

Abstract
DNA not only transmits genetic information but can also serve as a versatile supramolecular scaffold. Here we describe a strategy for the synthesis and replication of DNA displaying hundreds of substituents using directed evolution of polymerase function by short-patch compartmentalized self-replication (spCSR) and the widely used fluorescent dye labeled deoxinucleotide triphosphates Cy3-dCTP and Cy5-dCTP as substrates. In just two rounds of spCSR selection, we have isolated a polymerase that allows the PCR amplification of double stranded DNA fragments up to 1kb, in which all dC bases are substituted by its fluorescent dye-labeled equivalent Cy3- or Cy5-dC. The resulting “CyDNA” displays hundreds of aromatic heterocycles on the outside of the DNA helix and is brightly colored and highly fluorescent. CyDNA also exhibits significantly altered physicochemical properties compared to standard B-form DNA, including loss of silica and intercalating dye binding, resistance to cleavage by some endonucleases, an up to 40% increased apparent diameter as judged by atomic force microscopy and organic phase partitioning during phenol extraction. CyDNA also displays very bright fluorescence enabling significant signal gains in microarray and microfluidic applications. CyDNA represents a step toward a long-term goal of the encoded synthesis of DNA-based polymers of programmable and evolvable sequence and properties.
DNA is unique among polymers in that it can be synthetically and enzymatically manipulated with atomic precision, assembled according to well-defined rules into structures and devices with predictable geometry,(1) evolved to bind diverse compounds and catalyze chemical reactions,(2) and co-opted as a supramolecular scaffold to arrange chemical groups in space to promote chemical reactivity(3) or photophysical interactions.(4) The latter is particularly attractive as a means to expand the chemical and functional diversity of nucleic acids as the physicochemical and optical properties of the four canonical bases only span a narrow range. While a great deal of chemical diversity is accessible through solid-phase synthesis, the synthesis of longer oligomers (>100nts) remains challenging. This has spurred the development of substituted nucleotide triphosphates that can be incorporated and replicated by polymerases. Particularly successful have been substitutions at the C5-position of the pyrimidine bases (dU or dC). These retain Watson−Crick base-pairing and project substituents into the major groove with relatively minor interference with DNA structure and polymerase interaction especially for small substitutents.5−7 Consequently a great variety of C5-substitutions have been explored and found to be compatible with at least sporadic incorporation (reviewed in refs (5,8, and 9)). Of outstanding importance among this class of analogs are fluorescent dye-labeled nucleotides, which enable core technologies of modern biology, e.g., next-generation sequencing, microarrays, and single-molecule techniques. However, despite considerable work to optimize reaction conditions10−13 or substrate properties by variation of fluorophore and/or linker chemistry,13−17 they have remained challenging polymerase substrates especially at high density substitution, presumably due to steric clashes between the large heterocyclic dye substituents and the polymerase active site.
We reasoned that the tailoring of the polymerase active site for high-density incorporation of fluorescent dye labeled nucleotides would not only yield DNA probes that enhance the scope of imaging applications but provide a route to the synthesis, replication, and evolution of polymers with potentially novel properties due to the large array of aromatic heterocycles displayed on the DNA scaffold. Polymerases have previously been successfully engineered by design18−21 screening22−24 and various selection approaches.26−28 These studies have uncovered significant plasticity in the polymerase active site for the acceptance of noncognate chemistries (reviewed in refs (5 and 9)). Polymerases have also previously been engineered for the improved incorporation of fluorescent dye-labeled nucleotide triphosphates.(19) However, Cy-dye labeled nucleotide triphosphates have remained poor polymerase substrates at high substitution density.
We had previously developed a strategy for the evolution of polymerases, called “compartmentalized self-replication” (CSR).(28) CSR is based on a simple feedback loop, in which a polymerase replicates only its own encoding gene. Compartmentalization within the aqueous compartments of a water-in-oil (w/o) emulsion(29) serves to isolate individual self-replication reactions from each other. This segregation of self-replication into discrete, physically separate compartments is critical to ensure the linkage of genotype and phenotype during CSR and ensures that each polymerase replicates only its own encoding gene to the exclusion of those in other compartments. In such a system adaptive gains directly (and proportionally) translate into genetic amplification of the encoding gene. CSR has proven a powerful method for the directed evolution of polymerase function including thermostability, resistance to inhibitors, expanded substrate spectrum, and the ability to replicate damaged DNA or hydrophobic base analogues.28,30−32 However, CSR makes rigorous demands on the catalytic efficiency and processivity of polymerases precluding its direct application to chemically challenging substrates. In order to reduce the adaptive burden and increase the sensitivity and versatility of the method, we devised a modification of the CSR method called short-patch CSR (spCSR),(33) in which only a short, defined segment of the polymerase gene is replicated and evolved (Figure 1a) and which allows direct selection for the utilization of challenging polymerase substrates.
Figure 1.

