

Abstract
Protein phosphatases are believed to coordinate with kinases to execute biological functions and examples of such integrated activities however are still missing. In this report, we have identified PTPH1 (protein tyrosine phosphatase H1) as a specific phosphatase for p38γ MAPK (mitogen-activated protein kinase) and demonstrated their cooperative oncogenic activity through direct binding. p38γ, a Ras effector known to act independent of its phosphorylation, was first shown to require its unique PDZ-binding motif to increase Ras transformation. Yeast two-hybrid screening and in vitro and in vivo analysis further identified PTPH1 as a specific p38γ phosphatase through PDZ-mediated binding. Additional experiments showed that PTPH1 itself plays a role in Ras-dependent malignant growth in vitro and/or in mice by mechanism depending on its p38γ-binding activity. Moreover, Ras increases both p38γ and PTPH1 protein expression and there is a coupling of increased p38γ and PTPH1 protein expression in primary colon cancer tissues. These results reveal a coordinative oncogenic activity of a MAPK with its specific phosphatase and suggest that PDZ-mediated p38γ/PTPH1 complex may be a novel target for Ras-dependent malignancies.
Introduction
Mitogen-activated protein kinases (MAPKs) are major signaling pathways in regulating Ras oncogene activity, including ERKs (extracellular signal-regulated kinases), JNKs (Jun N-terminal kinases) and p38s. While the ERK pathway is generally required for Ras activity (1), a suppressive role has been proposed for the p38 (2, 3). The Ras inhibitory activity of the p38 pathway was first demonstrated by the fact that p38 activation leads to either an inhibition of Ras-dependent growth (4) or an induction of Ras-dependent cell death (5). This observation has been further consolidated by an increased Ras tumorigenesis through knocking out either p38 activating kinases MKK3/6 (6), p38α (7) or downstream p38 regulated/activated protein kinase (PRAK) (8). Studies of inhibitory p38 MAPK pathways may offer a great promise to control Ras oncogene activity.
The p38 family however consists of four proteins {p38α (also called p38), β, γ and δ} and our recent studies suggest that p38γ is required for Ras oncogenesis (9–11). p38γ is a 43 kDa protein with an unique carboxyl terminal sequence -ETXL that can dock with PDZ (PSD-95/Dlg/ZO-1 homology) domains of different proteins as substrates such as α1-syntrophin (12), SAP90/PSD-95 (13) and SAP97 (14). Among p38 family proteins p38γ is the only member whose expression is induced during cell differentiation (15) and Ras activation (9, 10). More interestingly, p38γ is dephosphorylated by Ras signaling inside cells by unknown mechanisms (9) and a non-phosphorylated p38γ has a greater potency in increasing Ras transformation (11). These results together indicate a potential involvement of protein phosphatases in p38γ regulating Ras transformation through its PDZ binding motif-mediated protein-protein interactions. In this report, we have identified PTPH1 as a p38γ-specific phosphatase and demonstrated that p38γ and PTPH1 cooperate to promote Ras oncogenesis through direct binding.
Materials and Methods
Gene expression, gene silencing, and viral infection
Flag-tagged wild-type (WT) p38γ and p38γΔ4 or Δ13 were stably expressed in IEC-6 cells through G418 selection and pooled resistant cells were infected with LZRS-K-Ras through a puromycin selection (9). PTPH1 and its mutant were similarly stably expressed in IEC-6/K-Ras cells. For gene silencing, the target sequence was cloned into a plenti6/Block-iT vector by including a sequence from luciferase gene as a control. To produce virus, retrovirus and Lentivirus were transfected into their respective packaging cells and supernatants were collected, filtered and used to infect target cells.
In vitro binding and p38γ/α dephosphorylation experiments
Flag-tagged p38γ and its Δ4/Δ13 mutants (together with p38α and its mutant) were expressed in 293T cells. Thereafter, cells were lysed, supernatants mixed with 8 μg of GST or GST-PTPH1 proteins, the mixtures incubated with reduced glutathione beads overnight, and precipitates were analyzed by Western blot. For in vitro p38γ/α dephosphorylation experiment, Flag-p38γ/α was co-expressed with an active MKK6/2E in 293T cells and purified by an anti-Flag antibody (M2-conjugated agarose beads). Precipitates were then incubated with GST-PTPH1 in a reaction buffer (50 mM Tris-HCl, pH7.5, 3 mM DTT, 30 mM MgCl2) at 37°C for 30 min and mixtures were analyzed by Western for p38 phosphorylations.
Cell growth, soft-agar assays, and mouse experiments
Cell proliferation was estimated by thymidine incorporation assays after the peptide incubation (9). For soft-agar assays, cells were plated on growth media containing 0.33% Sea-plaque-agarose and colonies photographed and counted about two weeks later. For animal experiments, HCT116 cells were infected with lenti-sh-PTPH1 or Lent-sh-Luc (luciferase), and selected with blasticidin (15 μg/ml) for 7 days. Cells (2 × 106) in 0.1 ml PBS were then s.c. injected into athymic nude mouse (Harlan) at both front flanks and the tumor volume (π abc/6) was measured every 2–3 days. The animal experimental procedures were performed in accordance with the approved IACUC protocol.
Human colon cancer specimens and immunohistochemistry
All human colon cancer tissues were collected by Department of Pathology, Medical College of Wisconsin with informed consent. Immuno-histochemistry analyses were conducted in accordance with Institutional Review Board approval from Medical College of Wisconsin. Briefly, sections of formalin-fixed and paraffin-embedded blocks were subjected to immuno-staining as described (16). An anti-p38γ (1:1200, R&D, Cat #: AF1644) and anti-PTPH1 (1:900, Santa Cruz, Cat #: SC-9789) were used as primary antibodies. Staining results (intensity × percentage of positive cells) were examined independently by two observers as previously described (16, 17) and a consensus score was assigned to each case. The intensity was scored according to the following scale: 0 (negative), 1 (weak), 2 (moderate), and 3 (strong), whereas the percentage was rated using the following categories: 0 (0%), 1 (<10%), 2 (11–50%), 3 (51–75%), and 4 (76–100%). The signal increases for PTPH1 in the malignant tissues after subtracting from the matched normal tissues were plotted against those from p38γ for their correlation.
