

Abstract
F1-ATPase is a rotary molecular motor in which the central γ subunit rotates inside a cylinder made of α3β3 subunits. To clarify how ATP hydrolysis in three catalytic sites cooperate to drive rotation, we measured the site occupancy, the number of catalytic sites occupied by a nucleotide, while assessing the hydrolysis activity under identical conditions. The results show hitherto unsettled timings of ADP and phosphate releases: starting with ATP binding to a catalytic site at an ATP-waiting γ angle defined as 0°, phosphate is released at ∼200°, and ADP is released during quick rotation between 240° and 320° that is initiated by binding of a third ATP. The site occupancy remains two except for a brief moment after the ATP binding, but the third vacant site can bind a medium nucleotide weakly.
Introduction
F1-ATPase, a water-soluble portion of ATP synthase, has been predicted (1,2) and proved (3) to be a rotary molecular motor in which the central γ subunit rotates inside a hexameric cylinder made of alternately arranged three α and three β subunits (4). The rotation is powered by ATP hydrolysis in three catalytic sites and proceeds in steps of 120° (5). The three catalytic sites reside at β-α interfaces and are hosted primarily by a β subunit, whereas the other three α-β interfaces provide noncatalytic nucleotide binding sites (4). Nucleotide binding to the noncatalytic sites renders the enzyme less susceptible to the MgADP inhibition where F1 stops catalysis for minutes when product (or medium) MgADP is tightly bound to a catalytic site(s) and fails to dissociate (6–8).
Single-molecule observations have shown basic features of this chemo-mechanical energy-converter (9,10). Reverse rotation by application of an external force to the γ subunit has been shown (11,12) to result in net synthesis of ATP from ADP and inorganic phosphate (Pi), implicating a γ-dictator (γ-controlled) mechanism (10) in which the rotary angle of γ determines which of the chemical reaction steps, binding/release of ATP, ADP, or Pi and hydrolysis/synthesis of ATP, are to occur in the three catalytic sites. The coupling scheme, or the precise relationship between the γ angle and chemical reaction steps, that summarizes our experimental findings is shown in Fig. 1. The 120° step (per ATP hydrolyzed) is split into 80–90° and 40–30° substeps (13), which we refer to here as 80° and 40° substeps. A 80° step is initiated by ATP binding to a catalytic site, and ATP binding drives (confers energy to) the 80° rotation (13); the catalytic site that is to bind the ATP is dictated by the γ angle (14). After the 80° rotation, F1 pauses for ∼2 ms, during which the ATP that was bound 200° ago is hydrolyzed into ADP and Pi (14–16). The Pi produced by the hydrolysis, or alternatively the one that was produced 120° ago and has remained on that site (scheme B in Fig. 1), is released in the 80° interim, and the Pi release drives the last 40° of rotation (17). At 120°, a next ATP binds, which triggers the release of the ADP that has been produced at 80° (17). That is, ATP binding and ADP release are nearly simultaneous, and ADP is released after 240° of rotation since it was bound as ATP. ADP release is expected to drive the 80° substep in collaboration with ATP binding.
Figure 1.

Experimental scheme for coupling between catalysis and rotation in F1-ATPase. Circles at bottom represent three catalytic sites (β subunits), with the central arrow showing γ orientation (the orientation in the first ATP-waiting dwell is defined as 0° and the arrow is arbitrarily chosen upward for this orientation). (A and B) Two possible schemes that differ in the release timing of Pi formed by hydrolysis (17). This study points to A, with asterisks indicating the low affinity open site that is in rapid equilibrium with medium nucleotide; at high [ATP], an ATP molecule that happens to be in this site at the Pi release stage may engage in the next rotation. Colors in the time course at top indicate the site in which the rate-limiting reaction is to occur in A: ATP binding at 0°, and hydrolysis and Pi release at 80°.
The scheme above obtained with F1 of thermophilic origin (TF1) implies that the catalytic site occupancy, the number of catalytic sites occupied by a nucleotide, remains two for most of the time except for a short moment between ATP binding and subsequent ADP release (Fig. 1). Early studies on mitochondrial F1-ATPase (MF1), on the other hand, have indicated a bi-site mechanism where the occupancy alternates between one and two, remaining mostly one at low ATP concentrations (18,19). In contrast, Weber et al. (20) and Weber and Senior (21) have directly assessed the site occupancy in Escherichia coli F1 (EF1) by introducing a reporter tryptophan in the catalytic sites, and have shown that the occupancy rises to three at high ATP concentrations ([ATP]) and that the hydrolysis activity parallels the occupancy of the third site, implying a tri-site mechanism where the occupancy alternates between two and three (22). Tryptophan studies on TF1 have led to a similar conclusion (23–25). Crystal structures of F1 with a clearly resolved γ subunit mostly contain two catalytic nucleotide (26), but one binds three nucleotides and γ in this structure is twisted clockwise, possibly mimicking the 80° intermediate (27). The three-nucleotide structure supports the tri-site scheme, connoting that the other two-nucleotide structures resemble the ATP-waiting state (28). At least some of the two-nucleotide structures, however, represent the MgADP inhibited state (26), where γ is at an 80° position (8). Our fluorescence study (29) has also indicated that the two-nucleotide structures mimic the 80° rotation intermediate rather than the ATP-waiting state at 0°. We initiated this study to solve these apparent inconsistencies.
A problem in our coupling scheme was that the timing of ADP release was assessed with a fluorescent substrate analog Cy3-ATP, of which the kinetics may be different from unlabeled ATP. Also, the release timing was resolved by artificially slowing kinetics, and thus, in unrestricted rapid rotation, ADP may fail to dissociate until the end of the 320° interim (orange ADP in Fig. 1 A remaining in the asterisk site), consistent with the tryptophan results. Previous tryptophan studies, on the other hand, have been made in the absence of an ATP regeneration system, and thus ATP may have been largely exhausted during fluorescence assay. In addition, accumulation of ADP likely brings a significant portion of F1 into the MgADP-inhibited state. Whether the fluorescence signal is proportional to the site occupancy has also been challenged (30). In this work, we have assessed the site occupancy of a mutant TF1 from tryptophan quenching whereas the F1 was confirmed to be active, by using an ATP regeneration system lacking tryptophan and monitoring hydrolysis activity under identical conditions. The TF1 mutant we used lacks noncatalytic nucleotide binding sites, allowing unambiguous estimation of the catalytic site occupancy by equilibrium dialysis. We calibrated the tryptophan signal with the dialysis data. The occupancy results we obtained have, unexpectedly revealed the timings of ADP and Pi releases, in addition to showing that the catalysis occurs in the tri-site mode and that apparent bi-site behaviors observed under certain conditions are due to transient heterogeneity in the enzyme population (coexistence of active and inactive forms).
