

Abstract
Magnetic nanoparticles (MNP) have been widely studied for use in targeted drug delivery. Analysis of MNP biodistribution is essential to evaluating the success of targeting strategies and the potential for off-target toxicity. This work compared the applicability of inductively-coupled plasma optical emission spectroscopy (ICP-OES) and electron spin resonance (ESR) spectroscopy in assessing MNP biodistribution. Biodistribution was evaluated in 9L-glioma bearing rats administered with MNP (12-25 mg Fe/kg) under magnetic targeting. Ex vivo analysis of MNP in animal tissues was performed with both ICP-OES and ESR. A cryogenic method was developed to overcome the technical hurdle of loading tissue samples into ESR tubes. Comparison of results from the ICP-OES and ESR measurements revealed two distinct relationships for organs accumulating high or low levels of MNP. In organs with high MNP accumulation such as liver and spleen, data were strongly correlated (r = 0.97, 0.94 for liver and spleen, respectively), thus validating equivalency of the two methods in this high concentration range (> 1000 nmol Fe/g tissue). The two sets of measurements, however, differed significantly in organs with lower levels of MNP accumulation such as brain, kidney, and the tumor. Whereas ESR resolved MNP to 10-55 nmol Fe/g tissue, ICP-OES failed to detect MNP due to masking by endogenous iron. These findings suggest that ESR coupled to cryogenic sample handling is more robust than ICP-OES, attaining better sensitivity in analyses. Such advantages render ESR the method of choice for accurate profiling of MNP biodistribution across tissues with high variability in nanoparticle accumulation.
Keywords: iron oxide nanoparticles, magnetic nanoparticle biodistribution, electron spin resonance spectroscopy, inductively-coupled plasma optical emission spectroscopy, targeted drug delivery, magnetic targeting
1. INTRODUCTION
Iron oxide-based magnetic nanoparticles (MNP) have gained widespread interest in the biomedical field. Comprised of a superparamagnetic iron oxide core coated with a biocompatible polymer shell, MNP have recently been exploited for both diagnostic and therapeutic applications1. Biomedical utilization of MNP includes the delineation of pathology with MRI2-4; thermal ablation of tumors with magnetic hyperthermia5, 6; and the delivery of chemotherapeutic agents to tumor tissue1, 7-10. Therapeutic compounds can be associated with nanoparticles by chemical conjugation, electrostatic interaction, or encapsulation within the nanoparticle polymeric coating8, 11, 12. Therapeutic-loaded MNP can then be selectively accumulated in tumors. Due to their nano-scale size, MNP were shown to passively penetrate compromised tumor vasculature and accumulate in tumor interstitium by the so-called enhanced permeability and retention (EPR) effect13-15. In addition, active targeting of MNP has been achieved with the coupling of tumor-specific ligands (peptides, small molecules, antibodies, etc.) 12. Furthermore, MNP can also be accumulated in tumor tissue via active magnetic targeting with the aid of external magnetic field owing to MNP magnetic responsiveness1, 7. Indeed, we recently demonstrated a significantly improved tumor accumulation of MNP in glioma-bearing rats following magnetic targeting10.
In order to quantitatively evaluate the success of any targeting strategy, biodistribution studies are essential. Nanoparticle accumulation at the target site gives insight to efficacy. Moreover, an important goal of targeting is to minimize toxicity. Although unmodified MNP have been shown to exhibit a generally favorable biocompatibility profile16-18, off-target MNP accumulation still can raise toxicity concerns. Local elevation of low molecular weight iron species due to MNP biodegradation may be implicated in tissue damage via oxidative stress19. In addition, off-target deposition of MNP loaded with highly toxic pharmaceuticals may compromise healthy tissues. Organs of elimination (liver, spleen, lung, and kidney) and tissue immediately adjacent to the target tumor are at a particular risk for such toxicity. Therefore, exposure of these tissues must be thoroughly assessed when using MNP as a drug delivery vehicle.
The ideal method to study tissue biodistribution of MNP would require minimal sample preparation and possess high detection sensitivity. A variety of methods have been exploited to study nanoparticle content in tissues and cells, including radiologic and spectroscopic techniques. Radiologic methods require modification of MNP with a radiolabel followed by analysis via nuclide counting9, 20-22. While radioactive nuclides provide for high sensitivity and low detection limits, they are biohazardous and require special safety precautions. As a result, spectroscopic methods are usually preferred. Inductively coupled plasma optical emission spectroscopy (ICP-OES) is one of the most commonly used techniques23-30 for MNP quantification. However, it must be pointed out that this technique determines total tissue iron. In addition to the exogenous iron, animal tissues are also rich in endogenous iron in the form of iron-containing proteins (e.g. hemoglobin, transferrin, ferritin). These endogenous iron species contribute to non-MNP iron background, which may reduce the ability of ICP-OES to detect low tissue concentrations of MNP.
Another technique, electron spin resonance (ESR) spectroscopy, may provide a more sensitive alternative for MNP determination. ESR is used to study chemical species with unpaired electrons including paramagnetic (e.g. Fe(III)-transferrin), ferri-/ferromagnetic (e.g. magnetite) and anti-ferromagnetic (e.g. ferritin) materials31, 32. Both endogenous iron-protein complexes (i.e. hemoglobin, transferrin, ferritin, hemosiderin) and the exogenous MNP can be detected with ESR. However, unlike ICP-OES, which cannot distinguish between endogenous and exogenous sources of iron, ESR shows greater sensitivity for MNP (per weight Fe) than for endogenous iron species. Indeed, this technique has been successfully utilized for quantification of MNP concentrations in biological samples. Our previous studies revealed that ESR is able to detect MNP in brain tumors of magnetically targeted rats at concentrations as low as approximately 30 nmol Fe/g tissue10.
Although the viability of ESR to detect MNP in tissues has been demonstrated, the technique has not been widely utilized for biodistribution studies31, 33, 34. This limited use could be attributed to ESR’s unexplored relationship with more commonly used ICP-OES, as well as the technical challenge of loading tissues into a long (190 mm), narrow (3 mm ID) ESR tube for sample analysis. In order to validate ESR as a choice methodology for MNP biodistribution studies, the goals of this work were two-fold. First, we sought to develop a simple and convenient protocol for ESR analysis with minimal tissue processing. Second, we aimed to compare the utility of ESR and ICP-OES methods for biodistribution studies. Given the critical importance of biodistribution assessment in evaluating the success of MNP-mediated tumor targeting, we have chosen to study the robustness of ESR and ICP-OES methods in a magnetically targeted 9L glioma-bearing rat model.
2. EXPERIMENTAL
2.1. Iron oxide nanoparticles
Iron oxide nanoparticles coated with starch (fluidMAG-D) were generously contributed by Chemicell® (Berlin, Germany). The physical properties of fluidMAG-D were previously analyzed 10 and are summarized in Table 1.
Table 1.