Statistical analysis
Colony numbers, tumor volume, and tumor weight were analyzed by student’s t test or ANOVA for statistical difference. A Pearson’s analysis was used to determine the correlation between increased p38γ and PTPH1 protein expression. Increased protein expression (p38γ and PTPH1) in colon cancer tissues versus matched normal tissues was analyzed by ANOVA.
Results
p38γ requires its PDZ-binding motif to increase Ras transformation and to bind PTPH1
To investigate roles of p38γ PDZ-binding motif in Ras tumorigenesis, two Flag-tagged C-terminal truncated p38γ mutants lacking 4 (Δ4) and 13 (Δ13) amino acids were generated by PCR and stably expressed in rat intestinal epithelial IEC-6 cells through G418 selection together with a wild-type (WT) p38γ. These cells were further expressed with K-Ras oncogene via retroviral infection and Ras oncogenic activity was assessed by anchorage-independent growth on soft-agar (9). Results in Fig. 1A showed that expression of p38γ but not its mutants increases the soft-agar growth. Since both p38γΔ4 and p38γΔ13 lack the PDZ motif ETPL, these results suggest that p38γ require its PDZ protein-binding activity to increase Ras transformation.
Figure 1.
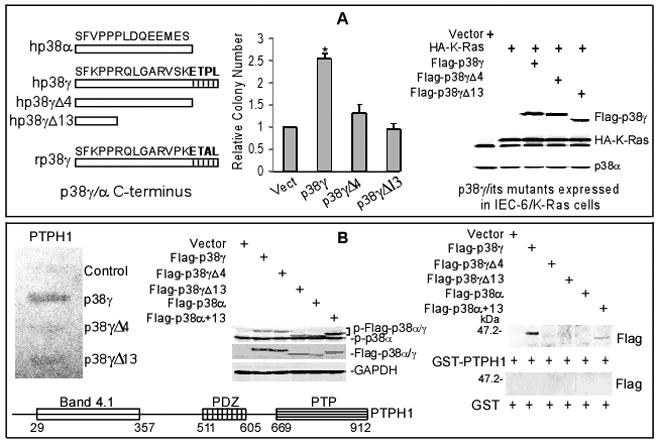
p38γ requires C-terminus to increase Ras transformation and to bind PTPH1. A, p38γ requires its C-terminus to increase Ras transformation. IEC-6 cells were stably expressed with p38γ or its mutants (see left for structures) and then transduced with retrovirus containing K-Ras, which were analyzed for soft-agar growth (middle) and protein expression (right). Colony numbers from 20 fields per 60 mm dish are counted and results shown are relative over vector controls (from three separate experiments with each in triplicate, * p < 0.05 by ANOVA). B, yeast two-hybrid screening and GST pull-down reveal a PDZ-dependent interaction of p38γ with PTPH1. The detail procedure and full results of the screening were described in Supplemental data and Fig. S1A/B. The β-Gal activity assay with PTPH1 confirmed a positive reaction with p38γ (left panel) and structure of human PTPH1 protein was depicted on bottom (left). GST pull-down results were given on right panel with the input control in the middle (see Fig. S1C for similar GST and GST-PTPH1 proteins used).
To search for candidate PDZ proteins, we performed a yeast two-hybrid screen of a human adult colon cDNA library using human p38γ and p38γΔ13 as baits. This screen yielded 13 individual clones encoding residues 410-912 of PTPH1, a protein tyrosine phosphatase H1, when p38γ was used as bait, but no PTPH1 was detected in all 5 clones assayed with p38γΔ13 (Fig. S1A). Since this region contains a single PDZ domain and only p38γ but not p38γΔ13 binds PTPH1, these results indicate that p38γ interacts with PTPH1 through its PDZ motif ETPL. To confirm this speculation, p38γΔ4 (lacking ETPL only) was fused to GAL4 DNA binding domain, β-galactosidase (β-Gal) activity assay was performed after re-transformation with isolated PTPH1 by including GAL4-p38γ and GAL4-p38γΔ13 for comparison. Results in Fig. 1B (left) shows that PTPH1 only interacts with p38γ but not with the Δ4 or Δ13 mutant, indicating a necessary role of the PDZ motif for the interaction (full results of this screening are given in Fig. S1B). Previous two-hybrid screening revealed that rat p38γ interacts with PDZ protein α1-syntrophin via the PDZ motif (ETAL), which was not observed in our analyses, likely as a result of their utilization of rat p38γ as a bait to screen a human brain cDNA library (12). These results together indicate that human p38γ requires C-terminal PDZ binding motif to interact with PTPH1.
To further demonstrate PDZ-dependent p38γ-PTPH1 interaction, PTPH1 was expressed as a GST fusion protein that was incubated with 293T lysates containing transfected p38γ and its mutants, and GST pull-down assays were then performed. To examine if the PDZ motif confers a binding activity towards PTPH1, the last C-terminal 13 amino acid sequence of p38γ was fused to the corresponding portion of a human p38α and this modified p38α (p38α+13) also included in the analysis as compared with WT p38α. Results in Figs. 1B (middle and right panel) and S1C show that GST-PTPH1 binds p38γ and p38α+13, but not p38γΔ4, p38γΔ13 or WT p38α, indicating an important role of the PDZ motif in p38α/γ interacting with PTPH1.
