

Abstract
We present a method that harnesses massively parallel DNA synthesis and sequencing for the high-throughput functional analysis of regulatory sequences at single-nucleotide resolution. As a proof of concept, we quantitatively assayed the effects of all possible single-nucleotide mutations for three bacteriophage promoters and three mammalian core promoters in a single experiment per promoter. The method may also serve as a rapid screening tool for regulatory element engineering in synthetic biology.
A broad range of methods exist for annotating functional regulatory elements in genomes. These include comparative and ab initio prediction algorithms1–3 and high-throughput assays such as ChIP-Seq4 and CAGE5,6. Despite much progress, the architectures of the vast majority of regulatory elements have yet to be systematically and quantitatively dissected at high resolution. Effective methods for this include classical saturation mutagenesis7 and combinatorial promoter shuffling8,9, but these have been applied only at low throughput. Furthermore, the effects of promoter modification are measured using techniques that are not always sufficiently sensitive to detect subtle changes in transcription.
Here we present a high-throughput method to systematically analyze the effect in a single experiment of mutations at every position in a core promoter (Fig. 1a). Mutant promoters are synthesized in parallel as DNA oligonucleotides on a programmable microarray and released into solution10, resulting in a complex library. Each oligonucleotide in the library is designed to include a unique barcode sequence downstream of the promoter's transcription start site (TSS). The oligos are transcribed in vitro, and the resulting transcripts are sequenced. The relative abundance of each programmed barcode provides a digital readout of the transcriptional efficiency of its cis-linked mutant promoter.
Figure 1.
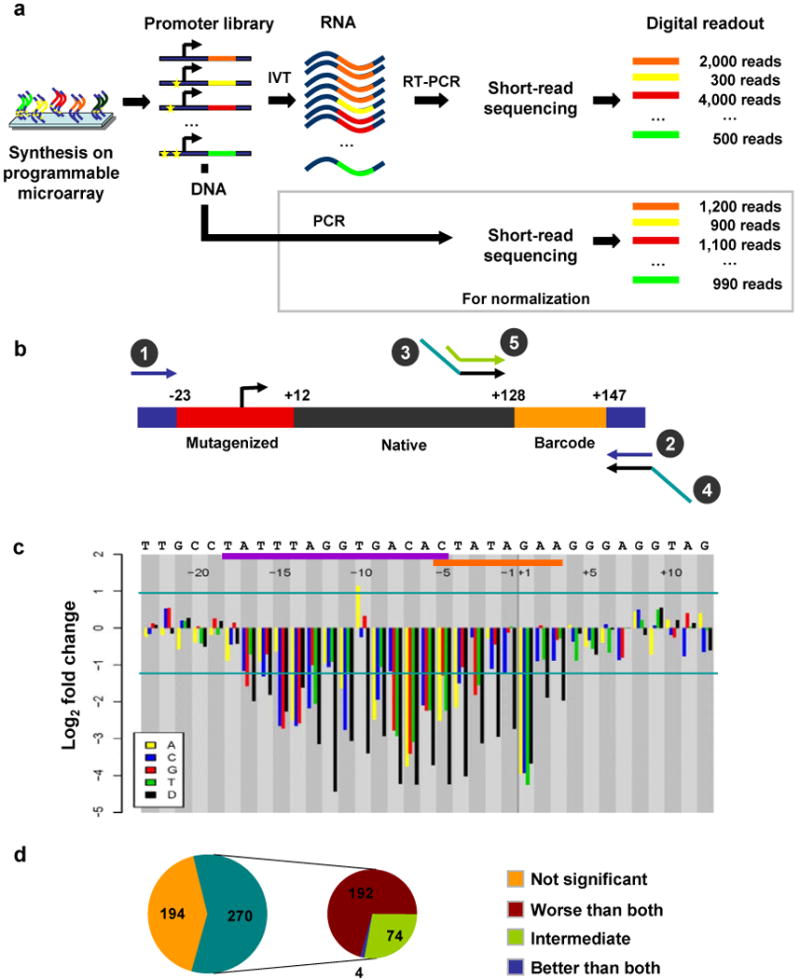
Synthetic saturation mutagenesis of a bacteriophage promoter. (a) Promoter templates are synthesized on a programmable microarray, released into solution and amplified by PCR (primers 1 and 2 in b). One fraction of the amplified promoter library is subjected to in vitro transcription followed by reverse transcription PCR (primers 3 and 4 in b). Another fraction is simply PCR amplified using the same primers. Barcodes within RNA- and DNA-derived amplicons are sequenced separately (primer 5 in b). RNA-derived barcode counts provide a digital readout of the transcriptional efficiency of associated promoters. DNA-derived barcode counts are used to normalize for any nonuniformity in the initial oligonucleotide concentrations. (b) For bacteriophage promoters, each 200-nt oligonucleotide consists of the promoter (red), 115-nt of the native downstream sequence (black), a variable 20-nt barcode sequence (orange) and 15-nt PCR primers (blue) on either side. (c) Changes in transcriptional efficiency (average of six barcodes) for each single-nucleotide substitution or deletion (D) relative to the native promoter for bacteriophage promoter SP6. Horizontal lines mark significance cutoffs (P < 0.01). Horizontal axis denotes the position of the mutation relative to TSS, from −23 to +12, with naive nucleotides specified above. Polymerase binding (purple bar) and melting/initiation (orange bar) regions are also indicated above. (d) Classification of SP6 double-mutant templates based on their effect on transcription. The templates where either the double mutant or at least one of the corresponding single mutants have a significant effect on transcription relative to the native promoter are further classified based on the effect of the double mutant as compared to the two single mutants.