Physical properties of fluidMAG-D magnetic nanoparticles as previously determined10. Data expressed as MEAN ± SD
| Property | Value | Units |
|---|---|---|
| Hydrodynamic diameter | 110 (± 22) | nm |
| Saturation magnetization (Ms) | 94 | Emu/g Fe |
| R2 relaxivity | 43.8 (± 2.6) | s−1 mM−1 |
2.2. Animal preparation
Cell Culture
Rat 9L-glioma cells were cultured and prepared for tumor induction as previously described.10 Briefly, 9L-glioma cells (Brain Tumor Research Center, University of California, San Francisco) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum, 100 IU/mL penicillin, 100 μg/mL streptomycin and 0.29 mg of L-glutamine at 37°C in a humidified atmosphere of 5% CO2. Prior to implantation, cells were grown to confluency in 100 mm culture dishes and harvested using 0.25% trypsin/0.1% ethylene-diamine-tetra-acetic acid (EDTA) solution. Cells were pelleted by centrifugation at 1000g for 5 min, resuspended in serum free DMEM at a concentration of ~105 cells/μL and kept on ice until use.
Induction of Brain Tumors in Rat Model
Intracerebral 9L tumor induction was carried out as previously reported.35 Male Fisher 344 rats (125–150 g, Harlan Sprague Dawley Inc., Indianapolis, IN) were anesthetized by intraperitoneal injection of ketamine/xylazine mixture (87/13 mg/kg body weight). A small skin incision was made over the right hemisphere and the tissue was carefully removed until the bregma was identified. A 1-mm-diameter burr hole was drilled through the skull 1mm anterior to the bregma and approximately 5 mm lateral from the midline. Ten microliters of 9L cell suspension was injected through the burr hole at a depth of 3 mm beneath the skull. The surgical field was cleaned with 70% ethanol and the burr hole was filled with bone wax (Ethicon Inc., Summerfield, NJ) to prevent extracerebral extension of the tumor and the skin incision was closed with tissue adhesive (3M Vetbond, Animal Care products, St. Paul, MN). Animals were imaged using MRI (described below) beginning at 10 days post-cell implantation to select tumors between 50 and 70 μL in volume as previously described.35
All animal experiments were conducted according to protocols approved by the University of Michigan Committee on Use and Care of Animals (UCUCA).
2.3. Magnetic Targeting of Starch Coated Iron-Oxide Nanoparticles and Tissue Excision
Starch-coated, iron oxide (magnetite) nanoparticles (fluidMAG-D) were magnetically targeted to brain tumors as described.10 Briefly, nanoparticle suspensions were diluted with PBS and filtered through a 0.2 μm disposable syringe filter to obtain a preparation of about 6 mg Fe/mL, as determined by ICP-EOS described below. Tumor-bearing animals were anesthetized with an inhaled mixture of 1.5% isoflurane/air and tail veins cannulated using a 26-gauge angiocatheter (AngiocathTM, Becton Dickinson, Sandy, UT). The animals were then placed ventrally on a platform with their head positioned between the poles of an electromagnet. The magnetic field density within the air gap between the poles was adjusted to 0.4 T. Animals were then injected with nanoparticle suspension (12-25 mg Fe/kg) through the catheter placed in the tail vein and retained in the magnetic field for thirty minutes. Fifty minutes after nanoparticle administration, animals were sacrificed and organs (brain, liver, spleen, lung, and kidney) were immediately excised for ex vivo analysis. Tumors were carefully dissected from the right brain hemisphere. Animals not exposed to nanoparticles or magnetic targeting served as controls. All tissues were stored at −80°C prior to analysis.
2.3. Magnetic Resonance Imaging (MRI)
MRI experiments were performed on an 18-cm horizontal-bore, 7 Tesla Varian Unity Inova imaging system (Varian, Palo Alto, CA). Animals were anesthetized with 1.5 % isoflurane/air mixture and imaged using a 35-mm-diameter quadrature RF head coil (USA Instruments Inc, OH). To visualize the tumor localization within the rat brain, 13 axial sections of the brain were acquired with a T2-weighted fast spin echo sequence using the following parameters: repetition time (TR) = 4 s, echo time (TE) = 60 ms, field of view = 30 × 30 over 128 × 128 matrix, slice thickness = 1mm, slice separation = 2 mm, four signal averages per phase encoding step. To determine nanoparticle distribution in the brain, 13 gradient echo (GE) axial slices of the brain were collected before the nanoparticle administration (baseline scans) and immediately following magnetic targeting. GE images were acquired with the following parameters: TR = 20 ms, TE = 5 ms, field of view = 30 × 30 over 128 × 128 matrix, slice thickness = 1mm.
2.4. Ex vivo ESR Analysis of Tissues
Excised tissue samples (n = 7-8) were first subjected to ESR spectroscopy to determine MNP content. Samples from animals (n = 5) not exposed to nanoparticles were also analyzed to assess background. Frozen tissue samples (~30 mg) were sectioned into 2 mm × 2 mm cubes using a razor blade. Sections were applied to the top of an ESR tube, pre-cooled on dry ice, and quickly pushed to the tube bottom using a thin glass rod (see Figure 2). Tubes were weighed before and after packing to determined tissue sample mass. For each excised organ, triplicate tissue samples were packed for analysis. It is important to note that magnetic susceptibility of MNP, and consequently magnitude of the ESR signal (per unit iron), varies across different MNP preparations due to differences in structural attributes (e.g. core morphology, size distribution, composition of surface coating). For accurate quantitative analysis, it is thus important to calibrate the ESR signal with the investigated MNP species. Therefore, calibration standards were prepared, in triplicate, from the same MNP stock used for in vivo studies. Iron concentration of the stock was determined by ICP-OES. Stock dilutions (100 μL) were carefully applied to the bottom of ESR tubes (to prevent sample loss on tube walls) with a micro-syringe coupled to a 12 in. teflon needle (Wilmad Labglass, Buena, NJ). Tissues samples and calibration standards were analyzed on an EMX ESR Spectrometer (Bruker Instruments, Billerica, MA) equipped with a liquid nitrogen cryostat. Spectra were acquired using the following parameters: resonant frequency = ~9.3 GHz; microwave power = 20 mW; and temperature = −128°C.33 Receiver gain and modulation amplitude varied depending on the tissue (see Table 2). Spectra were obtained as the first derivative (dP dB) of absorbed microwave power (P) with respect to applied magnetic field (B).
Figure 2.

Advantage of the developed cryo-handling methodology for introduction of tissue samples into ESR tubes. (A,B) Pushing of the sample into the tube using glass rod under ambient temperature (A) leads to sample loss due to smearing of the tissue along the tube walls (B). (C.D) In contrast, cryogenically handled sample is pushed to the bottom of the tube without any tissue loss.
Table 2.
Receiver gain and modulation amplitude values used in ESR analyses of different tissues
| Tissue | Receiver Gain | Modulation Amplitude (G) |
|---|---|---|
| Liver | 5 × 103 | 1 |
| Spleen | 5 × 103 | 1 |
| Lung | 5 × 103 | 1 |
| Brain | 5 × 104 | 5 |
| Tumor | 5 × 104 | 5 |
| Kidney | 5 × 104 | 5 |
The double integral of the first derivative signal is known to be proportional to the number of resonating electronic spins in a measured sample. Double integrals of acquired spectra (DI) were calculated using MathCAD software package (PTC Corp, Needham, MA). DI values of MNP-containing tissues were normalized by sample weight and background corrected by subtracting weight-normalized DI of corresponding blank tissues (obtained from animals not exposed to MNP). Tissue MNP concentrations (nmol Fe/ g tissue) were recalculated from weight-normalized background-corrected DI values using calibration curves constructed with MNP standards.
