

Abstract
High-intensity interval training (HIT) induces skeletal muscle metabolic and performance adaptations that resemble traditional endurance training despite a low total exercise volume. Most HIT studies have employed ‘all out’, variable-load exercise interventions (e.g. repeated Wingate tests) that may not be safe, practical and/or well tolerated by certain individuals. Our purpose was to determine the performance, metabolic and molecular adaptations to a more practical model of low-volume HIT. Seven men (21 ± 0.4 years, 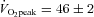 ml kg−1 min−1) performed six training sessions over 2 weeks. Each session consisted of 8–12 × 60 s intervals at ∼100% of peak power output elicited during a ramp
ml kg−1 min−1) performed six training sessions over 2 weeks. Each session consisted of 8–12 × 60 s intervals at ∼100% of peak power output elicited during a ramp 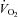 peak test (355 ± 10 W) separated by 75 s of recovery. Training increased exercise capacity, as assessed by significant improvements on both 50 kJ and 750 kJ cycling time trials (P < 0.05 for both). Skeletal muscle (vastus lateralis) biopsy samples obtained before and after training revealed increased maximal activity of citrate synthase (CS) and cytochrome c oxidase (COX) as well as total protein content of CS, COX subunits II and IV, and the mitochondrial transcription factor A (Tfam) (P < 0.05 for all). Nuclear abundance of peroxisome proliferator-activated receptor γ co-activator 1α (PGC-1α) was ∼25% higher after training (P < 0.05), but total PGC-1α protein content remained unchanged. Total SIRT1 content, a proposed activator of PGC-1α and mitochondrial biogenesis, was increased by ∼56% following training (P < 0.05). Training also increased resting muscle glycogen and total GLUT4 protein content (both P < 0.05). This study demonstrates that a practical model of low volume HIT is a potent stimulus for increasing skeletal muscle mitochondrial capacity and improving exercise performance. The results also suggest that increases in SIRT1, nuclear PGC-1α, and Tfam may be involved in coordinating mitochondrial adaptations in response to HIT in human skeletal muscle.
peak test (355 ± 10 W) separated by 75 s of recovery. Training increased exercise capacity, as assessed by significant improvements on both 50 kJ and 750 kJ cycling time trials (P < 0.05 for both). Skeletal muscle (vastus lateralis) biopsy samples obtained before and after training revealed increased maximal activity of citrate synthase (CS) and cytochrome c oxidase (COX) as well as total protein content of CS, COX subunits II and IV, and the mitochondrial transcription factor A (Tfam) (P < 0.05 for all). Nuclear abundance of peroxisome proliferator-activated receptor γ co-activator 1α (PGC-1α) was ∼25% higher after training (P < 0.05), but total PGC-1α protein content remained unchanged. Total SIRT1 content, a proposed activator of PGC-1α and mitochondrial biogenesis, was increased by ∼56% following training (P < 0.05). Training also increased resting muscle glycogen and total GLUT4 protein content (both P < 0.05). This study demonstrates that a practical model of low volume HIT is a potent stimulus for increasing skeletal muscle mitochondrial capacity and improving exercise performance. The results also suggest that increases in SIRT1, nuclear PGC-1α, and Tfam may be involved in coordinating mitochondrial adaptations in response to HIT in human skeletal muscle.
Introduction
Endurance exercise training induces numerous morphological and metabolic adaptations in skeletal muscle, including mitochondrial biogenesis and an enhanced capacity to oxidize fuels such as glucose and fats (Holloszy, 1967; Holloszy & Booth, 1976). These adaptations to exercise training have significant scientific and clinical relevance, as increased physical activity is linked with improved metabolic health and reduced risk for many chronic disorders, including obesity, insulin resistance and type 2 diabetes (Warburton et al. 2006; Hawley, 2004). We (Burgomaster et al. 2005, 2006, 2007, 2008; Gibala et al. 2006) and others (Parra et al. 2000; Babraj et al. 2009) have demonstrated that high-intensity interval training (HIT) induces numerous physiological adaptations that resemble traditional endurance training, despite a low total exercise volume. For example, 2 weeks of HIT was similar to 2 weeks of endurance training in leading to increases in exercise performance as well as the maximal activity and protein content of the mitochondrial enzyme cytochrome c oxidase (COX) (Gibala et al. 2006). Low-volume HIT has also been shown to promote improvements in markers of metabolic control and vascular endothelial function which are comparable to endurance training (Burgomaster et al. 2008; Rakobowchuk et al. 2008). What is most intriguing about these findings is that the volume of exercise and time spent training were ∼90% and ∼75% lower, respectively, with HIT compared to ET. This suggests that HIT is a potent and time-efficient strategy to induce skeletal muscle metabolic adaptations and improve functional exercise capacity. Given that ‘lack of time’ is the most commonly cited barrier to performing regular exercise in a variety of populations (Godin et al. 1994; Trost et al. 2002), low-volume HIT may represent an alternative to endurance training to improve metabolic health and reduce the risk for chronic diseases.
Our previous studies examining adaptations to low-volume HIT have used a training protocol that consists of repeated ‘all out’ maximal cycling efforts (i.e. repeated Wingate tests). This type of training is extremely demanding and requires a specialized cycle ergometer, and thus may not be safe or practical for some individuals. In light of this, our primary purpose in this study was to examine the exercise performance and muscle metabolic adaptations to a more practical model of HIT that is nonetheless still time efficient. Specifically, we kept total training time low in the present study (<30 min per session), decreased the absolute intensity of the intervals but increased interval duration, and reduced the recovery period between intervals. Despite these modifications, the training was still relatively ‘time efficient’ in that only ∼10–15 min of exercise was performed over a 20–30 min period during each training session. Other groups have shown that lower intensity HIT may be effective for inducing metabolic adaptations (Talanian et al. 2007; Perry et al. 2008), but the protocols involved interval training sessions lasting ≥ 60 min in duration. We hypothesized that our training model would provide a sufficient stimulus to improve muscle oxidative capacity and functional exercise performance, similar to that observed after our all-out Wingate-based training protocols (Burgomaster et al. 2005, 2006, 2007, 2008; Gibala et al. 2006). In addition, we also examined the effect of low-volume HIT on several proposed mediators of mitochondrial biogenesis and metabolic adaptation in skeletal muscle, including peroxisome proliferator-activated receptor γ co-activator (PGC)-1α, silent information regulator T1 (SIRT1), nuclear respiratory factor (NRF)-1, and mitochondrial transcription factor A (Tfam).
Methods
Subjects
Seven healthy young men (21 ± 1 years, 83 ± 4 kg) took part in the study. The subjects were recreationally active 2–3 times per week but none were engaged in a structured exercise training programme. The study was approved by the Hamilton Health Sciences/Faculty of Health Sciences Research Ethics Board and conformed to the Declaration of Helsinki. Following medical screening to rule out any conditions that might have precluded their participation, all subjects provided written informed consent.
Experimental protocol
The experimental protocol consisted of familiarization procedures, baseline testing, a 2 week exercise training intervention, and post-training measurements.