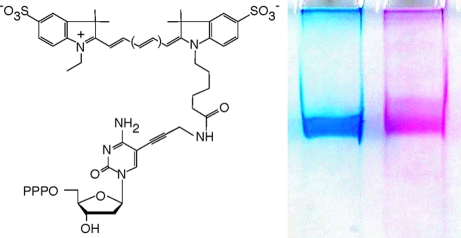
(a) Principle of short-patch CSR (spCSR). spCSR is based on a simple feedback loop, in which a polymerase replicates a short segment (a patch) of its own encoding gene as defined by the flanking primer selection (half arrows). Compartmentalization in a water-in-oil emulsion serves to isolate individual self-replication reactions from each other. In such a system adaptive gains translate into genetic amplification of the encoding gene. Two independent emulsion compartments are shown. Polymerases (Pol1 (left compartment)) that are capable of utilizing Cy5-dCTP are able to replicate, i.e., produce “offspring”, while polymerases like Pol2 (right compartment) that are unable to utilize it disappear from the gene pool. (b) Structures of Cy3-dCTP (left) and Cy5-dCTP (right), showing the cyanine dyes attached to the 5′ position of cytidine via a flexible linker. Cy3-dCTP differs from Cy5-dCTP only by the length of the polymethine bridge separating the indolene heterocycles. PPP stands for triphosphate.
Here we describe the application of spCSR for the rapid generation of polymerases capable of efficient incorporation and replication of fluorescent dye substituted nucleotides into DNA. Starting from the widely used cyanine dye (Cy3 and Cy5) substituted deoxinucleotide triphosphates (Figure 1b) as model compounds and a repertoire of mutants of the polB-family polymerase from Pyrococcus furiousus (Pfu), we isolated E10, a polymerase capable of complete substitution of dCTP with Cy3- or Cy5-dCTP in PCR in just two rounds of spCSR selection. The resulting highly Cy-dye substituted DNA, decorated with hundreds of fluorophores per DNA molecule (“CyDNA”), displayed strikingly altered physicochemical properties as well as bright fluorescence. We also describe the characterization of the properties of CyDNA by atomic force microscopy and gel electrophoresis and explore its utility as fluorescent probes in microarray and microfluidics applications.
Results
The Cyanine-dye labeled nucleotide triphosphates Cy3- and Cy5-dCTP are among the standard labeling reagents used in microarray and other multicolor hybridization applications due to their bright, photostable and spectrally resolved fluorescence. However, Cy3- and Cy5-dCTP (Figure 1b) proved exceedingly difficult polymerase substrates at full substitution, and initial attempts at selection from previously described polymerase repertoires31−33 were unsuccessful (not shown). B-family polymerases had previously been identified as having favorable properties with respect to the incorporation of substituted nucleotides.10,12,34,35 We chose the polB polymerase from Pyrococcus furiosus (Pfu),(36) which is widely used in PCR applications as it proved relatively straightforward to clone and express in E. coli K12 strains with only mild toxicity (unlike, e.g., other polBs such as VENT or 9°N (not shown)). Due to the relative ease of high efficiency transformation of K12 strains compared to B-strains (like the commonly used BL21), the latter is an important parameter expediting library construction and selection. Pfu also displayed favorable properties in model CSR selections but only after inclusion of several additives (DTT, BSA, formamide (FA), RNaseA, see the Supporting Information, Materials & Methods) to improve compatibility with classical Span80-based w/o-emulsions.28,29 Presumably these serve i.a. to prevent oxidation of free cysteine thiols in the Pfu structure by peroxide contaminants present in the Span80 surfactant (DTT),(37) to enhance replication and reduce interference from bulk cellular nucleic acids (FA, RNase) and to buffer interaction of the polymerase with the hydrophobic surfactant shell (BSA). To enhance incorporation of unnatural nucleotides and increase processivity we also disabled the 3′-5′ exonuclease-(38) and uracil-stalling functions(39) yielding Pfu variant (V93Q, D141A, E143A), henceforth referred to as Pfuexo-, as a scaffold for library construction and spCSR seicction.
We anticipated that accommodation of the bulky Cy-labeled nucleotide analogues would require remodeling of the polymerase active site. In order to improve accommodation of the dye-labeled substrate we initially targeted the A-motif(40) (and adjacent residues), a conserved sequence motif in the polymerase active site (Figure 2a) involved in both catalysis and recognition of the incoming deoxinucleotide triphosphate (dNTP) substrate. We diversified the sequence region of E399-I415 using a combination of random mutagenesis (avoiding the catalytic aspartate D405) of the central A-motif and synthetic shuffling(41) (i.e., encoding phylogenetic diversity) of the immediate flanking sequences (Figure 2b). Polymerases were selected for self-replication with complete replacement of dCTP with Cy5-dCTP by spCSR, requiring the replication of 460 nucleotides (nts) for each strand and the incorporation and replication of 191 Cy5-dCs in round 1.
Figure 2.