Materials and Methods
See the Supporting Material.
Results
Concentration of ΔNCβY341W subcomplex
In this study we used the minimal subcomplex of TF1 that is active in hydrolysis and rotation, comprising α3β3γ subunits (9,10). Our standard subcomplex is an HC95 mutant (13) (α-W463F to remove the sole tryptophans in the original wild-type TF1, β-His10 at amino terminus to attach the subcomplex to a surface in rotation assay, and γ-S107C/I210C to attach a probe on γ for observation of rotation) that we regard as the wild-type for rotation assay. We made a ΔNCβY341W mutant (24) that lacks noncatalytic nucleotide binding sites and that has a reporter tryptophan (20) in the three catalytic sites by introducing two mutations in HC95: α-K175A/T176A to suppress binding of a nucleotide to the noncatalytic sites (7), and β-Y341W as the reporter tryptophan. We refer to this mutant also as F1 unless ambiguity arises.
For the estimation of site occupancy, the concentration of F1 must be determined accurately. We routinely determine F1 concentrations from absorbance using the molar extinction coefficient ɛ280 nm = 154,000 M−1cm−1 (7). For the mutant ΔNCβY341W, we prepared four independent samples at 1.7–4.1 μM based on the ɛ280 nm above and subjected the samples to amino-acid analysis. The wild-type HC95 was also analyzed as a control. Results on 11 amino acids that could be analyzed reliably are summarized in Table S1. The recovery was 97.9 ± 2.1% for HC95 (mean ± SD for the 11 amino acids) and 93.5 ± 2.7% for ΔNCβY341W. We thus use ɛ280 nm = 165,000 M−1cm−1 for the determination of the concentration of tryptophan-containing ΔNCβY341W in this study.
Fluorescence quenching is proportional to site occupancy
The sole three tryptophan residues in ΔNCβY341W are in the three catalytic sites. Unlike the original EF1 mutant βY331W developed for the tryptophan-quenching assay (20), the fluorescence of ΔNCβY341W was completely quenched at high [ADP] (Fig. 2 A) or high [ATP], as has been reported for the same mutant (24) or a βY341W mutant of TF1 (23). We therefore define percent quenching by 100% × (F0 − F)/(F0 − Fbuf) in all experiments below, where F0 is the fluorescence intensity in the absence of added nucleotide, F the intensity at a given nucleotide concentration, and Fbuf the intensity without F1. In samples containing the ATP regeneration system with tryptophan-less (W-less) pyruvate kinase (PK), the background intensity Fbuf was considerably high, but the fluorescence originating from F1, F0 − Fbuf, remained the same. Quenching in the presence of ATP or ADP reached a constant level well within 100 s (see below), and thus we take 100-s data as steady-state values. As seen in Fig. 2 A, quenching was proportional to [ADP] up to [ADP]/[F1] = 2 where the degree of quenching was close to 67%. At steady-state, ATP and ADP produced indistinguishable results (Fig. 2 B). This is reasonable because the enzyme mixed with ATP falls into the MgADP inhibited state by 100 s even in the presence of the ATP regenerating system (Fig. 3 A below). Note that Mg was 2 mM in excess over MgATP or MgADP in all experiments in this work, to ensure that the nucleotides are in the form of MgATP or MgADP that are the substrates of this enzyme or the whole ATP synthase (22).
Figure 2.
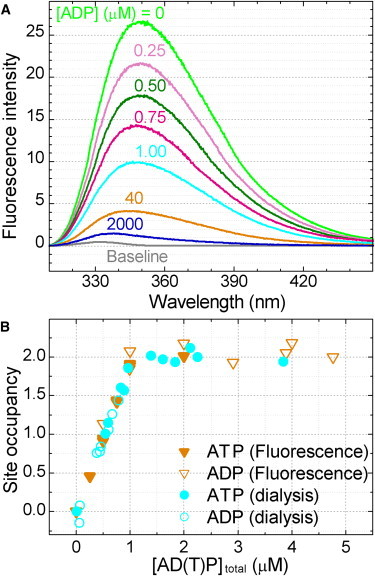
Relation between quenching of tryptophan fluorescence and catalytic site occupancy. (A) Quenching of the fluorescence of reporter tryptophan. Fluorescence spectra of 470 nM ΔNCβY341W measured 100 s after the addition of ADP. The spectrum of the medium alone (baseline), mainly the water Raman emission, has been subtracted from each spectrum. (B) Catalytic site occupancy estimated from fluorescence quenching and micro equilibrium dialysis. To 470 nM ΔNCβY341W (fresh for each measurement) in buffer M, MgATP, or MgADP was added. Horizontal axis is the total nucleotide concentration in the cuvette (fluorescence) or in the central chamber containing F1 (dialysis). For fluorescence, the occupancy was assumed to be proportional to the degree of quenching, full quenching to the buffer level taken as occupancy of three.
Figure 3.
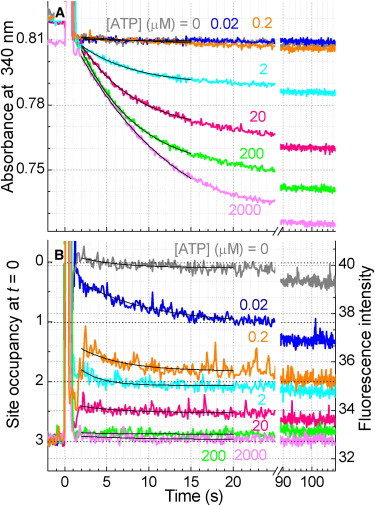
Time courses of (A) ATP hydrolysis activity and (B) nucleotide binding at various [ATP]s. 19 nM F1 was added at time 0 to buffer M containing an ATP regeneration system (1.0 mM PEP, 0.5 mg mL−1 W-less PK, 0.15 mM NADH, 0.05 mg mL−1 LDH). NADH and LDH were omitted in B. The slight decrease at 0 μM ATP in B is due to photobleaching of tryptophan. The baseline fluorescence of ∼32.8 in B comes mostly from the W-less PK. Smooth black curves show fits from which 3-s values were calculated; the values are included in Fig. 4, B and C.