Familiarization
Subjects initially performed an incremental cycling test to volitional fatigue on an electronically braked cycle ergometer (Lode Excalibur v2.0, Groningen, the Netherlands) to determine peak oxygen uptake (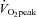 ) using an online gas collection system (Moxus modular oxygen uptake system, AEI technologies, Pittsburgh, PA, USA). Subjects began pedalling at 50 W for 2 min and the workload was increased by 1 W every 2 s thereafter until volitional fatigue. Mean
) using an online gas collection system (Moxus modular oxygen uptake system, AEI technologies, Pittsburgh, PA, USA). Subjects began pedalling at 50 W for 2 min and the workload was increased by 1 W every 2 s thereafter until volitional fatigue. Mean 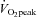 , based on the highest value averaged over 30 s for each subject, was 46 ± 2 ml kg−1 min−1. On two separate days, subjects performed 50 kJ and 750 kJ cycling time trials on an electronically braked ergometer to become familiar with these performance tests. For these familiarization tests, conditions were identical to baseline and post-testing time trials. The ergometer was set to linear mode so that resistance increased proportional to cadence and force. Subjects were instructed to complete the tests as quickly as possible with no verbal, temporal or physiological feedback as previously described (Gibala et al. 2006). The only feedback provided was work completed, which was presented to subjects as ‘distance covered’ on a computer monitor. Feedback was presented in units of distance rather than work completed (i.e. 50 kJ was equated to 2 km, and 750 kJ was equated to 30 km). Exercise duration and average power were recorded upon completion of each test.
, based on the highest value averaged over 30 s for each subject, was 46 ± 2 ml kg−1 min−1. On two separate days, subjects performed 50 kJ and 750 kJ cycling time trials on an electronically braked ergometer to become familiar with these performance tests. For these familiarization tests, conditions were identical to baseline and post-testing time trials. The ergometer was set to linear mode so that resistance increased proportional to cadence and force. Subjects were instructed to complete the tests as quickly as possible with no verbal, temporal or physiological feedback as previously described (Gibala et al. 2006). The only feedback provided was work completed, which was presented to subjects as ‘distance covered’ on a computer monitor. Feedback was presented in units of distance rather than work completed (i.e. 50 kJ was equated to 2 km, and 750 kJ was equated to 30 km). Exercise duration and average power were recorded upon completion of each test.
Baseline testing
Prior to training, a resting needle muscle biopsy sample was obtained from the vastus lateralis of one thigh under local anaesthesia (1% xylocaine) as previously described (Gibala et al. 2006). The muscle sample was immediately frozen in liquid nitrogen and stored at −80°C until further analyses. One hour following the biopsy, subjects performed a 50 kJ cycling time trial, followed ∼48 h later by a 750 kJ time trial.
Training
Three days following the 750 kJ time trial, subjects initiated the training protocol, which consisted of six sessions over 2 weeks (Monday, Wednesday, Friday). Each training session consisted of repeated 60 s efforts of high-intensity cycling at a workload that corresponded to the peak power achieved at the end of the ramp 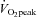 test (355 ± 10 W). These intervals were interspersed by 75 s of cycling at a low intensity (30 W) for recovery. Subjects completed eight high-intensity intervals during the first two training sessions, 10 intervals during the third and fourth sessions, and 12 intervals on the final two sessions. A 3 min warm-up at 30 W was performed each day prior to training. The total time commitment during each training session therefore ranged from ∼20 to 29 min, including warm-up and recovery, for a total commitment of 2 h and 25 min of exercise over 2 weeks. All subjects completed all training sessions without complications. We did not specifically evaluate psychosocial variables of exercise tolerance. However, the modified HIT protocol was generally well tolerated and subjects did not report any feelings of dizziness, light-headedness, or nausea that is occasionally experienced by subjects after Wingate-based training.
test (355 ± 10 W). These intervals were interspersed by 75 s of cycling at a low intensity (30 W) for recovery. Subjects completed eight high-intensity intervals during the first two training sessions, 10 intervals during the third and fourth sessions, and 12 intervals on the final two sessions. A 3 min warm-up at 30 W was performed each day prior to training. The total time commitment during each training session therefore ranged from ∼20 to 29 min, including warm-up and recovery, for a total commitment of 2 h and 25 min of exercise over 2 weeks. All subjects completed all training sessions without complications. We did not specifically evaluate psychosocial variables of exercise tolerance. However, the modified HIT protocol was generally well tolerated and subjects did not report any feelings of dizziness, light-headedness, or nausea that is occasionally experienced by subjects after Wingate-based training.
Post-training measurements
Approximately 72 h following the last training session, a resting muscle biopsy was obtained from the same leg as used for the first biopsy, separated by 2–3 cm from the original incision site. Subjects also performed 50 and 750 kJ time trials, the nature and timing of which were identical in all respects to baseline testing.
Muscle analyses
The biopsy samples were initially sectioned under liquid nitrogen into several pieces for analyses of enzyme activity and protein content as detailed below. One piece was freeze-dried, powdered and dissected free of all non-muscle elements for analyses of muscle glycogen (see below).
Preparation of whole muscle lysates
Approximately 30–40 mg of frozen wet muscle was homogenized on ice in 25 volumes of buffer (70 mm sucrose, 220 mm mannitol, 10 mm Hepes) supplemented with protease inhibitors (Complete Mini, Roche Applied Science, Laval, PQ, Canada) using 50 strokes of a glass-on-glass homogenizer. Homogenates were centrifuged at 700 g for 10 min and the supernatant was taken as the whole muscle lysate for enzyme activity assays and Western blotting. Homogenates were subjected to two freeze–thaw cycles to help release mitochondrial proteins prior to enzyme activity measurement.
Preparation of nuclear extracts
Nuclear fractions were prepared from 30–40 mg of wet muscle using a commercially available nuclear extraction kit (NE-PER no. 78833, Pierce, Rockford, IL, USA). Briefly, samples were homogenized in CER-I buffer containing protease inhibitors using an electronic homogenizer (Pro 250, Pro Scientific, Oxford, CT, USA). Pellets containing nuclei were obtained by centrifugation at 16,000 g for 10 min at 4°C and were subsequently washed four times in PBS to remove cytosolic contaminating proteins. Nuclear proteins were extracted in nuclear extraction reagent (NER) supplemented with protease inhibitors according to the manufacturer's instructions. Enrichment and purity of nuclear fractions was confirmed by the abundance of nuclear matrix protein p84 and absence of the cytosolic protein lactate dehydrogenase (LDH) in Western blot analyses (Wright et al. 2007) (Supplementary Fig. 1).
Mitochondrial enzyme activity
The maximal activities of citrate synthase (CS) and cytochrome c oxidase (COX) were determined in whole muscle lysates using a spectrophotometer (Cary Bio-300, Varion, Inc., Palo Alto, CA, USA) as we have previously described (Gianni et al. 2004). Enzyme activities were expressed in mmol (kg protein)−1 h−1 wet weight (w.w.).