(a) Structural model of the active site of a polB-family polymerase (RB69, PDB: 1IG9(42)). The polypeptide chain is shown as a ribbon diagram overlaid with a transparent surface model. Primer and template strands are shown in orange and purple, incoming nucleotide triphosphate in elemental colors, and gray spheres represent the two catalytic Mg2+ ions. Regions corresponding to the A-motif are colored yellow, and those for the C-motif cyan. Also shown is the conserved B-motif (green). (b) For round 1 selection, diversity was focused on the A-motif and vicinity (399−415) comprising random mutation spike at the core of the A-motif (lime) as well as phylogenetic diversity in the adjacent sequences (purple). For round 2, successful clones from round 1 (e.g., 15) were diversified in the C-motif region (cyan), and selected for replication of A- and C-motif (399−546) yielding polymerase E10 (selected mutations in red).
380 selected variants from round 1 were screened by Polymerase-ELISA(34) and ranked for their ability to incorporate 4 consecutive Cy5-dCTPs or Cy3-dCTPs. Polymerase-ELISA identified 4 mutant polymerases with significantly enhanced ability to incorporate either Cy3-dCTPs or Cy5-dCTPs compared with wild-type Pfuexo-: A23 (N400D, I401L, R407I), AH12 (E399D, N400G, I401L, V402A, R407I, Q572H), 55 (N400G, R407I) and in particular 15 (V337I, E399D, N400G, R407I) (Figure 3a).
Figure 3.

(a) Polymerase ELISA. Shown are the hairpin substrate and activities of round 1 clones (15, A23, 55, AH12) and round 2 clones (9, 10, E10, 23). Clones were chosen i.a. on the their ability to incorporate both Cy3- and Cy5-dCTPs with comparable efficiency. Synthesis through the G8 template stretch was detected by incorporation of Digoxigenin-11-dUTP.(34) (b) PAGE gel of a 0.4kb PCR amplification (70% GC) with complete replacement of dCTP with either Cy5- or Cy3-dCTP comparing selected clones to wild-type Pfuexo- (Pfu). Only E10 (and to a lesser extent A3) are able to perform PCR amplification with both dyes. Visible spectrum absorption by the 287 cyanine dyes incorporated (on both strands) colors the DNA fragment brightly pink (Cy3) or blue (Cy5).
These four clones were diversified in another region of the polymerase active site in proximity to the incoming dNTP substrate (V537−Y546) comprising the conserved C-motif region,(40) which together with the A-motif harbors the catalytic triad of aspartates (D405, D541, D543), which are essential for catalysis and substrate binding (Figure 2). We again used a combination of spiked random mutation of conserved residues (excluding the catalytically important aspartates (D541, D543)) and synthetic shuffling(41) of phylogenetically diverse flanking sequences to optimize the percentage of active clones in our library (Figure 2b). For round 2, polymerase variants were selected for the ability to amplify a much larger, more challenging 780bp fragment encompassing both A- and C-motif diversity and requiring the incorporation and replication of 312 Cy5-dCs on both strands. Selected clones were ranked by Polymerase-ELISA for their ability to incorporate 8 consecutive Cy5-dCTPs or Cy3-dCTPs. Twenty positive clones were isolated and analyzed in more detail. These were found to be highly uniform, with mutations at a single residue (Y546) predominating (either Y546H or Y546L (20/20)).
In both ELISAs we not only screened for the ability to incorporate Cy5-dCTP or Cy3-dCTP but also for the ability to use them both with comparable efficiency. As with all directed evolution experiments, spCSR also yielded a significant number of “specialists”, i.e. polymerases that had selectively adapted only to the selection substrate Cy5-dCTP and were unable to incorporate Cy3-dCTP with comparable efficiency (not shown).
Only clones fulfilling these criteria (e.g., 9, 10, E10, 23, Figure 3a) were expressed, purified, and screened for the ability to PCR amplify DNA fragments with full replacement of dCTP by Cy3- or Cy5-dCTP in the PCR mix. The best clones were challenged with the amplification of a 70% GC 0.4kb fragment comprising 289 fluorophore dyes on both strands and resulting products were resolved by polyacrylamide gel electrophoresis (PAGE) (Figure 3b). This screen identified Pfu variant E10 (V337I, E399D, N400D, R407I, Y546H) (Figure 2b) as the most promising polymerase capable of processive synthesis of brightly colored, highly fluorescent DNA in PCR, in which every dC is replaced by Cy3- or Cy5-dC (henceforth called CyDNA).
Expansion of the substrate spectrum of a polymerase can be accompanied by reduced fidelity, in particular when it involves the accommodation of a bulky, distorting substrate in the active site.(43) We therefore determined the fidelity of E10 in PCR both with standard dNTPs as well as with complete replacement of dCTP by either Cy3- or Cy5-dCTP by sequencing of amplification products and compared it to the fidelity of the parent Pfuexo- enzyme (Table 1).