As a proof of concept, this method was applied to three well-characterized bacteriophage promoters: T3 (class 3, phi13), T7 (class 3, phi10) and SP6 (SP6p32). We focused on a 35-nt region, spanning 23-nt upstream and 12-nt downstream of each promoter's TSS (Fig. 1b). At each position, we mutated the native nucleotide to every other nucleotide or introduced a single-nucleotide deletion. We also included several double mutation promoters, allowing us to compare the single mutants to their combination. To guard against the potential influence of the barcode itself on transcriptional activity, we represented each mutant variant of each native promoter by six distinct 20-nt barcodes (Supplementary Methods). Native promoters with no mutations were also included and were each represented by 270 different barcodes. These served as positive controls and provided a baseline against which to compare transcriptional efficiencies of mutant promoters. Templates with random sequence in place of the promoter were included as negative controls (Supplementary Tables 1 and 2).
The promoter library was transcribed in vitro with one of three RNA polymerases (T7, T3 or SP6). The resulting RNA pools were reverse transcribed, PCR amplified and sequenced on an Illumina GAII system. Reads were then mapped back to the 20-nt barcodes that we had programmed in cis with each synthetic promoter. To control for potentially non-uniform representation of synthesized oligos (e.g., owing to differential synthesis efficiencies, systematic biases in PCR efficiency or biases inherent to the sequencer itself), we also PCR amplified the DNA library that served as input to the in vitro transcription reaction and sequenced it in a separate lane. A comparison between counts of DNA- and RNA-derived barcodes associated with each native (unmutated) promoter found that although synthetic promoter concentrations varied, they maintained a linear relationship with transcription efficiency (Supplementary Methods and Supplementary Fig. 1). The RNA-based counts associated with each barcode were therefore normalized by dividing by the corresponding DNA-based counts.
Counts of barcodes corresponding to the native promoter established the baseline activity of the native promoter and an empirical null distribution for assessing significance. The effect of each mutation was measured as a fold-change in transcription relative to the native promoter. Based on the variation observed within each set of 270 barcodes associated with each native promoter, we were able to call changes of twofold or greater as statistically significant (P < 0.01) (Supplementary Methods and Supplementary Fig. 2).
The observed transcriptional profiles clearly delineated a core ‘footprint’ for each promoter, within which substitutions and deletions caused a drastic drop in efficiency of transcription (Fig. 1c and Supplementary Fig. 3). We also observed a range of site and mutation-specific effects. For example, the −10 site within the SP6 promoter core region could be substituted without decreasing activity. In fact, a T→A substitution at this position caused a significant increase in transcriptional efficiency, consistent with previous studies of this promoter11. At certain sites, substitution of the native nucleotide by a specific nucleotide was tolerated whereas other nucleotides were not. For instance, the change from A→G at position −1 on the T3 promoter was deleterious, whereas A→C or A→T was benign. In general, the SP6 native promoter was more efficient than T7 and T3, and correspondingly more sensitive to the disruptions we introduced. An activity logo created using data from the SP6 mutants is included (Supplementary Fig. 4) for comparison with results from a previous saturation mutagenesis study11.