To see if quenching is proportional to the catalytic site occupancy, we directly measured the amount of nucleotide bound to F1 by equilibrium dialysis. The dialysis apparatus consisted of three 250-μL chambers separated by membranes with a cutoff molecular weight of 10 kDa. We put an ATP (or ADP) solution at 0-5 μM in chamber 1, 0.47 μM F1 in chamber 2, and nucleotide-free solution in chamber 3. After equilibration, evidenced by the concentration balance between chambers 1 and 3 to within 0.04 μM, we precipitated F1 in chamber 2 with perchloric acid and measured the total nucleotide concentration in chamber 2. The number of bound nucleotide per F1 was calculated by assuming that the free nucleotide concentration in chamber 2 was equal to that in chambers 1 and 3. This number is the catalytic site occupancy itself, because ΔNCβY341W lacks noncatalytic binding sites. In Fig. 2 B we compare the occupancy and steady-state fluorescence quenching in the same graph. Clearly the quenching is proportional to the occupancy, whether the starting nucleotide is ATP or ADP (long dialysis with ATP should also lead to MgADP inhibition). Occupancy above two could not be measured reliably by the dialysis method, but the rest involves only one catalytic site and thus the quenching above 67% is also expected to be proportional to the site occupancy. Below we calculate the catalytic site occupancy as 0.03 × the percent quenching. Fig. 2 B also shows that binding of the first and second nucleotides is quite tight at steady-state: with F1 at 0.47 μM, free nucleotide concentration remained <0.1 μM until the occupancy exceeded two.
Active and inactive F1 show similar site occupancy
The ATPase activity of ΔNCβY341W was monitored in a coupling assay (7) where ADP produced by F1 was converted back to ATP by the W-less PK and where the pyruvate produced in this ATP-regenerating reaction was converted to lactate by lactate dehydrogenase (LDH). NADH was consumed in the last reaction, which we monitored as a decrease in the absorbance at 340 nm (Fig. 3 A). The coupled reactions were started by adding nucleotide-free F1 at time zero. The rate of hydrolysis (slope of curves in Fig. 3 A) decreased with time, as MgADP inhibition set in, and eventually reached a negligible value (∼1 s−1 or less). Nucleotide binding to noncatalytic sites would relieve part of F1 of the inhibition (6,7), but ΔNCβY341W lacks the noncatalytic sites. Significant activity, however, persisted for tens of seconds: time constant for the activity decay in the initial 20 s was between 7–12 s (Fig. 3 A, [ATP] ≥ 2 μM; Fig. 5 A below, [ATP] ≥ 0.3 μM, or [ATP]free ≥ 50 nM).
Figure 5.
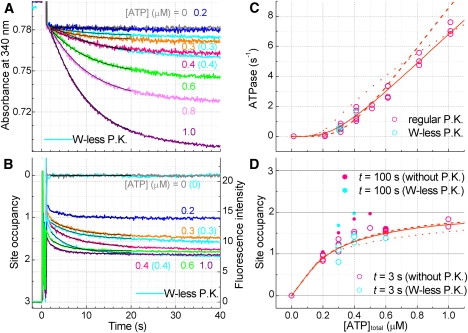
ATPase activity and ATP binding at low ATP/F1 ratios. 190 nM F1 was added to an ATP regenerating system consisting of 2.5 mM PEP, 0.5 or 0.75 mg mL−1 W-less (cyan plots in A–D) or 0.5 mg mL−1 regular PK (other colors), 0.075 mM NADH and 0.025 mg mL−1 LDH. (A) Time courses of ATP hydrolysis. (B) Time courses of ATP binding in the presence of W-less PK and 2.5 mM PEP (cyan) or 2.5 mM PEP alone (other colors). NADH and LDH were omitted. The right-hand scale does not apply to the W-less data, for which the intensity at full quenching was ∼2. (C) ATP dependence of the initial hydrolysis activity. [ATP]total here has been corrected for contaminant nucleotides in NADH (<30 nM). (D) Catalytic site occupancy at 3 s (open symbols) or 100 s (solid). Cyan plots, with W-less PK; pink, without PK. Black curves in A and B are fit from which 3-s values were obtained. Solid, dashed, and dotted lines in C and D are fit with kcat2 = 2.1 s−1 (best fit), 0 (fixed), and 10 s−1 (fixed), respectively (see Fig. 6).
Time courses of tryptophan fluorescence, measured under identical conditions except that LDH and NADH were omitted, are shown in Fig. 3 B. Quenching was not immediate at low [ATP], but this was largely due to the presence of phosphoenolpyruvate (PEP) needed for the coupling assay of hydrolysis activity (Figs. 4 C and 6 below; creatine phosphate showed a similar effect). Whether 1 mM PEP was present or not, the degree of quenching reached a constant value in a few seconds at [ATP] above μM, where significant hydrolysis activity was observed. Immediately after F1 addition, most of F1 must have been active, but almost all were inhibited at the end. The constant degree of quenching thus suggests that the catalytic site occupancy is the same for active and inactive F1 at [ATP] above μM.
Figure 4.

ATPase activity and site occupancy at different [ATP]s. Solid symbols show individual data and open symbols, where shown, their average. Where data points overlap, they are slightly displaced for clarity. [F1] = 19 nM. (A) ATPase activity measured as in Fig. 3A with 1.0 mM PEP and 0.5 or 0.7 mg mL−1 W-less PK (cyan) or 0.7 mg mL−1 regular PK (orange and blue). Orange and cyan, the initial activity at 3 s after the beginning of F1 mixing; blue, the activity at 100–300 s. [ATP]free was calculated from the binding parameters in B. Orange line, Michaelis-Menten fit with Vmax = 59 s−1 and Km = 6.8 μM, which is reproduced on a linear scale in B–E as a reference. (B) Catalytic site occupancy estimated from fluorescence quenching under conditions identical to cyan symbols in A except that NADH and LDH were omitted. Pink and green plots show occupancy at t = 3 s and 100 s, respectively. [ATP]free was calculated from the measured occupancy. Lines show fits with [S]/([S] + Kd1) + [S]/([S] + Kd2) + [S]/([S] + Kd3) where S represents free ATP (Eq. S1), with parameter values at bottom right (SE in smaller fonts). These lines are reproduced as broken lines in C–E. (C) Catalytic site occupancy in the absence of the ATP regenerating system. Symbols and lines as in B. The differences between B and C are due to the presence of 1 mM PEP in B (also see time courses in Fig. S1). (D and E) Occupancy with (D) ADP and (E) AMP-PNP in buffer M in the absence of an ATP regenerating system. Horizontal axes show [ADP] or [AMP-PNP].
