

Abstract
Phosphoribosylpyrophosphate synthetases (PRSs) catalyze the first step of nucleotide synthesis. Nucleotides are central to cell function, being the building blocks of nucleic acids and serving as cofactors in cellular signaling and metabolism. With this in mind, it is remarkable that mutations in phosphoribosylpyrophosphate synthetase 1 (PRPS1), which is the most ubiquitously expressed gene of the three PRS genes, are compatible with life. Mutations described thus far in PRPS1 are all missense mutations that result in PRS-I superactivity or in variable levels of decreased activity, resulting in X-linked Charcot-Marie-Tooth disease-5 (CMTX5), Arts syndrome, and X-linked nonsyndromic sensorineural deafness (DFN2). Patients with PRS-I superactivity primarily present with uric acid overproduction, mental retardation, ataxia, hypotonia, and hearing impairment. Postlingual progressive hearing loss is found as an isolated feature in DFN2 patients. Patients with CMTX5 and Arts syndrome have peripheral neuropathy, including hearing impairment and optic atrophy. However, patients with Arts syndrome are more severely affected because they also have central neuropathy and an impaired immune system. The neurological phenotype in all four PRPS1-related disorders seems to result primarily from reduced levels of GTP and possibly other purine nucleotides including ATP, suggesting that these disorders belong to the same disease spectrum. Preliminary results of S-adenosylmethionine (SAM) supplementation in two Arts syndrome patients show improvement of their condition, indicating that SAM supplementation in the diet could alleviate some of the symptoms of patients with PRPS1 spectrum diseases by replenishing purine nucleotides (J.C., unpublished data).
Main Text
Introduction
Missense mutations in phosphoribosylpyrophosphate synthetase 1 (PRPS1) (MIM 311850) result in four syndromes: PRS-I superactivity (MIM 300661),1 Charcot-Marie-Tooth disease-5 (CMTX5, or Rosenberg-Chutorian syndrome) (MIM 311070),2,3 Arts syndrome (MIM 301835),4,5 and X-linked nonsyndromic sensorineural deafness (DFN2) (MIM 304500).6 PRPS1 belongs to a family that consists of three very similar and highly conserved genes: PRPS1, PRPS2 (MIM 311860), and PRPS1L1 (MIM 611566). In contrast to PRPS1, PRPS2 and PRPS1L1 have not been implicated in disease. PRPS1 codes for PRS-I, which catalyzes the synthesis of phosphoribosyl pyrophosphate (PRPP) from ATP and ribose-5-phosphate.7 PRPP is essential for the de novo synthesis of purine,8 pyrimidine,9 and pyridine nucleotides.10 Thus, mutations in PRPS1 affect vital cell functions, such as nucleic acid synthesis, energy metabolism, and cellular signaling.
For purine synthesis, PRPP is utilized as a substrate for PRPP amidotransferase (PPAT), which is the first step in the de novo purine synthesis pathway, producing purine nucleotides such as ATP and GTP, and which serves specifically as the rate-limiting reaction for purine nucleotide synthesis in vivo.11 PRPP is also essential for pyrimidine nucleotide synthesis, where it acts as cofactor for uridine monophosphate synthetase (UMPS), which converts orotic acid into UMP,9 the precursor of all other pyrimidine nucleotides. Finally, PRPP is utilized for pyridine nucleotide synthesis by nicotinate phosphoribosyl transferase (NAPRT) and nicotinamide phosphoribosyl transferase (NAMPT) to add a ribonucleotide moiety to nicotinic acid and nicotinamide, respectively, which in turn are converted into the important cofactors nicotinamide adenine dinucleotide (NAD) and nicotinamide adenine dinucleotide phosphate (NADP).10
Apart from de novo synthesis of purine, pyrimidine, and pyridine nucleotides, PRPP is also used for the salvage of purine bases by hypoxanthine guanine phosphoribosyl transferase (HGPRT) and adenine phosphoribosyl transferase (APRT). This mechanism ensures efficient reutilization of purine bases and nucleosides, because de novo purine nucleotide synthesis with PRPP as the substrate requires in total seven moles of ATP for generation of each mole of nucleotide, whereas salvage requires only one mole of ATP for the synthesis of PRPP.12
The enzymatic activity of PRS-I is regulated by cofactors, metabolites, and interacting proteins. Inorganic phosphate regulates enzyme activity by allosteric competition with adenosine diphosphate, which is an inhibitor of enzyme activity.13,14 In addition, inorganic phosphate promotes the accessibility of the active site by stabilizing a flexible loop involved in ATP binding.15 Bivalent metal cations, most potently Mg2+, are needed to bind to β- and γ-phosphates of ATP, forming a Mg-ATP complex that is the actual substrate of the enzyme.13,16 In addition, the activity of PRS-I is regulated by its interaction with PRS-II, 39 kDa phosphoribosylpyrophosphate synthetase-associated protein (PAP39), and 41 kDa phosphoribosylpyrophosphate synthetase-associated protein (PAP41). Removal of the two PAP proteins from the PRS complex results in increased PRS enzyme activity,17 which suggests a negative regulatory role for these proteins, most likely by sequestration of PRPP synthetases.18
PRPS1 transcript levels are controlled by miR-376. miRNAs are ∼21-nucleotide-long transacting RNA molecules that repress activity of genes at the mRNA level by complementary base pairing, thereby inhibiting protein synthesis or causing mRNA degradation.19 The transcript of PRPS1 contains two binding sites for the edited version of miR-376 within 600 bp from the termination codon.20 These binding sites are not present in the 3′ untranslated region of PRPS2. The miR-376 cluster of miRNA genes maps to chromosome 14q32.3121 and is expressed in multiple tissues during embryonic development and in adult brain, heart, pancreas, and kidney.20,22 All of the cluster members are transcribed into one long primary RNA transcript that can be subject to extensive and simultaneous editing by adenosine deaminase at one or two specific sites20 during the processing of primary transcripts to mature miRNA.23–25 This editing is tissue specific for miR-376. In the brain cortex most transcripts are edited, whereas in the liver almost no editing occurs. Because only the edited miR-376 silences the expression of PRPS1, PRS-I activity is regulated by miR-376 in the cortex, but not in the liver.20 Notably, patients with PRS-I superactivity or impaired activity display neurological symptoms such as ataxia and mental retardation, whereas the liver functions normally.2,5,26 Taken together, these data indicate that the activity of PRS-I is normally tightly regulated in neuronal cells and that imbalances in nucleotide metabolism are particularly detrimental to neuronal function.
PRPS-1 Related Disorders
PRS-I Superactivity
PRS-I superactivity was first described by Sperling et al. in 19721 as an X-linked disorder associated with excessive uric acid production and gout. Since then, it has become clear that inherited PRS-I superactivity results in a variable clinical presentation characterized by early adult onset of purine overproduction that causes hyperuricemia and hyperuricosuria (Table 1)27,28 accompanied in some kindreds by neurological problems, most notably mental retardation, hypotonia, and sensorineural deafness.29–31 In one family, progressive axonal neuropathy with demyelination has been described.32
Table 1.
General Clinical Description of Purine and Pyrimidine Disorders in Which PRPP Is Involved as Substrate and AHCY Deficiency
| Clinical Symptom | PRS-I Superactivity | Arts Syndrome | CMTX5 | DFN2 | HGPRT Deficiency | APRT Deficiency | UMPS Deficiency | AHCY Deficiency |
|---|---|---|---|---|---|---|---|---|
| Neurological | ||||||||
| Mental retardation | ± | + | − | − | ± | − | − | + |
| Ataxia | ± | + | + | − | − | − | − | − |
| Hypotonia | ± | + | + | − | ± | − | − | + |
| Delayed motor development | ± | + | − | − | + | − | − | + |
| Areflexia | ± | + | − | − | − | − | − | − |
| Loss of deep tendon reflexes | − | + | + | − | − | − | − | + |
| Hearing impairment | ± | + | + | + | − | − | − | − |
| Optic atrophy | − | + | + | − | − | − | − | − |
| Uric acid overproduction | ||||||||
| Gout | + | − | − | − | + | − | − | − |
| Kidney stones | + | − | − | − | ± | +∗ | − | − |
| Renal failure | + | − | − | − | − | +∗ | − | − |
| Hematopoietic | ||||||||
| Recurrent infections | − | + | − | − | − | − | + | − |
| Anemia | − | − | − | − | ± | − | + | − |
| Other | ||||||||
| Short stature | − | − | − | − | ± | − | − | − |
| Self injury | − | − | − | − | ± | − | − | − |
| Early death | − | + | − | − | ± | − | ± | − |
The urolithiasis and renal failure in APRT deficiency are not caused by increased uric acid production but by accumulation of 2,8 dihydroxy-adenine.
PRS-I superactivity can be caused by two different mechanisms: (1) point mutations in the open reading frame (ORF) of PRPS1 that result in regulatory defects (Table 2),28,29,33–35 and (2) increased expression of PRS-I that has normal kinetic enzyme properties.36–41 In total, there are more than 30 patients reported with PRS-I superactivity.42 In seven of these, a point mutation in PRPS1 has been identified.
Table 2.
