

Abstract
The Helicobacter pylori babA gene encodes an outer membrane protein that mediates binding to fucosylated ABH antigens of the ABO blood group. We recently demonstrated that BabA expression is lost during experimental infection of rhesus macaques with H. pylori J166. We sought to test the generality of this observation by comparison of different H. pylori strains and different animal hosts. Challenge of macaques with H. pylori J99 yielded output strains that lost BabA expression, either by selection and then expansion of a subpopulation of J99 that had a single-base-pair mutation that encoded a stop codon or by gene conversion of babA with a duplicate copy of babB, a paralog of unknown function. Challenge of mice with H. pylori J166, which unlike J99, has 5′ CT repeats in babA, resulted in loss of BabA expression due to phase variation. In the gerbil, Leb binding was lost by replacement of the babA gene that encoded Leb binding with a nonbinding allele that differed at six amino acid residues. Complementation experiments confirmed that change in these six amino acids of BabA was sufficient to eliminate binding to Leb and to gastric tissue. These results demonstrate that BabA expression in vivo is highly dynamic, and the findings implicate specific amino acid residues as critical for binding to fucosylated ABH antigens. We hypothesize that modification of BabA expression during H. pylori infection is a mechanism to adapt to changing conditions of inflammation and glycan expression at the epithelial surface.
Helicobacter pylori is a gastric bacterial pathogen of humans that chronically infects an estimated 50% of the world's population (19). The consequence of long-term infection with H. pylori is typically asymptomatic gastritis, but ca. 10 to 15% of those infected will develop peptic ulcer disease or gastric adenocarcinoma (19). Several bacterial virulence factors are found more commonly in H. pylori strains that are associated with disease, the best studied of which are the cytotoxin-associated gene pathogenicity island (cag PAI) and certain alleles of the vacuolating cytotoxin, VacA. Another bacterial factor that is associated with disease, rather than asymptomatic infection, is the capacity for adherence. Approximately 4% of the H. pylori genome encodes a diverse repertoire of outer membrane proteins (OMPs), the largest of which is the 21-gene Hop family (1). BabA is a member of the Hop family that binds Lewis b (Leb) and related terminal fucose residues found on blood group O (H antigen), A, and B antigens that are expressed on the gastric epithelium (3, 6). Some studies suggest that patients infected with strains that express BabA are more likely to present with ulcer or gastric cancer, particularly if they also have the cag PAI and the s1m1 allele of vacA (12, 35).
DNA sequence analysis suggests that among the genes encoding Hop family proteins there is considerable potential for both antigenic variation, in which combinatorial DNA shuffling creates antigenically distinct proteins, and phase variation, in which there is reversible on/off switching of gene expression. For example, there is extensive 5′ and 3′ homology between babA and two other H. pylori OMPs of unknown function called babB and babC (1). The greater similarity among bab paralogues (within a genome) than among orthologues (across genomes) suggests that there is frequent recombination and concerted evolution among these genes (27). Regions of dinucleotide CT repeats in the 5′ coding region of babA, babB, and other Hop genes may promote phase variation by slipped-strand mispairing during DNA replication. Finally, polynucleotide (A or T) tracts in babA and other Hop promoters might alter expression by subtle changes in the spacing between −35 and −10 hexamers. BabA proteins are also highly polymorphic in the variable midregion (26, 27), and they differ by more than 1,500-fold in their affinity for Leb (3).
We recently showed that H. pylori strains recovered from experimentally challenged rhesus macaques had lost expression of BabA and the capacity to attach to Leb (33). In some cases the strains underwent a gene conversion event in which the babA gene was replaced by a duplicate copy of babB. In other cases the babA gene was present in H. pylori strains recovered from macaques, but it was not expressed due to phase variation that resulted from gain or loss of one CT repeat in the 5′ coding region of babA. These results demonstrate that H. pylori regulates OMP expression in vivo using both antigenic variation and phase variation. Here we sought to extend these observations by comparison of different animal hosts, including primate, mouse, and gerbil, and different H. pylori strains. The results indicate that loss of BabA expression and Leb attachment is a robust phenomenon that occurs via multiple genetic mechanisms in different H. pylori strains and across diverse animal models. Furthermore, analysis of subtle BabA variants that emerge during colonization permitted the identification of six amino acid changes that are sufficient to eliminate binding to Leb.
MATERIALS AND METHODS
Bacterial strains and culture.
H. pylori strains J166, J99, and 7.13, which have all been described (2, 11, 33), contain the cag PAI, express BabA, and attach to Leb. One strain recovered from a gerbil challenged with 7.13 was studied intensively and designated 8.31. Agar cultures were performed on brucella agar (BBL/Becton Dickinson, Sparks, MD) supplemented with 5% heat-inactivated newborn calf serum (NCS; Invitrogen, Carlsbad, CA) and ABPNV antibiotics (amphotericin B, 20 mg/liter; bacitracin, 200 mg/liter; polymyxin B, 3.3 mg/liter; nalidixic acid, 10.7 mg/liter; vancomycin, 100 mg/liter [all from Sigma]). H. pylori liquid cultures for inoculation were grown in brucella broth with 5% NCS and ABPNV for ca. 24 h (optical density at 600 nm [OD600], 0.35 to 0.45), pelleted by centrifugation, and suspended in brucella broth.
Animals and experimental challenge. (i) Mice.
Specific-pathogen (Helicobacter)-free, female C57BL/6 (Taconic, Germantown, NY), C3H/HeJ (Jackson Laboratories, Bar Harbor, ME), and FVB/N (National Cancer Institute; Frederick, MD) mice (n = 4 to 6/strain) were used. Mice were housed in microisolator cages and provided with irradiated food and autoclaved water ad libitum. At 10 to 12 weeks of age the mice were challenged three times at 2-day intervals with 2.5 × 109 CFU of H. pylori J166 suspended in 0.25 ml of brucella broth; the challenge was administered by oral gavage with a ball-end feeding needle. All mice were fasted 3 to 4 h before and after inoculation and were euthanized 2 weeks later with an overdose of pentobarbital sodium injection (50 mg/ml). The stomach was cut longitudinally, and half was placed in brucella broth, weighed, ground with a sterile glass rod until the mucosal cells were homogenized, and then plated quantitatively by serial dilution on brucella agar supplemented with 5% NCS and ABPNV.