Figure 6.
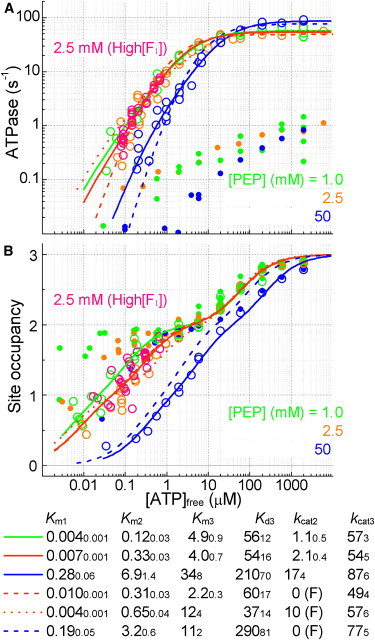
Effect of PEP on ATPase activity and catalytic site occupancy. (A) ATPase activity at indicated [PEP]. Data for 1.0 mM PEP are reproduction of Fig. 4A, and data for 2.5 mM PEP at high [F1] are from Fig. 5C. Other data are for 9–93 nM F1 in buffer M containing 0.5–0.75 mg mL−1 regular PK, 0.15 mM NADH, and 0.05 mg mL−1 LDH. Open symbols, the initial rate at 3 s; solid symbols, the rate at 100–300 s. (B) Catalytic site occupancy with indicated [PEP] without an ATP regenerating system. Data for 2.5 mM PEP at high [F1] are from Fig. 5D. [F1] = 19 nM in others. Open symbols, at 3 s; solid symbols, at 100 s. Data for 1.0 mM PEP here, obtained without PK, do not significantly differ from those in Fig. 4B with PK. Lines are global fit to the ATPase activity and occupancy with Eqs. 3 and 4. Parameter values are shown at bottom (SE in smaller fonts); Km and Kd3 in μM, kcat in s−1, and (F) indicates a fixed value.
ADP is released after 240° of rotation but before the end of the 320° interim
In Fig. 4, A and B, we compare the hydrolysis activity and site occupancy under identical conditions at various [ATP]. Comparison is made at an initial point (3 s from the addition of nucleotide-free F1) where F1 remains active and at ∼100 s where F1 is mostly inhibited. At [ATP] > 0.1 μM where we observed significant initial activity, the occupancy was about two or more except for the PEP-dependent lag below 1 μM ATP (compare Fig. 4, B and C; also Fig. S1), indicating a tri-site mode of catalysis where ADP release occurs after at least 240° of rotation.
The initial hydrolysis activity can be fitted with Michaelis-Menten kinetics with kcat = 59 s−1 and Km = 6.8 μM (Fig. 4 A, orange line). The fit is reproduced in subsequent panels, showing that the activity rises with [ATP] faster than the occupancy of the third site: the apparent dissociation constant for the third ATP, Kd3, was 40 μM at 3 s (Fig. 4 B), higher than the apparent Km of 6.8 μM for activity. At the Km, in particular, the occupancy of the third site is only 15%, indicating that the third site is mostly empty during the rate-limiting dwell at the 80° position. If the 15% occupancy at Km is ascribed to the product ADP, bound as ATP 320° ago, the ADP would be released in the first 30% of the 320° interim. In this case, the occupancy of the third site would saturate at 30% at high enough [ATP] where F1 stays at the rate-limiting interim position for most of the time (13,31).
The third site is open to medium nucleotides
In fact the occupancy rose to three as [ATP] was increased beyond Km. The fact that the occupancy curves at high [ATP], at occupancy above 2, are almost indistinguishable between t = 3 s and 100 s indicates that the nucleotide in the third catalytic site does not necessarily originate from catalysis (not necessarily the product ADP). We propose that medium ATP can enter and weakly bind to the third site (asterisks in Fig. 1 A) while F1 remains in the 80° interim.
This notion is corroborated by titration of F1 with ADP (Fig. 4 D): medium ADP bound to F1, which was not undergoing hydrolysis, with Kd3 similar to that for ATP (other Kd are also apparently similar but precision is low). Both in the MgADP-inhibited state where γ is at ∼80° position (8) and in the 80° interim during active rotation, one catalytic site is open to the medium, allowing loose and rapidly reversible binding of ATP or ADP with Kd ∼40 μM. The occupancy of the third site, characterized by a single Kd3, may almost entirely be due to a medium nucleotide. In this case, the product ADP is released mostly during the rotation from 240° to 320°.
The open site also accommodated an ATP analog AMP-PNP (Fig. 4 E), although the affinity was an order of magnitude lower. The difference between ATP and AMP-PNP seems genuine, because purification of commercial AMP-PNP by ion exchange chromatography did not alter the result. Prefilling of two catalytic sites with ATP or ADP did not alter the affinity of AMP-PNP for the third site (not shown), indicating that the structure of the third site is similar whether the other two sites bind ATP/ADP or AMP-PNP and that the third site discriminates AMP-PNP against ATP/ADP.
Signs of possible bi-site activity
In Fig. 4 B the site occupancy at 3 s decreases below two at [ATP] < 1 μM where the hydrolysis activity is not negligible. Although PEP is responsible for the low occupancy, the same amount of PEP was present in the hydrolysis assay, suggesting that bi-site activity may exist at low [ATP] at least in the presence of PEP. In the scheme in Fig. 1, bi-site hydrolysis would occur at low [ATP] if ADP is spontaneously released in the ATP-waiting state (at 0°) before the arrival of a next ATP. Spontaneous release of Cy3-ADP takes ∼10 s (17), whereas the release rate for unlabeled ADP is yet unknown.