Western blotting
Protein concentrations of whole muscle lysates and nuclear fractions were determined using a commercial assay (BCA Protein Assay, Pierce, Rockford, IL, USA). Equal amounts of protein (5–20 μg, depending on the protein of interest) were loaded onto 7.5–12.5% SDS-PAGE gels and separated by electrophoresis for 2–2.5 h at 100 V. Proteins were transferred to nitrocellulose membranes for 1 h at 100 V. Ponceau S staining was performed following the transfer and was used to control for equal loading and transfer between lanes. In order to verify the efficacy of this method we performed preliminary experiments which demonstrated a linear proportional relationship between protein loaded (5–20 μg) and whole lane Ponceau S quantification using NIH Image J software (R2= 0.99, n= 6, data not shown). Ponceau S quantification also did not differ between pre- and post-training samples (P= 0.88, data not shown). Membranes were blocked at room temperature (RT) by incubating in 5% fat-free milk Tris buffered saline 0.1% Tween-20 (TBS-T). Blots were then incubated with primary antibodies overnight at 4°C in 3% fat-free milk or fatty acid-free BSA. A rabbit monoclonal antibody from Cell Signaling Technology (Danvers, MA, USA) was used to detect PGC-1α. Polyclonal antibodies directed towards the C-terminus of SIRT1 and GLUT4 were from Millipore (Billerica, MA, USA). Tfam, NRF-1 and TATA box binding protein antibodies were from Abcam (Cambridge, MA, USA). COX subunit II and IV mouse monoclonal antibodies were from MitoSciences (Eugene, OR, USA). CS rabbit polyclonal antibody was a kind gift from Dr Brian Robinson (The Hospital for Sick Children, Toronto, ON, Canada). After incubation in appropriate secondary antibody for 1 h at RT, proteins were detected by chemiluminescence (Supersignal West Dura, Pierce) and quantified by spot densitometry using FluorChem SP Imaging system and software (Alpha Innotech Corp., San Leandro, CA, USA). TATA box binding protein was used to control for nuclear protein yield between pre- and post-training samples.
Muscle glycogen
Briefly, ∼2 mg of freeze-dried muscle was incubated in 500 μl 2.0 n HCl and heated for 2 h at 100°C to hydrolyse the glycogen to glucosyl units. The solution was subsequently neutralized with an equal volume of 2.0 n NaOH and analysed for glucose using an enzymatic assay adapted for fluorometry (Passoneau & Lowry, 1993).
Physical activity and nutritional controls
Subjects were instructed to maintain normal dietary and physical activity practices throughout the study. For 2 days prior to the biopsy procedures and exercise performance tests, subjects were asked to refrain from any exercise except for activities of daily living. In order to minimize variability in muscle metabolism attributable to diet, subjects were instructed to consume the same types and quantities of food for 2 days prior to the resting muscle biopsy and time trial tests. Subjects completed food diaries prior to baseline testing and these were collected, photocopied, and returned to subjects before post-training procedures so that individual diets could be replicated.
Statistical analyses
All data were analysed using Student's t test for paired data with significance set at P≤ 0.05 (Sigma Stat v3.10; Systat Software Inc., San Jose, CA, USA). All data is presented as mean ±s.d. unless otherwise indicated.
Results
Exercise performance
Time to complete the 50 kJ and 750 kJ time trials improved by 11% and 9%, respectively, after training (P= 0.04 and P= 0.005; Fig. 1). These changes were associated with significant increases in mean power output from 397 ± 27 to 436 ± 22 W in the 50 kJ test (P= 0.01) and 200 ± 7 to 221 ± 8 W in the 750 kJ test (P= 0.005). In order to confirm the effects of training, we also compared time trial performance on familiarization tests compared to baseline. Performance on the familiarization tests (∼14–16 days prior to training) were not different from pre-training values (50 kJ test: 129 ± 8 s vs. 130 ± 9 s, P= 0.89; 750 kJ test: 64 ± 3 min vs. 63 ± 2 min, P= 0.48).
Figure 1. Two weeks of high-intensity interval training improves cycling time trial performance.

A, time to complete 50 kJ cycling time trial before (pre) and after (post) training. B, time to complete 750 kJ cycling time trial before (pre) and after (post) training. *P < 0.05 vs. pre-training. Values are means ±s.e.m.
Mitochondrial enzymes
The maximal activity of COX increased by 29% after training (P= 0.02; Fig. 2A), which was similar to the 35% and 38% increase in protein content of COX subunits II (P= 0.01; Fig. 2B) and IV (P= 0.002; Fig. 2C), respectively. Training also increased the maximal activity and protein content of CS by ∼16% and 20%, respectively (both P= 0.01; Fig. 3).
Figure 2. High-intensity interval training increases activity and content of the mitochondrial enzyme cytochrome c oxidase.

A, maximal activity (mmol (kg protein)−1 h−1 w.w.) of cytochrome c oxidase (COX) measured in whole muscle homogenates prepared from muscle biopsy samples (v. lateralis) obtained before (pre) and after (post) training. B, protein content of COX subunit II before (pre) and after (post) training. C, protein content of COX subunit IV before (pre) and after (post) training. *P < 0.05 vs. pre-training. Values are means ±s.e.m. Representative Western blots from two subjects are shown.
Figure 3. High-intensity interval training increases activity and content of the mitochondrial enzyme citrate synthase.

A, maximal activity (mmol (kg protein)−1 h−1 w.w.) of citrate synthase (CS) measured in whole muscle homogenates prepared from muscle biopsy samples (v. lateralis) obtained before (pre) and after (post) training. B, protein content of CS before (pre) and after (post) training. *P < 0.05 vs. pre-training. Values are means ±s.e.m. Representative Western blots from two subjects are shown.
Regulators of mitochondrial biogenesis
The protein content of PGC-1α measured in nuclear fractions was elevated by ∼24% post-training (P= 0.02; Fig. 4A), but whole muscle PGC-1α protein was unchanged (P= 0.25; Fig. 4B). Total SIRT1 content increased by ∼56% following training (P= 0.03; Fig. 5). Tfam total protein content increased by ∼37% with training (P= 0.002; Fig. 6) whereas NRF-1 protein content remained unchanged (P= 0.52; data not shown).
Figure 4. High-intensity interval training increases nuclear but not whole muscle PGC-1α.

A, protein content of peroxisome proliferator-activated receptor γ co-activator (PGC)-1α measured in nuclear fractions prepared from muscle biopsy samples (v. lateralis) obtained before (pre) and after (post) training. B, protein content of PGC-1α measured in whole muscle homogenates before (pre) and after (post) training. *P < 0.05 vs. pre-training. Values are means ±s.e.m. PGC-1α antibody specificity is demonstrated in Supplementary Fig. 2. Representative Western blots from two subjects are shown.
Figure 5. High-intensity interval training increases SIRT1 protein content.

Protein content of SIRT1 measured in whole muscle homogenates prepared from muscle biopsy samples (v. lateralis) obtained before (pre) and after (post) training. *P < 0.05 vs. pre-training. Values are means ±s.e.m. Representative Western blots from two subjects are shown.
Figure 6. High-intensity interval training increases Tfam protein content.

Protein content of mitochondrial transcription factor A (Tfam) measured in whole muscle homogenates prepared from muscle biopsy samples (v. lateralis) obtained before (pre) and after (post) training. *P < 0.05 vs. pre-training. Values are means ±s.e.m. Representative Western blots from two subjects are shown.
To identify potential post-translational modifications which might promote or sustain the increase in nuclear PGC-1α, we attempted to immunoprecipitate PGC-1α and measure phosphorylation and/or acetylation status. Unfortunately, these attempts were unsuccessful using up to 150 μg of nuclear protein with both the Cell Signaling Technology antibody (no. 2178) and the H-300 antibody from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Immunoprecipitation of PGC-1α may require greater amounts of protein (Canto et al. 2009), which was not possible to obtain from nuclear fractions prepared from human muscle biopsy samples in the present study.
GLUT4 and muscle glycogen
Protein content of GLUT4 increased by 119% (P= 0.04; Fig. 7) and resting muscle glycogen increased by 17% following training (from 476.96 ± 29.28 to 558 ± 19 mmol (kg dry weight)−1, P= 0.05).
Figure 7. High-intensity interval training increases GLUT4 protein content.