To explore whether we could detect synergistic or antagonistic associations between point mutations, we also included templates with substitutions at two positions within the promoter. Because it was not practical to test all possible permutations of double mutations, we used results of a pilot experiment consisting of only single mutants (data not shown) to choose a subset that provided a robust sampling of mutation position and severity (Supplementary Methods). We compared the double-mutant outcomes against predictions based on the corresponding single mutants, assuming a log-additive model. Although 65–70% of the double mutants matched predicted values, the rest showed deviations from this model, hinting at synergistic and compensatory interactions (Fig. 1d, Supplementary Figs. 5 and 6). We filtered double mutants for the subset where at least one of either of the single mutants or the double mutant satisfied our significance threshold for fold-change relative to the native promoter (Supplementary Fig. 6).
As expected, the effect of most double mutants was greater than either of the corresponding single mutants. However, there were also a number of cases where the effect of the combination of mutations was intermediate to the effects of the two corresponding single mutants, suggesting varying degrees of partial rescue. Finally, there were four SP6 double mutants that were less harmful than either of their corresponding single mutants. Notably, each of these four involved an A→T substitution at −3 as one of the mutations (Supplementary Fig. 6c). In vitro binding assays have shown that this mutation leads to a twofold increase in the strength of polymerase binding11, which might explain the compensatory effect that we observe here. Although the single A→T mutation at −3 is associated with a decrease in transcriptional activity, we note that this is not necessarily inconsistent as we are measuring transcriptional activity rather than polymerase binding strength. For example, it may be that increased polymerase binding directly underlies the observed decrease in transcriptional efficiency associated with the single A→T mutation at −3 (Fig. 1c), whereas a second mutation occurring at any number of positions serves to reduce the strength of polymerase binding toward a more optimal level for transcription (Supplementary Fig. 6c).
In synthetic biology, the multiplex in vitro evaluation of large numbers of synthetic promoters would represent an efficient empirical strategy for identifying variants that adjust the in vivo activity of a promoter with predictable magnitude. We sought to evaluate whether activities of individual synthetic promoters determined within our multiplex in vitro assay were recapitulated in vivo. Six T7 promoter variants were individually inserted upstream of a bacterial luciferase reporter in pCS26, a low-copy number plasmid12, and the constructs were used to transform a T7 polymerase–expressing Eshcerichia coli strain. In vivo activities of the promoters as measured by luciferase luminescence correlated well with predictions based on the in vitro assay (r = 0.92) (Supplementary Fig. 7).
Next we evaluated whether this approach could be extended to promoters recognized by the mammalian transcriptional machinery. We assayed three core promoters: the immediate early promoter of the human cytomegalovirus (CMV), the promoter of the human beta globin gene (HBB) and the promoter of human S100 calcium binding protein A4 (S100A4/PEL98). The promoter region included on each oligonucleotide extended 100-nt upstream and 50-nt downstream of the TSS. For saturation mutagenesis, we focused on a 70-nt region spanning 45-nt upstream and 25-nt downstream of the TSS (Fig. 2a). As previously described, we included six barcode variants per mutation. Native promoters with no mutations were represented by 100 barcodes each (Supplementary Tables 3 and 4).
Figure 2.