To assess the hydrolysis activity reliably at low [ATP], we used a higher [F1] of 190 nM (and [PEP] of 2.5 mM) in Fig. 5. The activity was negligible at 200 nM total ATP (ATP/F1 ∼1), but significant activity was observed at 300 nM ATP and above. At the total [ATP] of 300 nM, the ATP/F1 ratio is only 1.6. The site occupancy measured under the same conditions is <1.5 at 3 s up to 400 nM ATP (Fig. 5 D). Apparently, some bi-site activity seems to exist. Similar results have been reported for MF1 to support its bi-site activity (32). Quantitative analyses below, however, show that the data can be explained without invoking bi-site activity.
PEP in the medium retards and suppresses binding of ATP (Fig. 6), which is the major reason for the apparent bi-site activity at low [ATP]. To assess the possible contribution of bi-site activity, we fitted each pair of activity and occupancy data in Fig. 6 and Fig. 5) simultaneously (global fit) with a reaction scheme that allows both bi-site (Km2 and kcat2) and tri-site (Km3 and kcat3) activities (Eq. S2, Eq. S3, and Eq. S4). The data at 1 mM PEP was fitted best with the saturating bi-site rate, kcat2, of 1.1 s−1, indicating an almost pure tri-site catalysis (Fig. 6, green solid lines). For the data at 2.5 mM PEP, the best fit was obtained with kcat2 of 2.1 s−1 (orange solid curves in Fig. 5, C and D, and Fig. 6), but the fit with kcat2 set to zero was only slightly worse (orange dashed lines). At 50 mM PEP, the occupancy at 3 s was considerably lower, and significant activity was observed with occupancy of ∼1 or below (∼1 μM ATP in Fig. 6). The global fit returned kcat2 of 17 s−1 (blue solid lines), apparently suggesting a non-negligible bi-site activity. We note, however, that the lower occupancy around μM ATP in the presence of 50 mM PEP was accompanied with lower activity. This is as expected if most of F1 initially binds PEP (due to the high [PEP]) and is unable to engage in hydrolysis until it releases the PEP and binds ATP. Binding of a substrate can be quite slow in the presence of a high concentration of a competitive inhibitor (Fig. S2). Indeed, the site occupancy of F1 rose with a time constant ∼10 s at 2 μM ATP and 50 mM PEP (data not shown but see Fig. S3 B where Pi shows a similar effect). At 3 s, only a small fraction of F1 that has bound ATP would hydrolyze ATP. That small, active fraction may have engaged in tri-site catalysis (Fig. S2). In this slow ATP binding scenario, active and PEP-inhibited F1 coexist for a long time, a situation not dealt with by the global fit that applies to active F1 (Eq. S3 and Eq. S4), and thus kcat2 of 17 s−1 above is irrelevant. The scenario may also apply to the data at 2.5 mM PEP. To summarize, the results up to this point apparently suggest some bi-site activity, but the data can also be explained without invoking a bi-site activity.
Bi-site activity is negligible
If bi-site activity exists (kcat2 > 0), the site occupancy must fall to one with a rate ∼kcat2 when the arrival of the next ATP is somehow delayed, e.g., by lowering [ATP]. In the scheme in Fig. 1, kcat2 is essentially the rate of ADP release in an ATP-waiting state. To see whether, or how fast, the occupancy falls, we added, when the occupancy reached between 1.5 and 2 (Fig. 7 B), a large excess of simple ΔNC mutant (without the βY341W mutation and hence lacking tryptophan) to the assay medium to let it bind and deplete free ATP (arrows). The hydrolysis reaction stopped immediately (Fig. 7 A), the slight residual activity being due to the added ΔNC that would hydrolyze the substoichiometric ATP very slowly (uni-site catalysis) as confirmed in a control experiment. Fluorescence immediately rose on the addition of ΔNC (Fig. 7 B), but this was due to the fluorescence of the added ΔNC (gray bars show the intensity of ΔNC measured independently), showing the absence of immediate ADP release. A slow ADP release (fluorescence increase) followed, but the rate was <0.01 s−1. Because this ADP release is a prerequisite for a bi-site activity, the bi-site activity, if it exists, must be <0.01 s−1. We conclude that the hydrolysis at low occupancy seen in Fig. 4 A, Fig. 5 and Fig. 6 also represents tri-site activity, and the low occupancy is due to the coexistence of F1 with occupancy of zero (some may be one), of which the fraction is greatly augmented by competitive binding of PEP.
Figure 7.

Time courses of hydrolysis and ADP release after ATP depletion. (A) Hydrolysis at 1.0 mM PEP. At time 0, 190 nM ΔNCβY341W was added to 1.0 mM PEP, 0.5 mg mL−1 W-less PK, 0.075 mM NADH, 0.05 mg mL−1 LDH, and indicated ATP in buffer M. At arrows, 2 μM ΔNC (0.022 volume of 91 μM ΔNC) was added to deplete ATP. The slight decline of absorbance after 6 s was due mostly to the hydrolysis by ΔNC, as confirmed by addition of ΔNC alone (not shown). (B) Occupancy at 1.0 mM PEP. Experiments as in A except NADH and LDH were omitted. Gray bars show the fluorescence intensity of added ΔNC measured separately, which accounts for the immediate rise in fluorescence. The slow increases in green and cyan curves between 5–35 s indicate ADP release with a rate <0.01 s−1. At the gray arrow, ΔNC was added again in both green and cyan curves. Brown is a control without ATP, showing the fluorescence level at zero occupancy.
We also found that Pi is a more potent competitive inhibitor, resulting in an apparently low (<2) occupancy with a significant hydrolysis activity (Results in Supporting Material). In the presence of Pi, too, bi-site activity was shown to be negligible.
Discussion
Bi-site versus tri-site
The simplest definition of the bi-site activity in F1 is that the catalytic site occupancy alternates between one and two, whereas the tri-site implies alternation between two and three. Here we have shown that the occupancy by catalytic nucleotides remains essentially two, only momentarily rises to three at low [ATP]. The occupancy of three at high [ATP], at least a large part of it, is superfluous in that it results from weak and reversible binding of a medium nucleotide. In this sense, our scheme may appear intermediate between genuine bi-site and tri-site. It is actually a tri-site mechanism because release of ADP requires binding of ATP and thus, for every catalytic cycle, there is a brief moment where the occupancy rises to three. If spontaneous ADP release occurred at low [ATP], simple promotion of ADP release by ATP binding might be regarded nonessential (33). In fact, the spontaneous release is very slow, at most 0.01 s−1. Also, release of ADP is not a passive event in that it drives, or confers torque for, rotation (17).