2.5. Ex vivo ICP-OES Analysis of Tissues
Tissue samples, initially analyzed by ESR, were then studied by ICP-OES, to minimize any variability that may arise from analyzing different tissue cuts. ESR tubes were scored with a diamond scribe and cracked open just above the tissue crown. Tube pieces containing tissue were placed on dry ice for one minute to refreeze thawed tissue. While still frozen, tissues were carefully extracted using forceps and weighed in 2 mL vials containing 1.0 mm high-density Zirconia beads (500 μL, Biospec Products, Bartlesville, OK). Tween 80 (0.1% in MilliQ water, 500 μL) was added, and the mixture was homogenized with a Mini-BeadBeater-8 homogenizer (Biospec Products, Bartlesville, OK). Concentrated hydrochloric acid (12M, 2 mL) was added to the homogenate (350 μL), and the mixture was digested for two hours at 70°C. MilliQ water and yttrium internal standard (GFS Chemicals, Columbus, OH) were then added for a total volume of 4 mL containing 1 mg/L yttrium. Digestion mixtures were then vortexed and filtered through a 0.45 μm syringe filter (Titan2 reversed cellulose, SunSri, Rockwood, TN) to remove solid content. Analysis of a high MNP content liver homogenate revealed no statistically significant difference in measured iron between filtered (11.9 ± 1.9 nmol Fe/mg tissue) and non-filtered (11.6 nmol ± 1.8 Fe/mg tissue) preparations. Iron content was determined from emission readings at Fe 238.204 nm emission line using an ICP-OES spectrometer (Optima DV 2000, PerkinElmer, Waltham, MA). The instrument was calibrated with dilutions of an iron standard (GFS Chemicals, Columbus, OH) also spiked with yttrium internal standard (1 mg/L).
Raw ICP results were calculated with WinLab32 for ICP software (Perkin Elmer, Waltham, MA) and used to back-calculate the amount of iron in the original homogenate. The mass of digested tissue was calculated based on the ratio of digested (350 μL) to total (500 μL) homogenate volume and the original mass of tissue. As with the ESR analysis, tissue iron amounts were normalized by weight and averaged to obtain a total iron concentration (nmol Fe/g tissue) for each organ. Data were then corrected for endogenous iron by subtracting a normalized average iron concentration determined for blank tissues (n=5) not exposed to MNP.
2.6. Statistical Analyses
All data are presented as mean ± SD unless otherwise noted. Statistical comparison of ICP-OES measured iron content in blank and targeted tumors (Figure 1B) was made using the Student’s t-test with a significance of p < 0.05. Correlations of liver and spleen data from ESR and ICP analyses (Figure 4A/4B) were determined as Pearson correlation coefficients (r) using SPSS 17.0 Software (SPSS, Chicago, IL).
Figure 1.
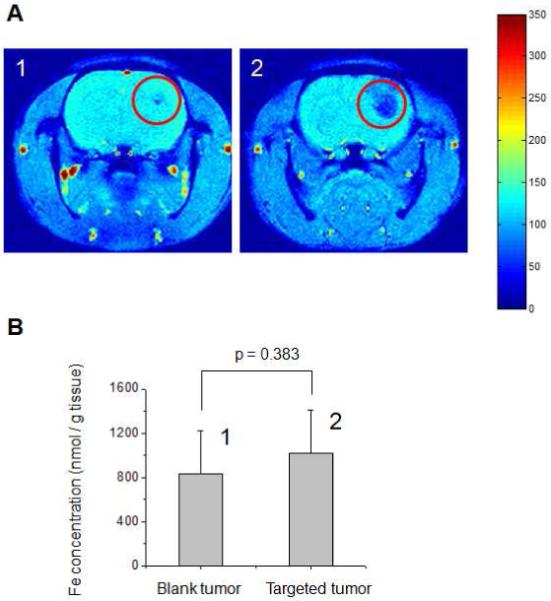
A. Representative brain MRI images (Gradient Echo (GE) axial scans) of 9L-glioma bearing rats (1) not exposed to iron oxide nanoparticles (blank) and (2) administered with iron oxide nanoparticles under a gradient of magnetic flux density (red circles indicate location of tumor lesion). B. ICP-OES analysis of the corresponding excised tumor tissues revealing no statistical difference (p=0.383) in Fe concentration between tumors of (1) the blank and (2) the nanoparticle-administered rats.
Figure 4.
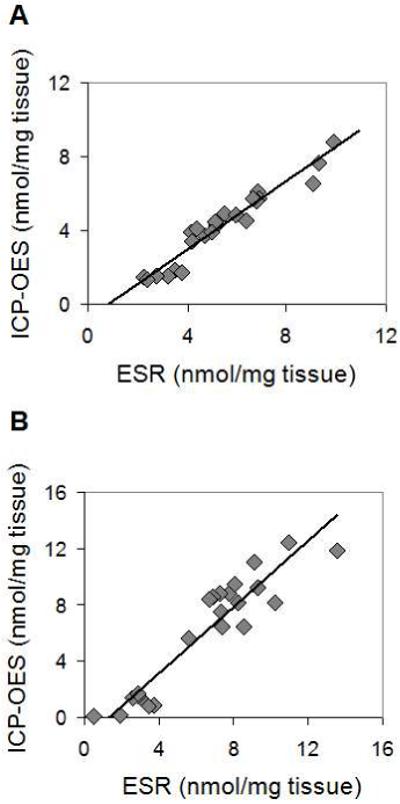
Comparison of ICP-OES and ESR methodologies for MNP concentration analysis of (A) liver and (B) spleen obtained from rats which were administered with different concentrations of magnetic nanoparticles within the range of 12-25 mg Fe/kg. ICP-OES and ESR data sets show strong positive correlation with r = 0.97 and r = 0.94 for the liver and the spleen tissues, respectively.
3. RESULTS
3.1. Initial ICP-OES Analysis of Excised Brain Tumors
ICP-OES is widely utilized to study the biodistribution of iron-oxide nanoparticles. We first aimed to explore the ability of this technique to accurately quantify nanoparticle concentration in tumor tissues after magnetic targeting. MR imaging of the animal brain was used as a point of reference for this assessment. Animals were imaged with MRI after magnetic targeting to visually confirm the presence of MNP in tumors prior to tissue excision. Figure 1A shows representative gradient echo (GE) MRI head scans of animals not exposed to MNP (Panel 1) and those magnetically targeted (Panel 2). Tumor lesions were identified with T2-weighted MRI and are highlighted with red circles. Magnetic nanoparticles are strong enhancers of T2/T2* proton relaxation and thus manifest themselves by pronounced hypointensity (negative contrast) on GE MRI scans. As seen in Figure 1, in contrast to blank tumors which show very little signal reduction, pronounced hypointensity can be observed in targeted tumors indicating nanoparticle accumulation.