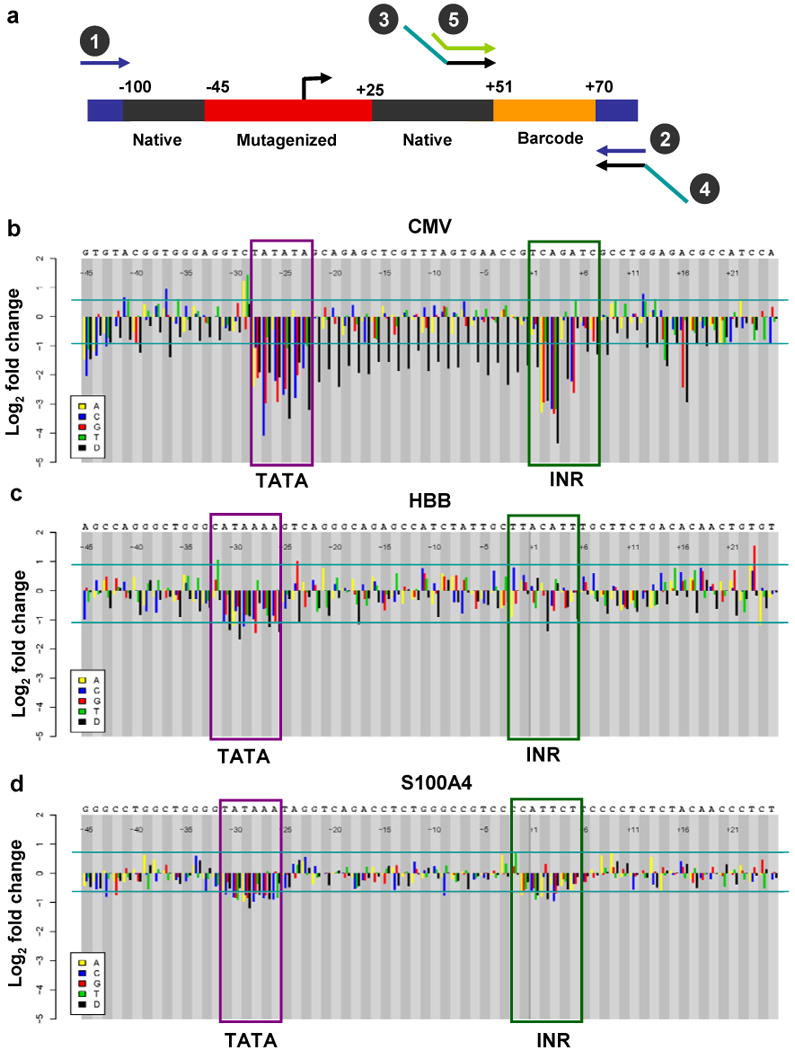
Mutagenesis of mammalian core promoters. (a) Mammalian Pol II promoter template design. Each 200-nt oligonucleotide consists of the promoter region from −100 to +50 (black), including the region subjected to saturation mutagenesis (red), followed by a variable 20-nt barcode sequence (orange) and 15-nt PCR primers (blue) on either side. (b–d) Impact of single-nucleotide substitutions and deletions on transcriptional efficiency. Transcriptional fold-change (average of six barcodes) for each single-nucleotide substitution or deletion (D) relative to the native promoter for CMV (b), HBB (c) and S100A4 (d). Horizontal lines mark significance cutoffs (P < 0.01). Horizontal axis denotes the position of the mutation relative to TSS, from −45 to +25 with native nucleotides specified above. Roles of the primers are as described in Figure 1b.
In vitro transcription was performed using HeLa nuclear extracts. Libraries were separately generated from RNA and DNA and sequenced separately, and analysis was carried out as above. In all three cases, we were able to detect changes in transcription that correlated with expectation (Fig. 2b–d). For example, mutations disrupting the AT-rich groove that defines the TATA box of the CMV promoter (TATATA, −28 to −23) led to a clear drop in transcriptional efficiency. Substitutions of C→A or C→T at −29 increased transcriptional efficiency, potentially secondary to the formation of a more optimal TATA box (−30 to −25) with respect to distance from the TSS (Fig. 2b). Mutations disrupting the initiator element (TCAGATC, +1 to +7; Supplementary Note) also caused significant drops in transcription. Single-nucleotide deletions at any position between the TATA box and the initiator sharply reduced transcription, likely a result of violation of spacing constraints13. The results also suggested the presence of two additional elements, one near +16 and another near the −45 region.
The HBB promoter has a noncanonical TATA box (CATAAA, −32 to −27)14, mutations in which have been documented in beta-thalassemia. As expected, our assay detected significant drops in transcription with changes to this motif (Fig. 2c). Notably, a C→T substitution at −32 (creating a canonical TATA box, TATAAA) increased the strength of the promoter. However, we did not observe any significant effects of initiator or E-box mutations, in contrast with previous studies in a different cell type15. With the S100A4 core promoter, mutations disrupting both the canonical TATA box (TATAAA, −31 to −26) and the initiator element (CCATTCT, −2 to +5) led to drops in transcriptional efficiency (Fig. 2d). Single-nucleotide deletions between the TATA box and the TSS did not show any significant effect on the HBB and S100A4 core promoters, in clear contrast with the CMV core promoter.