PTPH1 is a p38γ MAPK specific phosphatase through PDZ-mediated interaction
To further demonstrate the role of the PDZ motif in p38 interacting with PTPH1 in vivo, 293T cells were transiently expressed with indicated constructs and analyzed by co-immunoprecipitations (IP) and Western blotting (WB). Consistent with the GST pull-down results, analysis of Flag and HA precipitates showed an interaction of PTPH1 with both WT p38γ (but not its Δ4 and Δ13 mutants) and p38α+13 (Fig. 2A). Furthermore, PTPH1 binds p38γ and its kinase-dead mutant but not its non-phosphorable mutant p38γ/AGF, indicating a role of p38γ phosphorylation in this interaction (Figs. 2B and S2A and data not shown). In addition, a PDZ-domain deleted PTPH1 mutant (PTPH1ΔPDZ) fails to bind p38γ and stably transfected p38γ also requires the PDZ motif to bind endogenous PTPH1 protein in IEC-6/K-Ras cells (Fig. 2C/D). These results together indicate that it is the PDZ switch (the PDZ-binding motif of p38γ plus the PDZ domain of PTPH1) that controls their direct interaction.
Figure 2.
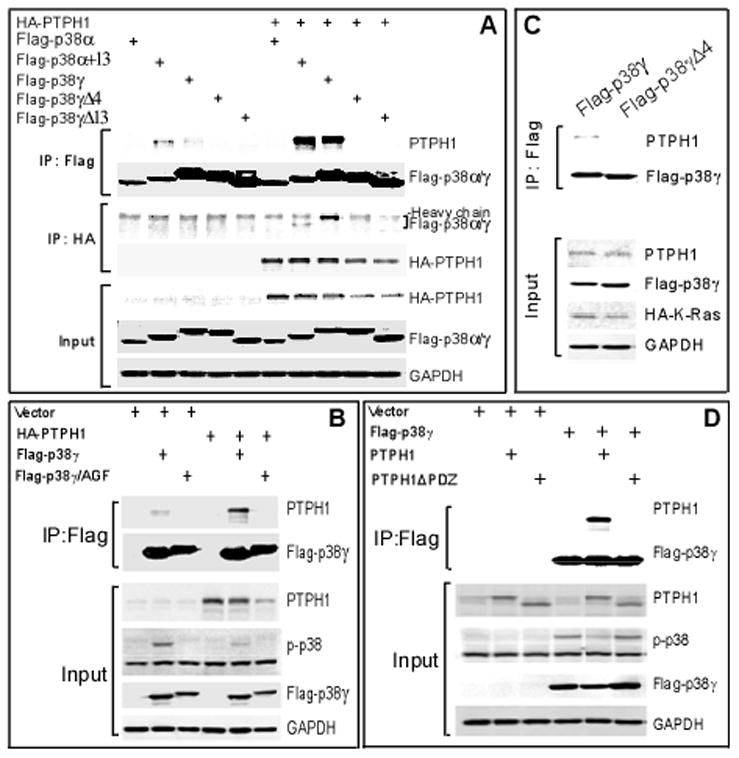
The PDZ dictates p38 interaction with PTPH1 in vivo. A, a role of the PDZ motif in p38 interacting with PTPH1. Flag-tagged p38γ/α constructs were expressed with and without HA-PTPH1 in 293T cells and Flag/HA precipitates were examined by Western. Please note that Flag p38γ and Flag p38α+13 also bind endogenous PTPH1 in the absence of HA-PTPH1 (lane 2 and 3 from left). B-D, p38γ depends on its phosphorylation and C-terminus to bind the PDZ domain of PTPH1 protein. Expressed proteins in 293T cells were isolated and examined for p38γ-PTPH1 binding. Please note that Flag-p38γ fails to bind the PTPH1ΔPDZ and the bound WT PTPH1 also leads to decreased p38γ phosphorylation from the input control (B/D). Results from C showed that stably expressed Flag-p38γ (but not its Δ4 mutant) also binds endogenous PTPH1 protein in IEC-6/K-Ras cells.
PTPH1 is a 120-kDa protein that belongs to the non-transmembrane PTP super-family (18). Previous studies have identified VCP (p97/CDC48) (19) and T cell receptor ζ subunit (20) as its substrates but mechanisms as well as biological consequences of these reactions remain unknown. The demonstration of PDZ-dependent p38γ-PTPH1 interaction prompted us to explore if PTPH1 may be a novel p38γ phosphatase. In this regard, different amounts of GST-PTPH1 proteins were incubated with Flag precipitates containing MKK6-activated Flag-p38γ or Flag-p38α through cotransfection in 293T cells. In vitro phosphatase assay was performed to assess p38 phosphorylation using a phosphor-specific p38 antibody that is reactive with all phosphorylated p38 (p-p38) family proteins. Results in Fig. 3A show that PTPH1 decreases p-p38γ without substantial effects on p-p38α. The co-transfection experiment further reveal that PTPH1 in vivo decreases phosphorylation of p38γ but not p38γΔ4, p38γΔ13 or p38α (Figs. 3B and S2B, left), indicating its specific p38γ phosphatase activity.
Figure 3.