Mutations Identified in PRPS1 in the Four Different PRPS1-Related Disorders
| Disorder | DNA Mutation | Protein Change | Functional Change | Reference |
|---|---|---|---|---|
| PRS-I superactivity | 154G>C | D52H | Gain of function | Becker et al.35 |
| 341A>G | N114S | Gain of function | Roessler et al.93 | |
| 385C>A | L129I | Gain of function | Becker et al.35 | |
| 547G>C | D182H | Gain of function | Roessler et al.93 | |
| 569C>T | A189V | Gain of function | Becker et al.35 | |
| 578A>T | H192L | Gain of function | Garcia-Pavia et al.94 | |
| 579C>G | H192Q | Gain of function | Becker et al.35 | |
| Arts syndrome | 398A>C | Q133P | Loss of function | de Brouwer et al.5 |
| 455T>C | L152P | Loss of function | de Brouwer et al.5 | |
| CMTX5 | 129A>C | E43D | Loss of function | Kim et al.2 |
| 344T>C | M115T | Loss of function | Kim et al.2 | |
| DFN2 | 193G>A | D65N | Loss of function | Liu et al.6 |
| 259G>A | A87T | Loss of function | Liu et al.6 | |
| 869T>C | I290T | Loss of function | Liu et al.6 | |
| 916G>A | G306R | Loss of function | Liu et al.6 |
In addition to a gain or loss of function, all mutations result in an unstable PRS-I protein. Depending on the cell type, gain-of-function mutations can then result in overactivity in actively dividing cells through loss of self-regulation or in a loss of activity in postmitotic cells, explaining the complete absence of activity in erythrocytes of patients with PRS-I superactivity.
(1) The severe PRS-I superactivity phenotypes with purine overproduction accompanied by neuropathy arise from PRPS1 ORF point mutations. These result in impaired nucleotide feedback inhibition and inorganic phosphate activation. In this respect, the enzyme is not truly “superactive” because it does not have a higher Vmax than the normal protein, but rather it lacks regulatory control of its activity,33 suggesting that these mutations can be considered as gain-of-function mutations. Accordingly, PRS activity and PRPP levels have been shown to be higher in fibroblasts and lymphoblasts of patients with PRPS1 ORF mutations.28,32,34,43 Remarkably, in erythrocytes—where PRS-I is the only PRS isoform present5—enzyme activity has been found to be completely absent, accompanied by reduced nucleotide levels, in particular low GTP and NAD.31,32,43 This suggests that the mutated protein may be more labile in postmitotic cells, such as erythrocytes and brain cells, whereas it may continue to overproduce purines in dividing tissues such as skin fibroblasts and liver.35 The latter is, perhaps not coincidently, together with the small intestine, the primary uric acid-producing tissue.44
(2) Increased expression of PRS-I accounts for the milder phenotypes that manifest as purine overproduction without any neuropathy. These patients have increased PRPS1 transcript levels, but no genetic defects have been identified in the ORF, the untranslated regions, or in 3 kb of the promoter region.40,41,45 In addition, PRPS1 was not duplicated.40 The overexpression has been shown to be accompanied by raised intracellular levels of PRPP in all cell types examined, i.e., fibroblasts, lymphoblasts, and erythrocytes.36–39 This does not result in raised nucleotide levels, but instead the increased de novo purine synthesis causes accelerated nucleotide degradation to uric acid.26
Because the kinetic properties of PRS-I in these patients is not affected, the superactivity may be caused by changes in a pretranslational mechanism of PRPS1 gene expression through miR-376-regulated changes in PRPS1 transcript levels. Theoretically, mutations in the miR-376 binding sites of PRPS1 or in miR-376 itself may result in PRS-I superactivity. However, no binding site mutations have been identified to date,41 miR-376 itself has not been sequenced, and the seven patients reported with PRPS1 overexpression were male, whereas autosomal miR-376 involvement would predict males and females to be equally affected.
Arts Syndrome
Arts syndrome is an X-linked disorder characterized by mental retardation, early-onset hypotonia, ataxia, delayed motor development, profound congenital sensorineural hearing impairment, and optic atrophy (Table 1).4,5 In males, liability to infections, especially of the upper respiratory tract, almost invariably has resulted in an early death before the age of five years in the original Arts syndrome family. Obligate carrier females may show isolated and milder manifestations of symptoms, such as perceptive hearing impairment, ataxia, hypotonia, and hyperreflexia.4 Indeed, there was no skewing of X inactivation in the carrier females (A.P.M.d.B., unpublished data), although PRPS1 is clearly subject to X inactivation.46
Linkage analysis in the Arts syndrome families suggested that the causative genetic defect mapped to Xq22.1-q24.47 Whole-genome expression profiling of fibroblasts from two probands of the Dutch family revealed a nonsignificant 1.8-fold reduction in PRPS1 RNA transcript levels.5 Subsequent sequencing of PRPS1 led to the identification of two missense mutations, c.455T>C (p.L152P) in the original Dutch Arts syndrome kindred and c.398A>C (p.Q133P) in an Australian family. PRPS1 expression in the Australian family was found to be normal. Both mutations result in a loss of PRS-I activity, as demonstrated by in vitro PRS enzyme assays in erythrocytes and fibroblasts from the Australian family and fibroblasts from the Dutch kindred. This was confirmed by undetectable urine hypoxanthine and reduced plasma uric acid levels in the two patients from the Australian family.5
A sural nerve biopsy from an affected boy of the Australian family with Arts syndrome showed mild paranodal demyelination, indicative of peripheral neuropathy.5 In addition, the central nervous system can be affected, as shown by the complete absence of myelin in the posterior columns of the spinal cord in the only patient from the original Arts syndrome kindred who was examined at autopsy.4 Conversely, MRI of the brains in the affected brothers in the Australian family showed no recognizable abnormalities, such as a reduction of white matter, which is indicative of demyelination.5 Taken together, these observations suggest that the demyelination is more apparent in the periphery than in the central nervous system.
CMTX5, or Rosenberg-Chutorian Syndrome
CMTX5, or Rosenberg-Chutorian syndrome, is defined by peripheral neuropathy, early-onset sensorineural hearing impairment, and optic neuropathy (Table 1).2,3 Hypotonia, gait disturbances, and loss of deep tendon reflexes with an onset from the age of 10 to 12 years have also been reported. Peripheral demyelination and axonal loss seem to be at the root of these late onset problems, as demonstrated in the oldest living patient by a sural nerve biopsy.2 In contrast to Arts syndrome, mental retardation and recurrent infections are not features of CMTX5.
CMTX5 was mapped in a Korean kindred to Xq21.31-q24.48 Analysis of 71 genes in the linkage interval revealed a c.344T>C mutation in PRPS1, which leads to the substitution of a threonine for a methionine residue at position 115 of PRS-I. PRS activity was decreased in fibroblasts from patients with this mutation.2 Subsequently, in a family with Rosenberg-Chutorian syndrome, a transversion of c.129A>C was identified that results in the substitution of an aspartic acid for a glutamic acid residue at position 43 of PRS-I. Serum uric acid levels in patients with either mutation were normal.2
DFN2
Patients with DFN2 have postlingual progressive nonsyndromic hearing loss,6,49,50 although in one family congenital profound nonsyndromic hearing loss was reported.51 Linkage analysis in a large Chinese family fine-mapped the DNF2 locus to Xq22.1-Xq23, which contains 119 genes.6 Subsequent DNA sequence analysis of the seven exons of PRPS1 showed a transition of c.193G>A in exon 2 that results in the substitution of an aspartic acid for an asparagine at amino acid residue position 65. The PRS-I activity in erythrocytes and fibroblasts was found to be half, compared to unaffected males in the family. In the other three DFN2 families, c.259G>A (p.A87T), c.869T>C (p.I290T), and c.916G>A (p.G306R) mutations were identified in PRPS1.6
Effect of the Disease-Causing Mutations on the Structure of PRS-I
The physiologically active PRS unit is a hexamer that consists of three homodimers arranged in a propeller-like shape (Figure 1A), each with an active site and two regulatory allosteric sites, I and II.15,52 The active site comprises binding sites for both ATP and ribose-5-phosphate and is located at the interface of two domains within one homodimer. Allosteric site I is located at the interface of the three homodimers of a hexamer, and allosteric site II is at the interface of two monomers within one homodimer.15,53
Figure 1.
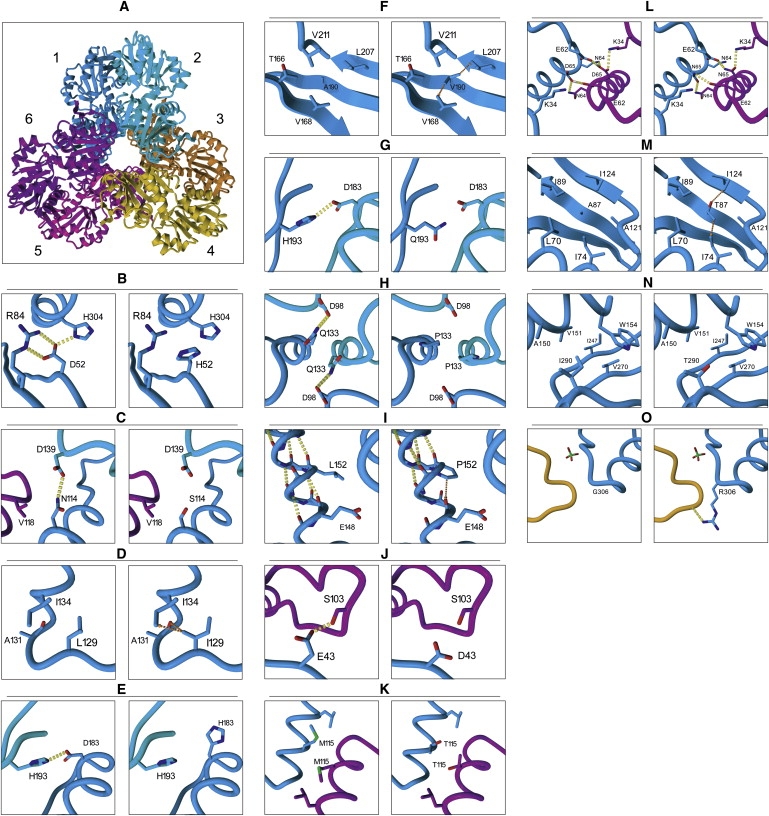
Three-Dimensional Model of PRS-I as a Hexamer and a Close-Up of the Regions that Contain the Amino Acid Substitutions
(A) Three-dimensional model of PRS-I as a hexamer, with the six monomers labeled with different colors.
