

Abstract

A new versatile synthetic methodology for the synthesis of enantiomerically pure natural phenanthroindolizidines and phenanthroquinolizidines has been established and described. Natural products R-antofine and R-cryptopleurine, as well as a novel E-ring expanded analog 13c (E7), 12-oxo-S-antofine (17), and 12N-methyl-12-aza-S-antofine (18) were synthesized with the new method. This strategy will greatly facilitate future SAR studies on the natural alkaloids with E-ring variations.
Phenanthroindolizidines and phenanthroquinolizidines are two series of natural alkaloids primarily found in the Asclepiadaceae and Moracea plant families. The leaves of these plants have been used since ancient times to treat asthma, bronchitis, rheumatism, etc.1 To date, over 60 compounds have been isolated and characterized, such as R-tylophorine, R-antofine, and R-cryptopleurine (Figure 1), which are well-known representatives reported to have potent antitumor activity. Due to low natural abundance and interesting anticancer activity, total synthesis has attracted much attention to assist the development of both the natural compounds and new analogs.
Figure 1.
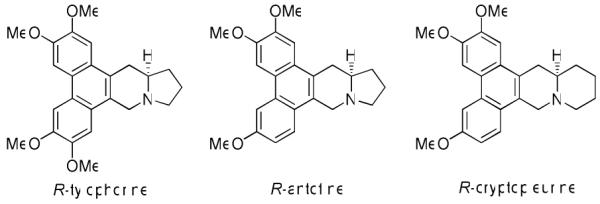
Representative structures of phenanthroindolizidines and phenanthroquinolizidines
To date, numerous synthetic strategies have been reported in the literature.2 One representative strategy, first reported by Rapoport and coworker, used building blocks such as proline, glutamic acid, aminoadipate, and pyroglutamate as the sources of the chiral center followed by intramolecular electrophilic addition.3-7 Other strategies include a chiral auxiliary approach,8 chiral allylic alcohol,9 enantioselective catalysis for intramolecular alkene carboamination,10 and enantioselective phase transfer alkylation;11 some of them have also been used in the synthesis of seco structures.
It has been reported that a major side effect prohibiting the therapeutic use of these natural alkaloids is their CNS toxicity, such as disorientation and ataxia.12 Analogs with higher polarity may be desirable to ameliorate such side effects by preventing the compounds from crossing the blood-brain barrier. However, only a few polar antofine analogs with a C-14 OH group have been synthesized.13 No E-ring substituted analogs (at C11-C14) have been reported to date, despite the large pool of approaches presently available. As a result, current SAR study is still in a premature stage, and additional phenanthroindolizidine and phenanthroquinolizidine analogs with diverse structural features, especially polar functionalities on the E-ring, are urgently needed for a more extensive study of the biological properties.
In this paper, we report the design and synthesis of a key intermediate 7 that should prove to be a versatile precursor to a series of interesting E-ring modified analogs. Possible modifications include incorporation of heteroatoms, introduction of polar groups such as hydroxy and amino groups, and addition of multiple substitutions, all of which are not readily accessible by reported synthetic methods. Two natural products, R-antofine and R-cryptopleurine, were synthesized to verify the feasibility of this new strategy, and three new compounds, 7-membered analog E7 (13c), 12-oxo-S-antofine (17), as well as 12N-methyl-12-aza-S-antofine (18) were synthesized for the first time to further corroborate the versatility of this method.
In our innovative approach, the E-ring is constructed after the D-ring has been conjugated. This synthetic strategy has the following merits: the size of the E-ring can be easily altered via ring-closing metathesis and various functional groups can be readily introduced during E-ring formation and subsequent reactions. The resulting new analogs would provide useful SAR information about the alkaloid E-ring. In contrast to Rapoport’s lactam moiety, another novelty in our strategy is the use of an oxazolidinone moiety, which not only allows efficient formation of the D-ring, but also is easily cleaved to provide the useful synthetic precursor 7.
As shown in Scheme 1, compound 1 was obtained via three steps as reported in the literature in 45% yield.14 Subsequent reduction with LiAlH4 was followed by oxidization with Py•SO3/DMSO to yield an aldehyde, which was reductively aminated by using D-serine methyl ester hydrochloride to give 2 in an overall yield of 59% (three steps). Construction of the oxazolidinone ring system was accomplished by reaction of 2 with Im2CO in CH2Cl2 to afford 3 in 76% yield. The D-ring was formed by acylation of acid 4 to yield 5 in 79% yield. Compound 6 was obtained from 5 by reduction of ketone to methylene in two steps. Refluxing 6 with 6N NaOH (aq.) in MeOH successfully cleaved the oxazolidinone to give the key intermediate 7 in 95% yield, from which a series of interesting modifications could be achieved.
Scheme 1.