However, despite the visual differences seen in MR images, both blank and targeted tumors showed similar levels of iron according to quantitative ICP-OES analysis (Figure 1B). No statistically significant difference (p = 0.38) in iron content could be detected between the two groups. Additionally, the level of iron measured in blank tumors was more than 25 times greater than the amount we recently attributed to targeted MNP.10 This high background level appeared to render ICP-OES insensitive to the small expected increase in tumor iron from MNP.
3.2. Cryogenic Methodology for Tissue Packing of ESR Tubes
ESR is an attractive methodology for assaying iron-oxide in tissues due to its sensitivity for paramagnetic species. However, before comparing the ESR and ICP-OES analyses, an important technical problem to ESR needed to be addressed – the introduction of tissue to the bottom of a long, narrow ESR tube. We first attempted loading small tissue cuts at ambient temperature into the tubes. Cuts were easily applied to the tops of ESR tubes as shown in Figure 2A. Upon pushing cuts further into the tube (Figure 2B), however, tissue smeared along the tube walls. Only a small fraction of the tissue shown in Figure 2A could be delivered to the bottom of the tube. To overcome this problem, we developed a cryogenic method for tissue handling (see Section 2.4). Exhibited in Figure 2C, frozen tissue cuts were loaded into the tops of ESR tubes. When quickly pushed with the glass rod, nearly all tissue shown in Figure 2C could be delivered to the bottom of the tube (Figure 2D). No visible smearing was observed. The methodology was also non-destructive rendering samples available for additional analyses (e.g. ICP-OES) upon extraction from tubes.
3.3. ESR Analysis of Tissues for MNP Content
We next conducted ESR analysis of various tissues using the cryogenic sample handling procedure described above. Liver, spleen, lung, brain, tumor, and kidney were chosen for analysis as they were expected to differ significantly in nanoparticle concentration. It is known, for example, that 80-90% of administered MNP are distributed in the liver and 5-8% in the spleen giving each of these organs relatively high concentrations when compared to other tissues (e.g. tumor, brain, and kidney)18. Representative spectra for each organ are shown in Figure 3. The spectra of organs from MNP injected animals exhibit broad resonance signal centered at about g = 2.2 with a linewidth of about 1100 Gauss. The shape and field location of this signal are identical to those of the standard MNP suspensions10 and are consistent with ESR spectra of superparamagnetic iron oxide nanoparticles reported in the literature36, 37. As seen by the representative control tissue spectra, the signal intensity contributions from all background sources is negligible compared to that of MNP containing tissues. The only visible signal, seen in the kidney control, is a low-intensity, narrow peak (g = 2.0, linewidthp-p = 240 Gauss), which can be attributed to ferritin32. Of important note, the test spectra were distinct from blanks not only for relatively high, but also for relatively low accumulating tissues, revealing high ESR sensitivity to MNP in tissue samples.
Figure 3.
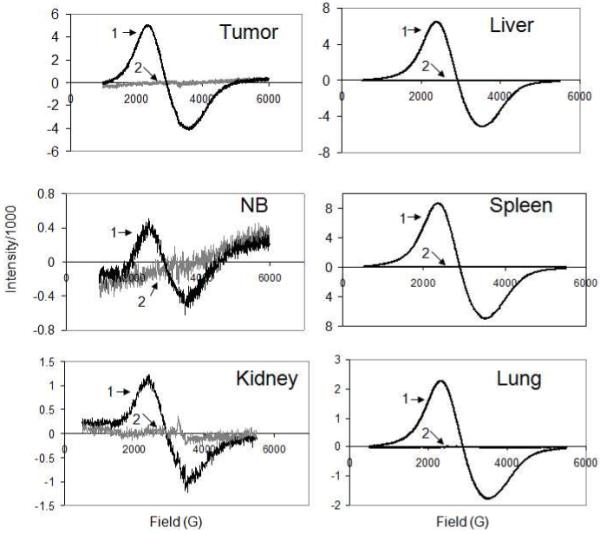
Representative ESR spectra of animal tissues excised from magnetically targeted animals (1) and animals not exposed to magnetic nanoparticles (2). Varied level of noise and different scale of the ordinate in spectra are primarily due to different receiver gain and modulation amplitude settings used in analyses (see Section 2.4).
3.4. Comparison of ESR and ICP-OES Methodologies
The ability of ESR and ICP-OES methods to quantify nanoparticle accumulation was next compared for two groups of organs with high or low MNP accumulation. Animals were dosed with varied amounts of MNP to produce a range of tissue nanoparticle concentrations in each of the different organs tested. In addition, single organ ICP-OES and ESR analyses were performed on the same tissue sample to minimize the effect of accumulation heterogeneity across an organ. Liver and spleen samples, expected to have high iron oxide accumulation, were first compared. As seen in Figure 4A, the ICP-OES and ESR data obtained from liver samples followed a strong, positive linear correlation (r = 0.97, p<0.01) with a negative intercept. Data obtained from spleen tissue (Figure 4B) were similarly correlated (r = 0.94, p<0.01), also with negative intercept. The high correlations shown in Figure 4 indicated that the two methods were equivalent in evaluating tissues that accumulated high levels of MNP. The negative intercepts shown in Figure 4 suggested that ICP-OES was insensitive to MNP when tissue accumulation became relatively low.
We next sought to better understand the discrepancy between the ESR and ICP-OES methodologies in analysis of low and high MNP accumulating tissues. To this regard, we examined the differences between the experimental (with administration of MNP) and background (without administration of MNP) signal for both ESR and ICP-OES. Figure 5 demonstrates that in high-MNP accumulating tissues the signal generated by MNP-exposed organs was significantly higher (p < 0.001) than the background from the corresponding “blank” organs (not exposed to MNP) with both the ICP-OES (Fig. 5A) and the ESR (Fig. 5B) methodologies. Thus, the experimental tissue signal could be corrected for background to calculate the fraction of the signal corresponding to exogenous MNP. In the case of high-accumulating tissues, this procedure could be reliably performed with either methodology, explaining the strong correlation between ESR and ICP-OES in MNP determination for liver and spleen (Figure 4).
Figure 5.
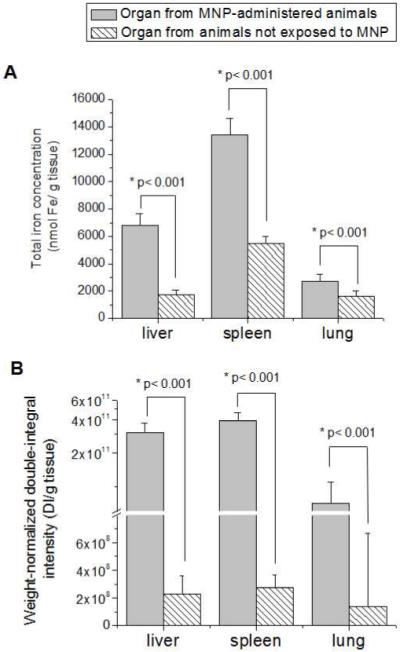
(A) ICP-OES and (B) ESR analysis of high MNP accumulating organs from animals administered with MNP under magnetic targeting (test) and animals not exposed to MNP (blank). Data of ICP-OES analysis are expressed as total iron concentration of the tissue. Data of ESR analysis are expressed as a double integral of acquired spectra normalized by the weight of tissue samples. Statistically significant difference in corresponding parameter between the nanoparticle-containing and blank organs is indicated by an asterix (*).