Milgrom et al. (32) have shown consistently that MF1 operates in the bi-site mode. Their experiment (32) similar to our Fig. 5 C, which was actually inspired by their work, gave a result close to the dotted line in Fig. 5 C, claiming bi-site stronger than our result would. In another report (19), site occupancy was measured in filter assays 5 s after mixing 0.85 μM MF1 with ATP. The occupancy was below 1.5 up to 60 μM ATP, whereas hydrolysis activity obeyed simple Michaelis-Menten kinetics with Km of 77 μM. The measurements were made in the presence of 10 mM (or 25 mM) PEP, and thus part of the sample may have bound PEP instead of ATP, as implied for our results. Also, analysis of filtrates may not have been very precise at [ATP] >> [MF1]. Thus, the claim that MF1 undergoes bi-site catalysis may not be solid.
ADP release
An outstanding question has been the timing of ADP release during free rotation. The scheme in Fig. 1 was based on the behavior of the fluorescent ATP during artificially slowed rotation (17). The possibilities remained that the fluorescent ADP may dissociate faster than unlabeled ADP, or that the controlled rotation was too slow, letting otherwise lingering ADP dissociate at ∼240°. This work narrows down the ADP release timing to immediately after ATP binding, within the 80° substep that the ATP binding initiates: Fig. 1 applies equally to the release of unlabeled ADP during unhindered, rapid catalysis. The product ADP might linger, but not beyond 30% of the next dwell. The number of “catalytic nucleotides” thus remains two even at saturating [ATP], except for the brief moment of the 80° substep.
The early release of product ADP allows medium ATP to fill the asterisked site in Fig. 1 A at high [ATP]. If an ATP molecule happens to stay at the moment of Pi release, that ATP molecule may initiate the next 80° rotation: entry of ATP in the catalytic site may precede Pi release. Early ATP binding has been proposed in a somewhat different context (31).
The scheme in Fig. 1 A may not apply to TF1 at extremely low temperatures: at 4°C a rate-limiting reaction other than ATP binding governs the 0° dwell, an obvious possibility being slowed ADP release that would block the next ATP binding (34). Different schemes have also been proposed for F1 under load (31).
Pi release
Of the two possibilities for the Pi release timing (Fig. 1, A or B) our results here point to Fig. 1 A with an open site (asterisk) freely accessible from the medium: medium ATP or AMP-PNP can bind to this site, and medium Pi competes with the nucleotide binding (Results in Supporting Material), denying the retention of product Pi in this site. An event in the asterisked site has been shown to be rate limiting in a particular TF1 mutant (16), but it is not Pi release, possibly protein isomerization or slowed ADP release. Note that the low affinity asterisked site is converted to an ATP-binding form on counterclockwise rotation of γ, whereas its affinity for ADP increases on clockwise rotation for ATP synthesis. The interchange between ATP-favoring and ADP-favoring forms through a fully open form has been proposed (33) as part of the binding change mechanism.
Oxygen exchange
Oxygen exchange studies have shown that, at low [ATP]s, bound ATP undergoes rounds of reversible hydrolysis/resynthesis before ADP and Pi are eventually released into the medium (18,35). The reversals must occur at ATP-waiting angles, possibly accompanying angular fluctuations of γ. In our scheme A in Fig. 1, it would be the cyan ATP at 0° that would undergo reversible hydrolysis. Our view is that, at 0°, the equilibrium is overwhelmingly toward synthesis (hence we designate the state simply as ‘ATP’), whereas it shifts to hydrolysis at 80°; momentary but frequent hydrolysis at 0° can account for the oxygen exchange results. This view is consistent with the finding in a mutant TF1 that, at low [ATP] where the ATP-waiting dwell at 0° is extremely long, ATP hydrolysis apparently proceeds to completion during the 0° dwell (36).
Crystal structures
All two-nucleotide structures in crystals reported so far are grossly similar to each other, and most are likely MgADP-inhibited with some exceptions (26). In the inhibited state, γ is oriented at 80° (8), suggesting that the crystal structures mimic the 80° intermediate during rotation. Our fluorescence study of rotating TF1 (29) has also indicated that the two-nucleotide structures resemble the 80° intermediate rather than the ATP-waiting state (we took this as evidence for a bi-site mechanism, a view we now abandon). This assignment, also shown for EF1 (37), has not been accepted widely, because, according to a simple tri-site scheme, the intermediate must bind three catalytic nucleotides and one three-nucleotide structure has been reported (27). This study resolves this difficulty by showing that the number of catalytic nucleotides is in fact two at 80°. Medium nucleotide can still bind to the third site, but binding is loose with Kd around 40 μM (Fig. 4, B–D). Thus, slight wobble of the nucleotide is sufficient to obscure its presence in the x-ray analysis, whereas fluorescence quenching, presumably due to direct contact between a nucleotide and the tryptophan as suggested by the crystal structures, does not require precise positioning. The failure to observe a third nucleotide in a crystal grown in 5 mM AMP-PNP (38) is not surprising in view of the weaker binding of AMP-PNP (Fig. 4 E). The three-nucleotide structure (27) may mimic a state during the 80° rotation, before ADP release. A recent report (39) that the structure of nucleotide-free MF1 is similar, including γ orientation, to the two-nucleotide structures may indicate that the MgADP-inhibited structure is the most stable form of the enzyme and F1, unless rotating, falls into this form irrespective of the number of bound nucleotides.
In the two-nucleotide structures solved at high resolutions, a phosphate (or sulfate) ion has often (but not always) been found in the empty catalytic site (26,39,40). This raises the possibility that Pi release lags behind the release of its companion ADP (40), as in scheme B in Fig. 1. Our results indicate that it would be a medium Pi (sulfate), not the product Pi, that fills the asterisked site in Fig. 1 A.