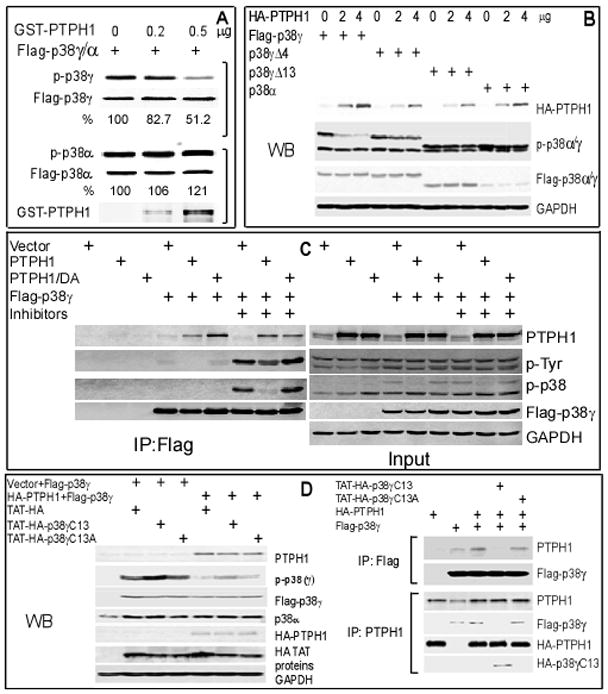
PTPH1 dephosphorylates p38γ but not p38α in vitro and in vivo. A, PTPH1 dephosphorylates p38γ but not p38α in vitro. Flag-tagged p38α/γ were co-expressed with MKK6 in 293T cells and activated p38s were isolated with a Flag antibody, which were examined for in vitro phosphorylation using a specific p-p38 antibody following incubation with GST-PTPH1. The percentage indicates the relative p-p38s (normalized to Flag-p38α/γ) over those in the absence of GST-PTPH1 (measured with ImageQuant 5.0 software). B, p38γ is dephosphorylated by PTPH1 in vivo dependent of its PDZ-binding motif. Different amounts of HA-PTPH1 were co-expressed with indicated constructs in 293T cells and examined for p38γ phosphorylation. C, there is an increased complex-formation between p38γ and PTPH1/DA. Flag-p38γ was transiently co-expressed with PTPH1 or PTPH1/DA and expressed proteins were isolated in the absence or the presence of phosphatase inhibitors (1mM Sodium Vanadate, 20mMβ-glycerophosphate, and 20mM ρ-nitrophenylphosphate) and analyzed by Western. D, PTPH1 binding and dephosphorylating p38γ are both inhibited by a peptide corresponding to the p38γ C-terminus. p38γ and PTPH1 were expressed in 293T cells for 24 hr, which were then subjected to the peptide treatment (10 μM, 5 hr) and Flag IP and/or Western analyses (see Fig. S2B for input control).
To further demonstrate if p38γ is a physiological substrate of PTPH1, a substrate-trapping PTPH1 mutant (PTPH1/DA) (19) was co-expressed with Flag-p38γ and their binding activity was analyzed in the absence and presence of phosphatase inhibitors (21). Results in Fig. 3C showed an increased p38γ-PTPH1/DA complex formation over PTPH1 transfection, consistent with an increased enzyme-substrate binding by PTP DA mutants (19, 21). The increased binding however was abolished in the presence of phosphatase inhibitors, which couples with increased p38γ phosphorylation as detected with either p-p38 or p-Tyr antibody, thus further confirming their direct interaction (22). Importantly, the p-p38γ signal is decreased after co-transfection with PTPH1 but not with its DA mutant. A stronger suppression of p-p38γ than p-p38γ-Tyr in Flag precipitates by PTPH1 (Fig. 3C) suggests that PTPH1 may specifically act on the tyrosine residue within the activation loop of p38γ kinase, a finding consistent with the notion that dephosphorylation of the tyrosine residue is a determinant step for MAPK inactivation (23).
To investigate if PTPH1 regulates p38γ phosphorylation via PDZ binding under physiological conditions, the last 13-amino-acids of p38γ C-terminus (p38γC13 or WT) and its mutant (p38γC13A or MT, with the last 4 amino-acids changed to Alanine) were cloned into a pTAT-HA vector (24) and expressed as HA-tagged TAT-fusion peptides to regulate in situ p38γ-PTPH1 interaction. Results in Figs. 3D and S2B (bottom and right) showed that PTPH1-induced p38γ dephosphorylation is substantially inhibited by p38γC13 while minimally affected by its mutant, which couples with its activity to disrupt the p38γ-PTPH1 binding, indicating that p38γ is a physiological substrate of PTPH1 through PDZ-mediated interaction.
PTPH1 signals downstream of Ras and p38γ to promote colon cancer growth
Previous genetic analysis suggested a tumor suppressor function of PTPH1 (also called PTPN3) along with its family members as a result of their somatic mutations in human colon cancer (25). Our results that p38γ requires its PTPH1-binding activity to increase Ras transformation however suggest that PTPH1 may play a positive role in Ras oncogenesis. To demonstrate if Ras signals to PTPH1, IEC-6/K-Ras and control cells were analyzed for protein expression by Western blotting. Results in Fig. 4A (left) showed that p38γ and PTPH1 protein expression are both induced by K-Ras. Furthermore, transient expression of either oncogenic Ras or p38γ protein in normal IEC-6 cells also stimulates PTPH1 expression (Fig. 4A, middle and right), suggesting PTPH1 signaling downstream of both Ras and p38γ. Since p38γ phosphorylation is inhibited by Ras and promotes Ras transformation independent of phosphorylation (9), Ras-induced p38γ and PTPH1 proteins may cooperate to promote its oncogenesis through PDZ-mediated binding and resultant p38γ dephosphorylation.
Figure 4.
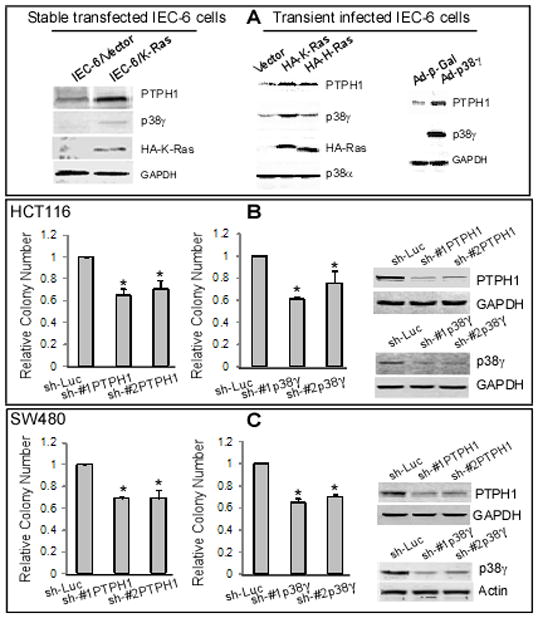
PTPH1 signals downstream of Ras and p38γ and is required for Ras-dependent colon cancer growth. A, Ras increases p38γ and PTPH1 protein expression. Ras transformed IEC-6 cells were examined for protein expression (left). In addition, normal IEC-6 cells were transiently infected with LZRS retrovirus (expressing H-Ras or K-Ras) or adenovirus (p38γ) for 48 h, and examined for protein expression (middle and right). B, C, depletion of PTPH1 or p38γ protein expression inhibits soft-agar growth of HCT116 or SW480 human colon cancer cells. Cells were transiently infected with lentivirus (shLuc, #1 and #2 shRNA against PTPH1 or p38γ as indicated) and 72 h later assayed for the soft-agar growth and Western blot. The relative colony number is shown as in Fig. 1B from three separate experiments (* p < 0.05 vs. sh-Luc).