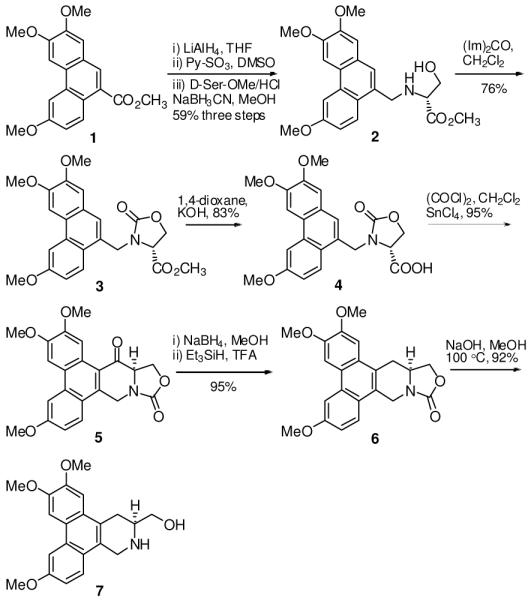
Synthesis of the key intermediate 7
E7 (13c) was first synthesized along with two natural products, R-antofine (13a) and R-cryptopleurine (13b), from 7 as described below. The amino group was protected with a Boc group to furnish 8 in 96% yield (Scheme 2). The hydroxy group was then oxidized by Py•SO3 to give an aldehyde, which was converted to an alkene 9 by Wittig reagent in 68% yield over two steps. After the Boc group was removed, an appropriate unsaturated acid (e.g., 10a: acrylic acid; 10b: 3-butenoic acid; 10c: 4-pentenoic acid) was introduced to afford 10 in an average yield of 80% using EDC or DEPC (for 10b, double-bond isomerization occurred on the N-amide side-chain using EDC/HOBt). Cycloalkene analogs (11a-c) were obtained by cyclization with Grubb’s 2nd generation (G2) catalyst, and then underwent hydrogenation with H2/Pd/C in MeOH to afford 12a-c in high yields. The desired target alkaloids R-antofine (13a) and R-cryptopleurine (13b) as well as an E-ring expanded analog (13c) were obtained by reduction of amide to amine in an average yield of 70%. The analytical information obtained from our synthesized 13a and 13b agreed with the data reported in the literature.4,9
Scheme 2.

Synthesis of antofine, cryptopleurine, and E7
The versatility and usefulness of this strategy were further demonstrated by the synthesis of two new analogs: 12-oxo-S-antofine (17, Scheme 3) and 12N-methyl-12-aza-S-antofine (18, Scheme 4). Compounds 17 and 18 contain a ketone and an additional nitrogen, respectively, in the E-ring.
Scheme 3.
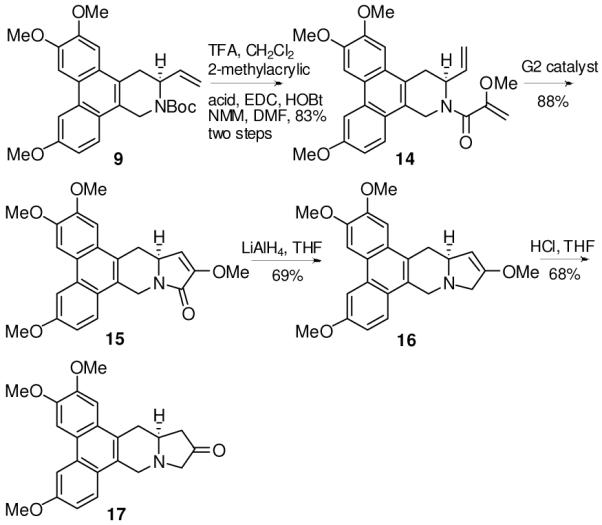
Synthesis of 12-oxo-S-antofine
Scheme 4.
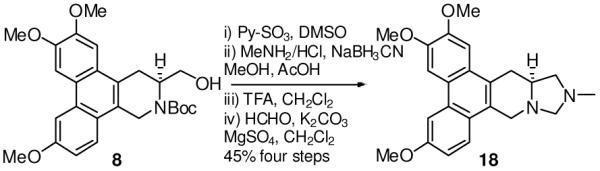
Synthesis of 12N-methyl-12-aza-S-antofine
Compound 14 was obtained by reacting 9 with 2-methoxyacrylic acid after removal of the Boc group in 83% yield. The ring closure was accomplished using G2 catalyst to give compound 15 in 88% yield. Subsequent reduction by LiAlH4 in THF and hydrolysis with HCl afforded 17 in 50% yield over two steps to generate a carbonyl functionality at C12 position. For the synthesis of compound 18, the alcohol 8 was oxidized to the aldehyde, which underwent reductive amination to give an intermediate amine. After removal of the Boc group, both nitrogens were connected by HCHO to produce 18 in 45% yield over four steps. By virtue of similar strategies, we are able to synthesize a series of cryptopleurine analogs with different functional groups on the E ring for further derivatization (unpublished data). Moreover, this novel strategy is also applicable for other natural products in this family, such as tylophorine, which has different substitution patterns on the phenanthrene scaffold.