This study shows what we believe to be the chemo-mechanical coupling scheme for the major reaction pathway of TF1 at room temperature over a broad range of ATP concentrations. F1 of different origin, or TF1 in different conditions, may behave differently. Also, actual kinetics must be stochastic, and thus the angle and chemical states in the scheme fluctuate in reality. At present, though, we are unaware of a compelling piece of evidence that is totally unreconcilable with our scheme. A major task that remains is to explain the coupling scheme in terms of atomic structures. A structure resembling the ATP-waiting conformation is highly awaited. Also, a recent discovery that the penetrating portion of the γ subunit is not necessary for rotation (41) calls for rethinking of possible mechanisms by which conformational changes in the β (and α) subunits are converted to γ rotation and, conversely, the γ angle controls the conformations of β and α subunits.
Acknowledgments
We thank members of Kinosita lab for help and advice; and H. Umezawa, M. Fukatsu, and K. Sakamaki for lab management and encouragement.
This work was supported by Grants-in-Aid for Specially Promoted Research and for Young Scientists (B) from the Ministry of Education, Sports, Culture, Science, and Technology of Japan.
Footnotes
Dr. Masasuke Yoshida's present address is Department of Biotechnology, Kyoto Sangyo University, Kamigamo-Motoyama, Kita-ku, Kyoto, Japan.
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Supporting Material
References
- 1.Boyer P.D., Kohlbrenner W.E. The present status of the binding-change mechanism and its relation to ATP formation by chloroplasts. In: Selman B.R., Selman-Reimer S., editors. Energy Coupling in Photosynthesis. Elsevier; Amsterdam: 1981. pp. 231–240. [Google Scholar]
- 2.Oosawa F., Hayashi S. The loose coupling mechanism in molecular machines of living cells. Adv. Biophys. 1986;22:151–183. doi: 10.1016/0065-227x(86)90005-5. [DOI] [PubMed] [Google Scholar]
- 3.Noji H., Yasuda R., Kinosita K., Jr. Direct observation of the rotation of F1-ATPase. Nature. 1997;386:299–302. doi: 10.1038/386299a0. [DOI] [PubMed] [Google Scholar]
- 4.Abrahams J.P., Leslie A.G.W., Walker J.E. Structure at 2.8 Å resolution of F1-ATPase from bovine heart mitochondria. Nature. 1994;370:621–628. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- 5.Yasuda R., Noji H., Yoshida M. F1-ATPase is a highly efficient molecular motor that rotates with discrete 120° steps. Cell. 1998;93:1117–1124. doi: 10.1016/s0092-8674(00)81456-7. [DOI] [PubMed] [Google Scholar]
- 6.Jault J.M., Dou C., Allison W.S. The α3β3γ subcomplex of the F1-ATPase from the thermophilic Bacillus PS3 with the βT165S substitution does not entrap inhibitory MgADP in a catalytic site during turnover. J. Biol. Chem. 1996;271:28818–28824. doi: 10.1074/jbc.271.46.28818. [DOI] [PubMed] [Google Scholar]
- 7.Matsui T., Muneyuki E., Yoshida M. Catalytic activity of the α3β3γ complex of F1-ATPase without noncatalytic nucleotide binding site. J. Biol. Chem. 1997;272:8215–8221. doi: 10.1074/jbc.272.13.8215. [DOI] [PubMed] [Google Scholar]
- 8.Hirono-Hara Y., Noji H., Yoshida M. Pause and rotation of F1-ATPase during catalysis. Proc. Natl. Acad. Sci. USA. 2001;98:13649–13654. doi: 10.1073/pnas.241365698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoshida M., Muneyuki E., Hisabori T. ATP synthase—a marvelous rotary engine of the cell. Nat. Rev. Mol. Cell Biol. 2001;2:669–677. doi: 10.1038/35089509. [DOI] [PubMed] [Google Scholar]
- 10.Kinosita K., Jr., Adachi K., Itoh H. Rotation of F1-ATPase: how an ATP-driven molecular machine may work. Annu. Rev. Biophys. Biomol. Struct. 2004;33:245–268. doi: 10.1146/annurev.biophys.33.110502.132716. [DOI] [PubMed] [Google Scholar]
- 11.Itoh H., Takahashi A., Kinosita K., Jr. Mechanically driven ATP synthesis by F1-ATPase. Nature. 2004;427:465–468. doi: 10.1038/nature02212. [DOI] [PubMed] [Google Scholar]
- 12.Rondelez Y., Tresset G., Noji H. Highly coupled ATP synthesis by F1-ATPase single molecules. Nature. 2005;433:773–777. doi: 10.1038/nature03277. [DOI] [PubMed] [Google Scholar]
- 13.Yasuda R., Noji H., Itoh H. Resolution of distinct rotational substeps by submillisecond kinetic analysis of F1-ATPase. Nature. 2001;410:898–904. doi: 10.1038/35073513. [DOI] [PubMed] [Google Scholar]
- 14.Nishizaka T., Oiwa K., Kinosita K., Jr. Chemomechanical coupling in F1-ATPase revealed by simultaneous observation of nucleotide kinetics and rotation. Nat. Struct. Mol. Biol. 2004;11:142–148. doi: 10.1038/nsmb721. [DOI] [PubMed] [Google Scholar]
- 15.Shimabukuro K., Yasuda R., Yoshida M. Catalysis and rotation of F1 motor: cleavage of ATP at the catalytic site occurs in 1 ms before 40° substep rotation. Proc. Natl. Acad. Sci. USA. 2003;100:14731–14736. doi: 10.1073/pnas.2434983100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ariga T., Muneyuki E., Yoshida M. F1-ATPase rotates by an asymmetric, sequential mechanism using all three catalytic subunits. Nat. Struct. Mol. Biol. 2007;14:841–846. doi: 10.1038/nsmb1296. [DOI] [PubMed] [Google Scholar]
- 17.Adachi K., Oiwa K., Kinosita K., Jr. Coupling of rotation and catalysis in F1-ATPase revealed by single-molecule imaging and manipulation. Cell. 2007;130:309–321. doi: 10.1016/j.cell.2007.05.020. [DOI] [PubMed] [Google Scholar]
- 18.Boyer P.D. The ATP synthase—a splendid molecular machine. Annu. Rev. Biochem. 1997;66:717–749. doi: 10.1146/annurev.biochem.66.1.717. [DOI] [PubMed] [Google Scholar]