Protein content of glucose transporter 4 (GLUT4) measured in whole muscle homogenates prepared from muscle biopsy samples (v. lateralis) obtained before (pre) and after (post) training. *P < 0.05 vs. pre-training. Values are means ±s.e.m. Representative Western blots from two subjects are shown.
Discussion
The present study demonstrates that 2 weeks of low-volume, constant-load interval exercise is a practical, time-efficient strategy to induce mitochondrial biogenesis in skeletal muscle and improve functional exercise capacity. The changes we observed in maximal enzyme activity, exercise performance, as well as resting muscle glycogen were comparable to changes we have previously measured after 2 weeks of ‘all out’ Wingate-type training, as well as a much larger volume of traditional endurance training (Burgomaster et al. 2005, 2006; Gibala et al. 2006). The present study also extends our previous work by providing mechanistic insight into the molecular events that potentially mediate the skeletal muscle adaptive response to HIT. To our knowledge, for the first time in humans we show that the nuclear abundance of PGC-1α was increased after exercise training. The protein content of SIRT1, an NAD+-dependent deacetylase which has been shown to deacetylate and activate PGC-1α (Cantóet al. 2009; Gerhart-Hines et al. 2007) was also elevated post-training, as was content of Tfam, a mitochondrial transcription factor which is induced by PGC-1α (Wu et al. 1999).
A practical model of HIT is a time-efficient training stimulus
We have previously shown that low-volume HIT consisting of 4–6 repeated 30 s ‘all-out’ Wingate cycling tests with 4 min recovery is an effective stimulus for improving mitochondrial enzyme capacity and exercise performance (Burgomaster et al. 2005, 2006, 2007, 2008; Gibala et al. 2006). Similar to the present study, Wingate-based HIT also leads to elevated levels of resting muscle glycogen (Burgomaster et al. 2005, 2006, 2008; Gibala et al. 2006). Although Wingate-based HIT is a very potent and time-efficient training strategy (Burgomaster et al. 2005, 2006, 2007, 2008; Gibala et al. 2006), training necessitates an all-out effort on a specialized cycle ergometer against a high resistance. This requires a high level of subject motivation and can result in feelings of nausea and discomfort due to the extreme physical exertion. For these reasons, Wingate-based HIT may not be practical or suitable for the general population (Coyle, 2005), especially obese individuals and older adults who may benefit most from the adaptations to interval exercise training.
The training protocol in the current study was designed to involve lower-intensity intervals at a constant workload without the need for specialized equipment. Interval intensity, duration and training volume were based on several a priori assumptions. In our previously published HIT studies, recreationally active males typically achieve a maximal aerobic power of ∼350 W during a ramp 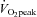 test and produce a mean power output of ∼700 W over each 30 s sprint interval (Burgomaster et al. 2005; Gibala et al. 2006). Thus, to closely match the amount of external work completed in a 30 s Wingate, intervals were set at 100% maximal aerobic power (i.e. ∼350 W) for 60 s. However, the relationship between training intensity and duration is not linear (Dudley et al. 1982). That is, simply matching work using a lower training intensity may not have the same training effect (Dudley et al. 1982). Therefore, the prescribed number of intervals per session was doubled compared to Wingate-based HIT (Burgomaster et al. 2005, 2006, 2007, 2008; Gibala et al. 2006) such that subjects performed 8–12 intervals at 100% maximal aerobic power, resulting in a concomitant doubling of total external energy expenditure to ∼210 kJ per training session. This volume of exercise is still considerably lower than traditional endurance-type training (Gibala et al. 2006; Burgomaster et al. 2008). In addition to a low training volume, one of the most attractive features of HIT is that metabolic and performance adaptations are achieved with a very low time commitment (Coyle, 2005; Gibala et al. 2006). In keeping with the time efficiency of HIT, total training time commitment was kept low in the present study. Training sessions involved only 8–12 min of exercise spread out over a ∼18–26 min training session. Therefore, total weekly time commitment averaged only ∼60 min. Given that lack of time is the number one perceived barrier to performing regular exercise (Godin et al. 1994; Trost et al. 2002), a low-volume HIT program similar to the one employed in this study may be a potent, practical, and time-efficient exercise strategy for increasing muscle mitochondrial content and functional exercise capacity. Since increases in skeletal muscle mitochondrial capacity and physical fitness are associated with improved metabolic health (Bruce et al. 2003; Booth & Roberts, 2008) low-volume HIT may represent an alternative exercise strategy for promoting important health benefits. Although we did not include a control group in the current investigation, there were clearly no differences when comparing exercise performance on tests conducted ∼2 weeks apart prior to the training intervention. In addition, we have previously established that the muscle metabolic and performance adaptations after short-term HIT are attributable to the training per se, since controls subjects show no change when tested 2 weeks apart with no exercise intervention (Burgomaster et al. 2005, 2006, 2007). While the current investigation was conducted in young, healthy male subjects, there is growing appreciation for the benefits of interval-based exercise training in several chronic disease states. For example, higher volume interval training at a similar intensity (4 × 4 min at 90% maximal heart rate interspersed with 3 min rest periods) has been shown to be more effective than traditional continuous endurance-type training for improving muscle metabolic parameters and clinical outcomes in patients with obesity (Schjerve et al. 2008), metabolic syndrome (Tjønna et al. 2008), and heart failure (Wisløff et al. 2007). Our findings indicate that the HIT protocol utilized in this study may lead to muscle and performance adaptations which are linked with improved metabolic health despite a lower volume and time commitment. It will be interesting to evaluate whether the current low-volume HIT protocol leads to health benefits in conditions of metabolic disease and we are currently conducting studies in this regard.
test and produce a mean power output of ∼700 W over each 30 s sprint interval (Burgomaster et al. 2005; Gibala et al. 2006). Thus, to closely match the amount of external work completed in a 30 s Wingate, intervals were set at 100% maximal aerobic power (i.e. ∼350 W) for 60 s. However, the relationship between training intensity and duration is not linear (Dudley et al. 1982). That is, simply matching work using a lower training intensity may not have the same training effect (Dudley et al. 1982). Therefore, the prescribed number of intervals per session was doubled compared to Wingate-based HIT (Burgomaster et al. 2005, 2006, 2007, 2008; Gibala et al. 2006) such that subjects performed 8–12 intervals at 100% maximal aerobic power, resulting in a concomitant doubling of total external energy expenditure to ∼210 kJ per training session. This volume of exercise is still considerably lower than traditional endurance-type training (Gibala et al. 2006; Burgomaster et al. 2008). In addition to a low training volume, one of the most attractive features of HIT is that metabolic and performance adaptations are achieved with a very low time commitment (Coyle, 2005; Gibala et al. 2006). In keeping with the time efficiency of HIT, total training time commitment was kept low in the present study. Training sessions involved only 8–12 min of exercise spread out over a ∼18–26 min training session. Therefore, total weekly time commitment averaged only ∼60 min. Given that lack of time is the number one perceived barrier to performing regular exercise (Godin et al. 1994; Trost et al. 2002), a low-volume HIT program similar to the one employed in this study may be a potent, practical, and time-efficient exercise strategy for increasing muscle mitochondrial content and functional exercise capacity. Since increases in skeletal muscle mitochondrial capacity and physical fitness are associated with improved metabolic health (Bruce et al. 2003; Booth & Roberts, 2008) low-volume HIT may represent an alternative exercise strategy for promoting important health benefits. Although we did not include a control group in the current investigation, there were clearly no differences when comparing exercise performance on tests conducted ∼2 weeks apart prior to the training intervention. In addition, we have previously established that the muscle metabolic and performance adaptations after short-term HIT are attributable to the training per se, since controls subjects show no change when tested 2 weeks apart with no exercise intervention (Burgomaster et al. 2005, 2006, 2007). While the current investigation was conducted in young, healthy male subjects, there is growing appreciation for the benefits of interval-based exercise training in several chronic disease states. For example, higher volume interval training at a similar intensity (4 × 4 min at 90% maximal heart rate interspersed with 3 min rest periods) has been shown to be more effective than traditional continuous endurance-type training for improving muscle metabolic parameters and clinical outcomes in patients with obesity (Schjerve et al. 2008), metabolic syndrome (Tjønna et al. 2008), and heart failure (Wisløff et al. 2007). Our findings indicate that the HIT protocol utilized in this study may lead to muscle and performance adaptations which are linked with improved metabolic health despite a lower volume and time commitment. It will be interesting to evaluate whether the current low-volume HIT protocol leads to health benefits in conditions of metabolic disease and we are currently conducting studies in this regard.