(ii) Monkeys.
Two male rhesus macaques (7 and 8 years old) were raised at the California National Primate Research Center from the day of birth using methods previously described (31, 32) to ensure that they had normal gastric histology and were free of H. pylori infection. Animals were housed individually and fed commercial primate chow (Purina) and fruit, with water available ad libitum. The rhesus macaques were orogastrically inoculated with 109 CFU of H. pylori J99 suspended in 2 ml of brucella broth. Endoscopy was performed with ketamine anesthesia (10 mg/kg given intramuscularly) after an overnight fast at 1, 4, 8, 12, and 18 weeks postinoculation. Four mucosal biopsy specimens each were obtained from the gastric antrum and corpus and were cultured as described for the mice.
(iii) Gerbils.
Male Mongolian gerbils (n = 2; Harlan Labs, Indianapolis, IN) at 4 to 8 weeks of age were orogastrically challenged three times at 2-day intervals with 109 CFU of H. pylori 7.13 suspended in 0.8 ml of brucella broth. Gerbils were anesthetized with CO2 and euthanized with cervical dislocation at 12 weeks postinoculation. The stomachs were removed, and the glandular portions were divided in half. One half was ground with a sterile glass rod until the mucosal cells were homogenized and then plated quantitatively by serial dilution on brucella agar supplemented with 5% NCS and ABPNV.
Culture plates from mice, monkeys, and gerbils were incubated for up to 6 days at 37 C° in 5% CO2. H. pylori was identified in the conventional manner by colony morphology (pinhead-sized translucent colonies), microscopy (Gram-negative curved organisms), and biochemistry (oxidase, catalase, and urease positive). Single colonies were expanded and frozen at −80°C in brucella broth containing 20% (vol/vol) glycerol. All animals were housed under protocols approved by ALAAC and the Institutional Animal Care and Use Committee.
Leb adherence assay.
Attachment of H. pylori to the Leb blood group antigen was measured in duplicate by using an enzyme-linked immunosorbent assay as described previously (12). Briefly, Leb conjugated to human serum albumin (Isosep, Tullinge, Sweden) was immobilized on polystyrene 96-well microtiter plates (Nalgene; Nunc) and incubated with digoxigenin-labeled (Roche Molecular Biochemicals) H. pylori harvested from 48-h plate cultures. Bound bacteria were detected using anti-digoxigenin antibody conjugated to horseradish peroxidase and ABTS [2,2′azinobis(3-ethylbenzthiazolinesulfonic acid)] solution (Roche Molecular Biochemicals). Extinction was quantified in a microplate reader (Bio-Rad) using a dual wavelength (405/490 nm), and the Leb adherence ratio was expressed as the ratio of the OD of wells coated with Leb to that of uncoated control wells.
DNA sequence analysis of babA and babB.
DNA was prepared from single colony isolates by using the CTAB (cetyltrimethylammonium bromide) extraction method (4). In H. pylori J166, babA (JHP0833) is downstream of hypD (JHP0835) and babB (JHP1164) is downstream of s18 (JHP1165). Both babA and babB contain eight pairs of CT repeats in the 5′ end of the gene. Phase variation by slipped-strand mispairing that deletes or adds a CT repeat may result in loss of the open reading frame (ORF) and, in the case of babA, loss of the Leb attachment. To determine the ORF status of babA and babB in H. pylori J166, PCR was performed using standard conditions and gene-specific primers (Table 1) to amplify a 1,500- to 2,000-bp fragment that contained babA (primers HypDF1 and BabAR1) or babB (primers S18F1 and BabBR1) as previously described (7). Primers P160R and P178R were then used to sequence ∼450 bp of J166 babA and babB, respectively, which contained the CT repeats. PCR of H. pylori J99 was performed with primers (HypDF1 and BabAR10) to amplify and sequence the first 1,281 bp of babA from rhesus output strains that did not attach to Leb (Leb adherence ratio < 1.5). If the babA gene was not detected, we used primers (HypDF1 and BabBR1) to amplify and sequence 772 bp of babB at the babA locus. H. pylori 7.13 contains two babA alleles (and no babB): babA2 downstream of hypD and babA1 downstream of s18. The full-length babA2 gene from H. pylori 7.13 and from gerbil output strain 8.31 was amplified and sequenced in two fragments (primer pair HypDF1 and BabAR129 and primer pair BabAF4 and BabAR927). Full-length babA1 from 7.13 and 8.31 was amplified and sequenced in two fragments (primer pair S18F1 and BabAR129 and primer pair BabAF4 and 1162R). All DNA sequencing was performed on both strands of purified PCR fragments (QIAquick PCR purification kit; Qiagen, Valencia, CA) using dye terminator sequencing chemistry.
TABLE 1.
Primers used for amplification and sequencing
| Primer | Designationa | Gene | Positionb | Sequence (5′-3′)c |
|---|---|---|---|---|
| P160R | JHP0833 | babA | 444-424 | ATACCCTGGCTCGTTGTTGAA |
| BabAF4 | JHP0833 | babA | 856-876 | GCGAGCACGCTCATTAACACC |
| BabAF1253 | JHP0833 | babA | 2193-2216 | AACGGATCCCAGGCTTTATAGCGTGTATTTGAA |
| BabAR1 | JHP0833 | babA | 723-705 | TTTGCCGTCTATGGTTTGG |
| BabAR129 | JHP0833 | babA | 1002-983 | CATCTTTTGGATCGCGCTGA |
| BabAR10 | JHP0833 | babA | 1281-1261 | ACCAGTACCAGCGGTTGATGG |
| BabAR1252 | JHP0833 | Intergenic | −17 to −46* | AACGAGCTCGGATTAAATTGGATTAGTATTAGGGATTTC |
| BabAR927 | JHP0833 | Intergenic | 997-977† | GTTCATTATAGCACAAGCGAT |
| HypDF1 | JHP0835 | hypD | 675-697 | TTTTGAGCCGGTGGATATATTAG |
| HypDF2 | JHP0835 | hypD | 627-648 | AATGCGGCCGCCTATGCTCCTTTAGTGGATCGC |
| 0895R | NAd | Predicted | 288-267 | AATCTCGAGCACATCTCTCATGGCTTGTTGC |
| 1162R | JHP1162 | Predicted | 247-228 | CTCCCTTGCGGTATAACCG |
| BabBR1 | JHP1164 | babB | 772-750 | TCGCTTGTTTTAAAAGCTCTTGA |
| S18F1 | JHP1165 | s18 | 197-218 | CTTTAATCCCCTACATTGTGGA |
| P178R | JHP1164 | babB | 481-459 | CATGTCCTGGCTCATAATACGAA |
| UF1 | NA | cat | NA | CGACTTTGGTTAACCCGCAAATCCCAT |
| CamF | NA | cat | NA | GATATAGATTGAAAAGTGGATAGATTTA |
| C2CamR | NA | cat | NA | AACGGATCCTTATCAGTGCGACAAACTGGGAT |
| RpsLCamR | NA | rpsL | NA | TAAATCTATCCACTTTTCAATCTATATCATCTAACGGATTTGTCTGTATG |
| UR6 | NA | rpsL | NA | GGGCGTGGTGGATTATGTGTATTA |
| RpsLF | NA | rpsL | NA | AACGAGCTCGATGCTTTATAACTATGGATTAAACAC |
The designation corresponds to H. pylori strain J99.