To evaluate reproducibility, we replicated the entire experiment for all six promoters. The distribution of observed fold-changes in transcriptional efficiency for each mutation as compared to the native promoter was reproducible, with correlation coefficients of 0.98, 0.97, 0.96, 0.99, 0.87 and 0.70 for the SP6, T7, T3, CMV, S100A4 and HBB core promoters respectively (Supplementary Fig. 8). The lower reproducibility of S100A4 and HBB core promoters appears to be related to lower levels of transcriptional activity relative to the bacteriophage and CMV core promoters. The current experimental design required fitting the promoter, barcode and other common sequences to the maximum available length of synthetic oligos (200 nt), whereas longer promoter fragments would have been likely to yield higher levels of activity16. The extension of this approach beyond moderately active core promoters—for example, to interrogate full proximal promoters or other types of regulatory elements—may therefore be dependent on the ability of array-based oligonucleotides synthesis technologies to achieve longer maximal lengths.
Synthetic saturation mutagenesis with quantitative readout by deep sequencing of cis-linked barcodes enables the measurement of the relative activities of thousands of core promoter variants in a single experiment. The use of programmable synthetic oligonucleotides also allows precise combinations of mutations to be studied in a directed fashion. Sequence barcodes eliminate the need for reporter genes or other cumbersome quantification techniques while allowing for a high level of multiplexing. Synthetic saturation mutagenesis may represent a useful and scalable tool for both regulatory element analysis and forward engineering of gene networks.
A full list of the variant promoter sequences, associated counts and estimated relative expression values are provided as Supplementary Data. Raw Illumina sequencing reads have been submitted to the NCBI Short Read Archive under center name UWGS-JS.
Supplementary Material
Acknowledgments
The authors would like to thank M.G. Surette (Univ. of Calgary) for the generous gift of the pCS26 plasmid used for the luciferase assays; E. LeProust and W. Woo (Agilent Technologies) for array-derived oligonucleotides libraries and E. Turner, J.B. Hiatt, S. Ng, J. Kitzman, R. Monnat, B. Stone, A. Dudley and N. Goddard for helpful discussions. D.P. holds a Career Award at the Scientific Interface from the Burroughs Wellcome Fund.
References
- 1.Wang X, Xuan Z, Zhao X, Li Y, Zhang M. Genome Res. 2009;19:266–275. doi: 10.1101/gr.081638.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jin VX, Singer GA, Agosto-Perez FJ, Liyanarachchi S, Davuluri RV. BMC Bioinformatics. 2006;7:114. doi: 10.1186/1471-2105-7-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abeel T, Saeys Y, Bonnet E, Rouzé P, Van de Peer Y. Genome Res. 2008;18:310–323. doi: 10.1101/gr.6991408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson DS, Mortazavi A, Myers RM, Wold B. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 5.de Hoon M, Hayashizaki Y. Biotechniques. 2008;44:627–628. 630, 632. doi: 10.2144/000112802. [DOI] [PubMed] [Google Scholar]
- 6.Carninci P, et al. Nat Genet. 2006;38:626–635. doi: 10.1038/ng1789. [DOI] [PubMed] [Google Scholar]
- 7.Baliga NS. Biol Proced Online. 2001;3:64–69. doi: 10.1251/bpo24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kinkhabwala A, Guet CC. PLoS ONE. 2008;3:e2030. doi: 10.1371/journal.pone.0002030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gertz J, Siggia E, Cohen B. Nature. 2008;457:215–218. doi: 10.1038/nature07521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cleary MA, et al. Nat Methods. 2004;1:241–248. doi: 10.1038/nmeth724. [DOI] [PubMed] [Google Scholar]
- 11.Shin I, Kim J, Cantor CR, Kang C. Proc Natl Acad Sci USA. 2000;97:3890–3895. doi: 10.1073/pnas.97.8.3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goh EB, et al. Proc Natl Acad Sci USA. 2002;99:17025–17030. doi: 10.1073/pnas.252607699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ponjavic J, et al. Genome Biol. 2006;7:R78. doi: 10.1186/gb-2006-7-8-r78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wobbe CR, Struhl K. Mol Cell Biol. 1990;10:3859–3867. doi: 10.1128/mcb.10.8.3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leach KM, et al. Nucleic Acids Res. 2003;31:1292–1301. doi: 10.1093/nar/gkg209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cooper SJ, Trinklein ND, Anton ED, Nguyen L, Myers RM. Genome Res. 2006;16:1–10. doi: 10.1101/gr.4222606. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.