(B–G) Seven substitutions that result in PRS-I superactivity: p.D52H (B), p.N114S (C), p.L129I (D), p.D183H (E), p.A190V (F), and p.H193L (G). Note that pH193Q has a similar effect on the PRS-I structure as p.H193L.
(H–K) Four substitutions that cause Arts syndrome and CMTX5: p.Q133P (H), p.L152P (I), p.E43D (J), and p.M115T (K).
(L–O) Four substitutions that result in DFN2: p.D65N (L), p.A87T (M), p.I290T (N), and p.G306R (O). All substitutions are shown for monomer 1. Homodimer interactions are shown between monomer 1 (blue) and monomer 2 (cyan). Trimer interactions are shown between monomer 1, monomer 2, and monomer 6 (purple). Hydrogen bonds are indicated with yellow dashed lines. Strong interatomic steric hindrance is indicated with orange dashed arrows. The effect of PRPS1 mutations on the structure of PRS-I was examined with the crystal structures of the human protein from Li et al.15 The altered amino acid side chains in the model were positioned with a backbone-dependent rotamer library, as implemented in the YASARA program.
Mutations that Cause PRS-I Superactivity
Two of the amino acid residues' substitutions, p.D52H and p.L129I, which result in PRS-I superactivity, disturb the local structure near one of the two allosteric sites, hindering feedback inhibition. The p.D52H substitution results in disruption of the hydrogen bonds in which the Asp52 side chain is engaged (Figure 1B), which is predicted to destabilize the local structure around Asp52, disturbing allosteric site I. Leu129 is in the vicinity of allosteric site II, where the leucine-to-isoleucine change results in moderate steric hindrance of the isoleucine side chain with both the protein backbone at Ala131 and the side chain of Ile134 (Figure 1D). This is predicted to destabilize the local structure around both of these amino acids and hence disrupts allosteric site II.
The other substitutions described that cause PRS-I superactivity, p.N114S, p.A190V, p.D183H, p.H193L, and p.H193Q are all located in the homodimer interface. Asn114 is located in an α helix and interacts with the second monomer via a hydrogen bond with Asp139 (Figure 1C). The p.N114S change is predicted to break this hydrogen bond and destabilize the homodimer. In addition, the Asn114 interaction with Val118 in the trimer interface is also impaired by the p.N114S change. Ala190 is located in the center of a β sheet that plays a prominent role near the top of the homodimer interface (Figure 1F). The p.A190V change results in the substitution of the small alanine side chain by a more bulky valine side chain, which is predicted to disturb the tight hydrophobic packing environment in the homodimer interface. Asp183 and His193 are both located in the upper region of the dimer interface (Figures 1E and 1G). The PRS-I crystal structure shows that Asp183 of one PRS-I subunit is involved in a hydrogen bond to His193 of the adjacent PRS-1 subunit in the homodimer. The p.D183H and p.H193L/Q substitutions break this interaction, resulting in a destabilization of the homodimer interface. Destabilization of the homodimer is likely to disturb both PRS-I allosteric sites, because these consist of amino acids originating from either two (allosteric site II) or three (allosteric site I) PRS-I molecules. Based on its central location in the dimer, we predict allosteric site II to be the foremost site affected by the above changes that disturb the homodimer interface. The disturbance of allosteric site II would consequently result in loss of feedback inhibition and in higher PRS enzyme activity, as seen in fibroblasts and lymphoblasts.28,32,34,43 Moreover, decreased stability of the PRS unit could then result in a gradual loss of enzyme activity, which could explain the absence of activity observed in postmitotic cells such as erythrocytes and presumably brain cells.31,32,43
Mutations that Cause Arts Syndrome
PRS-I loss of activity observed in Arts syndrome has been associated with p.Q133P and p.L152P. Gln133 is positioned in the homodimer interface and forms a hydrogen bond to Asp98 from the second subunit in the homodimer (Figure 1H). The p.Q133P change is predicted to disrupt two intermolecular hydrogen bonds in the dimer interface, destabilizing both the homodimer and the nearby allosteric site II. In addition, Gln133 and especially its hydrogen-bonding partner Asp98 are in very close proximity to the ATP-binding pocket. The amino acid residues neighboring Asp98, namely Arg96, Gln97, Lys99, and Asp101, all interact with the incoming ATP molecule. Loss of the hydrogen bonding capacity at position 98 is predicted to destabilize the ATP-binding pocket and thus impair PRS-I activity. Leu152 is positioned in the middle of one of the α helixes (Figure 1I). Replacement of this leucine with a proline breaks the α-helical configuration and may consequently result in severe local distortion of the tertiary structure. Leu152 is positioned directly above Glu148, which interacts in the homodimer interface with Lys102, originating from the other monomer. The p.L152P substitution introduces interatomic interference with the backbone of Glu148, which is predicted to severely destabilize the local structure. This probably also affects both Lys102 and the adjacent Tyr146, which both participate in allosteric site II. Because Lys102 neighbors the above-mentioned ATP-binding domain, ranging from Arg96 to Asp101, the p.L152P substitution is predicted to impair PRS-I activity.
Mutations that Cause CMTX5
The two amino acid residue changes that result in PRS-I loss of function in patients with CMTX5 are both involved in the PRS-I trimer interface. Glu43 is located in the so-called flag region (residues Val30–Ile44) and interacts via a hydrogen bond with Ser103 located in the adjacent dimer in the PRS-I hexamer (Figure 1J). The p.E43D change is predicted to alter the way the flag region interacts with the adjacent PRS-I homodimer. Because this region of the structure plays a critical role in ATP binding by the complete hexamer, the substitution should result in decreased synthetase activity. Met115 is also positioned in the trimer interface (Figure 1K). Two Met115 side chains originating from two different PRS-I dimers interact intimately in the hydrophobic trimer interface. Rearrangement of the side chains in the p.M115T substitution is predicted to disturb the hydrophobic packing in the trimer interface, destabilizing the ATP-binding site and the allosteric site I, because both consist of amino acids of different PRS-I units coming together at the trimer interface.
Mutations that Cause DFN2
The four mutations described for DFN2 can be divided into two classes: those that foremost affect local structure (p.A87T and p.I290T) and those that affect the trimer interface (p.D65N and p.G306R). Ala87 is located in a β sheet in the hydrophobic core of the N-terminal domain (Figure 1L). A change of Ala87 to a more bulky and more polar threonine side chain will destabilize the surrounding tightly packed hydrophobic environment. Ile290 is located at the surface of the PRS-I protein at the edge of the central β sheet of the C-terminal domain (Figure 1M). Here, changing to a smaller, but more polar, threonine side chain will result in a less-optimal hydrophobic packing, effectively destabilizing this region of the protein.
Asp65 is located in the vicinity of the so-called flag region of the trimer interface but interacts in the hexamer interface with Glu62 and Asn64 from another subunit (Figure 1N). Hence, this change is predicted to lead to an altered affinity between the different subunits in the hexamer. The p.G306R change is located near the center of the hexamer, at the surface of the protein (Figure 1O). Modeling shows that the introduction of the much larger arginine side chain can most likely be accommodated by PRSP1, because the arginine side chain will be pointing outward. However, stability of PRSP1 might be affected to some degree by the introduction of three charged arginine side chains near the center of the hexamer.
Comparison of the Structural Effect of PRPS1 Mutations
When comparing the protein structural effects of the PRPS1 mutations involved in all four syndromes, the mutations resulting in the mildest symptoms are predicted to have the least impact on the PRS-I structure (Table 3). Mutations that result in DFN2 either disturb local stability of PRS-I or moderately affect interactions in the trimer interface. Mutations causing PRS-I superactivity mainly perturb the dimer interface and thereby affect allosteric sites I and II. Those mutations that result in Arts syndrome and CMTX5 not only affect the allosteric sites but also directly impact the ATP-binding site. CMTX5 mutations appear to affect the stability of the interactions in the trimer interface, whereas the mutations in Arts syndrome are predicted to more severely disrupt the overall PRS-I structure. Additional studies in erythrocytes of patients or expression of mutant human PRS-I in, for example, Escherichia coli are needed to confirm these predictions on the basis of computer-assisted molecular modeling.
Table 3.