The position corresponds to H. pylori strain J99. *, This position is in the intergenic region upstream from the babA start codon in H. pylori strain 7.13; †, this position is in the intergenic region downstream from the babA stop codon in H. pylori strain 7.13.
Boldface nucleotides are complementary to the primer CamF.
NA, not applicable.
Immunoblotting.
Bacterial cells were sonicated and the supernatants were quantitated by using a Bradford assay (Bio-Rad, Hercules, CA). Control proteins were prepared from wild-type H. pylori J166, and its isogenic babA knockout was created by allelic exchange using methods similar to those described previously (16). For each blot, 10 μg of total protein was used for SDS-PAGE, followed by transfer to a polyvinylidene difluoride membrane (Hybond-P; GE Healthcare, Piscataway, NJ). BabA was detected with polyclonal antibody (AK277) kindly provided by Stefan Odenbreit (25), with detection by enhanced chemiluminescence peroxidase anti-rabbit antibodies (GE Healthcare) and visualization by chemiluminescent signal on a Typhoon Imager 9410 (GE Healthcare).
Contraselection for genetic exchange of babA.
Contraselectable streptomycin susceptibility was used essentially as described previously (9) to exchange babA genes between H. pylori 7.13 (gerbil input) and 8.31 (gerbil output), except that the erm gene was replaced with a cat resistance gene. To make the cat-rpsL cassette, cat was amplified (primers UF1 and CamF, Table 1) from H. pylori containing a cat insert in the urease gene (34), and rpsL was amplified (primers RpsLCamR and UR6) from the erm-rpsL cassette (9). Because the primer RpsLCamR (from the rpsL gene) overlaps at its 5′ end with the primer CamF (from the cat gene), the cat and rpsL fragments could be ligated by amplification with primers UF1 and UR6. The 1.4-kb cat-rpsL cassette was then amplified by primers RpsLF and C2CamR, which contained SacI and BamHI restriction sites at the 5′ ends, and ligated to amplified fragments of DNA upstream (1.8 kb, primers HypDF2 and BabAR1252) and downstream (1.5 kb, primers 0895R and BabAF1253) of babA2 that contained complementary restriction sites. H. pylori 7.13 was made streptomycin resistant by transformation with genomic DNA from a mutant of strain 26695, which contained an A-to-G change at codon 43 of rpsL, and selection on streptomycin (10 μg/ml). babA2 was then deleted from streptomycin-resistant 7.13 by transformation with the cat-rpsL cassette, with its flanking arms from babA2, and selection on chloramphenicol (5 μg/ml). The babA2 gene was reinserted by transformation of the babA2 knockout of 7.13 with babA2 amplified from either 7.13 or 8.31, and selection on streptomycin. Streptomycin-resistant, chloramphenicol-sensitive colonies were fully sequenced at the babA2 locus to confirm that they had undergone the desired genetic exchange and then tested for Leb adherence as described above.
In situ histosection adherence assay.
Gastric tissue from H. pylori-infected and uninfected gerbils was used to detect H. pylori binding to tissue sections using methods previously described (6, 10). Briefly, bacterial cells were washed three times in phosphate-buffered saline (PBS), resuspended in 0.1 M carbonate buffer (0.15 M NaCl, pH 9) at an OD600 of 1.0, and then labeled by incubation with 10 μl of fluorescein isothiocyanate (FITC; 10 mg of 0.1 M carbonate buffer/ml). Bacteria were then incubated in the dark for 10 min, washed three to five times in 900 ml of blocking buffer (1% bovine serum albumin in PBS-Tween 20) or until the supernatant was clear, and adjusted to an OD600 of 1.0 in blocking buffer. FITC-labeled bacteria were diluted (20 μl in 80 μl of blocking buffer) and incubated for 2 h in the dark with deparaffinized tissue that had been rehydrated and blocked with 100 μl of blocking buffer overnight. Slides were washed in PBS-Tween 20, protected with a coverslip applied with mounting medium (ProLong Gold Antifade Reagent; Invitrogen, Eugene, OR), and dried overnight. Labeled bacteria were viewed with a fluorescence microscope (Zeiss Axioskop). Quantitation of bound fluorescent bacteria was performed on images of tissue sections using ImageJ software (http://rsbweb.nih.gov/ij/ [National Institutes of Health, Bethesda, MD]). Images were cropped to retain only gastric mucosa and split into red, green, and blue channels using the RGB split command. The green channel was used to calculate the area of bound fluorescent bacteria because it provided maximal contrast between the fluorescence and the rest of the mucosa. The area of fluorescence was divided by the total area of the mucosal epithelium for each image.
Accession numbers.
The DNA sequences for babA2 and babA1 in H. pylori 7.13 and 8.31 were deposited in GenBank under accession numbers GQ272324 to GQ272327.
RESULTS
H. pylori J99 recovered from monkeys shows loss of BabA expression and Leb binding by both mutation and by gene conversion.