In contrast, the analysis outcomes with ESR and ICP-OES for low accumulating tissues did not show the same agreement (Figure 6). Total iron concentrations determined for MNP-exposed tumor, normal brain and kidney tissues with ICP-OES did not significantly differ (p>0.05) from the background total iron in the corresponding blank organs (Figure 6A). High levels of background iron masked exogenous content, thus, compromising MNP detection. However, ESR analysis of low accumulating tissues revealed a different pattern. Weight-normalized double integrated intensities of the ESR signal from MNP-exposed tissues were found to be significantly higher (p<0.001) than the corresponding background values (Figure 6B). The discrepancy in ESR and ICP-OES analyses of low accumulating tissues clearly demonstrates that contribution of endogenous iron species to the ESR signal does not scale with their elemental iron content. Thus, despite high background of endogenous elemental iron, ESR successfully resolved MNP accumulation in brain, tumor, and kidney tissues of targeted animals.
Figure 6.
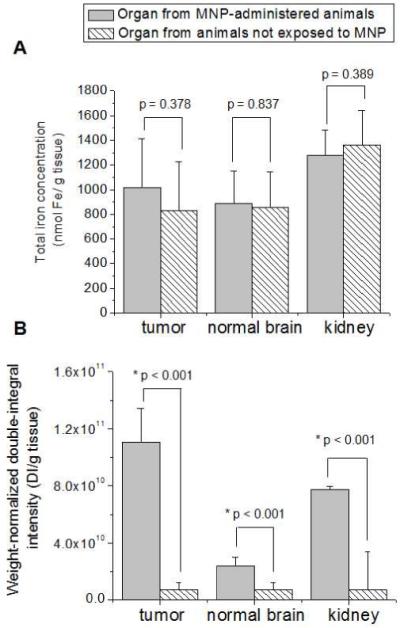
(A) ICP-OES and (B) ESR analysis of low MNP accumulating organs from animals administered with MNP under magnetic targeting (test) and animals not exposed to MNP (blank). Data of ICP-OES analysis are expressed as total iron concentration of the tissue. Data of ESR analysis are expressed as a double integral of acquired spectra normalized by the weight of tissue samples. Statistically significant difference in corresponding parameter between the nanoparticle-containing and blank organs is indicated by an asterix (*).
The impact of performance differences between the ESR and ICP-OES methods on biodistribution analysis is shown in Figure 7. While MNP were successfully detected in high accumulating organs (liver, spleen, and lung) by both methodologies, ICP-OES was insensitive to MNP in tumor, brain, and kidney (Figure 7A), yielding an incomplete profile of biodistribution. This shortcoming, however, was rectified when the analysis was performed with ESR (Figure 7B). Not only the MNP content of the tumor (39.2 ± 10.3 nmol Fe/g tissue), but also of the kidney (6.1 ± 2.4 nmol Fe/g tissue) and normal brain (5.6 ± 0.8 nmol Fe/g tissue) could be successfully determined with ESR.
Figure 7.
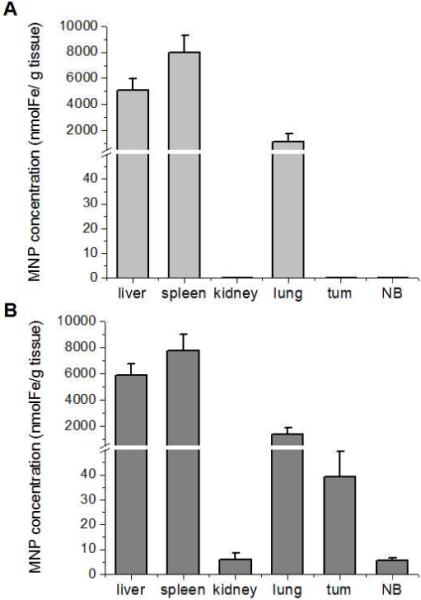
Biodistribution profiles of MNP in magnetically targeted animals obtained with (A) ICP-OES and (B) ESR methodologies. With ICP-EOS, plotted MNP concentrations (expressed in units of iron) were calculated from the data in Figures 5A and 6A by subtracting iron content of blank organs (background) from the iron content of MNP-exposed organs as described in Section 2.5. With ESR, plotted MNP values were calculated from the data in Figures 5B and 6B using a two-step analysis. First, weight-normalized double integral intensities of experimental tissues were corrected for the background values determined in tissues of non-MNP exposed animals. Then, background-corrected weight-normalized double integral values were recalculated for MNP concentrations using calibration curves with MNP standards as described in Section 2.4.
4. DISCUSSION
Due to the breadth of potential biomedical applications for MNP, establishment of a reliable protocol for MNP biodistribution studies is essential. Spectroscopy is favored over radiologic methods primarily due to its safety advantages. This work aimed to compare the applicability of two spectroscopic techniques, ICP-OES and ESR, to study biodistribution of MNP. Before carrying out the comparison we also sought to address a technical challenge of introducing tissues to the bottom of long and narrow ESR tubes, which is perhaps a serious drawback to ESR application.
Tissue at ambient temperature smears when pushed into the tube, with little delivered to its bottom (Figure 2B). Since only the tissue at the bottom of the tube contributes to the ESR signal, tissue loss on the tube walls results in an underreported MNP accumulation. To address this issue, we developed a simple and fast method for cryogenic tissue loading into ESR tubes. Cryogenic handling of tissue samples for ESR has been previously described in the literature. However, most described methods for sample preparation utilize specialized custom-made tools, which are not readily available. In addition, these methods expose tissue samples to sources of potential contamination that can affect the ESR spectra. For example, Shuter et. al. describe a freeze-clamp method which requires “specially constructed freeze-clamps” to compress frozen tissue into cylindrical pellets which can be loaded into ESR tubes38. While the process seems to be straight-forward at first glance, rigorous cleaning of the clamp’s pellet-forming voids would be required after each use to prevent cross-contamination of samples. Although feasible for few samples, the cleaning procedure itself is likely to consume an inordinate amount of time for studies requiring multiple-sampling (min. of 3), of multiple organs (e.g. heart, lung, brain, tumor, etc), from multiple animals (typically n > 3). Another method (Mulsch et al) describes the loading of samples into Teflon tubes, which are then immersed into liquid nitrogen for freezing39. The frozen pellet is then pushed out with a glass rod to produce a solid cylinder which can be used for ESR measurements. Our experience has shown that the loading of non-frozen tissues into thin tubes, such as the described Teflon tube, creates smearing all along the inner tube wall – a potential source of both sample loss and cross-contamination. Therefore, utilization of this method for sample preparation would require availability of large amounts of tissue to account for sample loss and necessitate that the tube is thoroughly cleaned, which can be difficult with small diameter tubes. Single use of these tubes is unlikely due to the relatively high cost of Teflon. On the other hand, the method we present is superior as it both minimizes sample processing time and tissue loss while also reducing the chances of sample contamination. First, tissue sections are pre-frozen to eliminate smearing and allow “clean”, rapid loading of samples. Elimination of tissue loss is of critical importance since it allows analysis of scarcely available tissue samples such as the tumor, which can be less than 30 mg in mass. Second, the sample is loaded directly into disposable ESR tubes – preventing contamination and eliminating the need for tube cleaning between samples. Overall, our method can be used to efficiently prepare a large number of contamination-free samples for study with as little as 20-30 mg tissue.
