-bridge compliance. Biophys J. 2009;97:806–824. doi: 10.1016/j.bpj.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohki T, Mikhailenko SV, Morales MF, Onishi H, Mochizuki N. Transmission of force and displacement within the myosin molecule. Biochemistry. 2004;43:13707–13714. doi: 10.1021/bi048954f. [DOI] [PubMed] [Google Scholar]
- Lowey S, Waller GS, Trybus KM. Skeletal muscle myosin light chains are essential for physiological speeds of shortening. Nature. 1993;365:454–456. doi: 10.1038/365454a0. [DOI] [PubMed] [Google Scholar]
- van Duffelen M, Chrin LR, Berger CL. Nucleotide dependent intrinsic fluorescence changes of W29 and W36 in smooth muscle myosin. Biophys J. 2004;87:1767–1775. doi: 10.1529/biophysj.104.044388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita-Becker S, Tsiavaliaris G, Ohkura R, Shimada T, Manstein DJ, Sutoh K. Functional characterization of the N-terminal region of myosin-2. J Biol Chem. 2006;281:36102–36109. doi: 10.1074/jbc.M605171200. [DOI] [PubMed] [Google Scholar]
- Lowey S, Saraswat LD, Liu H, Volkmann N, Hanein D. Evidence for an interaction between SH3 domain and the N-terminal extension of the essential light chain in class II myosins. Journal of Molecular Biology. 2007;371:902–913. doi: 10.1016/j.jmb.2007.05.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashibara T, Miyanishi T. Binding of the amino-terminal region of myosin alkali l light chain to actin and its effect on actin-myosin interaction. Biochemistry. 1994;33:12821–12827. doi: 10.1021/bi00209a013. [DOI] [PubMed] [Google Scholar]
- Schaub MC, Hefti MA, Zuellig RA, Morano I. Modulation of contractility in human cardiac hypertrophy by myosin essential light chain isoforms. Cardiovascular Research. 1998;37:381–404. doi: 10.1016/S0008-6363(97)00258-7. [DOI] [PubMed] [Google Scholar]
- Wagner PD, Weeds AG. Studies on the role of myosin alkali light chains. Recombination and hybridization of light chains and heavy chains in subfragment one preparations. J Mol Biol. 1977;109:455–470. doi: 10.1016/S0022-2836(77)80023-5. [DOI] [PubMed] [Google Scholar]
- Milligan RA. Protein-protein interactions in the rigor actomyosin complex. Proc Natl Acad Sci USA. 1996;93:21–26. doi: 10.1073/pnas.93.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayment I, Holden HM, Whittaker M, Yohn CB, Lorenz M, Holmes KC, Milligan RA. Structure of the actin-myosin complex and its implications for muscle contraction. Science. 1993;261:58–65. doi: 10.1126/science.8316858. [DOI] [PubMed] [Google Scholar]
- Mendelson RA, Morris EP. The structure of the acto-myosin subfragment 1 complex: Results of searches using data from electron microscopy and x-ray crystallography. Proc Natl Acad Sci USA. 1997;94:8533–8538. doi: 10.1073/pnas.94.16.8533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Root DD. A computational comparison of the atomic models of the actomyosin interface. Cell Biochemistry and Biophysics. 2002;37:97–110. doi: 10.1385/CBB:37:2:097. [DOI] [PubMed] [Google Scholar]
- Liu Y, Scolari M, Im W, Woo H-J. Protein-protein interactions in actin-myosin binding and structural effects of R405Q mutation: a molecular dynamics study. Proteins: Structure, Function, and Bioinformatics. 2006;64:156–166. doi: 10.1002/prot.20993. [DOI] [PubMed] [Google Scholar]
- Murphy CT, Spudich JA. The sequence of the myosin 50-20 K loop affects myosin's affinity for actin throughout the actin-myosin ATPase cycle and its maximum ATPase activity. Biochemistry. 1999;38:3785–3792. doi: 10.1021/bi9826815. [DOI] [PubMed] [Google Scholar]
- Murphy CT, Spudich JA. Dictyostelium myosin 25-50 K loop substitutions specifically affect ADP release rates. Biochemistry. 1998;37:6738–6744. doi: 10.1021/bi972903j. [DOI] [PubMed] [Google Scholar]
- Murphy CT, Spudich JA. Variable surface loops and myosin activity: Accessories to a motor. Journal of Muscle Research and Cell Motility. 2000;21:139–151. doi: 10.1023/A:1005610007209. [DOI] [PubMed] [Google Scholar]
- Sata M, Ikebe M. Functional analysis of the mutations in the human cardiac betamyosin that are responsible for familial hypertrophic cardiomyopathy. J Clin Invest. 1996;98:2866–2873. doi: 10.1172/JCI119115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita H, Sugiura S, Momomura S, Omata M, Sugi H, Sutoh K. Characterization of mutant myosins of Dictyostelium discoideum equivalent to human familial hypertrophic cardiomyopathy mutants. J Clin Invest. 1997;99:1010–1015. doi: 10.1172/JCI119228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knetsch MLW, Uyeda TQP, Manstein DJ. Disturbed communication between actin and nucleotide-binding sites in a myosin II with truncated 50/20-kDa junction. J Biol Chem. 1999;274:20133–20138. doi: 10.1074/jbc.274.29.20133. [DOI] [PubMed] [Google Scholar]