Predicted Structural Effects of the Known PRSP1 Disease-Causing Mutations
| Mutation | Disturbing Local Structure | Affecting Dimer Interface | Affecting Trimer Interface | Disturbing ATP-Binding Site | Disturbing Allosteric Site I | Disturbing Allosteric Site II | Neuropathy∗ |
|---|---|---|---|---|---|---|---|
| PRS-I superactivity | |||||||
| p.D52H | + | − | + | − | + | − | − |
| p.N114S | − | + | + | − | ± | ± | + |
| p.L129I | + | − | − | − | − | + | + |
| p.D183H | − | + | − | − | ± | ± | + |
| p.A190V | + | + | − | − | ± | ± | + |
| p.H193L∗∗ | − | + | − | − | ± | ± | − |
| p.H193Q | − | + | − | − | ± | ± | + |
| Arts syndrome | |||||||
| p.Q133P | + | + | − | + | ± | + | + |
| p.L152P | + | + | − | + | ± | + | + |
| CMTX5 | |||||||
| p.E43D | − | − | + | + | + | − | + |
| p.M115T | − | − | + | + | + | − | + |
| DFN2 | |||||||
| p.D65N | − | − | + | − | − | − | ? |
| p.A87T | + | − | − | − | − | − | ? |
| p.I290T | + | − | − | − | − | − | ? |
| p.G306R | − | − | + | − | − | − | ? |
This mutation is found in a female patient.
Genotype-Phenotype Correlations in PRPS1 Spectrum Diseases
Aberrations in PRS activity can in principle affect all of the enzymatic conversions catalyzed by phosphoribosyltransferase enzymes with PRPP as substrate, i.e., PPAT, HGPRT, APRT, UMPS, NAPRT, and NAMPT. Consequently, a number of symptoms that are present in patients with deficiencies in phosphoribosyltransferase enzymes overlap with those present in patients with PRPS1-related disorders. For example, patients with HGPRT deficiency or Lesch-Nyhan disease (MIM 300322) have purine overproduction similar to PRS-I superactivity and can also have mental retardation and hypotonia, as found in patients with Arts syndrome.54 Recurrent infections and early death occur in patients with Arts syndrome and were recorded originally for UMPS deficiency (MIM 258900).55 In general, the symptoms in PRPS1-related disorders can be classified into three main groups: neurological, hematopoietic, and purine overproduction (Table 1). The purine overproduction symptoms manifest primarily as uric acid stones, hyperuricemia, and gout and are associated only with PRS-I superactivity. The mechanisms of the neurological and hemapoietic symptoms appear more complex, and phenotypic variation remains perplexing.
Neurological Symptoms
The major peripheral and central nervous system neuropathies in the four PRPS1 spectrum diseases are ataxia, hypotonia, optic atrophy, and hearing impairment. Remarkably, neuropathy is observed in patients with gain-of-function PRS-I superactivity,29–32 despite the predicted higher levels of purine and pyrimidine nucleotides. This apparent paradox can be explained by a more labile PRS-I in case of the superactivity mutations, as predicted by computer-assisted molecular modeling and supported by low enzyme activity and low nucleotide levels in erythrocytes.31,32,43 Thus, postmitotic cells, including neuronal cells, appear to have impaired PRS-I activity in all four syndromes caused by PRPS1 mutations.
The neuropathies could be a result of the reported demyelination of neurons. Membrane and myelin synthesis are dependent on the lipid esters of pyrimidine nucleotides (e.g., CDP-choline, CDP-neuraminic acid, and CDP-ethanolamine).56 In addition, myelin synthesis requires the methylation cofactor S-adenosylmethionine (SAM) that is made from methionine and the purine nucleotide adenosine. Accordingly, impaired PRS-I function may cause a reduction in purine and pyrimidine nucleotides, resulting in demyelination.
A second mechanism for the neuropathy, the involvement of impaired pyrimidine nucleotide synthesis in neuropathy, seems to be contradicted by the apparent lack of neuropathology in UMPS deficiency. However, the natural course of UMPS deficiency is not completely known, because for the past 40 years patients have been successfully treated with uridine supplementation,54,55 thus preventing possible long-term neuropathological consequences, although one UMPS patient in Brisbane on uridine has a long-term and progressive weakness (J. McGill, personal communication). In addition, pyrimidine nucleotide salvage, unlike purine salvage, is not dependent upon PRPP, because pyrimidine nucleosides (including dietary pyrimidines) are salvaged by kinases. As a result, the major pyrimidine nucleotide in erythrocyte, uridine diphosphate-glucose, is normal in PRS-I superactivity patients, whereas purine nucleotides are depleted.54 Taken together, however, these observations indicate that impaired pyrimidine synthesis may not be the primary cause of the neuropathy observed in PRPS1-related disorders.
A third model to explain the neuropathy in PRPS1-related disorders involves the high-energy demand of neuronal cells. De novo purine synthesis requires seven ATP molecules for the formation of each mole of purine nucleotide.12 Therefore, HPRT and APRT normally salvage purine nucleotides by using PRPP and purine bases as substrate. These enzymatic conversions need only one ATP,12 thus conserving much of the energy that might otherwise be consumed in de novo purine synthesis. Ninety percent of the free purine bases in humans are recycled, stressing the importance of purine base salvage.57 Impaired PRS-I function reduces PRPP levels, affecting both purine nucleotide de novo synthesis and salvage. Because PPAT is the rate-limiting enzyme of de novo purine nucleotide synthesis, specific upregulation of PPAT may be expected in patients with loss-of-function mutations in PRS-I to compensate for the reduced purine nucleotide levels. Indeed, preliminary experimental results indicate that in fibroblasts from Arts syndrome patients, mRNA transcript levels of PPAT (MIM 172450) are significantly increased by 1.6-fold (p = 0.028 with a two-sided Student's t test; A.P.M.d.B. and H.v.B., unpublished data). This would then result in a higher energy consumption for purine nucleotide synthesis in patients with PRS-I loss-of-function mutations, because PPAT upregulation commits the cell to energy-demanding de novo synthesis rather than to the energy-saving salvage pathway. In addition, the brain appears to maintain its major purine nucleotides largely via HGPRT salvage of hypoxanthine. Free adenosine, guanosine, or adenine levels are usually low, less than 1 μM.58,59 When HGPRT deficiency in Lesch Nyhan syndrome cuts purine salvage, the result is a severe neuropathy similar to that in patients with CMTX5 and Arts syndrome.54 Furthermore, the lack of any neurological phenotype in patients with APRT deficiency60 supports the assumption that salvage of adenine is not essential for ATP synthesis in the brain. Dependence of ATP salvage on HGPRT only would render neuronal cells even more sensitive to a reduction in PRPP levels caused by a loss of PRS-I activity compared to other cell types. In this energy-depletion model, gain-of-function PRPS1 mutations would simply result in the use of more ATP than normal through stimulation of de novo synthesis of purine nucleotides. This hypothesis is supported by the high degree of PRS-I activity regulation in the brain, at least on an RNA level, by miR-376.
A fourth model for the PRS-I neuropathy proposes the involvement of reduced GTP levels in neurons.61 GTP is essential for the activation of the Rho family of small (20–30 kDa) GTPases,62 of which the major function in neurons is to regulate the assembly and organization of the actin cytoskeleton.63 In the nervous system, these GTPases are essential regulators of neuronal growth cone motility, axonal migration, and dendritic spine morphogenesis.64–66 Mutations in several regulators or effectors of Rho GTPases, such as ARHGEF6 (MIM 300267), OPHN1 (MIM 300127), PAK3 (MIM 300142), FGD1 (MIM 300546), and GDI1 (MIM 300104), result in mental retardation.67 Patients with HGPRT deficiency have low GTP levels in erythrocytes and fibroblasts61,68 and a neuronal phenotype that can include mental retardation and that in general resembles that of the PRPS1-related disorders.54 Specifically, HGPRT deficiency is associated with abnormal dopamine function that is thought to delay neuronal development,69–71 and maintenance of dopamine levels is totally dependent upon GTP72 through the first step in dopamine synthesis catalyzed by GTP cyclohydrolase I. Inherited deficiency of GTP cyclohydrolase I (MIM 233910) results in a severe neurological syndrome similar to both HGPRT deficiency and PRPS1-related disorders.2,5,26,31,73 Because erythrocytes of patients with PRS-I superactivity also contain low levels of GTP,31 reduced GTP levels might play a role in the neurological dysfunction in these patients as well. Supporting the concept of a GTP-related neurological disease group, a number of other genes involved in the Charcot-Marie-Tooth spectrum of diseases are also GTP dependent, for example, ARGHEF10 (MIM 608136), DNM2 (MIM 602378), FGD4 (MIM 611104), MFN2 (MIM 608507), and RAB7 (MIM 602298).74,75 Thus, depletion in GTP levels, affecting central neurological functions such as GTPases and dopamine synthesis, may explain an important part of the neurological phenotype of Arts syndrome and CMTX5.
Finally, the occurrence of PRPS1-related neuropathy may relate to the loss of pyridine nucleotide synthesis, for example, the energy cofactors NAD and NADP. In particular, NAD is required for oxidoreductive cell processes and is essential for mitochondrial oxidative phosphorylation and ATP generation. Production of NAD is PRPP dependent, and these pyridine nucleotides have been shown to be severely reduced in erythrocytes of patients with PRS-I superactivity and a severe neuropathy.31 Further studies of the status of pyridine nucleotides in patients with Arts syndrome or CMTX5 are required to establish mitochondrial functionality, especially in neuronal cells.
Hematopoietic Symptoms
Arts syndrome differs from DFN2, CMTX5, and PRS-I superactivity in that Arts syndrome patients are liable to severe infections of the respiratory tract that often result in early death.4 A role for PRS-I in hematopoietic differentiation is supported by the high expression of PRPS1 in erythrocytes, B- and T-lymphocytes, and natural killer cells (BioGPS),76 as well as normal lymphatic tissues, such as lymph, spleen, and thymus (UniGene expression profile). In Arts syndrome patients, the total number of B- and T-lymphocytes and natural killer cells was normal, but IgA was found repeatedly in the low-normal range.4 This may point to a defect in the differentiation of B-lymphocytes to plasma cells and memory cells, because IgA plays a crucial role in the first line of immune defense at mucosal surfaces, such as the respiratory and urinary tract.77 A specific role for PRS-I in hematopoiesis is further supported by the direct association of PRS-I with p300,78 a histone acetyltransferase that is essential for hematopoietic differentiation, as shown in murine embryonic stem cells lacking p300 expression.79 In humans, heterozygous mutations in p300 cause an atypical form of the autosomal-dominant inherited Rubinstein-Taybi syndrome,80,81 which is characterized by mental retardation, typical dysmorphic features, and recurrent infections in the respiratory or urinary tract.