Molecular clues regarding the adaptive response to HIT
In order to gain a better understanding of the mechanisms by which low-volume HIT promotes the muscle adaptive response, we examined several regulators of mitochondrial biogenesis in resting muscle biopsy samples obtained before and after training. We first examined the total protein content of PGC-1α, a transcriptional co-activator which plays a crucial role in co-ordinating mitochondrial gene transcription (Lin et al. 2005; Hood & Saleem, 2007; Wright et al. 2007). A role for PGC-1α in the adaptive response to exercise training is highlighted by the findings that acute endurance (Pilegaard et al. 2003) and interval (Gibala et al. 2009) exercise increase skeletal muscle PGC-1α mRNA and that muscle-specific over-expression of PGC-1α in mice results in increased mitochondrial content and prolonged endurance exercise capacity (Calvo et al. 2008). Perhaps somewhat surprisingly, and in contrast to longer term endurance and/or interval training (Russell et al. 2003; Burgomaster et al. 2008; Morton et al. 2009), PGC-1α measured at the whole muscle level was unchanged following training. However, total PGC-1α protein content may not be entirely indicative of PGC-1α activation; rather PGC-1α activity may be primarily determined by its subcellular location (Rim et al. 2004; Wright et al. 2007; Cowell et al. 2007; Sano et al. 2007; Anderson et al. 2008; Olson et al. 2008) and several post-translational modifications (Knutti et al. 2001; Jäger et al. 2007; Cantóet al. 2009). Therefore, we prepared nuclear fractions from skeletal muscle biopsy samples and examined the nuclear abundance of PGC-1α to further examine the potential role of PGC-1α in the adaptive response. Results indicated that training resulted in a significant increase in nuclear PGC-1α protein. Thus, our novel findings suggest that the total amount of PGC-1α may not be affected by short-term training but that its subcellular location, an indicator of PGC-1α activation (Wright et al. 2007), may be altered such that more PGC-1α protein is present in the nucleus in the initial adaptive response to training. Greater nuclear PGC-1α would presumably be more conducive for promoting or maintaining an increase in mitochondrial biogenesis via increased co-activation of transcription factors linked to mitochondrial gene expression. Accordingly, we found increases in several classical markers of mitochondrial biogenesis, including the maximal activity and protein contents of CS and COX. Alternatively, measuring PGC-1α in nuclear fractions may be a more sensitive technique than measuring the content of this protein at the whole muscle level, increasing the ability to detect changes following training. Regardless, our findings suggest that subcellular location of PGC-1α may be an important consideration when examining the role of this critical regulator of mitochondrial biogenesis in response to exercise training.
We have recently demonstrated that acute Wingate-based HIT leads to activation of AMPK and p38 MAPK as well as increased mRNA expression of PGC-1α (Gibala et al. 2009). These responses are comparable to the effects of acute endurance exercise (Yu et al. 2001; Pilegaard et al. 2003; Lee-Young et al. 2008) and suggest that both training methods may act through similar pathways to induce mitochondrial biogenesis. The low-volume HIT protocol in the current study may therefore have promoted adaptation partly through repeated activation of signalling pathways which activate PGC-1α and mitochondrial biogenesis. Additional cellular factors promoting the increase in nuclear PGC-1α and mitochondrial biogenesis cannot be fully elucidated in the present study, but the training-induced increase in SIRT1 protein content may be involved. SIRT1 is a NAD+-dependent type III deacetylase which, in exercising mouse muscle and C2C12 myotubes, deacetylates PGC-1α and appears critical for PGC-1α activation (Cantóet al. 2009). In rats, it was recently shown that exercise training increases in SIRT1 protein in some muscles in the absence of increases in total PGC-1α (Suwa et al. 2008). These authors (Suwa et al. 2008) suggested that the increase in SIRT1 may serve to increase PGC-1α activation, circumventing a need to increase total PGC-1α content to drive mitochondrial biogenesis. Similarly, Canto and colleagues (2009) have recently shown that acute exercise activates PGC-1α and mitochondrial gene transcription via a pathway involving SIRT1-mediated deacetylation of PGC-1α. Data from human skeletal muscle is scarce, but SIRT1 and PGC-1α mRNA expression have been shown to increase in parallel following caloric restriction and caloric restriction combined with exercise, two interventions conclusively demonstrated to improve mitochondrial function (Civitarese et al. 2007). Our results seem to agree with these studies and may extend previous findings to suggest that an increase in SIRT1 protein, in human skeletal muscle, coincides with an increase in the nuclear abundance of PGC-1α. Together, our findings indicate that 2 weeks of low-volume HIT may induce skeletal muscle mitochondrial biogenesis by a mechanism involving co-ordinated induction and/or activation of SIRT1 and PGC-1α. The exact nature of SIRT1-mediated mitochondrial biogenesis has not been fully elucidated, but deacetylation of PGC-1α by SIRT1 may increase PGC-1α nuclear abundance, sequester or stabilize PGC-1α in the nucleus, or influence formation of different nuclear PGC-1α protein complexes (Rodgers et al. 2008; Cantóet al. 2009; Cantó & Auwerx, 2009). However, it must be noted that total SIRT1 protein content may not be entirely reflective of its deacetylase activity; rather SIRT1 activity may be primarily regulated by metabolic factors such as the NAD+:NADH ratio, intracellular nicotinamide levels, and several post-translational modifications (Cantó & Auwerx, 2009). Nonetheless, SIRT1 activity and protein content are highly correlated (Gurd et al. 2009) and an increase in total SIRT1 protein would theoretically increase the potential for interaction with its targets, such as PGC-1α.
Although SIRT1 has been primarily recognized as a positive regulator of mitochondrial biogenesis (Rodgers et al. 2005; Civitarese et al. 2007; Canto et al. 2009; Gerhart-Hines et al. 2007; Suwa et al. 2008), recent data from Gurd and colleagues (2009) has challenged this view. Electrotransfection of the SIRT1 gene into an isolated hindlimb muscle of rats increased SIRT1 protein content by ∼250% and was accompanied by a decrease in PGC-1α and mitochondrial protein content (Gurd et al. 2009). Thus, SIRT1 may not always be associated with increased mitochondrial biogenesis. It is possible that the large increase in SIRT1 protein following electrotransfection produces different effects from the more modest increase in SIRT1 protein following training. Furthermore, although the electrotransfection technique is a powerful approach for increasing SIRT1 protein in an isolated muscle, it may not be representative of the in vivo metabolic environment which promotes the adaptive response to exercise training. Clearly, more research is required to clarify the effects of exercise training and other manipulations on SIRT1 protein and mitochondrial biogenesis in skeletal muscle in vivo.