The new compound E7 was screened in vitro against a panel of human tumor cell lines including A549 (lung), KB (nasopharyngeal), DU-145 (prostate), and HCT-8 (colon), using R-antofine and R-cryptopleurine as a comparison. The results (GI50) are listed in Table 1. Interestingly, compound E7 showed significant cytotoxic activity and was as potent as R-antofine against A549 cell growth but with improved selectivity relative to KB and DU145 tumor cell lines. This result suggests that the E-ring size may affect the interaction between the molecule and the binding site in the target, and the expansion of the E-ring may play a role in the anticancer selectivity. Although moderate to significant reduction in cytotoxicity was observed for compounds 17 and 18, further modifications on the ketone and substituents on the imidazolidine nitrogen are of great interest to potentially improve the anticancer activity.
Table 1.
Anticancer activities (GI50) of R-antofine, R-cryptopleurine, E7, 17, and 18
| compound | A549 (nM) |
DU145 (nM) |
KB (nM) |
HCT-8 (nM) |
|---|---|---|---|---|
| R-antofine | 22 | 25 | 36 | ND |
| R-cryptopleurine | 1.38 | 1.59 | 1.51 | 1.09 |
| E7 | 25 | 179 | 102 | 10 |
| 17 | 290 | 710 | 480 | 420 |
| 18 | 660 | 2030 | 1720 | 1000 |
In conclusion, we have established a novel strategy to synthesize new phenanthroindolizidine and phenanthroquinolizidine analogs, which provides a useful tool for managing CNS toxicity and exploring the SAR profile, especially on the E-ring, for which no analogs have been reported so far, despite substantial progress in the development of their syntheses. Three new E-ring modified analogs E7, 17, and 18 were first reported through our novel strategy detailed above to verify our method’s feasibility and apparent versatility. Other modifications using R-cryptopleurine as template are being studied in our laboratories, aiming to discover interesting molecules with strong anticancer activity and reduced CNS toxicity. The detailed synthetic pathways to other analogs, their biological activity, and mechanistic study results will be reported in due time.
Supplementary Material
Figure 2.
Ring and position numbering of phenanthroindozlidines and phenanthroquinolizidines
Acknowledgement
This study was supported by grant CA17625-32 from National Cancer Institute awarded to K. H. Lee.
References
- (1).Gellert E. J. Nat. Prod. 1982;45:50–73. [Google Scholar]
- (2).For reviews, see: Li Z, Jin Z, Huang R. Synthesis. 2001;16:2365–2378. Chemler SR. Current Bioactive Compounds. 2009;5:2. doi: 10.2174/157340709787580928.
- (3).Buckley TF, Rapoport H. J. Org. Chem. 1983;48:4222–4232. [Google Scholar]
- (4).Nordlander JE, Njoroge FG. J. Org. Chem. 1987;52:1627–1630. [Google Scholar]
- (5).Furstner A, Kennedy JWJ. Chem. Eur. J. 2006;12:7398–7410. doi: 10.1002/chem.200600592. [DOI] [PubMed] [Google Scholar]
- (6).Jin Z, Li SP, Wang QM, Huang RQ. Chinese Chem. letts. 2004;15:1164. [Google Scholar]
- (7).Faber L, Wiegrebe W. Helv. Chim. Acta. 1976;59:2201–2212. doi: 10.1002/hlca.19760590633. [DOI] [PubMed] [Google Scholar]
- (8).(a) Comins DL, Chen X, Morgan LA. J. Org. Chem. 1997;62:7435–7438. doi: 10.1021/jo9711495. [DOI] [PubMed] [Google Scholar]; (b) Ihara M, Takino Y, Tomotake M, Fukumoto K. J. Chem. Soc. perkin Trans. 1990;1:2287–2292. [Google Scholar]; (c) Suzuki H, Aoyagi S, Kibayashi C. J. Org. Chem. 1995;60:6114–6122. [Google Scholar]
- (9).Kim S, Lee T, Lee E, Lee J, Fan GJ, Lee SK, Lee D. J. Org. Chem. 2004;69:3144–3149. doi: 10.1021/jo049820a. [DOI] [PubMed] [Google Scholar]
- (10).Zeng W, Chemler SR. J. Org. Chem. 2008;73:6045–6047. doi: 10.1021/jo801024h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kim SH, Lee J, Lee T, Park HG, Kim D. Org. Lett. 2003;5:2703–2706. doi: 10.1021/ol0349007. [DOI] [PubMed] [Google Scholar]
- (12).Suffness M, Douros J. Anticancer Agents Based on Natural Product Models. Academic press; 1980. pp. 465–487. [Google Scholar]
- (13).(a) Gao W, Busson S, Grill SP, Gullen EA, Hu YC, Huang X, Zhong S, kaczmarek C, Gutierrez J, Francis S, Baker DC, Yu S, Cheng YC. Bioorg. & Med. Chem. Lett. 2007;17:4338–4342. doi: 10.1016/j.bmcl.2007.05.021. [DOI] [PubMed] [Google Scholar]; (b) Gao W, Lam W, Zhong S, kaczmarek C, Baker DC, Cheng YC. Cancer Res. 2004;64:678–688. doi: 10.1158/0008-5472.can-03-1904. [DOI] [PubMed] [Google Scholar]
- (14).Su CR, Damu AG, Chiang PC, Bastow KF, Morris-Natschke SL, Lee KH, Wu TS. Bioorg. & Med. Chem. 2008;16:6233–6241. doi: 10.1016/j.bmc.2008.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.