Recurrent infections are also a symptom of one pyrimidine disorder, UMPS deficiency,82 and two purine disorders, purine nucleoside phosphorylase (PNP) deficiency (MIM 164050)83 and adenosine deaminase (ADA) deficiency (MIM 102700).84 Patients with PNP deficiency suffer from severe neuropathy accompanied by a late-onset T-cell immunodeficiency. Patients with ADA deficiency present neonatally with severe combined T- and B-cell deficiency. In both diseases, the impaired immune system is proposed to result from reduced nucleic acid synthesis caused by inhibition of ribonucleotide reductase through abnormal accumulation of the deoxynucleotides deoxyGTP or deoxyATP,84–86 rather than from nucleotide depletion. Furthermore, UMPS deficiency would also result in impaired nucleic acid synthesis. However, in contrast to patients with UMPS deficiency, patients with Arts syndrome are not anemic (W.F.A., unpublished data). In summary, we propose that the immune deficiency in patients with Arts syndrome is predominantly caused by impaired nucleic acid synthesis in hematopoietic cells, resulting in flawed hematopoietic differentiation.
Inter- and Intrafamilial Phenotypic Variability
The phenotype among patients with PRS-I superactivity and Arts syndrome varies considerably.4,26 This phenotypic variation could be the result of redundancy in the production of PRPP among the three PRS isoforms, encoded by PRPS1, PRPS2, and PRPS1L1. However, PRPS2 transcript levels were not found to be elevated to compensate for PRS-I deficiency in patients with Arts syndrome.5 Second, the clinical variability may result from polymorphisms in genes encoding regulators of PRS-I synthesis, such as miR-376. However, no miR-376 sequence variants have been identified in the original Arts syndrome kindred (W. Chen, personal communication). Third, differences in PPAT expression levels between individuals might result in an attenuation of the phenotype. This last possibility is yet to be pursued.
SAM Treatment of Patients with PRS-I Deficiency
Dietary pyrimidine nucleotides are readily absorbed by the gut55 so that symptoms caused by impaired de novo pyrimidine synthesis can be moderated by the addition of dietary pyrimidines, such as uridine. In contrast, dietary purines are usually oxidized to uric acid by the gut rather than being absorbed into the body.87 However, unlike most purines, SAM can cross both the gut and the blood-brain barrier and has been successfully used in the treatment of depression and osteoarthritis.88 Importantly, side effects are few. In addition, SAM is also a source of methionine, which could overcome a possible deficiency of methylation-dependent processes, including myelin synthesis. Theoretically, SAM can replenish both ATP and GTP independently of PRPP (Figure 2). Methyltransferases convert SAM into S-adenosylhomocysteine. Subsequently, S-adenosylhomocysteine hydrolase (AHCY) catalyzes the conversion of S-adenosylhomocysteine to mainly adenosine, although adenine can be formed by AHCY, or SAM can be converted directly to adenine, via the polyamine pathway.89,90 Adenine can subsequently be turned into AMP by APRT, which still requires PRPP as substrate. Alternatively, adenosine can be salvaged to form purine nucleotides via ubiquitous adenosine kinase, which does not require PRPP as cofactor. AMP can then be converted to ATP, or via AMP deaminase to IMP, which can then be used to synthesize GTP.
Figure 2.
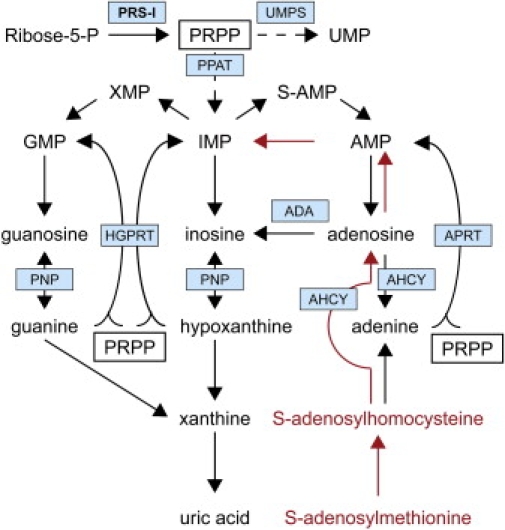
Simplified Overview of the Purine Metabolism Pathway
The scheme is derived from KEGG Pathways hsa00230 (purine metabolism) and hsa00240 (pyrimidine metabolism).12 Indicated in red is the alternative pathway to replenish purine nucleotides via SAM. Enzymes central in this review are highlighted by blue boxes. PRS-I is printed in bold. Boxes show the essential role of PRPP in the purine metabolism. Dashed arrows indicate multiple intermediate steps that are not shown. PRPP is also used for the de novo synthesis of pyrimidines, as indicated by the arrow to UMP, the central intermediate in pyrimidine pathway. In addition, PRPP is essential for pyridine nucleotide (NAD/NADP) synthesis (not shown).
AHCY is ubiquitously and highly expressed throughout the human body (BioGPS;76 UniGene expression profile). Notably, the phenotype of a patient with two complementary mutations that result in AHCY deficiency (MIM 180960) resembles that of PRPS1-related disorders (Table 1).91 The patient presented with delayed psychomotor development accompanied by hypotonia and a loss of deep tendon reflexes that is likely the result of the reported abnormal myelination. However, the patient also exhibited symptoms that are not present in patients with PRPS1-related disorders, namely progressive myopathy and mild chronic hepatitis. This difference may be explained by the low PRPS1 expression levels in muscle and liver. Taken together, the phenotype suggests that AHCY activity is particularly important for energy-requiring tissues, such as the nervous system, muscle, and liver. This may be because AHCY plays an important role in ATP provision, independent of PRS-I or PRS-II, by recycling adenosine.
Recently, a patient with HPRT deficiency was shown to clearly benefit neurologically from SAM administration without untoward side effects,92 which supports the hypothesis that brain purine nucleotide levels can be elevated and clinical symptoms alleviated by SAM therapy. We have begun oral supplementation with SAM at a dose of 20 mg/kg/day in the two affected boys with Arts syndrome from the Australian family (J.C., unpublished data; trial is approved by the Ethics and Drug Committees of the Children's Hospital at Westmead, Sydney, Australia). They appear to have benefited from this therapy. Prior to commencing SAM treatment, the older of the two boys spent a total of 180 days over a time span of 84 months in the hospital, including two admissions that required mechanical ventilation in a pediatric intensive care unit. In the 33 months since the onset of the treatment, he only had one 5 day admission to a general ward. His younger brother was admitted to the hospital for 82 days in the 84-month period prior to commencement of the therapy but has required no admissions over the last 33 months since starting SAM treatment. In addition, the progression of other symptoms like the ataxia and hearing impairment appear to have been stabilized. Until now, there has been no apparent need for supplementation with uridine to compensate for theoretically reduced de novo synthesis of pyrimidine nucleotides.
Conclusions
Both gain-of-function and loss-of-function mutations have now been identified in PRPS1. Gain-of-function mutations result primarily in a loss of feedback regulation and hence PRS-I “superactivity,” as assayed in fibroblasts and lymphoblasts. However, these mutations also result in a loss of activity in postmitotic cells, such as erythrocytes, and perhaps neuronal cells.26 Predictions of the effect of the gain-of-function mutations on the structure of the PRS-I protein by computer-assisted molecular modeling suggest that this loss of activity may be caused by a lower stability of the PRS hexamer that surpasses the loss of feedback regulation in these cells. Loss-of-function mutations cause DFN2, CMTX5, and Arts syndrome.2,5,6 DFN2 and CMTX5 seem to be milder forms of PRS-I deficiency, because only hearing loss and/or peripheral neuropathy are reported. In contrast, patients with Arts syndrome have neuropathy of the central nervous system as well as a defective immune system. Taken together, the clinical and molecular findings suggest that the four PRPS1 disorders discovered to date are part of the same disease spectrum. Additional neurophysiological investigations may reveal further phenotypical overlap or differences between the PRPS1 spectrum diseases. In general, the neurological phenotype in PRPS1 disorders may result primarily from GTP depletion and possibly reduced levels of other nucleotides, including ATP. We propose that the immune deficiency in patients with Arts syndrome is predominantly caused by impaired nucleic acid synthesis in hematopoietic cells, resulting in flawed hematopoietic differentiation. Because PRPS1 loss-of-function mutations in Arts syndrome give rise to a very severe disorder with early death, deletions of the gene may be embryonically lethal.
SAM supplementation in the diet may alleviate some of the symptoms of the patients with PRPS1 loss-of-function mutations by replenishing purine nucleotides independent of PRPP production. An open-label clinical trial in the two affected Australian brothers is currently under way and appears to have improved the health of the patients, although it is too early to draw significant conclusions. Patients with DFN2 and CMTX5 and mildly affected carrier females from the original Arts syndrome may also benefit from SAM supplementation in their diet.