Table 1. Polymerase Fidelity.
| polymerase | PCR substrates | mutations/kb | mutation rate/bp/doublinga |
|---|---|---|---|
| Pfuexo- | dNTPs | 1.1 | 4.4 × 10−5 |
| (4.7 × 10−5)b | |||
| E10 | dNTPs | 0.4 | 1.6 × 10−5 |
| (2.6 without additivesc) | (1.04 × 10−4)c | ||
| Pfuexo- | dNTPsb | 2.7 | 6.0 × 10−5 |
| E10 | dATP, dGTP, dTTP, Cy3-dCTP | 4.3 | 9.6 × 10−5 |
| E10 | dATP, dGTP, dTTP, Cy5-dCTP | 4.9 | 1.1 × 10−4 |
Corrected for the number of doublings (PCR cycles).
As determined by a lacZ reversion assay.(44)
In the absence of additives (1% formamide, 10% glycerol, 10 μg/mL RNase, 1 mM DTT).
Sequencing of amplification products from both E10 and Pfuexo- revealed that the enhanced incorporation and replication of bulky fluorescent dye substituted nucleotides by E10 does not appear to significantly impair fidelity with standard dNTPs. The error rate of E10 (4 × 10−4 per base-pair (bp)) was comparable to that measured for Pfuexo- (1.1 × 10−3 per bp). Similar results were independently obtained using a well-established lacZ forward mutation assay (M. Arana, PH, T. Kunkel, unpublished results). However, unlike Pfuexo-, the E10 error rate was dependent on the inclusion of the additives (formamide, glycerol, RNase, DTT) used in the selection of E10 and increased more than 6-fold when they were omitted.
Replacement of dCTP by Cy3- or Cy5-dCTP in PCR resulted in an error rate (4.9 × 10−3 per bp (Cy5-dCTP)) approximately 2-fold higher than that of Pfuexo- (2.4 × 10−3 per bp) utilizing standard dNTPs under the same conditions (with dC → dA or dT mutations predominating (not shown)). When corrected for the number of total PCR cycles, the error rates for both Pfuexo- and E10 for PCR with standard dNTPs and for E10 for PCR with replacement dCTP by Cy3- or Cy5-dCTP are in excellent agreement with previously obtained measurements for the error rate of Pfuexo- using a lacI reversion assay (Table 1).(44)
These mild effects on polymerase fidelity are not unexpected as CSR inherently selects for polymerase fidelity. All self-replication must proceed with a minimal fidelity, as first recognized by Manfred Eigen and formulated as the “error threshold”, defining the minimal level of replication fidelity below which genetic information would be irretrievably corrupted.(45) In other words, polymerases that are too error-prone will introduce too many mutations into their own gene during CSR with mostly deleterious effects on the fitness of their “offspring”. It is clear that this error threshold is to some degree context dependent. In the present case, where self-replication includes regions in the polymerase active site that are absolutely essential for catalysis, particularly stringent selection for fidelity would be expected.
High-density display of the large cyanine dye heterocycles on the DNA scaffold significantly altered the physicochemical properties of resulting DNA (CyDNA). While CyDNA could still be by resolved by gel electrophoresis (Figure 3b), unlike normal DNA it displayed almost no binding to silica resins,(46) did not stain with ethidium bromide, and partitioned to the organic phase in a phenol extraction (Figure 4a).
Figure 4.

(a) Organic phase partitioning of CyDNA is shown for Cy3-DNA (left) and Cy5-DNA (right). Essentially 100% partioning occurs in the presence of 150 mM NaCl (the yellow color of the phenol phase is due to addition of 8-hydroxyquinoline to prevent oxidation). (b) Agarose gel electrophoresis of CyDNA restriction digests. Both Cy3- and Cy5-DNA are resistant to cleavage by the restriction endonuclease DdeI but are cut by MseI. Cy5-DNA displays no fluorescence despite ethidium bromide staining, while Cy3-DNA fluorescence is due to UV excitation (and seen to the same extent in ethidium bromide free gels (not shown)). m is ϕX HaeIII marker.
We therefore wondered if CyDNA would still adopt a canonical double helical structure or if the presence of the heterocyclic dyes had promoted a structural transition. To probe CyDNA structure we initially used restriction endonucleases, which are sensitive probes of noncanonical DNA conformations such as those which occur under torsional strain.(47) Indeed, some like DdeI (C’TNAG) failed to cut CyDNA, presumably due to steric hindrance by bulky Cyanine dyes, while others, notably MseI (T’TAA), cut CyDNA efficiently (Figure 4b). This indicates that at least the local regions of AT-sequence in Cy-DNA adopt a canonical B-form conformation. To get a more global picture of CyDNA conformation, we used atomic force microscopy (AFM) imaging of single CyDNA molecules. AFM unequivocally revealed contours consistent with a double-stranded conformation over the whole length of the CyDNA fragments with only minor deviations (<7%) in both average contour length and stiffness (persistence length) compared to canonical B-form DNA (Figure 5, Supporting Information, Table 1). However, AFM also revealed a substantially increased contour height of CyDNA that was increased by up to 40% compared to native DNA for Cy5-DNA (Figure 5), presumably due to dense packing of fluorophore heterocycles on the outside of the DNA helix.
Figure 5.