We previously reported that infection of specific-pathogen-free (SPF) rhesus monkeys with H. pylori J166 resulted in the accumulation of babA-null variants, either by gene conversion or by phase variation in a poly(CT) tract in the 5′ region of babA (33). To test the generality of this finding, we challenged SPF macaques with normal gastric histology using H. pylori J99, which does not have CT repeats in babA and has not been used previously to challenge monkeys. Although single colonies recovered 1 week after challenge bound Leb, binding decreased significantly with time (Kruskal-Wallis, two tailed, P < 0.001), and by 8 weeks postinoculation Leb binding was typically lost (Fig. 1). To characterize the mechanism for loss of Leb binding, we sequenced babA from all output strains that had lost Leb binding. In one monkey (animal 30364), 11 output strains that lost Leb binding had acquired a single base-pair mutation at position 853 in the babA ORF that encoded a stop codon. In comparison, eight output strains that lost Leb binding from a second monkey (animal 31105) underwent a gene conversion event in which babA was replaced with a second copy of babB. Immunoblot analysis of representative output colonies that acquired a stop codon in babA or that underwent gene conversion of babA by babB showed that in each case the BabA protein was not expressed (data not shown). These results confirm our previous observation that BabA expression is lost in vivo and extend the results to different monkeys challenged with a different H. pylori strain.
FIG. 1.

Leb adherence ratio for individual H. pylori colonies isolated from two rhesus macaques (30364 and 31105) 1 to 18 weeks after inoculation with strain H. pylori J99. H. pylori attachment to Leb decreased significantly with time after inoculation (Kruskal-Wallis test, two tailed, P < 0.001).
H. pylori J166 undergoes loss of BabA expression by phase variation during colonization of mice.
To determine whether loss of BabA expression seen in monkeys is characteristic of other mammalian hosts, we inoculated H. pylori J166 into C57BL/6, C3H/HeJ, and FVB/N mice (n = 4 to 6 mice per strain) and recovered H. pylori colonies when the animals were sacrificed 2 weeks later. The mean colonization density of H. pylori was ∼104 CFU/g of stomach tissue in all mouse strains. H. pylori J166 babA and babB each contain eight pairs of CT repeats at the 5′ end of the gene. To determine the ORF status of babA and babB, PCR and DNA sequence analysis were performed on 13 to 32 single colonies recovered from each mouse strain. In colonies recovered from C57BL/6 and FVB/N mice, there was frequently loss or gain of a dinucleotide CT repeat, so that a babA ORF was present in only 50 and 41% of output colonies, respectively (Fig. 2). In contrast, frameshift mutations were uncommon in colonies recovered from C3H/HeJ mice, in which 93% of bacteria had an intact babA ORF (Fig. 2). Since babB also has CT repeats, we next sequenced the 5′ portion of babB to determine whether an ORF was present. In contrast to babA, every colony recovered from each of the three mouse strains contained an intact babB ORF (Fig. 2). Sequence analysis of babA from 22 single colonies of the input J166 demonstrated that an ORF was present in 100%, which suggests that a frameshift mutation with loss of BabA expression was selected during colonization of C57BL/6 and FVB/N mice.
FIG. 2.
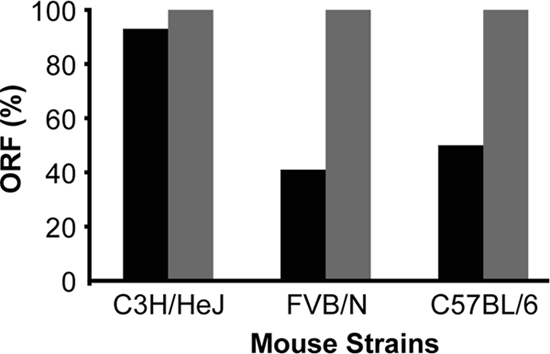
Percentage of H. pylori output colonies with an ORF for babA (▪) and babB (░⃞) 2 weeks postinoculation with strain J166 in three strains of mice (n = 4 to 6 mice/strain). Percentages were calculated as a fraction of total colonies (13 to 32 per strain) isolated from each mouse strain. An ORF was present if DNA sequencing demonstrated the presence of eight CT dinucleotide repeats in the 5′ end of babA. Out of frame sequences had seven or nine CT repeats, which resulted in a stop codon at bp 78 (addition of CT) or bp 48 (loss of CT).
To confirm the significance of number of CT repeats, we analyzed protein expression and binding to Leb for representative single colony isolates from mouse output strains that contained seven (loss of one CT), eight (wild type), or nine (gain of one CT) pairs of CT repeats. Output strains that had not undergone phase variation expressed BabA protein and had high Leb binding affinity (Fig. 3). In contrast, output strains that had lost or gained a CT repeat did not express BabA and had significantly less binding to Leb (Mann-Whitney U test, two tailed, P < 0.001). These results confirm that analysis of the babA ORF reflects BabA expression and attachment to Leb.
FIG. 3.
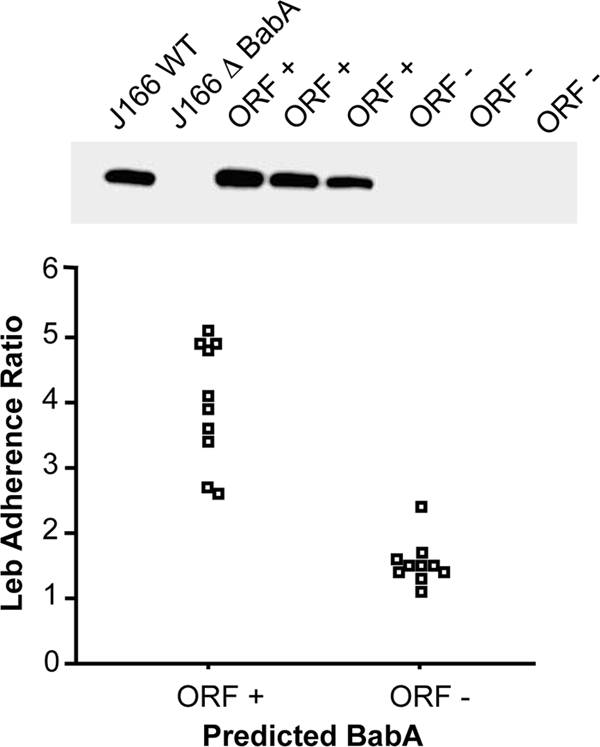
Verification of expression and Leb adherence in H. pylori J166 with a babA ORF. Immunoblot of representative single-colony isolates from mouse output strains with an in frame (ORF+) or out of frame (ORF−) babA gene probed with antibody to BabA (top panel). Wild-type H. pylori J166 (J166WT) and an isogenic babA knockout (J166rbabA) served as controls. Only strains with the babA ORF expressed protein. Analysis of Leb binding (bottom panel) of H. pylori J166 single-colony mouse output strains showed that those with a babA ORF showed significantly greater attachment to Leb (Mann-Whitney U test, two tailed, P < 0.001).