Web Resources
The URLs for data presented herein are as follows:
BioGPS: The Gene Portal Hub, http://biogps.gnf.org/
Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathways, http://www.genome.jp/kegg/
Online Mendelian Inheritance in Man, http://www.ncbi.nlm.nih.gov/Omim/
UniGene Expression Profile, http://www.ncbi.nlm.nih.gov/unigene/
Yet Another Scientific Artificial Reality (YASARA) Program, http://www.yasara.org/
Acknowledgments
We are grateful to Christian Gilissen (Department of Human Genetics, Radboud University Nijmegen Medical Centre, The Netherlands) for the expression profiling data analysis. S.B.N. is supported by VENI grant 700.58.410 from the Netherlands Organization for Scientific Research.
References
- 1.Sperling O., Eilam G., Sara-Persky-Brosh, De Vries A. Accelerated erythrocyte 5-phosphoribosyl-1-pyrophosphate synthesis. A familial abnormality associated with excessive uric acid production and gout. Biochem. Med. 1972;6:310–316. doi: 10.1016/0006-2944(72)90017-8. [DOI] [PubMed] [Google Scholar]
- 2.Kim H.J., Sohn K.M., Shy M.E., Krajewski K.M., Hwang M., Park J.H., Jang S.Y., Won H.H., Choi B.O., Hong S.H. Mutations in PRPS1, which encodes the phosphoribosyl pyrophosphate synthetase enzyme critical for nucleotide biosynthesis, cause hereditary peripheral neuropathy with hearing loss and optic neuropathy (cmtx5) Am. J. Hum. Genet. 2007;81:552–558. doi: 10.1086/519529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg R.N., Chutorian A. Familial opticoacoustic nerve degeneration and polyneuropathy. Neurology. 1967;17:827–832. doi: 10.1212/wnl.17.9.827. [DOI] [PubMed] [Google Scholar]
- 4.Arts W.F., Loonen M.C., Sengers R.C., Slooff J.L. X-linked ataxia, weakness, deafness, and loss of vision in early childhood with a fatal course. Ann. Neurol. 1993;33:535–539. doi: 10.1002/ana.410330519. [DOI] [PubMed] [Google Scholar]
- 5.de Brouwer A.P., Williams K.L., Duley J.A., van Kuilenburg A.B., Nabuurs S.B., Egmont-Petersen M., Lugtenberg D., Zoetekouw L., Banning M.J., Roeffen M. Arts syndrome is caused by loss-of-function mutations in PRPS1. Am. J. Hum. Genet. 2007;81:507–518. doi: 10.1086/520706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lui X., Han D., Li J., Han B., Ouyang X., Cheng J., Li X., Jin Z., Wang Y., Bitner-Glindzicz M. Loss-of-function mutations in the PRPS1 gene cause a type of nonsyndromic X-linked sensorineural deafness, DFN2. Am. J. Hum. Genet. 2009;86:65–71. doi: 10.1016/j.ajhg.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kornberg A., Lieberman I., Simms E.S. Enzymatic synthesis and properties of 5-phosphoribosylpyrophosphate. J. Biol. Chem. 1955;215:389–402. [PubMed] [Google Scholar]
- 8.Hartman S.C., Buchanan J.M. Biosynthesis of the purines. XXI. 5-Phosphoribosylpyrophosphate amidotransferase. J. Biol. Chem. 1958;233:451–455. [PubMed] [Google Scholar]
- 9.Lieberman I., Kornberg A., Simms E.S. Enzymatic synthesis of pyrimidine nucleotides; orotidine-5′-phosphate and uridine-5′-phosphate. J. Biol. Chem. 1955;215:403–451. [PubMed] [Google Scholar]
- 10.Preiss J., Handler P. Biosynthesis of diphosphopyridine nucleotide. II. Enzymatic aspects. J. Biol. Chem. 1958;233:493–500. [PubMed] [Google Scholar]
- 11.Yen R.C., Raivio K.O., Becker M.A. Inhibition of phosphoribosylpyrophosphate synthesis in human fibroblasts by 6-methylthioinosinate. J. Biol. Chem. 1981;256:1839–1845. [PubMed] [Google Scholar]
- 12.Kanehisa M., Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fox I.H., Kelley W.N. Human phosphoribosylpyrophosphate synthetase. Distribution, purification, and properties. J. Biol. Chem. 1971;246:5739–5748. [PubMed] [Google Scholar]
- 14.Switzer R.L., Sogin D.C. Regulation and mechanism of phosphoribosylpyrophosphate synthetase. V. Inhibition by end products and regulation by adenosine diphosphate. J. Biol. Chem. 1973;248:1063–1073. [PubMed] [Google Scholar]
- 15.Li S., Lu Y., Peng B., Ding J. Crystal structure of human phosphoribosylpyrophosphate synthetase 1 reveals a novel allosteric site. Biochem. J. 2007;401:39–47. doi: 10.1042/BJ20061066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roth D.G., Shelton E., Deuel T.F. Purification and properties of phosphoribosyl pyrophosphate synthetase from rat liver. J. Biol. Chem. 1974;249:291–296. [PubMed] [Google Scholar]
- 17.Kita K., Ishizuka T., Ishijima S., Sonoda T., Tatibana M. A novel 39-kDa phosphoribosylpyrophosphate synthetase-associated protein of rat liver. Cloning, high sequence similarity to the catalytic subunits, and a negative regulatory role. J. Biol. Chem. 1994;269:8334–8340. [PubMed] [Google Scholar]
- 18.Tatibana M., Kita K., Taira M., Ishijima S., Sonoda T., Ishizuka T., Iizasa T., Ahmad I. Mammalian phosphoribosyl-pyrophosphate synthetase. Adv. Enzyme Regul. 1995;35:229–249. doi: 10.1016/0065-2571(94)00017-w. [DOI] [PubMed] [Google Scholar]
- 19.Pillai R.S., Bhattacharyya S.N., Filipowicz W. Repression of protein synthesis by miRNAs: How many mechanisms? Trends Cell Biol. 2007;17:118–126. doi: 10.1016/j.tcb.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 20.Kawahara Y., Zinshteyn B., Sethupathy P., Iizasa H., Hatzigeorgiou A.G., Nishikura K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science. 2007;315:1137–1140. doi: 10.1126/science.1138050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seitz H., Royo H., Bortolin M.L., Lin S.P., Ferguson-Smith A.C., Cavaillé J. A large imprinted microRNA gene cluster at the mouse Dlk1-Gtl2 domain. Genome Res. 2004;14:1741–1748. doi: 10.1101/gr.2743304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poy M.N., Eliasson L., Krutzfeldt J., Kuwajima S., Ma X., Macdonald P.E., Pfeffer S., Tuschl T., Rajewsky N., Rorsman P., Stoffel M. A pancreatic islet-specific microRNA regulates insulin secretion. Nature. 2004;432:226–230. doi: 10.1038/nature03076. [DOI] [PubMed] [Google Scholar]
- 23.Blow M.J., Grocock R.J., van Dongen S., Enright A.J., Dicks E., Futreal P.A., Wooster R., Stratton M.R. RNA editing of human microRNAs. Genome Biol. 2006;7:R27. doi: 10.1186/gb-2006-7-4-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luciano D.J., Mirsky H., Vendetti N.J., Maas S. RNA editing of a miRNA precursor. RNA. 2004;10:1174–1177. doi: 10.1261/rna.7350304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang W., Chendrimada T.P., Wang Q., Higuchi M., Seeburg P.H., Shiekhattar R., Nishikura K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 2006;13:13–21. doi: 10.1038/nsmb1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Becker M.A. Phosphoribosylpyrophosphate synthetase and the regulation of phosphoribosylpyrophosphate production in human cells. Prog. Nucleic Acid Res. Mol. Biol. 2001;69:115–148. doi: 10.1016/s0079-6603(01)69046-9. [DOI] [PubMed] [Google Scholar]
- 27.Sperling O., Boer P., Persky-Brosh S., Kanarek E., De Vries A. Altered kinetic property of erythrocyte phosphoribosylpsyrophosphate synthetase in excessive purine production. Rev. Eur. Etud. Clin. Biol. 1972;17:703–706. [PubMed] [Google Scholar]
- 28.Zoref E., De Vries A., Sperling O. Mutant feedback-resistant phosphoribosylpyrophosphate synthetase associated with purine overproduction and gout. Phosphoribosylpyrophosphate and purine metabolism in cultured fibroblasts. J. Clin. Invest. 1975;56:1093–1099. doi: 10.1172/JCI108183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Becker M.A., Puig J.G., Mateos F.A., Jimenez M.L., Kim M., Simmonds H.A. Inherited superactivity of phosphoribosylpyrophosphate synthetase: Association of uric acid overproduction and sensorineural deafness. Am. J. Med. 1988;85:383–390. doi: 10.1016/0002-9343(88)90591-8. [DOI] [PubMed] [Google Scholar]
- 30.Nyhan W.L., James J.A., Teberg A.J., Sweetman L., Nelson L.G. A new disorder of purine metabolism with behavioral manifestations. J. Pediatr. 1969;74:20–27. doi: 10.1016/s0022-3476(69)80004-1. [DOI] [PubMed] [Google Scholar]