Atomic force microscopy (AFM) imaging of mica-adsorbed identical DNA (left), Cy3-DNA (middle), and Cy5-DNA (right) fragments. A putative DNA or CyDNA molecule can be seen at the center of each image. Histogram of AFM contour heights for n = 40 molecules for DNA (black), Cy3-DNA (red), and Cy5-DNA (green). Contour heights (width) of Cy3-DNAs or Cy5-DNAs were 20% or 40% respectively larger than DNA, while contour lengths were unchanged. Average contour lengths and heights were obtained by fitting a Gaussian to the histogram.
High-density fluorophore labeling of DNA probes should translate into high photon yields, increasing the sensitivity and expanding the dynamic range in fluorescent hybridization applications. Having established a methodology for the synthesis of CyDNA displaying a fluorophore at every dC base, we went on to assess whether such high-density fluorophore labeling of DNA probes was still compatible with specific target hybridization. We first performed model microarray experiments with randomly spotted targets interspersed with nonspecific targets (salmon-sperm DNA). These experiments revealed highly specific binding to target DNA with bright fluorescence and no nonspecific binding to either the glass surface or salmon-sperm DNA (Figure 6).
Figure 6.

Specific binding of CyDNA measured by a model array comprising a dilution series (200−12.5 ng/μL) of probe molecules (Pfu sequence and sheared salmon sperm DNA. Specific hybridization of CyDNA is evident with no binding to the slide surface or salmon genomic DNA.
Next we examined if the high-density labeling of DNA probes using E10 would translate into a higher fluorescence signal. When we compared fluorescent Cy-probes synthesized by E10 with those synthesized by Pfuexo- with 10% Cy3 and Cy5-dCTP (the maximum tolerated by Pfuexo-), we obtained an up to 4.5-fold increased fluorescence signal from the E10 probes. Surprisingly, we found that fluorescence was affected by labeling densities and was, in fact, improved by reducing labeling densities from 100% to 50% for Cy5-dCTP (Figure 7) and by white-light excitation (not shown). The effect is likely to be due to reabsorption and re-emission effects, causing red-shifting of the excitation and emission maxima in CyDNA probes.
Figure 7.

Scatter plot of fluorescence signals from cohybridization of DNA fragments labeled with Cy3- or Cy5-dCTP using Pfuexo- (red) or E10 (blue, green) on the model array (Figure 6). Fragments labeled using E10 display an up to 4.5-fold higher fluorescence signal for Cy3 (blue) or up to 2.5-fold higher fluorescence signal for Cy5 (green). High-density labeling (100%) quenches Cy5 (but not Cy3) fluorescence and reverses signal gains.
Melting temperature analysis of CyDNA revealed a slightly lower Tm (0.1kb CyDNA fragment (70% GC) 70 Cy-dyes): Cy3-DNA: 90 °C, Cy5-DNA: 88 °C) compared to native DNA (DNA: 91 °C)—as previously observed with highly Cy3-dU modified oligonucleotide probes(48)—as well as slightly less cooperative melting behavior (Supporting Information Figure 1). We therefore modified hybridization temperatures and wash stringencies for future microarray experiments accordingly (see Materials and Methods). This improved CyDNA fluorescence signal without compromising specificity (not shown).
When we compared fluorescence signals of state-of-the art commercial microarray probes (generated by random priming with Klenow polymerase) with CyDNA probes generated using E10, we discovered probe length as another critical parameter affecting fluorescence signals. Standard labeling protocols using Klenow yield very short probe fragments (<0.15kb) due to aborted elongation at homopolymeric dC runs. In contrast, due to its proficiency at incorporating Cy labeled dNTPs, E10 yields CyDNA probes up to 3kb in length (Supporting Information, Figure 2a). To determine the influence of probe length on relative fluorescence signal, we prepared DNA probes of defined length and labeled them with Cy3- and Cy5-dCTP using either Klenow or E10. Then we compared them in cohybridization experiments, whereby equivalent amounts of the same length Cy3-Klenow and Cy5-E10 probes (and vice versa) are hybridized to the same array targets. Indeed, we found that for short fragments E10 labeled probes gave on average an up to 32-fold higher fluorescence signal than Klenow labeled probes, whereas for longer probes E10 yielded only a 2−4-fold gain (Figure 8).
Figure 8.

Box plot showing the influence of fragment length on fluorescence signal. The fluorescence signal of E10 labeled CyDNA (blue/teal) was measured relative to Klenow labeled DNA (lime/red) prepared from templates of the same length (270bp/1.3kb) for Cy3 (left panel) and Cy5 (right panel). E10270 (teal) yielded a 34-fold higher Cy3 signal (or 20-fold higher Cy5 signal than Klenow270 (red)) whereas the longer E101300 fragment (blue) resulted in only a 3.6-fold higher Cy3 signal (2.5-fold higher Cy5 signal) than Klenow1300 (lime). Boxes describe the interquartile range, lines across the box the median and central cross the average. Error bars define the signal spread. While E10 labeled probes can provide enormous signal gains, fragment length is a crucial parameter.