To directly examine roles of PTPH1 and p38γ in Ras-dependent growth, their gene expression was silenced by shRNA in Ras-activated HCT116 and SW480 human colon cancer cells and resultant effects on soft-agar growth were next determined. Results in Fig. 4B/C show that depletion of either PTPH1 or p38γ protein expression by two separate shRNAs inhibits the malignant growth of both cell lines, indicating their growth-promoting roles in Ras-activated colon cancer. To further demonstrate the PTPH1 oncogenic activity, HCT116 cells were stably depleted of PTPH1 protein (Figs. S3A/B and S4A), which were then injected into right and left front flanks of nude mouse and examined for their tumor-forming activities. Results in Figs. 5A and S3A/B showed that the intra-tumor PTPH1 depletion significantly inhibits tumor growth as revealed either by a lowered growth-curve over a 2–3 week period or by a decreased tumor weight by the end of experiment. Comparative analyses of tumor weight with PTPH1 protein expression from the number 1 and 5 mouse further revealed a coupling of the growth inhibition with the PTPH1 depletion (Fig. 5A insert and S4B). These results indicate that both p38γ and PTPH1 are required for Ras-dependent colon cancer growth in vitro and/or in mice.
Figure 5.
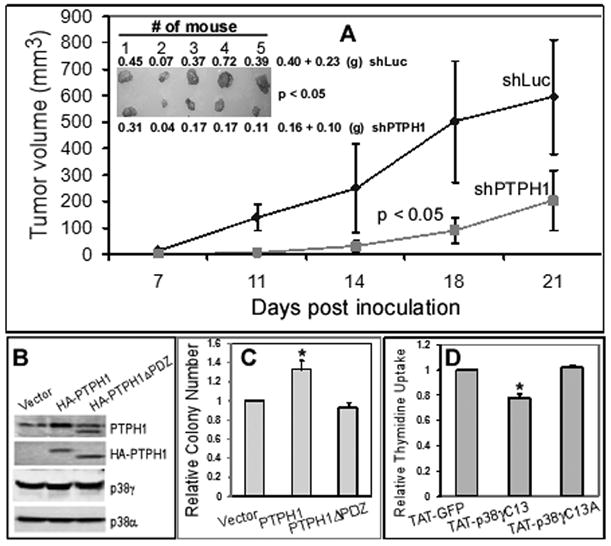
PTPH1 promotes colon cancer growth in vivo and requires its p38γ-binding activity to increase Ras-dependent growth in vitro. A, PTPH1 depletion inhibits the tumor growth in mice. HCT116 cells were stably infected with Lenti-shLuc or Lenti-#1shPTPH1 in cell culture (see Fig. S4A for Western) and 2 × 106 of these cells s.c. injected into both front flanks of nude mouse (right, shLuc; left, shPTPH1) and the tumor growth was monitored. Moreover, tumors were excised, photographed, and weighed at the end of experiment (insert, p < 0.05 between two groups for either tumor weight or volume in all time point) and similar results were obtained from additional two experiments (Fig. S3A/B). B, C, PTPH1 requires its PDZ domain to increase Ras transformation. IEC-6/K-Ras cells were stably expressed with PTPH1 or its PDZ-deleted mutant PTPH1ΔPDZ, and subjected to Western (B) and Soft-agar assays (C) (means of three separate experiments, * p < 0.05). D, disruption of the p38γ-PTPH1 interaction inhibits colon cancer cell proliferation. HCT116 cells were incubated with the wild-type and mutant peptide as described in Fig. 3D with a TAT-GFP as a separate control and cell growth was estimated by thymidine incorporation as previously described (10). Results shown (relative to TAT-GFP control) are mean of three separate experiments (*, p < 0.05, p38γC13 versus p38γC13A).
PTPH1 depends on its interaction with p38γ to increase the malignant growth
We have shown that p38γ requires its PDZ-binding motif to increase Ras transformation (Fig. 1A). To demonstrate if PTPH1 depends on its p38γ binding activity to promote malignant growth, IEC-6/K-Ras cells were stably expressed with WT and PDZ-deleted PTPH1 proteins and their colony forming activities were then compared. Results in Fig. 5B/C showed that the stable expression of PTPH1 but not its mutant increases the soft-agar growth and similar results were also obtained with another p38γ-binding deficient PTPH1 mutant (data not shown). Since PTPH1ΔPDZ fails to bind/dephosphorylate p38γ (Fig. 2D), these results suggest that PTPH1 requires its p38γ binding/dephosphorylating activity to promote Ras tumorigenesis. Together with the role of the p38γ C-terminus in Ras transformation, these results indicate that it may be the PDZ-mediated p38γ-PTPH1 complex that acts to promote malignant growth through resultant p38γ dephosphorylation.