- Rovner AS, Freyzon Y, Trybus KM. Chimeric substitutions of the actin-binding loop activate dephosphorylated but not phosphorylated smooth muscle heavy meromyosin. J Biol Chem. 1995;270:30260–30263. doi: 10.1074/jbc.270.51.30260. [DOI] [PubMed] [Google Scholar]
- Luther HP, Podlowski S, Hetzer R, Baumann G. Analysis of sense and naturally occurring antisense transcripts of myosin heavy chain in the human myocardium. Journal of Cellular Biochemistry. 2001;80:596–605. doi: 10.1002/1097-4644(20010315)80:4<596::AID-JCB1014>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Yamashita H, Tyska MJ, Warshaw DM, Lowey S, Trybus KM. Functional consequences of mutations in the smooth muscle myosin heavy chain at sites implicated in familial hypertrophic cardiomyopathy. J Biol Chem. 2000;275:28045–28052. doi: 10.1074/jbc.M005485200. [DOI] [PubMed] [Google Scholar]
- Ajtai K, Garamszegi SP, Park S, Velazquez Dones AL, Burghardt TP. Structural characterization of β-cardiac myosin subfragment 1 in solution. Biochemistry. 2001;40:12078–12093. doi: 10.1021/bi0112098. [DOI] [PubMed] [Google Scholar]
- Bálint M, Sréter FA, Wolf I, Nagy B, Gergely J. The substructure of heavy meromyosin. The effect of Ca2+ and Mg2+ on the tryptic fragmentation of heavy meromyosin. J Biol Chem. 1975;250:6168–6177. [PubMed] [Google Scholar]
- Bobkov AA, Bobkova EA, Lin SH, Reisler E. The role of surface loops (residues 204-216 and 627-646) in the motor function of the myosin head. Proc Natl Acad Sci USA. 1996;93:2285–2289. doi: 10.1073/pnas.93.6.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joel PB, Trybus KM, Sweeney HL. Two conserved lysines at the 50/20-kDa junction of myosin are necessary for triggering actin activation. J Biol Chem. 2001;276:2998–3003. doi: 10.1074/jbc.M006930200. [DOI] [PubMed] [Google Scholar]
- Furch M, Geeves MA, Manstein DJ. Modulation of actin affinity and actomyosin adenosine triphosphatase by charge changes in the myosin motor domain. Biochemistry. 1998;37:6317–6326. doi: 10.1021/bi972851y. [DOI] [PubMed] [Google Scholar]
- Mornet D, Pantel P, Audemard E, Kassab R. The limited tryptic cleavage of chymotryptic S-1: An approach to the characterization of the actin site in myosin heads. Biochem Biophys Res Commun. 1979;89:925–932. doi: 10.1016/0006-291X(79)91867-9. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Sekine T. Interaction of myosin subfragment-1 with actin.III.Effect of cleavage of the heavy chain on its interaction with actin. J Biochem. 1979;86:1869–1881. doi: 10.1093/oxfordjournals.jbchem.a132710. [DOI] [PubMed] [Google Scholar]
- Applegate D, Azarcon A, Reisler E. Tryptic cleavage and substructure of bovine cardiac myosin subfragment 1. Biochemistry. 1984;23:6626–6630. doi: 10.1021/bi00321a053. [DOI] [PubMed] [Google Scholar]
- Coureux P-D, Wells AL, Menetrey J, Yengo CM, Morris CA, Sweeney HL, Houdusse A. A structural state of the myosin V motor without bound nucleotide. Nature. 2003;425:419–422. doi: 10.1038/nature01927. [DOI] [PubMed] [Google Scholar]
- Ajtai K, Garamszegi SP, Watanabe S, Ikebe M, Burghardt TP. The myosin cardiac loop participates functionally in the actomyosin interaction. J Biol Chem. 2004;279:23415–23421. doi: 10.1074/jbc.M310775200. [DOI] [PubMed] [Google Scholar]
- Volkmann N, Hanein D, Ouyang G, Trybus KM, DeRosier DJ, Lowey S. Evidence for cleft closure in actomyosin upon ADP release. Nature Structural Biology. 2000;7:1147–1155. doi: 10.1038/82008. [DOI] [PubMed] [Google Scholar]
- Onishi H, Mikhailenko SV, Morales MF. Towards understanding actin-activation of myosin ATPase: The role of surface loops. Proc Natl Acad Sci USA. 2006;103:6136–6141. doi: 10.1073/pnas.0601595103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmann N, Lui H, Hazelwood L, Trybus KM, Lowey S, Hanein D. The R403Q myosin mutation implicated in familial hypertropic cardiomyopathy causes disorder at the actomyosin interface. Plos One. 2007;e1123 doi: 10.1371/journal.pone.0001123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, Lin Y, MacDonald JR, Pang AWC, Shago M, Stockwell TB, Tsiamouri A, Bafna V, Bansal V, Kravitz SA, Busam DA, Beeson KY, McIntosh TC, Remington KA, Abril JF, Gill J, Borman J, Rogers YH, Frazier ME, Scherer SW, Strausberg RL, Venter JC. The Diploid Genome Sequence of an Individual Human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šali A, Blundell TL. Comparative protein modeling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. Journal of Molecular Graphics. 1999;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Instructions for retrieving SNPs. Two perl programs provide automated retrieval and organization of SNP data from the NCBI database. Additional file 1 contains the perl programs and instructions for their use.