H. pylori 7.13 recovered from gerbils loses attachment to Leb and undergoes gene conversion of a functional babA with a second babA copy.
We next extended these observations to the gerbil model of H. pylori, which is unique in that animals develop adenocarcinoma that resembles that seen in humans. Male Mongolian gerbils (n = 2) were inoculated with H. pylori strain 7.13 and sacrificed 12 weeks later. Input 7.13 binds Leb (adherence ratio = 2.258) and expresses BabA protein on immunoblot (not shown). Analysis of four output colonies from each animal showed that Leb binding was lost after 12 weeks of colonization, with a mean adherence ratio of 1.101 (standard error [SE] ± 0.03) and 1.374 (SE ± 0.07), respectively. To examine the mechanism by which Leb attachment was lost, we performed immunoblots on one output colony from each animal that did not attach to Leb. Surprisingly, both output strains expressed BabA protein (data not shown). DNA sequence analysis showed that the predicted BabA protein from both output colonies differed at six amino acid residues compared to that in strain 7.13 (Fig. 4). Since these changes would be unlikely to arise independently in two animals, we suspected that a recombination event may have occurred, similar to what we observed previously when babB converted babA (33). Previous studies showed that some strains of H. pylori have a second babA allele at the babB locus (7, 17). Therefore, we amplified and sequenced the babB locus of strain 7.13 and compared it to that in the gerbil output strain 8.31. The results showed that strain 7.13 has two copies of babA: one at the J99 babA locus that has an ORF and a second at the J99 babB locus that does not have an ORF due to addition of a single adenine in a short poly(A) tract at the 5′ end of the gene (Fig. 4). We designate these genes babA2 and babA1, respectively, by analogy with the initial description in H. pylori 17875, which has an expressed gene designated babA2 and a silenced gene designated babA1 (17). DNA sequence analysis in output strain 8.31 that did not bind Leb showed that it had undergone a gene conversion event in which a portion of babA1 replaced babA2 (Fig. 4). Identical results were obtained in a second output strain (data not shown). The recombination event in the output strains occurred so as to retain the poly(A) tract length of babA2 (six adenines) rather than babA1 (seven adenines), which is consistent with the immunoblot that showed expression of BabA protein. At amino acid 280 the gerbil output strain has Q in BabA2 and not K as in BabA1. This may represent a simple mutation, since the codon for K (lysine) and Q (glutamine) differ only at the first nucleotide. Alternatively, this might be a gap in integration, in which a single-nucleotide-polymorphism (SNP) unique to babA2 in 7.13 (codon for Q, glutamine) is flanked by SNPs unique to babA1 (20). babA1 is identical in strains 7.13 and 8.31. BabB was not detected in strain 7.13 by DNA sequence analysis or by immunoblot (data not shown). This is consistent with the observation that strain B128, which is the parent strain to 7.13 (11), does not contain babB (24).
FIG. 4.

Schematic representation of the gene conversion event that occurred in H. pylori strain 7.13 after 12 weeks of colonization in gerbils. Input strain 7.13 has an expressed BabA2 and a silent BabA1, which has one additional adenine residue in a polynucleotide tract (7A versus 6A) that begins immediately after the ATG start codon and results in a stop codon at bp 61. Shown are the 11 amino acid residues (single-letter designations numbered according to the mature protein after removal of the signal peptide) for BabA1 that differ from those predicted in BabA2 if the protein were expressed. Gerbil output strain 8.31 has undergone gene conversion of babA2 by babA1. The recombination occurred so as to retain the 6A polynucleotide tract, so BabA2 is expressed, but the replacement of 6 amino acids from BabA2 with those from BabA1 (shaded) is sufficient to eliminate binding to Leb.
Six amino acid changes in BabA2 are sufficient to eliminate binding to Leb and to gastric tissue.
Gene conversion of babA2 with babA1 yielded output strain 8.31 that no longer attached to Leb, and that contained a BabA2 that was modified by a change in six amino acid residues (Fig. 4). To determine whether these six amino acid changes were sufficient to eliminate Leb binding, we used contraselection (9) to move babA alleles between strains 7.13 and 8.31. The babA2 gene of streptomycin-resistant H. pylori 7.13 was deleted by homologous recombination with the cat-rpsL cassette (chloramphenicol resistance, dominant streptomycin susceptibility) and then transformed with the desired babA2 gene amplified from 7.13 or 8.31. Transformants that were chloramphenicol susceptible and streptomycin resistant (due to loss of the cassette) were screened for the appropriate babA gene by full-length DNA sequence analysis of babA2, and the desired clones were tested for adherence to Leb (Table 2). As expected, attachment to Leb was observed for the ancestral H. pylori 7.13 but not for ΔbabA2::cat-rpsL derivatives of 8.31 or 7.13. Replacement of babA2 in 7.13 with babA2 from 8.31 eliminated attachment to Leb, which demonstrated that the six amino acid differences (Fig. 4) between these two alleles were sufficient to eliminate Leb binding. In a control experiment, replacement of ΔbabA2::cat-rpsL with the original babA2 from 7.13 restored the wild-type phenotype as expected (Table 2). These results demonstrated that one or more of the six amino acid differences (Fig. 4) between these two alleles were critical for binding of this BabA to its Leb receptor.
TABLE 2.
Exchange of babA2 alleles in H. pylori strains 7.13 and 8.31
| H. pylori strain | babA2 donor strain | Mean Leb adherence ratio ± SEa |
|---|---|---|
| 7.13 | None | 3.63 ± 0.18 |
| 7.13ΔbabA2 | None | 1.11 ± 0.03 |
| 7.13ΔbabA2 | 8.31 | 1.02 ± 0.03 |
| 7.13ΔbabA2 | 7.13 | 3.08 ± 0.43 |
| 8.31 | None | 1.19 ± 0.10 |
Data are means of three replicates performed on independent colonies for wild-type strains or independent transformants for complemented strains. Nonparametric one-way analysis of variance (Kruskal-Wallis) results were significant (P = 0.01, one tailed), as were pairwise comparisons between 7.13 (or 7.13ΔbabA2 complemented with 7.13 babA2) and each of the other strains (P = 0.05, one tailed, Mann-Whitney U test).