To directly test this hypothesis, HCT116 cells were incubated with TAT-p38γC13 and its mutant TAT-p38γC13A peptide with TAT-GFP as a separate control and cell proliferation was assessed by thymidine incorporation. Results in Fig. 5D showed that the wild-type peptide significantly inhibits DNA synthesis as compared with the mutant and GFP control. Since this peptide blocks the p38γ/PTPH1 interaction as well as p38γ dephosphorylation as compared to its mutant (Fig. 3D), its cell-growth suppressive effect strongly indicates a required role of the p38γ/PTPH1 complex and resultant p38γ dephosphorylation in colon cancer growth. Because the mutant peptide sequence only differs from its wild-type counterpart at the ETPL, these results suggest that it is this four amino-acids PDZ motif that integrates p38γ oncogenic activity with its phosphatase PTPH1.
There is a correlation of hyper-expressed p38γ with PTPH1 in primary colon cancer tissues and a coupling of decreased PTPH1 expression with increased p38γ phosphorylation
The cooperative oncogenic activity of p38γ and PTPH1 promoted us to examine if both proteins are over-expressed in primary colon cancer tissues. In this regard, 142 cases of human colon cancer specimens (invasive carcinoma) were analyzed by immuno-staining for their protein expression. Two slides of each specimen were processed for staining with a specific antibody against p38γ and PTPH1 respectively, and their signals in the tumor and the nearby matched normal tissues were independently scored. Results from Figs. 6A (left) and S4C showed that levels of cytoplasmic p38γ and PTPH1 staining signals are both significantly increased in the malignant over the nearby normal tissues and overall, increased p38γ and PTPH1 protein expression in the malignant tissues was observed in 81% and 75% of samples, respectively. Importantly, when signals from tumors are subtracted by those from their matched normal tissues, there is a significant correlation between increased p38γ and PTPH1 protein expression (Fig. 6A, right). Since Ras induces both PTPH1 and p38γ expression and depletion of either of them or disruption of their binding inhibits the malignant growth, a coupling of hyper-expressed p38γ with PTPH1 in primary tissues further suggests their cooperative oncogenic activity under patho-physiological conditions.
Figure 6.
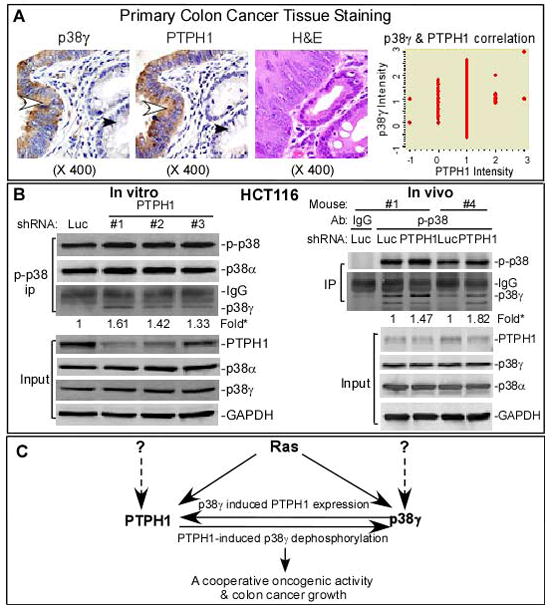
Roles of PTPH1 and p38γ in colon cancer. A, hyperexpressed p38γ correlates with increased PTPH1 expression in primary colon cancer tissues. p38γ and PTPH1 protein expression in invasive colon carcinomas were analyzed by immuno-histochemistry staining. Representative pictures from the same patient were given in left two panels showing an increased positive brown signals for p38γ and PTPH1 in malignant tissues (indicated by an open arrow head) over those in normal gland (marked with a closed arrow head). A Pearson’s correlation was reached between increased p38γ and PTPH1 protein expression in this group of specimens (142 cases) after subtracting signals of the tumors from those of matched normal tissues (p < 0.05, right panel). Additional results about p38γ/PTPH1 protein expression in colon cancer specimens are shown in Fig. S4C and Table S1. B, levels of endogenous PTPH1 protein expression inversely couple with intrinsic p38γ phosphorylation. Cells were depleted of PTPH1 protein and subjected to IP/Western analysis for increased p-p38γ proteins (IP) (left). On right panel, protein samples were prepared from two pairs of tumors (#1 and #4 mouse, Fig. 5A insert) and subjected to the IP/WB analysis, which together with another set of experiments (Fig. S3C) showed a coupling of decreased PTPH1 protein expression with increased p38γ phosphorylation. * indicates a fold increase in p-p38γ over individual shLuc control. C, an experimental model shows a PDZ-mediated cooperative oncogenic activity of p38γ MAPK with its phosphatase PTPH1. Ras was shown to increase protein expression of p38γ and PTPH1 in which PTPH1 is also induced by its substrate p38γ and acts to dephosphorylate p38γ through PDZ-mediated binding. Experimental evidence is presented to indicate that it is the PDZ-mediated p38γ/PTPH1 complex that possesses cooperative oncogenic activity leading to increased colon cancer growth. The dotted lines indicate that p38γ and PTPH1 may be up-regulated by Ras-independent proliferative signals.
Since PTPH1 was shown as a p38γ MAPK isoform-specific phosphatase, we wished to explore if levels of endogenous PTPH1 protein expression negatively correlate with intrinsic p38γ phosphorylation. Because p-p38γ proteins are undetectable by direct Western, total phosphorylated p38s were isolated by IP and examined for the p38γ abundance by Western for its correlation with the residual PTPH1 after the shRNA-induced PTPH1 depletion. Results in Fig. 6B (left panel) showed that levels of decreased PTPH1 protein expression from the input control appear to be negatively correlated with increased p-p38γ phosphorylations from the precipitates. To further link this negative correlation with the growth regulatory activity of the p38γ/PTPH1 complex, protein samples were prepared from the HCT-116 tumors excised from the number 1 and 4 mouse, and further analyzed by p-p38 IP/Western. The in vivo tumor growth assay showed that the PTPH1 depletion exhibited a greater tumor-growth inhibition in the number 4 mouse (0.17/0.72; shPTPH1/shLuc) over the number 1 counterpart (0.31/0.45) (Fig. 5A, insert), which again couples with a greater PTPH1 depletion and a more substantial p-p38γelevation from p-p38 precipitates (Fig. 6B). These results thus provide further evidence to indicate the cooperative oncogenic activity of p38γ with its phosphatase PTPH1 through the resultant p38γ dephosphorylation (Fig. 6C).