To realize more of the potential signal gains of CyDNA for microarray applications, we therefore had to develop a strategy to reduce probe length. However, CyDNA proved resistant to DNase I digestion. Fragmentation using shearing forces only yielded fragments of 300−500 nts (not shown). We therefore developed an alternative enzymatic fragmentation method based on sporadic dUTP (10%) incorporation, followed by uracil excision using Uracil DNA Glycosylase (UDG) and strand cleavage using human apurinic/apyrmidinic endonuclease I (APE-1), which cleaves the phosphodiester backbone of DNA immediately 5′ to the abasic site generated by UDG. Although still larger than Klenow probes, the resulting CyDNA fragments were now consistently smaller than 200 nts and showed 6−8 fold higher fluorescence (compared to standard Klenow labeled probes) in a model microarray experiment (Figure 9a).
Figure 9.

(a) Model array showing cohybridization of fragmented CyDNA (E10) with Klenow labeled DNA (K): K Cy3/E10 Cy5 (left); K Cy3/K Cy5 (middle); E10 Cy3/K Cy5 (right). E10 yields 8- (Cy3) or 6-fold (Cy5) higher signals as evident by the red (E10 Cy5 (left)) and green (E10 Cy3 (right))-shifted fluorescence signal. (b) Comparative genome hybridization array (aCGH) of human genomic DNA (m (male) vs f (female)). Average fluorescence signal of aCGH probes for Cy3m vs Cy5f DNA as well as dye-swap control (Cy3f vs Cy5m) for Klenow labeling, (Klenow*, blue), Klenow or E10 labeling of adaptor linked DNA (white (K), orange (E10)). E10 labeling yields an up to 4-fold higher signal.
Next we applied our combined strategies to a 30′000 feature comparative genome hybridization (CGH) array experiment. Fragmentation of human genomic DNA labeled with E10 proved more challenging and yielded larger probes (average ca. 300 nts (Supporting Information, Figure 2b)) and consequently smaller yet still significant signal gains of up to 4-fold in the CGH array compared to state-of-the-art Klenow labeling (Figure 9b, Supporting Information, Tables 2 and 3, and SI Figure 5). We anticipate that further optimization of CyDNA fragmentation protocols together with customized filter optics and light sources (see above) should realize significant further signal gains in microarray applications.
We also attempted to observe single CyDNA molecules in motion within a microfluidic flowcell device calibrated using single-particle detection of quantum dots. CyDNA PCR products (0.27kb) labeled with either 100% Cy5-dCTP (102 Cy5) or 50% Cy3-dCTP (±51 Cy3), at near single molecule dilution, were coinjected into a fused silica capillary and imaged at a fixed point inside the lumen of the capillary. The bright fluorescence of CyDNA allowed us to observe CyDNA molecules as discrete peaks of Cy3 and Cy5 fluorescence, comparable in signal to that observed from single quantum dots (Supporting Information Figure 3). The frequency of events was consistent with the detection of single CyDNA molecules. While we cannot completely exclude the possibility that we observe the simultaneous detection of multiple CyDNA molecules or aggregates thereof, we note that peaks with mixed Cy3/Cy5 intensity ratios were not observed. We further note that signal intensities were in accordance with the detection of molecules carrying the expected number of Cy fluorophores consistent with major peaks corresponding to single CyDNA molecules in motion.
Discussion
DNA modifications in which substituents are placed at the 5′-position of either dU or dC are well tolerated by polymerases. This places the substituent in the mayor groove of the double helix, where interference with helix structure or minor groove hydrogen bonding to the polymerase is comparatively minimal. These design principles have been exploited both by chemists and by nature. Indeed, the majority of the known natural DNA modifications occur at the 5′-position of the pyrimidine bases ranging from the well-known epigenetic markers 5-methylcytosine (and the recently discovered 5-hydroxymethylcytosine(49)) to bases bearing sugar, amino, or carbamoyl substituents (reviewed in ref (50)). They mostly occur sporadically but, remarkably, can completely replace the canonical bases in the case of some viral genomes, such as the T-even bacteriophages (T2, T4, T6) of E. coli, where dC is completely replaced by hexosylated 5-hydroxymethylcytosine, or some B. subtilis phages (e.g., SP8, SPO1, ϕ25), where dT is completely replaced by 5-hydroxymethyluracil. Some of these modifications (e.g., α-Putrescinylthymine) are thought to improve packaging of the phage DNA (phage ϕW14, P. acidovorans(51)) into viral capsids by reducing charge density, in analogy to unnatural modifications such as zwitterionic DNA.(52) Others may serve to allow preferential replication of the modified genome by viral replicases,(50) but most alter the physicochemical properties of the viral DNA and protect it from nuclease degradation. By analogy, CyDNA also displays altered properties, which render is resistant to cleavage by some endonucleases including bovine pancreatic DNase I.