Hematoxylin and eosin stains demonstrated that H. pylori infection was associated with a marked inflammatory infiltrate and loss of normal epithelial architecture (Fig. 5A and E) that is typical of H. pylori-induced gastritis in gerbils (28). To determine whether the six amino acid replacements were also sufficient to eliminate binding to tissue sections, we FITC labeled strains 7.13, 8.31, and the 7.13 derivative that contained the 8.31 babA2 allele. Each strain was applied to gastric tissue sections from uninfected (Fig. 5A to D) and infected (Fig. 5E to H) gerbils, and binding was assessed by quantitative analysis of fluorescence microscopy. Consistent with the results of binding in the Leb adherence assay, H. pylori 7.13 showed binding to ca. 14% of the surface epithelium of uninfected gastric tissue (Fig. 5B), whereas 8.31 and 7.13 bearing the 8.31 babA2 bound only ca. 1% (Fig. 5C and D). Little or no binding was seen to tissue from infected gerbils (Fig. 5F to H). Together, these results indicate that in H. pylori 7.13, six amino acid changes are sufficient to eliminate binding to Leb and to gastric tissue and that receptors recognized by BabA may be lost during infection in the gerbil model.
FIG. 5.

Gastric histosections from gerbils that were uninfected (A to D) or infected (E to H) with H. pylori. Hematoxylin and eosin stained sections (A and E) demonstrate the marked cellular infiltrate and loss of normal epithelial architecture that is characteristic of H. pylori gastritis. Incubation of uninfected tissue sections with FITC-labeled bacteria showed that H. pylori 7.13, which binds Leb, also attached to gastric tissue (B), whereas strains 8.31 (C) and 7.13 expressing the 8.31 babA (D) did not. Interestingly, none of the strains bound appreciably to infected gerbil tissue (F to H), suggesting that H. pylori infection reduces expression of the BabA receptor. The percentage of epithelium with bound bacteria was determined as described in Materials and Methods and is shown in the upper right of each panel.
DISCUSSION
Studies in different human populations in many geographic locations have demonstrated the remarkable genomic diversity of H. pylori. Isolates from unrelated individuals represent a seemingly unlimited number of unique strains that differ in gene content, position, and allelic profile. Diversity is thought to develop through point mutation, genomic recombination, and genetic exchange, as well as phase variation, all of which are common in H. pylori (13, 29, 30), coupled with random genetic drift and selection for adaptation to particular environments. Animal models of H. pylori infection permit experimental observation of diversity that develops in vivo in real time after inoculation with a clonal population. We previously used the rhesus macaque model to analyze changes in the H. pylori genome over time within the same animal host (33). The results demonstrated that in rhesus macaques the expression of BabA was lost, either by phase variation or by nonreciprocal gene conversion, in which a duplicate copy of babB replaced babA.
Here we have replicated the analysis of BabA expression in macaques using a different H. pylori strain and extended the results to the mouse and gerbil, which are commonly used small-animal models of H. pylori. The results demonstrated the loss of BabA expression in macaques, mice, and gerbils, although the particular mechanisms differed. After challenge of mice with H. pylori J166, which has 5′ CT repeats in the babA gene, BabA expression, and Leb binding were lost by phase variation. In contrast, challenge of macaques with J99 yielded output strains that lost BabA expression either by selection and then expansion of a subpopulation of J99 that had a single base pair mutation that encoded a stop codon, or by nonreciprocal gene conversion of babA with a duplicate copy of babB as we observed previously (33). Gene conversion also explained loss of Leb binding activity in the gerbil model after colonization with strain 7.13, although in this case the babA gene that encoded Leb binding was converted by a nonbinding allele. This gene conversion was not present in strain 7.13, which was also recovered from a gerbil 3 weeks after challenge with strain B128 (11). This is likely explained by the short duration of infection, since loss of BabA expression is not uniform until 8 weeks of colonization (33).
Although most H. pylori strains recovered from humans express BabA (29, 35), strains similar to those recovered after animal challenge are sometimes seen in humans. For example, we previously examined 43 clinical isolates by PCR and DNA sequencing and showed that while most had babA and babB, in 16% of strains (7 of 43) babA was absent and babB was duplicated, like what is seen in macaques. Some strains had duplicate copies of babA (like strain 7.13), one of which was typically out of frame, while others had still different bab genotypes, such as babC (HopU, HP0317), at the locus where babA or babB are typically found (7). Others have found similar diversity in bab genotypes and demonstrated remarkable heterogeneity in Leb binding (3). Therefore, it is likely that whatever factors select for loss of BabA expression in animal models of H. pylori may sometimes also be found in humans.
The particular selective pressures that modulate expression of BabA and related proteins are unknown. One possibility is that BabA (but not BabB) induces an adaptive immune response, so that BabA expression is lost while BabB is maintained. This seems unlikely because we have been unable to demonstrate antibodies to BabA or BabB in macaques (33) and because other reports have failed to demonstrate BabA or BabB as immunodominant antigens in humans (14, 18). Furthermore, in the case of the gerbil, Leb-binding BabA expression is lost, but BabA expression per se is not. BabA expression is not lost in short-term infection with C3H/HeJ mice (Fig. 1), which are known to have a defect in TLR4 signaling, so innate immunity might also be important. The presence of Leb may also influence BabA expression. However, since Leb serves on the one hand as a cellular receptor for BabA and on the other hand as a decoy receptor that likely plays a role in innate immunity (21), the relationship between BabA and Leb expression is likely complex. BabA metastability and heterogeneity likely contribute to H. pylori fitness (5) and so are tightly regulated, with BabA sometimes expressed by most cells in the population and other times archived as “memory cells” ready to expand when conditions change.