Discussion
MAPKs function through coordinative phosphorylation and dephosphorylation to regulate dynamic cellular programs leading to various biological responses. Although extensive efforts have been made, there have been so far no reports about a cooperative activity of a MAPK with its phosphatase (26). Our studies reported here first identified a p38γ MAPK isoform-specific phosphatase PTPH1 and then provide evidence to indicate that it is the p38γ/PTPH1 complex that possesses an oncogenic activity through PDZ-mediated direct binding and resultant p38γ dephosphorylation. This conclusion is supported by the following: 1) removal of p38γ C-terminal PDZ-motif eliminates both its oncogenic activity and its interaction with/dephosphorylation by PTPH1; 2) deletion of the PDZ domain of PTPH1 also leads to a loss of both its p38γ-binding/dephosphorylating activity and its promoting effect on Ras transformation; 3) depletion of p38γ or PTPH1 alone inhibits the malignant growth, which can be mimicked by application of a specific peptide through inhibiting p38γ-PTPH1 interaction and resultant p38γ dephosphorylation; 4) Ras induces both p38γ and PTPH1 expression; and 5) there is a correlation of hyper-expressed p38γ with PTPH1 in primary colon cancer tissues and a coupling of decreased PTPH1 expression with increased intrinsic p38γ phosphorylation. These results together reveal a cooperative oncogenic activity of p38γ MAPK with its phosphatase PTPH1 through PDZ-mediated interaction (Fig. 6C).
MAPKs are best known to be inactivated by dual-specificity (Thr/Tyr) MAPK phosphatases (MKPs) with most of MKPs however acting on more than one MAPK member (27). Our results showed that p38γ but not p38α MAPK is a physiological PTPH1 substrate and this specificity appears to be determined by both of its C-terminal PDZ binding sequence ETPL and the conserved TGY motif within the kinase sub-domain. This is because either removal of p38γ C-terminus or mutation of the TGY disrupts its interaction with PTPH1 and fusion of the p38γ C-terminal fragment to p38α confers a PTPH1-binding activity. Among 10 classical and non-classical MAPK family members, p38γ (also called ERK6 or MAPK12) is the only kinase that meets this dual requirement (26). This unique p38γ structure together with the fact that PTPH1 is the only classical PTP that contains a single PDZ domain (28) may be the foundation for their specificity. We are currently investigating if the PDZ binding enables PTPH1 as a p38γ substrate that may additionally contribute to their cooperative oncogenic activity.
In contrast to the proliferative effect as shown in this study, PTPH1 transfection in mouse NIH3T3 cells was previously reported to inhibit cell-cycle progression (19), which may be a tissue- and/or species-specific effect. Besides PTPH1, several PTPs are known to be oncogenic, including SHP2 (PTPN11) (29), HePTP (30) and PTP1B (31), but none of these is shown to promote the malignant growth through cooperation with its substrate. Our results, on the other hand, indicate that PTPH1 may cooperate with its substrate p38γ to increase malignant growth. This finding is further highlighted by the fact that Ras induces both p38γ/PTPH1 protein expression (Fig. 4A) and p38γ dephosphorylation (9), and a non-phosphorylated p38γ is more potent in increasing Ras transformation (11). It remains unclear, however, if PTPH1 depends on its phosphatase activity to increase the malignant growth and whether PTPH1 is required for Ras-induced p38γ dephosphorylation. Future studies towards these goals may reveal the p38γ/PTPH1 complex as a noel target in Ras-dependent malignancies.
Supplementary Material
Acknowledgments
Grant Support: This study was in part supported by grants from National Institutes of Health (2R01 CA91576), Department of Veterans Affairs (Merit Review), and Breast Cancer Show-House (Cancer Center of Medical College of Wisconsin (G.C.).