- 31.Simmonds H.A., Webster D.R., Lingam S., Wilson J. An inborn error of purine metabolism, deafness and neurodevelopmental abnormality. Neuropediatrics. 1985;16:106–108. doi: 10.1055/s-2008-1052552. [DOI] [PubMed] [Google Scholar]
- 32.Christen H.J., Hanefeld F., Duley J.A., Simmonds H.A. Distinct neurological syndrome in two brothers with hyperuricaemia. Lancet. 1992;340:1167–1168. doi: 10.1016/0140-6736(92)93202-x. [DOI] [PubMed] [Google Scholar]
- 33.Becker M.A., Raivio K.O., Bakay B., Adams W.B., Nyhan W.L. Variant human phosphoribosylpyrophosphate synthetase altered in regulatory and catalytic functions. J. Clin. Invest. 1980;65:109–120. doi: 10.1172/JCI109640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Becker M.A., Losman M.J., Wilson J., Simmonds H.A. Superactivity of human phosphoribosyl pyrophosphate synthetase due to altered regulation by nucleotide inhibitors and inorganic phosphate. Biochim. Biophys. Acta. 1986;882:168–176. doi: 10.1016/0304-4165(86)90151-0. [DOI] [PubMed] [Google Scholar]
- 35.Becker M.A., Smith P.R., Taylor W., Mustafi R., Switzer R.L. The genetic and functional basis of purine nucleotide feedback-resistant phosphoribosylpyrophosphate synthetase superactivity. J. Clin. Invest. 1995;96:2133–2141. doi: 10.1172/JCI118267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Akaoka I., Fujimori S., Kamatani N., Takeuchi F., Yano E., Nishida Y., Hashimoto A., Horiuchi Y. A gouty family with increased phosphoribosylpyrophosphate synthetase activity: Case reports, familial studies, and kinetic studies of the abnormal enzyme. J. Rheumatol. 1981;8:563–574. [PubMed] [Google Scholar]
- 37.Becker M.A., Kostel P.J., Meyer L.J. Human phosphoribosylpyrophosphate synthetase. Comparison of purified normal and mutant enzymes. J. Biol. Chem. 1975;250:6822–6830. [PubMed] [Google Scholar]
- 38.Becker M.A., Losman M.J., Itkin P., Simkin P.A. Gout with superactive phosphoribosylpyrophosphate synthetase due to increased enzyme catalytic rate. J. Lab. Clin. Med. 1982;99:495–511. [PubMed] [Google Scholar]
- 39.Becker M.A., Losman M.J., Rosenberg A.L., Mehlman I., Levinson D.J., Holmes E.W. Phosphoribosylpyrophosphate synthetase superactivity. A study of five patients with catalytic defects in the enzyme. Arthritis Rheum. 1986;29:880–888. doi: 10.1002/art.1780290710. [DOI] [PubMed] [Google Scholar]
- 40.Ahmed M., Taylor W., Smith P.R., Becker M.A. Accelerated transcription of PRPS1 in X-linked overactivity of normal human phosphoribosylpyrophosphate synthetase. J. Biol. Chem. 1999;274:7482–7488. doi: 10.1074/jbc.274.11.7482. [DOI] [PubMed] [Google Scholar]
- 41.Becker M.A., Taylor W., Smith P.R., Ahmed M. Overexpression of the normal phosphoribosylpyrophosphate synthetase 1 isoform underlies catalytic superactivity of human phosphoribosylpyrophosphate synthetase. J. Biol. Chem. 1996;271:19894–19899. doi: 10.1074/jbc.271.33.19894. [DOI] [PubMed] [Google Scholar]
- 42.Becker M.A. Hyperuricemia and Gout. In: Valle D., editor. The Online Metabolic and Molecular Bases of Inherited Disease (OMMBID) McGraw-Hill; New York: 2008. chapter 205. [Google Scholar]
- 43.Becker M.A. Patterns of phosphoribosylpyrophosphate and ribose-5-phosphate concentration and generation in fibroblasts from patients with gout and purine overproduction. J. Clin. Invest. 1976;57:308–318. doi: 10.1172/JCI108282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wortmann R.L. Chapter 353: Disorders of purine and pyrimidine metabolism. In: Fauci A.S., Braunwald E., Kasper D.L., Hauser S.L., Longo D.L., Jameson J.L., Loscalzo J., editors. Harrison's Principles of Internal Medicine, 17e. McGraw-Hill Professional; New York: 2010. [Google Scholar]
- 45.Becker M.A., Ahmed M. Cell type-specific differential expression of human PRPP synthetase (PRPS) genes. Adv. Exp. Med. Biol. 2000;486:5–10. doi: 10.1007/0-306-46843-3_2. [DOI] [PubMed] [Google Scholar]
- 46.Carrel L., Willard H.F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434:400–404. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- 47.Kremer H., Hamel B.C., van den Helm B., Arts W.F., de Wijs I.J., Sistermans E.A., Ropers H.H., Mariman E.C. Localization of the gene (or genes) for a syndrome with X-linked mental retardation, ataxia, weakness, hearing impairment, loss of vision and a fatal course in early childhood. Hum. Genet. 1996;98:513–517. doi: 10.1007/s004390050250. [DOI] [PubMed] [Google Scholar]
- 48.Kim H.J., Hong S.H., Ki C.S., Kim B.J., Shim J.S., Cho S.H., Park J.H., Kim J.W. A novel locus for X-linked recessive CMT with deafness and optic neuropathy maps to Xq21.32-q24. Neurology. 2005;64:1964–1967. doi: 10.1212/01.WNL.0000163768.58168.3A. [DOI] [PubMed] [Google Scholar]
- 49.Cui B., Zhang H., Lu Y., Zhong W., Pei G., Kong X., Hu L. Refinement of the locus for non-syndromic sensorineural deafness (DFN2) J. Genet. 2004;83:35–38. doi: 10.1007/BF02715827. [DOI] [PubMed] [Google Scholar]
- 50.Manolis E.N., Eavey R.D., Sangwatanaroj S., Halpin C., Rosenbaum S., Watkins H., Jarcho J., Seidman C.E., Seidman J.G. Hereditary postlingual sensorineural hearing loss mapping to chromosome Xq21. Am. J. Otol. 1999;20:621–626. [PubMed] [Google Scholar]
- 51.Tyson J., Bellman S., Newton V., Simpson P., Malcolm S., Pembrey M.E., Bitner-Glindzicz M. Mapping of DFN2 to Xq22. Hum. Mol. Genet. 1996;5:2055–2060. doi: 10.1093/hmg/5.12.2055. [DOI] [PubMed] [Google Scholar]
- 52.Eriksen T.A., Kadziola A., Bentsen A.K., Harlow K.W., Larsen S. Structural basis for the function of Bacillus subtilis phosphoribosyl-pyrophosphate synthetase. Nat. Struct. Biol. 2000;7:303–308. doi: 10.1038/74069. [DOI] [PubMed] [Google Scholar]
- 53.Tang W., Li X., Zhu Z., Tong S., Li X., Zhang X., Teng M., Niu L. Expression, purification, crystallization and preliminary X-ray diffraction analysis of human phosphoribosyl pyrophosphate synthetase 1 (PRS1) Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006;62:432–434. doi: 10.1107/S1744309106009067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nyhan W.L. Disorders of purine and pyrimidine metabolism. Mol. Genet. Metab. 2005;86:25–33. doi: 10.1016/j.ymgme.2005.07.027. [DOI] [PubMed] [Google Scholar]
- 55.Ross B.M., Moszczynska A., Blusztajn J.K., Sherwin A., Lozano A., Kish S.J. Phospholipid biosynthetic enzymes in human brain. Lipids. 1997;32:351–358. doi: 10.1007/s11745-997-0044-x. [DOI] [PubMed] [Google Scholar]
- 56.Becroft D.M., Phillips L.I., Simmonds A. Hereditary orotic aciduria: Long-term therapy with uridine and a trial of uracil. J. Pediatr. 1969;75:885–891. doi: 10.1016/s0022-3476(69)80318-5. [DOI] [PubMed] [Google Scholar]
- 57.Lehninger A. The biosynthesis of nucleotides. In: Lehninger A., editor. Biochemistry. Worth Publishers; New York, NY: 1978. pp. 729–746. [Google Scholar]
- 58.Eells J.T., Spector R. Purine and pyrimidine base and nucleoside concentrations in human cerebrospinal fluid and plasma. Neurochem. Res. 1983;8:1451–1457. doi: 10.1007/BF00965000. [DOI] [PubMed] [Google Scholar]
- 59.Winn H.R., Park T.S., Curnish R.R., Rubio R., Berne R.M. Incorporation of adenosine and its metabolites into brain nucleotides. Am. J. Physiol. 1980;239:H212–H219. doi: 10.1152/ajpheart.1980.239.2.H212. [DOI] [PubMed] [Google Scholar]
- 60.Kelley W.N., Levy R.I., Rosenbloom F.M., Henderson J.F., Seegmiller J.E. Adenine phosphoribosyltransferase deficiency: A previously undescribed genetic defect in man. J. Clin. Invest. 1968;47:2281–2289. doi: 10.1172/JCI105913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sidi Y., Gelvan I., Brosh S., Pinkhas J., Sperling O. Guanine nucleotide metabolism in red blood cells: The metabolic basis for GTP depletion in HGPRT and PNP deficiency. Adv. Exp. Med. Biol. 1989;253A:67–71. doi: 10.1007/978-1-4684-5673-8_10. [DOI] [PubMed] [Google Scholar]
- 62.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 63.Hall A. G proteins and small GTPases: Distant relatives keep in touch. Science. 1998;280:2074–2075. doi: 10.1126/science.280.5372.2074. [DOI] [PubMed] [Google Scholar]