It is well established that exercise training results in an increase in mitochondrial content in skeletal muscle (Holloszy, 1967; Holloszy & Booth, 1976), yet the underlying mechanisms directing this adaptive response are not completely understood. A functional increase in mitochondrial content requires complex co-ordination of genes encoded by nuclear and mitochondrial DNA (Scarpulla, 2006, 2008). PGC-1α and Tfam appear to play integral roles regulating transcription of mitochondrial genes in the nucleus and mitochondria, respectively (Hood et al. 2006; Scarpulla, 2006, 2008). Our results suggest that an increase in nuclear PGC-1α and total Tfam protein content following training may play integral roles in co-ordinating the expression of mitochondrial proteins to drive an increase in mitochondrial biogenesis. Indeed, we found that COX subunit IV (nuclear encoded) and COX subunit II (mitochondrial encoded) were both increased following training, as was COX maximal activity. Further supporting a role for a co-ordinated increase in mitochondrial biogenesis was the finding that the protein content and maximal activity of the TCA cycle enzyme CS was increased with training. The training-induced increase in muscle oxidative capacity is likely to have played a role in improving exercise capacity as it would presumably allow subjects to exercise at a higher metabolic rate during the cycling time trials following training. Given the link between muscle oxidative capacity and insulin sensitivity (Bruce et al. 2003) and the important role that skeletal muscle mitochondria play in regulating whole body metabolism (Handschin et al. 2007), the adaptations promoted by low-volume HIT may also have important health implications. Interestingly, Babraj et al. (2009) have recently shown that 2 weeks of Wingate-based HIT improves insulin sensitivity in a sample of healthy young male subjects similar to the present study.
Despite the fact that we did not directly measure insulin sensitivity, the increase in muscle glycogen and GLUT4 following training provides some evidence that muscle insulin sensitivity may have been improved. Increases in resting muscle glycogen following training are common and have been linked with increased insulin sensitivity (Perseghin et al. 1996; Greiwe et al. 1999) and/or increased glucose transport capacity as a result of increased GLUT4 content (McCoy et al. 1996). Although less studied than its role in mitochondrial biogenesis, PGC-1α appears to be involved in regulating skeletal muscle glycogen content (Wende et al. 2007; Calvo et al. 2008). Over-expression of PGC-1α in mice increases resting muscle glycogen content (Calvo et al. 2008) and PGC-1α deficient animals have impaired muscle glycogen resynthesis following exercise (Wende et al. 2007). The regulation of skeletal muscle glycogen content by PGC-1α is not completely understood, but may involve PGC-1α mediated up-regulation of GLUT4 (Michael et al. 2001) and/or modulation of enzymes involved in glycogen synthesis and degradation (Wende et al. 2007). Therefore, the training induced increase in nuclear PGC-1α may have played a role in mediating the increase in GLUT4 and muscle glycogen.
Conclusions and significance
Previous low-volume HIT studies (Parra et al. 2000; Burgomaster et al. 2005, 2006, 2007, 2008; Gibala et al. 2006; Babraj et al. 2009) have utilized all-out exercise intervals on a specialized cycle ergometer and our findings are unique in that the HIT protocol was designed to be more practical and attainable for the general population. Six sessions of eight to twelve 60 s constant load intervals, completed in ∼18–26 min per session and requiring ∼1 h of total exercise time commitment per week resulted in significant improvements in functional exercise performance and skeletal muscle mitochondrial biogenesis. Although the molecular mechanisms promoting the muscle adaptive response could not be fully elucidated, we found changes in several proposed regulators of mitochondrial biogenesis following training. Resting levels of SIRT1 and Tfam were higher after training, as was the nuclear abundance of PGC-1α. Together, the results demonstrate that a practical low-volume HIT programme is effective for improving muscle metabolic capacity and functional performance and shed light on potential mechanisms by which exercise training promotes mitochondrial adaptations in skeletal muscle. Future research should examine whether practical low-volume HIT can improve markers of metabolic health in healthy individuals and those at risk of developing chronic inactivity-related diseases to determine whether this type of training is an effective health-enhancing exercise strategy.
Acknowledgments
The authors would like to thank Drs Kirsten Burgomaster and Krista Howarth for assistance with muscle glycogen measurements. We also thank Dr David Hood for providing skeletal muscle from PGC-1α knock-out mice. This work was funded by the Natural Sciences and Engineering Research Council (NSERC) of Canada. J.P.L. is supported by an NSERC Canada Graduate Scholarship. A.S. is supported by a Canadian Institutes of Health Research (CIHR) doctoral research award. N.C. is supported by an NSERC Postgraduate Scholarship (PGS-D).
Glossary
Abbreviations
- CS
citrate synthase
- ET
endurance training
- LDH
lactate dehydrogenase
- NRF-1
nuclear respiratory factor 1
- HIT
high-intensity interval training
- p38 MAPK
p38 mitogen activated protein kinase
- PGC-1α
peroxisome proliferator-activated receptor γ co-activator 1α
- SIRT1
sirtuin 1
- Tfam
mitochondrial transcription factor A
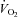
oxygen uptake
Author contributions
M.J.G., J.P.L., G.P.W. and M.A.T. conceived the study design. G.P.W. ran the exercise testing and supervised the training sessions. M.A.T. performed all the medical procedures. J.P.L. and A.S. performed the laboratory experiments and analysed the data. J.P.L., A.S. and M.J.G. interpreted the data and wrote the manuscript with input from all authors. Testing and experiments were performed in the laboratories of M.J.G. and M.A.T at McMaster University.