Chemists have studied a great variety of unnatural C5-substituted nucleotides and many have been successfully incorporated into DNA at low density.9,10 High-density incorporation, however, especially of deoxinucleotides bearing large (and charged) substituents, has remained challenging. To the latter group belong fluorescent dye-labeled deoxinucleotide triphosphates, which are essential components of several core technologies of modern biology including sequencing, microarrays, and fluorescent in situ hybridization (FISH). Despite systematic efforts to optimize substituent and linker chemistry, many dye labeled nucleotides have remained poor polymerase substrates, presumably due to steric incompatibility of the large substituents with the polymerase active site and/or primer-template duplex binding surface. Indeed, while high-density fluorophore labeling of DNA by polymerases has been reported in primer extension reactions,11−14 yields have generally been poor. A further challenge is presented by the replication of such polymers as the bulky substituents are now also present in the template strand, which can cause additional steric strain resulting in pausing or abortive synthesis.(16) In contrast, replication of nucleotide analogues functionalized with smaller substituents appears comparatively straightforward.53−57 The most advanced of these systems was described by Jäger and Famulok, who, through rigorous optimization of reaction conditions, achieved successful PCR amplification of short (<100bp) fragments in which all nucleotide analogues were functionalized with a range of small nonfluorescent groups.35,58 We substituted only one base (dC) but with a large, heterocyclic, charged cyanine dye substituent (Cy3-, Cy5-dC), which turned out to be a challenging substrate at full substitution. Nevertheless, two rounds of directed evolution of polymerase function by spCSR, targeting the conserved A- and C-motif regions in the polymerase active site, yielded E10 (Pfuexo-: V93Q, V337I, E399D, N400D, R407I, Y546H), a variant polymerase with a substantially enhanced capacity to utilize Cy3- and Cy5-dCTP as substrates. E10 is able to PCR amplify most sequences up to 1kb with complete substitution of dCTP by Cy3- or Cy5-dCTP. In the resulting “CyDNA” all dCs on both strands bear a large, hydrophobic heterocycle as substituent. The resulting display of hundreds of aromatic cyanine dyes on the DNA scaffold altered the physicochemical properties of CyDNA substantially and caused organic phase partitioning in phenol extractions commonly observed for proteins and lipids. Unlike “naked” DNA, but in analogy to many proteins, CyDNA is taken up by cells via the endocytic pathway (not shown). Although denatured DNA can also partition to the phenol/water interphase, we found no evidence that CyDNA is substantially denatured at ambient temperatures. On the contrary, restriction enzyme digests and, in particular, AFM strongly suggest a canonical B-form double-stranded configuration throughout.
We therefore favor an alternative explanation, whereby the presence of the hydrophobic cyanine heterocycles in the major groove disrupt hydration and DNA solvation, facilitating organic phase partitioning and the observed “salting-out” effect. The dense packing of fluorophores on the outside of the DNA helix rather than any unusual (non-B-form) conformation of CyDNA would also seem to be the most likely cause for the apparent increased diameter observed in AFM. The resulting shielding of the major groove by the organic heterocycles would be consistent with loss of binding to silica resins and of intercalating dyes such as ethidium bromide and with the protection against nuclease digestion.
π-arrays and clusters of organic chromophores and fluorophores have previously been examined because of their potential application in molecular devices and biosensors.(59) The spatial arrangement of the dye molecules on the DNA scaffold allows for novel photophysical interactions such as excimer fluorescence enhancement. In a striking example, Kool and colleagues examined a combinatorial array of chromophores displayed on a DNA backbone as C-nucleosides uncovering a wide range of emission colors.(60) We also observed significant interactions between fluorophores in CyDNA noticeable as significant red shifting of excitation and emission maxima (not shown) and a quenching of Cy5 (and to a lesser degree Cy3) fluorescence at 100% substitution. Such effects clearly depend on the precise arrangement of fluorophores in space and thus are a function of both labeling density and sequence.(61) It might therefore be possible to evolve optimal sequences for Cy-dye display on DNA to maximize photon yields. Such sequences might display brighter fluorescence than the CyDNA fragments analyzed herein, which, as single molecules displayed fluorescence yields comparable to those of single quantum dots. Thus, such sequences might be used as PCR templates to generate “homebrew” fluorescence beacons, e.g., for attachment to antibodies for fluorescence microscopy or other imaging applications.
In the context of microarrays, we found that shorter CyDNA probes exhibited a distinct signal advantage in cohybridization experiments. We suspect this to be due to two main effects. For one, the bulkier CyDNA probes display slower diffusion and hybridization kinetics than the shorter, less heavily labeled Klenow probes, thus competing ineffectively for target binding. Second, once bound, long probes provide further hybridization sites for the shorter probes directed against the same target therefore boosting their relative signal yield in cohybridization experiments (Supporting Information, Figure 4).
Given the increased bulk of the CyDNA helix, it is surprising that only four mutations (E399D, N400D, R407I, Y546H) are sufficient to give rise to the E10 phenotype (V337I does not contribute to the phenotype (not shown)). Inspection of the available polB structures indicates that residues E399 and E400 reside at the surface of the polymerase domain, in a surface loop that connects to the polymerase active site via a beta-strand also comprising R407. In previous polymerase evolution experiments e.g. for increased incorporation of ribonucleotide triphosphates,(34) distal mutations in the same loop and beta-strand motif in Taq, a polA-family polymerase, were found to fine-tune catalytic activities. Mutations at Taq S612 (polA equivalent of polB Pfu R407) in the same region were found to enhance incorporation of FITC-12-dATP,(62) a fluorescent dye-labeled nucleotide triphosphate, in which the fluorophore is attached to N7 of adenine, which also projects into the major groove.