- 64.Luo L. Actin cytoskeleton regulation in neuronal morphogenesis and structural plasticity. Annu. Rev. Cell Dev. Biol. 2002;18:601–635. doi: 10.1146/annurev.cellbio.18.031802.150501. [DOI] [PubMed] [Google Scholar]
- 65.Nikolic M. The role of Rho GTPases and associated kinases in regulating neurite outgrowth. Int. J. Biochem. Cell Biol. 2002;34:731–745. doi: 10.1016/s1357-2725(01)00167-4. [DOI] [PubMed] [Google Scholar]
- 66.Skaper S.D., Moore S.E., Walsh F.S. Cell signalling cascades regulating neuronal growth-promoting and inhibitory cues. Prog. Neurobiol. 2001;65:593–608. doi: 10.1016/s0301-0082(01)00017-x. [DOI] [PubMed] [Google Scholar]
- 67.Ropers H.H. X-linked mental retardation: Many genes for a complex disorder. Curr. Opin. Genet. Dev. 2006;16:260–269. doi: 10.1016/j.gde.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 68.Fairbanks L.D., Jacomelli G., Micheli V., Slade T., Simmonds H.A. Severe pyridine nucleotide depletion in fibroblasts from Lesch-Nyhan patients. Biochem. J. 2002;366:265–272. doi: 10.1042/BJ20020148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ernst M., Zametkin A.J., Matochik J.A., Pascualvaca D., Jons P.H., Hardy K., Hankerson J.G., Doudet D.J., Cohen R.M. Presynaptic dopaminergic deficits in Lesch-Nyhan disease. N. Engl. J. Med. 1996;334:1568–1572. doi: 10.1056/NEJM199606133342403. [DOI] [PubMed] [Google Scholar]
- 70.Lloyd K.G., Hornykiewicz O., Davidson L., Shannak K., Farley I., Goldstein M., Shibuya M., Kelley W.N., Fox I.H. Biochemical evidence of dysfunction of brain neurotransmitters in the Lesch-Nyhan syndrome. N. Engl. J. Med. 1981;305:1106–1111. doi: 10.1056/NEJM198111053051902. [DOI] [PubMed] [Google Scholar]
- 71.Wong D.F., Harris J.C., Naidu S., Yokoi F., Marenco S., Dannals R.F., Ravert H.T., Yaster M., Evans A., Rousset O. Dopamine transporters are markedly reduced in Lesch-Nyhan disease in vivo. Proc. Natl. Acad. Sci. USA. 1996;93:5539–5543. doi: 10.1073/pnas.93.11.5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Neve K.A., Seamans J.K., Trantham-Davidson H. Dopamine receptor signaling. J. Recept. Signal Transduct. Res. 2004;24:165–205. doi: 10.1081/rrs-200029981. [DOI] [PubMed] [Google Scholar]
- 73.Niederwieser A., Blau N., Wang M., Joller P., Atarés M., Cardesa-Garcia J. GTP cyclohydrolase I deficiency, a new enzyme defect causing hyperphenylalaninemia with neopterin, biopterin, dopamine, and serotonin deficiencies and muscular hypotonia. Eur. J. Pediatr. 1984;141:208–214. doi: 10.1007/BF00572762. [DOI] [PubMed] [Google Scholar]
- 74.Barisic N., Claeys K.G., Sirotković-Skerlev M., Löfgren A., Nelis E., De Jonghe P., Timmerman V. Charcot-Marie-Tooth disease: A clinico-genetic confrontation. Ann. Hum. Genet. 2008;72:416–441. doi: 10.1111/j.1469-1809.2007.00412.x. [DOI] [PubMed] [Google Scholar]
- 75.Stendel C., Roos A., Deconinck T., Pereira J., Castagner F., Niemann A., Kirschner J., Korinthenberg R., Ketelsen U.P., Battaloglu E. Peripheral nerve demyelination caused by a mutant Rho GTPase guanine nucleotide exchange factor, frabin/FGD4. Am. J. Hum. Genet. 2007;81:158–164. doi: 10.1086/518770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Su A.I., Wiltshire T., Batalov S., Lapp H., Ching K.A., Block D., Zhang J., Soden R., Hayakawa M., Kreiman G. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cerutti A. The regulation of IgA class switching. Nat. Rev. Immunol. 2008;8:421–434. doi: 10.1038/nri2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kaida A., Ariumi Y., Baba K., Matsubae M., Takao T., Shimotohno K. Identification of a novel p300-specific-associating protein, PRS1 (phosphoribosylpyrophosphate synthetase subunit 1) Biochem. J. 2005;391:239–247. doi: 10.1042/BJ20041308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rebel V.I., Kung A.L., Tanner E.A., Yang H., Bronson R.T., Livingston D.M. Distinct roles for CREB-binding protein and p300 in hematopoietic stem cell self-renewal. Proc. Natl. Acad. Sci. USA. 2002;99:14789–14794. doi: 10.1073/pnas.232568499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bartholdi D., Roelfsema J.H., Papadia F., Breuning M.H., Niedrist D., Hennekam R.C., Schinzel A., Peters D.J. Genetic heterogeneity in Rubinstein-Taybi syndrome: Delineation of the phenotype of the first patients carrying mutations in EP300. J. Med. Genet. 2007;44:327–333. doi: 10.1136/jmg.2006.046698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Roelfsema J.H., White S.J., Ariyürek Y., Bartholdi D., Niedrist D., Papadia F., Bacino C.A., den Dunnen J.T., van Ommen G.J., Breuning M.H. Genetic heterogeneity in Rubinstein-Taybi syndrome: Mutations in both the CBP and EP300 genes cause disease. Am. J. Hum. Genet. 2005;76:572–580. doi: 10.1086/429130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Girot R., Hamet M., Perignon J.L., Guesnu M., Fox R.M., Cartier P., Durandy A., Griscelli C. Cellular immune deficiency in two siblings with hereditary orotic aciduria. N. Engl. J. Med. 1983;308:700–704. doi: 10.1056/NEJM198303243081207. [DOI] [PubMed] [Google Scholar]
- 83.Giblett E.R., Ammann A.J., Wara D.W., Sandman R., Diamond L.K. Nucleoside-phosphorylase deficiency in a child with severely defective T-cell immunity and normal B-cell immunity. Lancet. 1975;1:1010–1013. doi: 10.1016/s0140-6736(75)91950-9. [DOI] [PubMed] [Google Scholar]
- 84.Van De Wiele C.J., Vaughn J.G., Blackburn M.R., Ledent C.A., Jacobson M., Jiang H., Thompson L.F. Adenosine kinase inhibition promotes survival of fetal adenosine deaminase-deficient thymocytes by blocking dATP accumulation. J. Clin. Invest. 2002;110:395–402. doi: 10.1172/JCI15683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moore E.C., Hurlbert R.B. Regulation of mammalian deoxyribonucleotide biosynthesis by nucleotides as activators and inhibitors. J. Biol. Chem. 1966;241:4802–4809. [PubMed] [Google Scholar]
- 86.Reichard P. Control of deoxyribonucleotide synthesis in vitro and in vivo. Adv. Enzyme Regul. 1972;10:3–16. doi: 10.1016/0065-2571(72)90003-9. [DOI] [PubMed] [Google Scholar]
- 87.Stow R.A., Bronk J.R. Purine nucleoside transport and metabolism in isolated rat jejunum. J. Physiol. 1993;468:311–324. doi: 10.1113/jphysiol.1993.sp019773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bottiglieri T. S-Adenosyl-L-methionine (SAMe): From the bench to the bedside—molecular basis of a pleiotrophic molecule. Am. J. Clin. Nutr. 2002;76:1151S–1157S. doi: 10.1093/ajcn/76/5.1151S. [DOI] [PubMed] [Google Scholar]
- 89.Palella T.D., Andres C.M., Fox I.H. Human placental adenosine kinase. Kinetic mechanism and inhibition. J. Biol. Chem. 1980;255:5264–5269. [PubMed] [Google Scholar]
- 90.Smolenski R.T., Fabianowska-Majewska K., Montero C., Duley J.A., Fairbanks L.D., Marlewski M., Simmonds H.A. A novel route of ATP synthesis. Biochem. Pharmacol. 1992;43:2053–2057. doi: 10.1016/0006-2952(92)90161-b. [DOI] [PubMed] [Google Scholar]
- 91.Baric I., Fumic K., Glenn B., Cuk M., Schulze A., Finkelstein J.D., James S.J., Mejaski-Bosnjak V., Pazanin L., Pogribny I.P. S-adenosylhomocysteine hydrolase deficiency in a human: A genetic disorder of methionine metabolism. Proc. Natl. Acad. Sci. USA. 2004;101:4234–4239. doi: 10.1073/pnas.0400658101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Glick N. Dramatic reduction in self-injury in Lesch-Nyhan disease following S-adenosylmethionine administration. J. Inherit. Metab. Dis. 2006;29:687. doi: 10.1007/s10545-006-0229-8. [DOI] [PubMed] [Google Scholar]
- 93.Roessler B.J., Golovoy N., Palella T.D., Heidler S., Becker M.A. Identification of distinct PRS1 mutations in two patients with X-linked phosphoribosylpyrophosphate synthetase superactivity. Adv. Exp. Med. Biol. 1991;309B:125–128. doi: 10.1007/978-1-4615-7703-4_28. [DOI] [PubMed] [Google Scholar]
- 94.García-Pavía P., Torres R.J., Rivero M., Ahmed M., García-Puig J., Becker M.A. Phosphoribosylpyrophosphate synthetase overactivity as a cause of uric acid overproduction in a young woman. Arthritis Rheum. 2003;48:2036–2041. doi: 10.1002/art.11058. [DOI] [PubMed] [Google Scholar]