With the developed cryogenic technique, sample preparation for ESR exhibits several important advantages over sample preparation for ICP-OES. Excised tissues require extensive processing before they can be analyzed with ICP-OES. This processing includes digestion of the sample, which may be preceded by homogenization, to liberate ionic iron, and incorporation of internal standards to account for consistency differences between digestion mixtures and calibration standards. Extreme care must be taken during the sample preparation to ensure that: (i) the homogenate is truly homogeneous and an aliquot represents its true fraction of the total mass homogenized; (ii) all iron in the tissue is released during digestion; (iii) sample volume remains constant during heating required for digestion; and (iv) internal standards accurately translate in computed results. Deviation from any of these criteria can lead to significant inaccuracy in obtained results.
In contrast to ICP-OES, ESR sample preparation with our cryohandling method only requires a reduction of tissue size before loading samples into tubes. This minimal processing requirement allows for more reliable data analysis. In addition, this methodology is non-destructive to tissues, rendering them available for additional analyses. Several uses could be envisioned for samples recovered from ESR tubes. For example, one could measure total tissue iron by ICP-OES as illustrated in our study. Such measurements could be important, for studying iron oxide metabolism and degradation kinetics in tissues. In addition, recovered tissue cuts could also be assayed for drug content and even enzymatic activity. This could facilitate characterization of drug/enzyme loaded iron oxide nanoparticles and enable evaluation of important metabolic enzymes that reflect toxicity. In contrast, when ICP-OES is used for nanoparticle quantification via iron analysis, no further measurements can be performed on assayed tissue fragments. Therefore, a “non-destructive” ESR methodology presents an important advantage over “destructive” ICP-OES.
The optimized methodology for ESR sample preparation allowed us to compare the applicability of ESR and ICP-OES to biodistribution analysis of MNP. The two methods were first examined when applied to high MNP accumulating tissues. Organs of elimination (liver, spleen) were chosen for this analysis as they were expected to possess a high content of MNP. Data sets collected from liver and spleen tissue samples exhibited strong ESR to ICP-OES correlation (Figure 4). These data validate ESR and ICP-OES as comparable options to study biodistribution in tissues of relatively high nanoparticle accumulation. In this case, the advantage of using ESR is its more efficient sample preparation procedure and non-destructive nature.
However, ESR outperforms ICP-OES for tissues having relatively minor MNP accumulation. The two techniques are based on different underlying phenomena. ICP-OES provides a measure of total tissue iron. Multiple endogenous iron-containing species (hemoglobin, ferritin, transferrin, hemosiderin, low molecular weight iron species) found in tissues contribute to the total iron content, thus generating relatively high ICP-OES background. While this background did not interfere with MNP determination in high MNP accumulating organs (liver, spleen, lung: Figure 5A), it masked MNP presence in low MNP accumulating organs (brain, tumor, kidney: Figure 6A). The consequence of reduced ability of ICP-OES to detect MNP, due to endogenous iron background, is masked distribution. Although MRI visually confirms accumulation of MNP in targeted tumors (Figure 1A), the conclusion from ICP-OES (Figure 1B) is that tumor accumulation does not occur. Analyses of targeted tumors and other tissues (normal brain, kidney) expected to have relatively low MNP accumulation all displayed no statistically significant difference in iron content over blanks. Figure 6A suggests that MNP do not distribute in these tissues. The obvious result is the incorrect, incomplete picture of biodistribution shown in Figure 7A.
In contrast to ICP-OES, where all sources of iron (endogenous or exogenous) are equal contributors (per unit iron) to the measured signal, the magnitude of the ESR signal (per unit iron) is determined by the order of interatomic arrangement in a particular iron species. Ferrimagnetically arranged cores of the MNP, composed of magnetite/maghemite, possess higher magnetic susceptibilities than antiferromagnetic (e.g. ferritin, hemosiderin) or paramagnetic (e.g. transferrin) endogenous iron species40. Consequently, the ESR signal of MNP (per unit iron) is known to be higher than that of endogenous iron species. In addition, the ESR signal of MNP can be further discriminated from endogenous contributors by judicial choice of measurement conditions, e.g. temperature. For example, it has been demonstrated that the ESR signal from hemoglobin (g = 5.8), transferrin (g = 4.3) and free radical species (g = 2.0) could be almost completely silenced at temperatures above 100K32 while MNP signal was found to be optimal above 100K36. In agreement with these findings, previous ESR analysis of tissue samples for MNP conducted by us10 and other investigators34 revealed a low background signal at 145K, the temperature utilized in the present study. Respectively, as seen in Figure 3, the ESR signal intensity of MNP (per unit iron) is significantly higher than that of endogenous iron species. The reduced background improved sensitivity of the method towards MNP, and allowed successful MNP quantification at a concentration as low as 5.6 ± 0.8 nmol Fe/g tissue. The impact of improved sensitivity is apparent when considering the differences between Figures 6A and 6B. ESR, in contrast to ICP-OES analysis, clearly shows that MNP are indeed distributed in the tumor, brain, and kidney. In short, the sensitivity of ESR for MNP enables a more accurate and complete picture of biodistribution as shown in Figure 7B.
A complete map of MNP accumulation is the ideal result of any biodistribution study and is essential to evaluating the success of any targeting strategy. In our model targeting strategy, the insensitivity of an ICP-OES analysis would have resulted in a conclusion against targeting feasibility and given no insight into potential toxicities. It is important to understand that the presented biodistribution study was simplified for proof-of-concept purposes. To more fully characterize MNP biodistribution in any specific organ, sampling across different sections of the organ would be necessary to account for any intra-organ variability. Nevertheless, our data suggest that an ESR analysis can simultaneously illustrate the efficacy of a targeting strategy and evaluate toxicity risks in healthy tissues. The method robustly profiles accumulation of MNP in both high and low accumulating tissues to produce a more complete picture of total biodistribution.
5. CONCLUSIONS
ESR tissue analysis of MNP biodistribution performed in conjunction with our optimized cryogenic sample handling technique achieved greater sensitivity than ICP-OES. The end product is a better understanding of targeting feasibility and any potential toxicity from off-target accumulation of MNP. These important advantages render ESR a choice method to characterize MNP biodistribution.