Although H. pylori strains differ markedly in binding affinity to Leb, no consensus amino acid sequences have been identified that are conserved among strains that bind Leb (3, 15). Since the strains recovered from gerbils did not bind Leb or gastric histosections and yet expressed BabA that differed at only six amino acids from the Leb-binding allele (Fig. 4), one or more of these six residues are likely important, either as binding sites or as key amino acids that alter the tertiary structure of BabA. The location of these residues within the midregion of BabA is consistent with this suggestion, since this portion of the protein is highly polymorphic (26, 27), is thought to be surface exposed, and is likely responsible for Leb binding. In most cases, the differences in amino acids between BabA2 and BabA1 at these positions represent a change in charge or hydrophobicity, such as E218K (negative to positive charge), K252E (positive to negative charge) and S302G (hydrophilic to hydrophobic), which might be expected to change the tertiary structure of the BabA protein.
The receptor for BabA in the gerbil is unknown, and Leb has generally been thought to be absent in rodents. However, recent studies in mice with anti-Leb antibodies, corroborated by antibody inhibition using specific synthetic saccharides, suggest that rodents may in fact express Leb or related Lewis type 1 structures (23). Loss of BabA-mediated binding to infected gerbil gastric tissue (Fig. 5) demonstrates that as H. pylori undergoes modification in surface adhesins during infection; so, too, does the host play an active role, probably with changes in Leb or closely related glycans expressed on the epithelial surface. This is consistent with recent studies that have demonstrated that H. pylori induces profound changes in surface glycoproteins, including the gastric mucins MUC5AC (8, 21) and MUC1 (22). We hypothesize that modification of BabA during H. pylori infection is a mechanism to adapt to changing conditions of inflammation and glycan expression at the epithelial surface.
Acknowledgments
Supported by Public Health Service Awards from the National Institutes of Health (NIH) to J.V.S. (AI42081 and AI070803), R.M.P. (DK58587, CA77955, and CA116087), and D.E.B. (DK63041) and by National Research Service Award from the NIH (T32 AI60555) to C.M.S. and C.L.C.
Editor: B. A. McCormick
Footnotes
Published ahead of print on 1 February 2010.
REFERENCES
- 1.Alm, R. A., J. Bina, B. M. Andrews, P. Doig, R. E. Hancock, and T. J. Trust. 2000. Comparative genomics of Helicobacter pylori: analysis of the outer membrane protein families. Infect. Immun. 68:4155-4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alm, R. A., L. L. Ling, D. T. Moir, B. L. King, E. D. Brown, P. C. Doig, D. R. Smith, B. Noonan, B. C. Guild, B. L. deJonge, G. Carmel, P. J. Tummino, A. Caruso, M. Uria-Nickelsen, D. M. Mills, C. Ives, R. Gibson, D. Merberg, S. D. Mills, Q. Jiang, D. E. Taylor, G. F. Vovis, and T. J. Trust. 1999. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 397:176-180. [DOI] [PubMed] [Google Scholar]
- 3.Aspholm-Hurtig, M., G. Dailide, M. Lahmann, A. Kalia, D. Ilver, N. Roche, S. Vikstrom, R. Sjostrom, S. Linden, A. Backstrom, C. Lundberg, A. Arnqvist, J. Mahdavi, U. J. Nilsson, B. Velapatino, R. H. Gilman, M. Gerhard, T. Alarcon, M. Lopez-Brea, T. Nakazawa, J. G. Fox, P. Correa, M. G. Dominguez-Bello, G. I. Perez-Perez, M. J. Blaser, S. Normark, I. Carlstedt, S. Oscarson, S. Teneberg, D. E. Berg, and T. Boren. 2004. Functional adaptation of BabA, the H. pylori ABO blood group antigen binding adhesin. Science 305:519-522. [DOI] [PubMed] [Google Scholar]
- 4.Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl. 1990. Current protocols in molecular biology. John Wiley & Sons, Inc., New York, NY.
- 5.Backstrom, A., C. Lundberg, D. Kersulyte, D. E. Berg, T. Boren, and A. Arnqvist. 2004. Metastability of Helicobacter pylori bab adhesin genes and dynamics in Lewis b antigen binding. Proc. Natl. Acad. Sci. U. S. A. 101:16923-16928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boren, T., P. Falk, K. A. Roth, G. Larson, and S. Normark. 1993. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science 262:1892-1895. [DOI] [PubMed] [Google Scholar]
- 7.Colbeck, J. C., L. M. Hansen, J. M. Fong, and J. V. Solnick. 2006. Genotypic profile of the outer membrane proteins BabA and BabB in clinical isolates of Helicobacter pylori. Infect. Immun. 74:4375-4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooke, C. L., H. J. An, J. Kim, D. R. Canfield, J. Torres, C. B. Lebrilla, and J. V. Solnick. 2009. Modification of gastric mucin oligosaccharide expression in rhesus macaques after infection with Helicobacter pylori. Gastroenterol. 137:1061-1071. [DOI] [PubMed] [Google Scholar]
- 9.Dailidiene, D., G. Dailide, D. Kersulyte, and D. E. Berg. 2006. Contraselectable streptomycin susceptibility determinant for genetic manipulation and analysis of Helicobacter pylori. Appl. Environ. Microbiol. 72:5908-5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Falk, P., K. A. Roth, T. Boren, T. U. Westblom, J. I. Gordon, and S. Normark. 1993. An in vitro adherence assay reveals that Helicobacter pylori exhibits cell lineage-specific tropism in the human gastric epithelium. Proc. Natl. Acad. Sci. U. S. A. 90:2035-2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Franco, A. T., E. Johnston, U. Krishna, Y. Yamaoka, D. A. Israel, T. A. Nagy, L. E. Wroblewski, M. B. Piazuelo, P. Correa, and R. M. Peek, Jr. 2008. Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. Cancer Res. 68:379-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gerhard, M., N. Lehn, N. Neumayer, T. Borén, R. Rad, W. Schepp, S. Miehlke, M. Classen, and C. Prinz. 1999. Clinical relevance of the Helicobacter pylori gene for blood-group antigen-binding adhesin. Proc. Natl. Acad. Sci. U. S. A. 96:12778-12783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gressmann, H., B. Linz, R. Ghai, K. P. Pleissner, R. Schlapbach, Y. Yamaoka, C. Kraft, S. Suerbaum, T. F. Meyer, and M. Achtman. 2005. Gain and loss of multiple genes during the evolution of Helicobacter pylori. PLoS Genet. 1:e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haas, G., G. Karaali, K. Ebermayer, W. G. Metzger, S. Lamer, U. Zimny-Arndt, S. Diescher, U. B. Goebel, K. Vogt, A. B. Roznowski, B. J. Wiedenmann, T. F. Meyer, T. Aebischer, and P. R. Jungblut. 2002. Immunoproteomics of Helicobacter pylori infection and relation to gastric disease. Proteomics 2:313-324. [DOI] [PubMed] [Google Scholar]