- 19.Milgrom Y.M., Cross R.L. Rapid hydrolysis of ATP by mitochondrial F1-ATPase correlates with the filling of the second of three catalytic sites. Proc. Natl. Acad. Sci. USA. 2005;102:13831–13836. doi: 10.1073/pnas.0507139102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weber J., Wilke-Mounts S., Senior A.E. Specific placement of tryptophan in the catalytic sites of Escherichia coli F1-ATPase provides a direct probe of nucleotide binding: maximal ATP hydrolysis occurs with three sites occupied. J. Biol. Chem. 1993;268:20126–20133. [PubMed] [Google Scholar]
- 21.Weber J., Senior A.E. Catalytic mechanism of F1-ATPase. Biochim. Biophys. Acta. 1997;1319:19–58. doi: 10.1016/s0005-2728(96)00121-1. [DOI] [PubMed] [Google Scholar]
- 22.Weber J., Senior A.E. ATP synthase: what we know about ATP hydrolysis and what we do not know about ATP synthesis. Biochim. Biophys. Acta. 2000;1458:300–309. doi: 10.1016/s0005-2728(00)00082-7. [DOI] [PubMed] [Google Scholar]
- 23.Dou C., Fortes P.A., Allison W.S. The α3(β Y341W)3γ subcomplex of the F1-ATPase from the thermophilic Bacillus PS3 fails to dissociate ADP when MgATP is hydrolyzed at a single catalytic site and attains maximal velocity when three catalytic sites are saturated with MgATP. Biochemistry. 1998;37:16757–16764. doi: 10.1021/bi981717q. [DOI] [PubMed] [Google Scholar]
- 24.Ono S., Hara K.Y., Muneyuki E. Origin of apparent negative cooperativity of F1-ATPase. Biochim. Biophys. Acta. 2003;1607:35–44. doi: 10.1016/j.bbabio.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 25.Ren H., Bandyopadhyay S., Allison W.S. The α3(βMet222Ser/Tyr345Trp)3γ subcomplex of the TF1-ATPase does not hydrolyze ATP at a significant rate until the substrate binds to the catalytic site of the lowest affinity. Biochemistry. 2006;45:6222–6230. doi: 10.1021/bi060232w. [DOI] [PubMed] [Google Scholar]
- 26.Bowler M.W., Montgomery M.G., Walker J.E. Ground state structure of F1-ATPase from bovine heart mitochondria at 1.9 Å resolution. J. Biol. Chem. 2007;282:14238–14242. doi: 10.1074/jbc.M700203200. [DOI] [PubMed] [Google Scholar]
- 27.Menz R.I., Walker J.E., Leslie A.G.W. Structure of bovine mitochondrial F1-ATPase with nucleotide bound to all three catalytic sites: implications for the mechanism of rotary catalysis. Cell. 2001;106:331–341. doi: 10.1016/s0092-8674(01)00452-4. [DOI] [PubMed] [Google Scholar]
- 28.Pu J., Karplus M. How subunit coupling produces the γ-subunit rotary motion in F1-ATPase. Proc. Natl. Acad. Sci. USA. 2008;105:1192–1197. doi: 10.1073/pnas.0708746105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yasuda R., Masaike T., Kinosita K., Jr. The ATP-waiting conformation of rotating F1-ATPase revealed by single-pair fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA. 2003;100:9314–9318. doi: 10.1073/pnas.1637860100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bulygin V.V., Milgrom Y.M. Studies of nucleotide binding to the catalytic sites of Escherichia coli βY331W-F1-ATPase using fluorescence quenching. Proc. Natl. Acad. Sci. USA. 2007;104:4327–4331. doi: 10.1073/pnas.0700078104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spetzler D., Ishmukhametov R., Frasch W.D. Single molecule measurements of F1-ATPase reveal an interdependence between the power stroke and the dwell duration. Biochemistry. 2009;48:7979–7985. doi: 10.1021/bi9008215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Milgrom Y.M., Murataliev M.B., Boyer P.D. Bi-site activation occurs with the native and nucleotide-depleted mitochondrial F1-ATPase. Biochem. J. 1998;330:1037–1043. doi: 10.1042/bj3301037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boyer P.D. Catalytic site occupancy during ATP synthase catalysis. FEBS Lett. 2002;512:29–32. doi: 10.1016/s0014-5793(02)02293-7. [DOI] [PubMed] [Google Scholar]
- 34.Furuike S., Adachi K., Kinosita K., Jr. Temperature dependence of the rotation and hydrolysis activities of F1-ATPase. Biophys. J. 2008;95:761–770. doi: 10.1529/biophysj.107.123307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O'Neal C.C., Boyer P.D. Assessment of the rate of bound substrate interconversion and of ATP acceleration of product release during catalysis by mitochondrial adenosine triphosphatase. J. Biol. Chem. 1984;259:5761–5767. [PubMed] [Google Scholar]
- 36.Shimabukuro K., Muneyuki E., Yoshida M. An alternative reaction pathway of F1-ATPase suggested by rotation without 80°/40° substeps of a sluggish mutant at low ATP. Biophys. J. 2006;90:1028–1032. doi: 10.1529/biophysj.105.067298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sielaff H., Rennekamp H., Junge W. Functional halt positions of rotary FOF1-ATPase correlated with crystal structures. Biophys. J. 2008;95:4979–4987. doi: 10.1529/biophysj.108.139782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Menz R.I., Leslie A.G., Walker J.E. The structure and nucleotide occupancy of bovine mitochondrial F1-ATPase are not influenced by crystallization at high concentrations of nucleotide. FEBS Lett. 2001;494:11–14. doi: 10.1016/s0014-5793(01)02302-x. [DOI] [PubMed] [Google Scholar]
- 39.Kabaleeswaran V., Shen H., Mueller D.M. Asymmetric structure of the yeast F1 ATPase in the absence of bound nucleotides. J. Biol. Chem. 2009;284:10546–10551. doi: 10.1074/jbc.M900544200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kabaleeswaran V., Puri N., Mueller D.M. Novel features of the rotary catalytic mechanism revealed in the structure of yeast F1 ATPase. EMBO J. 2006;25:5433–5442. doi: 10.1038/sj.emboj.7601410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Furuike S., Hossain M.D., Kinosita K., Jr. Axle-less F1-ATPase rotates in the correct direction. Science. 2008;319:955–958. doi: 10.1126/science.1151343. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.