ACKNOWLEDGEMENTS
This work was supported in part by NIH R01 Grants CA114612, NS066945, and the Hartwell Foundation Biomedical Research Award. In addition, this work was also partially sponsored by the World Class University (WCU) program through the Korea Science and Engineering Foundation funded by the Ministry of Education, Science and Technology (R31-2008-000-10103-01). Victor C. Yang is currently a Principal Investigator in the Department of Molecular Medicine and Biopharmaceutical Sciences, College of Medicine/College of Pharmacy, Seoul National University, South Korea. Beata Chertok was the recipient of Fred W. Lyons Jr and Rackham Pre-Doctoral Fellowships. Adam Cole was a recipient of a NIH Pharmacological Sciences and Bio-related Chemistry Training Program (GM007767 from NIGMS) grant and is currently an American Foundation for Pharmaceutical Education (AFPE) Pre-Doctoral Fellow. The authors would like to thank Dr. Christian Bergemann of Chemicell for his generosity in donating MNP and Roy Wentz in the University of Michigan Chemistry glass shop for assistance in working with ESR tubes.
REFERENCES
- 1.Pankhurst QA, Connolly J, Jones SK, Dobson J. Applications of magnetic nanoparticles in biomedicine. J. Phys. D: Appl. Phys. 2003;36(13):R167–R181. [Google Scholar]
- 2.Rodriguez I, Perez-Rial S, Gonzalez-Jimenez J, Perez-Sanchez JM, Herranz F, Beckmann N, Ruiz-Cabello J. Magnetic resonance methods and applications in pharmaceutical research. J. Pharm. Sci. 2008;97(9):3637–3665. doi: 10.1002/jps.21281. [DOI] [PubMed] [Google Scholar]
- 3.Weissleder R, Elizondo G, Wittenberg J, Rabito CA, Bengele HH, Josephson L. Ultrasmall superparamagnetic iron-oxide - characterization of a new class of contrast agents for MR imaging. Radiology. 1990;175(2):489–493. doi: 10.1148/radiology.175.2.2326474. [DOI] [PubMed] [Google Scholar]
- 4.Hong RY, Feng B, Chen LL, Liu GH, Li HZ, Zheng Y, Wei DG. Synthesis, characterization and MRI application of dextran-coated Fe3O4 magnetic nanoparticles. Biochem. Eng. J. 2008;42(3):290–300. [Google Scholar]
- 5.Hilger I, Hiergeist R, Hergt R, Winnefeld K, Schubert H, Kaiser WA. Thermal ablation of tumors using magnetic nanoparticles - An in vivo feasibility study. Invest. Radiol. 2002;37(10):580–586. doi: 10.1097/00004424-200210000-00008. [DOI] [PubMed] [Google Scholar]
- 6.Jordan A, Scholz R, Maier-Hauff K, van Landeghem FK, Waldoefner N, Teichgraeber U, Pinkernelle J, Bruhn H, Neumann F, Thiesen B, von Deimling A, Felix R. The effect of thermotherapy using magnetic nanoparticles on rat malignant glioma. J. Neurooncol. 2006;78(1):7–14. doi: 10.1007/s11060-005-9059-z. [DOI] [PubMed] [Google Scholar]
- 7.Dobson J. Magnetic nanoparticles for drug delivery. Drug Dev. Res. 2006;67(1):55–60. [Google Scholar]
- 8.Jain TK, Richey J, Strand M, Leslie-Pelecky DL, Flask CA, Labhasetwar V. Magnetic nanoparticles with dual functional properties: drug delivery and magnetic resonance imaging. Biomaterials. 2008;29(29):4012–21. doi: 10.1016/j.biomaterials.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Medarova Z, Pham W, Farrar C, Petkova V, Moore A. In vivo imaging of siRNA delivery and silencing in tumors. Nat. Med. 2007;13(3):372–7. doi: 10.1038/nm1486. [DOI] [PubMed] [Google Scholar]
- 10.Chertok B, Moffat BA, David AE, Yu F, Bergemann C, Ross BD, Yang VC. Iron oxide nanoparticles as a drug delivery vehicle for MRI monitored magnetic targeting of brain tumors. Biomaterials. 2008;29(4):487–96. doi: 10.1016/j.biomaterials.2007.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alexiou C, Arnold W, Hulin P, Klein RJ, Renz H, Parak F. G. n., Bergemann C, Ļbbe AS. Magnetic mitoxantrone nanoparticle detection by histology, X-ray and MRI after magnetic tumor targeting. J. Magn. Magn. Mater. 2001;225(12):187–193. [Google Scholar]
- 12.McCarthy JR, Weissleder R. Multifunctional magnetic nanoparticles for targeted imaging and therapy. Adv. Drug Del. Rev. 2008;60(11):1241–1251. doi: 10.1016/j.addr.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65(12):271–84. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 14.Corot C, Robert P, Idee JM, Port M. Recent advances in iron oxide nanocrystal technology for medical imaging. Adv. Drug Del. Rev. 2006;58(14):1471–1504. doi: 10.1016/j.addr.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 15.Larsen EKU, Nielsen T, Wittenborn T, Birkedal H, Vorup-Jensen T, Jakobsen MH, Ostergaard L, Horsman MR, Besenbacher F, Howard KA, Kjems J. Size-Dependent Accumulation of PEGylated Silane-Coated Magnetic Iron Oxide Nanoparticles in Murine Tumors. ACS Nano. 2009;3(7):1947–1951. doi: 10.1021/nn900330m. [DOI] [PubMed] [Google Scholar]
- 16.Marchal G, Vanhecke P, Demaerel P, Decrop E, Kennis C, Baert AL, Vanderschueren E. Detection of liver metastases with superparamagnetic iron-oxide in 15 patients - results of MR imaging at 1.5-T. Am. J. Roentgenol. 1989;152(4):771–775. doi: 10.2214/ajr.152.4.771. [DOI] [PubMed] [Google Scholar]
- 17.Weissleder R, Stark DD, Engelstad BL, Bacon BR, Compton CC, White DL, Jacobs P, Lewis J. Superparamagnetic iron-oxide - pharmacokinetics and toxicity. Am. J. Roentgenol. 1989;152(1):167–173. doi: 10.2214/ajr.152.1.167. [DOI] [PubMed] [Google Scholar]
- 18.Goya GF, Grazu V, Ibarra MR. Magnetic nanoparticles for cancer therapy. Current Nanoscience. 2008;4(1):1–16. [Google Scholar]
- 19.Crichton RR, Wilmet S, Legssyer R, Ward RJ. Molecular and cellular mechanisms of iron homeostasis and toxicity in mammalian cells. J. Inorg. Biochem. 2002;91(1):9–18. doi: 10.1016/s0162-0134(02)00461-0. [DOI] [PubMed] [Google Scholar]