- 15.Hennig, E. E., R. Mernaugh, J. Edl, P. Cao, and T. L. Cover. 2004. Heterogeneity among Helicobacter pylori strains in expression of the outer membrane protein BabA. Infect. Immun. 72:3429-3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hornsby, M., J. L. Huff, R. Kays, D. Canfield, C. Bevins, and J. Solnick. 2008. Helicobacter pylori induces an antimicrobial response in rhesus macaques in a Cag pathogenicity island-dependent manner. Gastroenterology 134:1049-1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ilver, D., A. Arnqvist, J. Ogren, I. M. Frick, D. Kersulyte, E. T. Incecik, D. E. Berg, A. Covacci, L. Engstrand, and T. Boren. 1998. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science 279:373-377. [DOI] [PubMed] [Google Scholar]
- 18.Kimmel, B., A. Bosserhoff, R. Frank, R. Gross, W. Goebel, and D. Beier. 2000. Identification of immunodominant antigens from Helicobacter pylori and evaluation of their reactivities with sera from patients with different gastroduodenal pathologies. Infect. Immun. 68:915-920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kusters, J. G., A. H. van Vliet, and E. J. Kuipers. 2006. Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 19:449-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin, E. A., X. S. Zhang, S. M. Levine, S. R. Gill, D. Falush, and M. J. Blaser. 2009. Natural transformation of Helicobacter pylori involves the integration of short DNA fragments interrupted by gaps of variable size. PLoS Pathog. 5:e1000337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Linden, S. K., J. Mahdavi, C. Semino-Mora, C. Olsen, I. Carlstedt, T. Boren, and A. Dubois. 2008. Glycan secretor phenotypes modulate mucosal innate immunity and affect Helicobacter pylori infection. PLoS Pathog. 4:e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Linden, S. K., Y. H. Sheng, A. L. Every, K. M. Miles, E. C. Skoog, T. H. Florin, P. Sutton, and M. A. McGuckin. 2009. MUC1 limits Helicobacter pylori infection both by steric hindrance and by acting as a releasable decoy. PLoS Pathog. 5:e1000617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Magalhaes, A., J. Gomes, M. N. Ismail, S. M. Haslam, N. Mendes, H. Osorio, L. David, J. Lependu, R. Haas, A. Dell, T. Boren, and C. A. Reis. 2009. Fut2-null mice display an altered glycosylation profile and impaired BabA-mediated Helicobacter pylori adhesion to gastric mucosa. Glycobiology 166:1793-1806. [DOI] [PMC free article] [PubMed]
- 24.McClain, M. S., C. L. Shaffer, D. A. Israel, R. M. Peek, Jr., and T. L. Cover. 2009. Genome sequence analysis of Helicobacter pylori strains associated with gastric ulceration and gastric cancer. BMC Genomics 10:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Odenbreit, S., H. Kavermann, J. Puls, and R. Haas. 2002. CagA tyrosine phosphorylation and interleukin-8 induction by Helicobacter pylori are independent from alpAB, HopZ and bab group outer membrane proteins. Int. J. Med. Microbiol. 292:257-266. [DOI] [PubMed] [Google Scholar]
- 26.Pride, D. T., and M. J. Blaser. 2002. Concerted evolution between duplicated genetic elements in Helicobacter pylori. J. Mol. Biol. 316:629-642. [DOI] [PubMed] [Google Scholar]
- 27.Pride, D. T., R. J. Meinersmann, and M. J. Blaser. 2001. Allelic variation within Helicobacter pylori babA and babB. Infect. Immun. 69:1160-1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rieder, G., J. L. Merchant, and R. Haas. 2005. Helicobacter pylori cag-type IV secretion system facilitates corpus colonization to induce precancerous conditions in Mongolian gerbils. Gastroenterology 128:1229-1242. [DOI] [PubMed] [Google Scholar]
- 29.Salama, N., K. Guillemin, T. K. McDaniel, G. Sherlock, L. S. Tompkins, and S. Falkow. 2000. A whole-genome microarray reveals genetic diversity among Helicobacter pylori strains. Proc. Natl. Acad. Sci. U. S. A. 97:14668-14673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salaun, L., B. Linz, S. Suerbaum, and N. J. Saunders. 2004. The diversity within an expanded and redefined repertoire of phase-variable genes in Helicobacter pylori. Microbiology 150:817-830. [DOI] [PubMed] [Google Scholar]
- 31.Solnick, J. V., D. R. Canfield, S. Yang, and J. Parsonnet. 1999. The rhesus monkey (Macaca mulatta) model of Helicobacter pylori: noninvasive detection and derivation of specific pathogen free monkeys. Lab. Anim. Sci. 49:197-201. [PubMed] [Google Scholar]
- 32.Solnick, J. V., L. M. Hansen, D. R. Canfield, and J. Parsonnet. 2001. Determination of the infectious dose of Helicobacter pylori during primary and secondary infection in rhesus monkeys (Macaca mulatta). Infect. Immun. 69:6887-6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Solnick, J. V., L. M. Hansen, N. Salama, J. K. Boonjakuakul, and M. Syvanen. 2004. Modification of Helicobacter pylori outer membrane protein expression during experimental infection of rhesus macaques. Proc. Natl. Acad. Sci. U. S. A. 101:2106-2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan, S., and D. E. Berg. 2004. Motility of urease-deficient derivatives of Helicobacter pylori. J. Bacteriol. 186:885-888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamaoka, Y., J. Souchek, S. Odenbreit, R. Haas, A. Arnqvist, T. Boren, T. Kodama, M. S. Osato, O. Gutierrez, J. G. Kim, and D. Y. Graham. 2002. Discrimination between cases of duodenal ulcer and gastritis on the basis of putative virulence factors of Helicobacter pylori. J. Clin. Microbiol. 40:2244-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]