Supplemental material
Supplementary Fig. 1
Supplementary Fig. 2
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Anderson RM, Barger JL, Edwards MG, Braun KH, O’Connor CE, Prolla TA, Weindruch R. Dynamic regulation of PGC-1α localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell. 2008;7:101–111. doi: 10.1111/j.1474-9726.2007.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babraj JA, Vollaard NB, Keast C, Guppy FM, Cottrell G, Timmons JA. Extremely short duration high intensity interval training substantially improves insulin action in young healthy males. BMC Endocr Disord. 2009;9:3. doi: 10.1186/1472-6823-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth FW, Roberts CK. Linking performance and chronic disease risk: indices of physical performance are surrogates for health. Br J Sports Med. 2008;42:950–952. doi: 10.1136/bjsm.2008.052589. [DOI] [PubMed] [Google Scholar]
- Bruce CR, Anderson MJ, Carey AL, Newman DG, Bonen A, Kriketos AD, Cooney GJ, Hawley JA. Muscle oxidative capacity is a better predictor of insulin sensitivity than lipid status. J Clin Endocrinol Metab. 2003;88:5444–5451. doi: 10.1210/jc.2003-030791. [DOI] [PubMed] [Google Scholar]
- Burgomaster KA, Howarth KR, Phillips SM, Rakobowchuk M, Macdonald MJ, McGee SL, Gibala MJ. Similar metabolic adaptations during exercise after low volume sprint interval and traditional endurance training in humans. J Physiol. 2008;586:151–160. doi: 10.1113/jphysiol.2007.142109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgomaster KA, Cermak NM, Phillips SM, Benton CR, Bonen A, Gibala MJ. Divergent response of metabolite transport proteins in human skeletal muscle after sprint interval training and detraining. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1970–R1976. doi: 10.1152/ajpregu.00503.2006. [DOI] [PubMed] [Google Scholar]
- Burgomaster KA, Heigenhauser GJF, Gibala MJ. Effect of short-term sprint interval training on human skeletal muscle carbohydrate metabolism during exercise and time-trial performance. J Appl Physiol. 2006;100:2041–2047. doi: 10.1152/japplphysiol.01220.2005. [DOI] [PubMed] [Google Scholar]
- Burgomaster KA, Hughes SC, Heigenhauser GJF, Bradwell SN, Gibala MJ. Six sessions of sprint interval training increases muscle oxidative potential and cycle endurance capacity in humans. J Appl Physiol. 2005;98:1985–1990. doi: 10.1152/japplphysiol.01095.2004. [DOI] [PubMed] [Google Scholar]
- Calvo JA, Daniels TG, Wang X, Paul A, Lin J, Spiegelman BM, Stevenson SC, Rangwala SM. Muscle-specific expression of PPARγ coactivator-1α improves exercise performance and increases peak oxygen uptake. J Appl Physiol. 2008;104:1304–1312. doi: 10.1152/japplphysiol.01231.2007. [DOI] [PubMed] [Google Scholar]
- Cantó C, Auwerx J. PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. 2009;20:98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civitarese AE, Carling S, Heilbronn LK, Hulver MH, Ukropcova B, Deutsch WA, Smith SR, Ravussin E. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007;4:e76. doi: 10.1371/journal.pmed.0040076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell RM, Blake KR, Russell JW. Localization of the transcriptional coactivator PGC-1α to GABAergic neurons during maturation of the rat brain. J Comp Neurol. 2007;502:1–18. doi: 10.1002/cne.21211. [DOI] [PubMed] [Google Scholar]
- Coyle EF. Very intense exercise-training is extremely potent and time efficient: a reminder. J Appl Physiol. 2005;98:1983–1984. doi: 10.1152/japplphysiol.00215.2005. [DOI] [PubMed] [Google Scholar]
- Dudley GA, Abraham WM, Terjung RL. Influence of exercise intensity and duration on biochemical adaptations in skeletal muscle. J Appl Physiol. 1982;53:844–850. doi: 10.1152/jappl.1982.53.4.844. [DOI] [PubMed] [Google Scholar]
- Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim S, Mostoslavsky R, Alt FW, Wu Z, Puigserver P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 2007;26:1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni P, Jan KJ, Douglas MJ, Stuart PM, Tarnopolsky MA. Oxidative stress and the mitochondrial theory of aging in human skeletal muscle. Exp Gerontol. 2004;39:1391–1400. doi: 10.1016/j.exger.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Gibala MJ, Little JP, van Essen M, Wilkin GP, Burgomaster KA, Safdar A, Raha S, Tarnopolsky MA. Short-term sprint interval versus traditional endurance training: similar initial adaptations in human skeletal muscle and exercise performance. J Physiol. 2006;575:901–911. doi: 10.1113/jphysiol.2006.112094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibala MJ, McGee SL, Garnham AP, Howlett KF, Snow RJ, Hargreaves M. Brief intense interval exercise activates AMPK and p38 MAPK signalling and increases the expression of PGC-1α in human skeletal muscle. J Appl Physiol. 2009;106:929–934. doi: 10.1152/japplphysiol.90880.2008. [DOI] [PubMed] [Google Scholar]
- Godin G, Desharnais R, Valois P, Lepage L, Jobin J, Bradet R. Differences in perceived barriers to exercise between high and low intenders: observations among different populations. Am J Health Promotion. 1994;8:279–284. [Google Scholar]
- Greiwe JS, Hickner RC, Hansen PA, Racette SB, Chen MM, Holloszy JO. Effects of endurance exercise training on muscle glycogen accumulation in humans. J Appl Physiol. 1999;87:222–226. doi: 10.1152/jappl.1999.87.1.222. [DOI] [PubMed] [Google Scholar]
- Gurd BJ, Yoshida Y, Lally J, Holloway GP, Bonen A. The deacetylase enzyme SIRT1 is not associated with oxidative capacity in rat heart and skeletal muscle and its overexpression reduces mitochondrial biogenesis. J Physiol. 2009;587:1817–1828. doi: 10.1113/jphysiol.2008.168096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschin C, Choi CS, Chin S, Kim S, Kawamori D, Kurpad AJ, Neubauer N, Hu J, Mootha VK, Kim Y, Kulkarni RN, Shulman GI, Spiegelman BM. Abnormal glucose homeostasis in skeletal muscle-specific PGC-1α knockout mice reveals skeletal muscle–pancreatic β cell crosstalk. J Clin Invest. 2007;117:3463–3474. doi: 10.1172/JCI31785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley JA. Exercise as a therapeutic intervention for the prevention and treatment of insulin resistance. Diabetes Metab Res Rev. 2004;20:383–393. doi: 10.1002/dmrr.505. [DOI] [PubMed] [Google Scholar]
- Holloszy JO. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J Biol Chem. 1967;242:2278–2282. [PubMed] [Google Scholar]
- Holloszy JO, Booth FW. Biochemical adaptations to endurance exercise in muscle. Annu Rev Physiol. 1976;38:273–291. doi: 10.1146/annurev.ph.38.030176.001421. [DOI] [PubMed] [Google Scholar]
- Hood DA, Irrcher I, Ljubicic V, Joseph A. Coordination of metabolic plasticity in skeletal muscle. J Exp Biol. 2006;209:2265–2275. doi: 10.1242/jeb.02182. [DOI] [PubMed] [Google Scholar]
- Hood DA, Saleem A. Exercise-induced mitochondrial biogenesis in skeletal muscle. Nutr Metab Cardiovasc Dis. 2007;17:332–337. doi: 10.1016/j.numecd.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Jäger S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc Natl Acad Sci U S A. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutti D, Kressler D, Kralli A. Regulation of the transcriptional coactivator PGC-1 via MAPK-sensitive interaction with a repressor. Proc Natl Acad Sci U S A. 2001;98:9713–9718. doi: 10.1073/pnas.171184698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Young RS, Koufogiannis G, Canny BJ, McConell GK. Acute exercise does not cause sustained elevations in AMPK signalling or expression. Med Sci Sports Exerc. 2008;40:1490–1494. doi: 10.1249/MSS.0b013e318173a037. [DOI] [PubMed] [Google Scholar]
- Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- McCoy M, Proietto J, Hargreaves M. Skeletal muscle GLUT-4 and postexercise muscle glycogen storage in humans. J Appl Physiol. 1996;80:411–415. doi: 10.1152/jappl.1996.80.2.411. [DOI] [PubMed] [Google Scholar]
- Michael LF, Wu Z, Cheatham RB, Puigserver P, Adelmant G, Lehman JJ, Kelly DP, Spiegelman BM. Restoration of insulin-sensitive glucose transporter (GLUT4) gene expression in muscle cells by the transcriptional coactivator PGC-1. Proc Natl Acad Sci U S A. 2001;98:3820–3825. doi: 10.1073/pnas.061035098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton JP, Croft L, Bartlett JD, Maclaren DP, Reilly T, Evans L, McArdle A, Drust B. Reduced carbohydrate availability does not increase training induced stress protein adaptations but up-regulates oxidative enzyme activity in skeletal muscle. J Appl Physiol. 2009;106:1513–1521. doi: 10.1152/japplphysiol.00003.2009. [DOI] [PubMed] [Google Scholar]
- Olson BL, Hock MB, Ekholm-Reed S, Wohlschlegel JA, Dev KK, Kralli A, Reed SI. SCFCdc4 acts antagonistically to the PGC-1α transcriptional coactivator by targeting it for ubiquitin-mediated proteolysis. Genes Dev. 2008;22:252–264. doi: 10.1101/gad.1624208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra J, Cadefau JA, Rodas G, Amigó N, Cussó R. The distribution of rest periods affects performance and adaptations of energy metabolism induced by high-intensity training in human muscle. Acta Physiol Scand. 2000;169:157–165. doi: 10.1046/j.1365-201x.2000.00730.x. [DOI] [PubMed] [Google Scholar]
- Passoneau J, Lowry O. Enzymatic Analysis: A Practical Guide. Totowa, NJ, USA: Humana; 1993. [Google Scholar]
- Perry CGR, Heigenhauser GJF, Bonen A, Spriet LL. High-intensity aerobic interval training increases fat and carbohydrate metabolic capacities in human skeletal muscle. Appl Physiol Nutr Metab. 2008;33:1112–1123. doi: 10.1139/H08-097. [DOI] [PubMed] [Google Scholar]
- Perseghin G, Price TB, Petersen KF, Roden M, Cline GW, Gerow K, Rothman DL, Shulman GI. Increased glucose transport-phosphorylation and muscle glycogen synthesis after exercise training in insulin-resistant subjects. N Engl J Med. 1996;335:1357–1362. doi: 10.1056/NEJM199610313351804. [DOI] [PubMed] [Google Scholar]
- Pilegaard H, Saltin B, Neufer PD. Exercise induces transient transcriptional activation of the PGC-1α gene in human skeletal muscle. J Physiol. 2003;546:851–858. doi: 10.1113/jphysiol.2002.034850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakobowchuk M, Tanguay S, Burgomaster KA, Howarth KR, Gibala MJ, MacDonald MJ. Sprint interval and traditional endurance training induce similar improvements in peripheral arterial stiffness and flow-mediated dilation in healthy humans. Am J Physiol Regul Integr Comp Physiol. 2008;295:R236–R242. doi: 10.1152/ajpregu.00069.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rim JS, Xue B, Gawronska-Kozak B, Kozak LP. Sequestration of thermogenic transcription factors in the cytoplasm during development of brown adipose tissue. J Biol Chem. 2004;279:25916–25926. doi: 10.1074/jbc.M402102200. [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Gerhart-Hines Z, Puigserver P. Metabolic adaptations through the PGC-1α and SIRT1 pathways. FEBS Lett. 2008;582:46–53. doi: 10.1016/j.febslet.2007.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell AP, Feilchenfeldt J, Schreiber S, Praz M, Crettenand A, Gobelet C, Meier CA, Bell DR, Kralli A, Giacobino J, Dériaz O. Endurance training in humans leads to fibre type-specific increases in levels of peroxisome proliferator-activated receptor-γ coactivator-1 and peroxisome proliferator-activated receptor-α in skeletal muscle. Diabetes. 2003;52:2874–2881. doi: 10.2337/diabetes.52.12.2874. [DOI] [PubMed] [Google Scholar]
- Sano M, Tokudome S, Shimizu N, Yoshikawa N, Ogawa C, Shirakawa K, Endo J, Katayama T, Yuasa S, Ieda M, Makino S, Hattori F, Tanaka H, Fukuda K. Intramolecular control of protein stability, subnuclear compartmentalization, and coactivator function of peroxisome proliferator-activated receptor γ coactivator 1α. J Biol Chem. 2007;282:25970–25980. doi: 10.1074/jbc.M703634200. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC. Nuclear control of respiratory gene expression in mammalian cells. J Cell Biochem. 2006;97:673–683. doi: 10.1002/jcb.20743. [DOI] [PubMed] [Google Scholar]
- Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–638. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- Schjerve IE, Tyldum GA, Tjønna AE, Stølen T, Loennechen JP, Hansen HEM, Haram PM, Heinrich G, Bye A, Najjar SM, Smith GL, Slørdahl SA, Kemi OJ, Wisløff U. Both aerobic endurance and strength training programmes improve cardiovascular health in obese adults. Clin Sci. 2008;115:283–293. doi: 10.1042/CS20070332. [DOI] [PubMed] [Google Scholar]
- Suwa M, Nakano H, Radak Z, Kumagai S. Endurance exercise increases the SIRT1 and peroxisome proliferator-activated receptor γ coactivator-1α protein expressions in rat skeletal muscle. Metabolism. 2008;57:986–998. doi: 10.1016/j.metabol.2008.02.017. [DOI] [PubMed] [Google Scholar]
- Talanian JL, Galloway SDR, Heigenhauser GJF, Bonen A, Spriet LL. Two weeks of high-intensity aerobic interval training increases the capacity for fat oxidation during exercise in women. J Appl Physiol. 2007;102:1439–1447. doi: 10.1152/japplphysiol.01098.2006. [DOI] [PubMed] [Google Scholar]
- Tjønna AE, Lee SJ, Rognmo Ø, Stølen TO, Bye A, Haram PM, Loennechen JP, Al-Share QY, Skogvoll E, Slørdahl SA, Kemi OJ, Najjar SM, Wisløff U. Aerobic interval training versus continuous moderate exercise as a treatment for the metabolic syndrome: a pilot study. Circulation. 2008;118:346–354. doi: 10.1161/CIRCULATIONAHA.108.772822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost SG, Owen N, Bauman AE, Sallis JF, Brown W. Correlates of adults’ participation in physical activity: review and update. Med Sci Sports Exerc. 2002;34:1996–2001. doi: 10.1097/00005768-200212000-00020. [DOI] [PubMed] [Google Scholar]
- Warburton DE, Nicol CW, Bredin SS. Health benefits of physical activity: the evidence. CMAJ. 2006;174:801–809. doi: 10.1503/cmaj.051351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wende AR, Schaeffer PJ, Parker GJ, Zechner C, Han D, Chen MM, Hancock CR, Lehman JJ, Huss JM, McClain DA, Holloszy JO, Kelly DP. A role for the transcriptional coactivator PGC-1α in muscle refueling. J Biol Chem. 2007;282:36642–36651. doi: 10.1074/jbc.M707006200. [DOI] [PubMed] [Google Scholar]
- Wisløff U, Støylen A, Loennechen JP, Bruvold M, Rognmo Ø, Haram PM, Tjønna AE, Helgerud J, Slørdahl SA, Lee SJ, Videm V, Bye A, Smith GL, Najjar SM, Ellingsen Ø, Skjaerpe T. Superior cardiovascular effect of aerobic interval training versus moderate continuous training in heart failure patients: a randomized study. Circulation. 2007;115:3086–3094. doi: 10.1161/CIRCULATIONAHA.106.675041. [DOI] [PubMed] [Google Scholar]
- Wright DC, Han D, Garcia-Roves PM, Geiger PC, Jones TE, Holloszy JO. Exercise-induced mitochondrial biogenesis begins before the increase in muscle PGC-1α expression. J Biol Chem. 2007;282:194–199. doi: 10.1074/jbc.M606116200. [DOI] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- Yu M, Blomstrand E, Chibalin AV, Krook A, Zierath JR. Marathon running increases ERK1/2 and p38 MAP kinase signalling to downstream targets in human skeletal muscle. J Physiol. 2001;536:273–282. doi: 10.1111/j.1469-7793.2001.00273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.