- 20.Bourrinet P, Bengele HH, Bonnemain B, Dencausse A, Idee JM, Jacobs PM, Lewis JM. Preclinical safety and pharmacokinetic profile of ferumoxtran-10, an ultrasmall superparamagnetic iron oxide magnetic resonance contrast agent. Invest. Radiol. 2006;41(3):313–324. doi: 10.1097/01.rli.0000197669.80475.dd. [DOI] [PubMed] [Google Scholar]
- 21.Kou G, Wang SH, Cheng CM, Gao J, Li BH, Wang H, Qian WZ, Hou S, Zhang DP, Dai JX, Gu HC, Guo YJ. Development of SM5-1-conjugated ultrasmall superparamagnetic iron oxide nanoparticles for hepatoma detection. Biochem. Biophys. Res. Commun. 2008;374(2):192–197. doi: 10.1016/j.bbrc.2008.06.126. [DOI] [PubMed] [Google Scholar]
- 22.Nataraian A, Gruettner C, Ivkov R, DeNardo GL, Mirick G, Yuan A, Foreman A, DeNardo SJ. NanoFerrite particle based radioimmunonanoparticles: Binding affinity and in vivo pharmacokinetics. Bioconjugate Chem. 2008;19(6):1211–1218. doi: 10.1021/bc800015n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun C, Sze R, Zhang MQ. Folic acid-PEG conjugated superparamagnetic nanoparticles for targeted cellular uptake and detection by MRI. J. Biomed. Mater. Res. Part A. 2006;78A(3):550–557. doi: 10.1002/jbm.a.30781. [DOI] [PubMed] [Google Scholar]
- 24.Moroz P, Pardoe H, Jones SK, St Pierre TG, Song S, Gray BN. Arterial embolization hyperthermia: hepatic iron particle distribution and its potential determination by magnetic resonance imaging. PMB. 2002;47(9):1591–1602. doi: 10.1088/0031-9155/47/9/312. [DOI] [PubMed] [Google Scholar]
- 25.Brillet PY, Gazeau F, Luciani A, Bessoud B, Cuenod CA, Siauve N, Pons JN, Poupon J, Clement O. Evaluation of tumoral enhancement by superparamagnetic iron oxide particles: comparative studies with ferumoxtran and anionic iron oxide nanoparticles. Eur. Radiol. 2005;15(7):1369–1377. doi: 10.1007/s00330-004-2586-8. [DOI] [PubMed] [Google Scholar]
- 26.Simberg D, Duza T, Park JH, Essler M, Pilch J, Zhang LL, Derfus AM, Yang M, Hoffman RM, Bhatia S, Sailor MJ, Ruoslahti E. Biomimetic amplification of nanoparticle homing to tumors. Proc. Natl. Acad. Sci. U. S. A. 2007;104(3):932–936. doi: 10.1073/pnas.0610298104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meng XX, Wan JQ, Jing M, Zhao SG, Cai W, Liu EZ. Specific targeting of gliomas with multifunctional superparamagnetic iron oxide nanoparticle optical and magnetic resonance imaging contrast agents. Acta Pharmacol Sin. 2007;28(12):2019–2026. doi: 10.1111/j.1745-7254.2007.00661.x. [DOI] [PubMed] [Google Scholar]
- 28.Xie J, Chen K, Lee HY, Xu CJ, Hsu AR, Peng S, Chen XY, Sun SH. Ultrasmall c(RGDyK)-coated Fe3O4 nanoparticles and their specific targeting to integrin alpha(v)beta(3)-rich tumor cells. J. Am. Chem. Soc. 2008;130(24):7542. doi: 10.1021/ja802003h. + [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wuang SC, Neoh KG, Kang ET, Pack DW, Leckband DE. Heparinized magnetic nanoparticles: In-vitro assessment for biomedical applications. Adv. Funct. Mater. 2006;16(13):1723–1730. [Google Scholar]
- 30.Huang B-R, Chen P-Y, Huang C-Y, Jung S-M, Ma Y-H, Wu T, Chen J-P, Wei K-C. Bioavailability of magnetic nanoparticles to the brain. J. Magn. Magn. Mater. 2009;321(10):1604–1609. [Google Scholar]
- 31.Gamarra L, Pontusckha W, Amaro E, Costa-Filho A, Brito G, Vieira E, Carneiro S, Escriba D, Falleiros A, Salvador V. Kinetics of elimination and distribution in blood and liver of biocompatible ferrofluids based on Fe3O4 nanoparticles: An EPR and XRF study. Mater. Sci. Eng., C. 2008;28:519–525. [Google Scholar]
- 32.Slawska-Waniewska A, Mosiniewicz-Szablewska E, Nedelko N, Galazka-Friedman J, Friedman A. Magnetic studies of iron-entities in human tissues. J. Magn. Magn. Mater. 2004;272-276(Part 3):2417–2419. [Google Scholar]
- 33.Mykhaylyk O, Cherchenko A, Ilkin A, Dudchenko N, Ruditsa V, Novoseletz M, Zozulya Y. Glial brain tumor targeting of magnetite nanoparticles in rats. J. Magn. Magn. Mater. 2001;225(12):241–247. [Google Scholar]
- 34.Mykhaylyk O, Dudchenko N, Dudchenko A. Doxorubicin magnetic conjugate targeting upon intravenous injection into mice: High gradient magnetic field inhibits the clearance of nanoparticles from the blood. J. Magn. Magn. Mater. 2005;293(1):473–482. [Google Scholar]
- 35.Ross BD, Zhao YJ, Neal ER, Stegman LD, Ercolani M, Ben-Yoseph O, Chenevert TL. Contributions of cell kill and posttreatment tumor growth rates to the repopulation of intracerebral 9L tumors after chemotherapy: an MRI study. Proc. Natl. Acad. Sci. U. S. A. 1998;95(12):7012–7. doi: 10.1073/pnas.95.12.7012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koseoglu Y, Aktas B. ESR studies on superparamagnetic Fe3O4 nanoparticles. Physica Stat. Solidi (c) 2004;1(12):3516–3520. [Google Scholar]
- 37.Mykhaylyk OR, Dudchenko AK, Pankratov YV, Dobrinsky EK, Sosnitsky VN, Bakai EA. Use of ESR, Mossbauer spectroscopy,and squid magnetometry for the characterization of magnetic nanoparticles on the base of metal iron and its implications in vivo. In: Hafeli U, editor. Scientific and clinical applications of magnetic carriers. Plenum Press; New York: 1997. pp. 177–204. ON. [Google Scholar]
- 38.Shuter SL, Davies MJ, Garlick PB, Hearse DJ, Slater TF. Myocardial Tissue Preparation for ESR Spectroscopy: Some Methods May Cause Artifactual Generation of Signals. Free Radical Res. 1990;9(1):55–63. doi: 10.3109/10715769009148573. [DOI] [PubMed] [Google Scholar]
- 39.Mulsch A, Mordvintcev P, Vanin A. Quantification of nitric oxide in biological samples by electron spin resonance spectroscopy. Neuroprotocols. 1992;1(2):165–173. [Google Scholar]
- 40.Lazaro FJ, Gutierrez L, Abadia AR, Romero MS, Lopez A, Munoz MJ. Whole tissue AC susceptibility after superparamagnetic iron oxide contrast agent administration in a rat model. J. Magn. Magn. Mater. 2007;311(1):460–463. [Google Scholar]