We would like to thank Drs Nicholas K. Tonks, Steven F. Dowdy, Jiahuai Han, Lawrence A. Donehower and Michael Dwindle for providing various reagents, Tarun Patel for discussions and Aniko Szabo for statistical analyses.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary data: Materials and Methods
References
- 1.Downward J. Targeting Ras signalling pathways in cancer therapy. Nature Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 2.Han J, Sun P. The pathways to tumor supression via route p38. Trends Biochem Sci. 2007;32:364–71. doi: 10.1016/j.tibs.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 3.Loesch M, Chen G. The p38 MAPK stress pathway as a tumor suppressor or more? Front Biosci. 2008;13:3581–93. doi: 10.2741/2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen G, Hitomi M, Han J, Stacey DW. The p38 pathway provides negative feedback to Ras proliferative signaling. J Biol Chem. 2000;275:38973–80. doi: 10.1074/jbc.M002856200. [DOI] [PubMed] [Google Scholar]
- 5.Qi X, Tang J, Pramanik R, et al. p38 MAPK activation selectively induces cell death in K-ras mutated human colon cancer cells through regulation of vitamin D receptor. J Biol Chem. 2004;279:22138–44. doi: 10.1074/jbc.M313964200. [DOI] [PubMed] [Google Scholar]
- 6.Brancho D, Tanaka N, Jaeschke A, et al. Mechanism of p38 MAP kinase activation in vivo. Genes & Dev. 2003;17:1969–78. doi: 10.1101/gad.1107303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dolado I, Swat A, Ajenjo N, De Vita G, Cuadrado A, Nebreda AR. p38α MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell. 2007;11:191–205. doi: 10.1016/j.ccr.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 8.Sun P, Yoshizuka N, New L, et al. PRAK is essential for ras-induced senescence and tumor suppression. Cell. 2007;128:295–308. doi: 10.1016/j.cell.2006.11.050. [DOI] [PubMed] [Google Scholar]
- 9.Tang J, Qi X, Mercola D, Han J, Chen G. Essential role of p38γ in K-Ras transformation independent of phosphorylation. J Biol Chem. 2005;280:23910–7. doi: 10.1074/jbc.M500699200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qi X, Tang J, Loesch M, Pohl N, Alkan S, Chen G. p38γ MAPK integrates signaling cross-talk between Ras and estrogen receptor to increase breast cancer invasion. Cancer Res. 2006;66:7540–7. doi: 10.1158/0008-5472.CAN-05-4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qi X, Pohl NM, Loesch M, et al. p38α antagonizes p38γ activity through c-Jun-dependent ubiquitin-proteasome pathways in regulating Ras transformation and stress response. J Biol Chem. 2007;282:31398–408. doi: 10.1074/jbc.M703857200. [DOI] [PubMed] [Google Scholar]
- 12.Hasegawa M, Cuenda A, Spillantini MG, et al. Stress-activated protein kinase-3 interacts with the PDZ domain of α1-syntrophin: a mechanism for specific substrate recognition. J Biol Chem. 1999;274:12626–31. doi: 10.1074/jbc.274.18.12626. [DOI] [PubMed] [Google Scholar]
- 13.Sabio G, Reuver S, Feijoo C, et al. Stress- and mitogen-induced phosphorylation of the synapse-associated protein SAP90/PSD-95 by activation of SAPK3/p38γ and ERK1/ERK2. Biochem J. 2004;380:19–30. doi: 10.1042/BJ20031628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sabio G, Simon J, Arthur C, et al. p38γ regulates the localisation of SAP97 in the cytoskeleton by modulating its interaction with GKAP. EMBO J. 2005;24:1134–45. doi: 10.1038/sj.emboj.7600578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cuenda A, Cohen P. Stress-activated protein kinase-2/p38 and a rapamycin-sensitive pathway are required for C2C12 myogenesis. J Biol Chem. 1999;274:4341–6. doi: 10.1074/jbc.274.7.4341. [DOI] [PubMed] [Google Scholar]
- 16.Zhi H, Yang XJ, Kuhnmuench J, et al. SmgGDS is up-regulated in prostate carcinoma and promotes tumour phenotypes in prostate cancer cells. J Pathol. 2009;217:389–97. doi: 10.1002/path.2456. [DOI] [PubMed] [Google Scholar]
- 17.Nagata Y, Lan K-H, Zhou X, et al. PTEN actiation, contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–27. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 18.Yang Q, Tonks NK. Isolation of a cDNA clone encoding a human protein-tyrosine phosphatase with homology to the cytoskeletal-associated proteins band 4.1, ezrin, and talin. Proc Natl Acad Sci USA. 1991;88:5949–53. doi: 10.1073/pnas.88.14.5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang S-H, Liu J, Kabayashi R, Tonks NK. Identification of the cell cycle regulator VCP (p97/CDC48) as a substrate of the band 4.1-related protein-tyrosine phosphatase PTPH1. J Biol Chem. 1999;274:17806–12. doi: 10.1074/jbc.274.25.17806. [DOI] [PubMed] [Google Scholar]
- 20.Sozio MS, Mathis MA, Young JA, et al. PTPH1 is a predominant protein-tyrosine phosphatase capable of interacting with and dephosphorylating the T cell receptor δ subunit. J Biol Chem. 2004;279:7760–9. doi: 10.1074/jbc.M309994200. [DOI] [PubMed] [Google Scholar]
- 21.Flint AJ, Tiganis T, Barford D, Tonks NK. Development of “substrate-trapping” mutants to identify physiological substrates of protein tyrosine phosphatases. Pro Natl Acad Sci USA. 1997;94:1680–5. doi: 10.1073/pnas.94.5.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tiganis T, Bennett AM. Protein tyrosine phosphatase function: the substrate perspective. Biochem J. 2007;402:1–15. doi: 10.1042/BJ20061548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barr AJ, Knapp S. MAPK-specific tyrosine phosphatases: new targets for drug discovery? Trends in Pharmacol Sci. 2006;27:525–30. doi: 10.1016/j.tips.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 24.Nagahara H, Vocero-Akbani AM, Snyder EL, et al. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27kip1 induces cell migration. Nat Med. 1998;4:1449–52. doi: 10.1038/4042. [DOI] [PubMed] [Google Scholar]
- 25.Wang Z, Shen D, Parsons DW, et al. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science. 2004;304:1164–6. doi: 10.1126/science.1096096. [DOI] [PubMed] [Google Scholar]
- 26.Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene. 2007;26:3100–12. doi: 10.1038/sj.onc.1210392. [DOI] [PubMed] [Google Scholar]
- 27.Owens DM, Keyse SM. Differential regulation of MAP kinase signaling by dual-specificity protein phosphatases. Oncogene. 2007;26:3203–13. doi: 10.1038/sj.onc.1210412. [DOI] [PubMed] [Google Scholar]
- 28.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–46. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 29.Tartaglia M, Niemeyer CM, Fragale A, et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34:148–50. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- 30.Saxena M, Williams S, Tasken K, Mustelin T. Crosstalk between cAMP-dependent kinase and MAP kinae through a protein tyrosine phosphatase. Nat Cell Biol. 1999;1:305–11. doi: 10.1038/13024. [DOI] [PubMed] [Google Scholar]
- 31.Julien SG, Dube N, Read M, et al. Protein tyrosine phosphatase 1B deficiency or inhibition delays ErB2-induced mammary tumorigenesis and protects from lung metastasis. Nat Gen. 2007;39:338–46. doi: 10.1038/ng1963. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.