The functional consequence of the Y546H mutation is less clear but is observed in phylogeny, for example, in the closely related polB-family polymerase 9°N, which displays a wide-ranging capacity to incorporate, extend and replicate unnatural nucleotide analogues, in particular in conjunction with the A485L mutation(20) (commercially available as TherminatorTM). However, neither Y546H, nor the combined A-motif mutations (E399D, N400D, R407I) by themselves provide a significant improvement in the synthesis and replication of Cy-dye labeled DNA (Figure 10a).
Figure 10.

To determine the relative phenotypic contributions of the selected mutations of Pfu E10 (V337I, E399D, N400G, R407I, Y546H), we determined activities of either wild-type Pfuexo- polymerase or Pfu variants comprising only A-motif mutations (A: Pfuexo- E399D, N400G, R407I)) or C-motif mutations (C: Pfuexo- Y546H) in (a) polymerase ELISA with Cy3- or Cy5-dCTP and (b) PCR with unmodified dNTPs. Both (a) Cy3- and Cy5-dCTP polymerase-ELISA and (b) PCR reveal that there is a strong synergistic epistasis between A- and C-motif mutations for the incorporation of Cy3- or Cy5-dCTP, which also results in an unexpected shortening of extension times in standard PCR compared to wild-type Pfuexo- polymerase.
Only their combination gives rise to the striking E10 phenotype allowing CyDNA synthesis and replication. Thus they display a strong and unusual epistatic interaction, whereby the selected mutations do not alter the polymerase phenotype independently but through a crucial functional interaction between the four residues during the polymerase catalytic cycle, which enables the synthesis and replication of Cy-dye labeled DNA. The same interaction also gives rise to a significant shortening of extension times in PCR amplification using standard dNTPs (Figure 10b). The iterative nature of directed evolution experiments, which has also been likened to a continuous “uphill” walk on the protein fitness landscape, commonly favors the acquisition of mutations that contribute to the phenotype in an incrementally additive way.63,64 Thus, epistatic interactions, whereby the phenotypic contribution of one mutation is significantly altered by the presence of another are usually not selected for. An exception are compensatory mutations that buffer some of the undesirable side-effects of adaptive mutations (e.g., loss of protein stability),65,66 which have been observed multiple times. Synergistic mutations, on the other hand, are very unusual and, to our knowledge, have not previously been observed in directed evolution experiments.
In conclusion, spCSR(34) allowed the evolution of a polymerase with a striking proficiency for the synthesis and replication of highly fluorophore-substituted DNA. High-density substitution of DNA with fluorophores is a potentially enabling feature for a number of applications in molecular genetics. We foresee many uses for CyDNA for example in hybridization applications such as microarrays or as fluorescent tags attached to antibodies or other proteins. E10 (or its descendants) should find applications outside fluorophore labeling such as the replication and evolution of DNA displaying a broad spectrum of chemical groups, which previously were poorly incorporated because of their bulky nature such as large organic heterocycles. We anticipate a variety of such DNA-based polymers tailored to display a wide range of such chemical functionalities on the nucleic acid scaffold. CyDNA offers a glimpse of the potential range of physicochemical and photophysical properties such polymers may exhibit.
The Cy3- and Cy5-dCTP structures were incorrect in Figure 1 in the version of this article published ASAP March 17, 2010. The corrected figure was published March 22, 2010.
Supporting Information Available
Additional data on CyDNA properties and performance in array CGH as well as Materials and Methods. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Seeman N. C. Trends Biochem. Sci. 2005, 30, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Liu J. Curr. Opin. Biotechnol. 2006, 17, 580–588. [DOI] [PubMed] [Google Scholar]
- Wrenn S. J.; Harbury P. B. Annu. Rev. Biochem. 2007, 76, 331–349. [DOI] [PubMed] [Google Scholar]
- Gao J.; Watanabe S.; Kool E. T. J. Am. Chem. Soc. 2004, 126, 12748–12749. [DOI] [PubMed] [Google Scholar]
- Brudno Y.; Liu D. R. Chem. Biol. 2009, 16, 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocek M.; Fojta M. Org. Biomol. Chem. 2008, 6, 2233–2241. [DOI] [PubMed] [Google Scholar]
- Weisbrod S. H.; Marx A. Chem. Commun. 2008, 44, 5675–5685. [DOI] [PubMed] [Google Scholar]
- Hocek M.; Fojta M. Org. Biomol. Chem. 2008, 6, 2233–2241. [DOI] [PubMed] [Google Scholar]
- Loakes D.; Holliger P. Chem. Commun. 2009, 4619–4631. [DOI] [PubMed] [Google Scholar]
- Anderson J. P.; Angerer B.; Loeb L. A. Biotechniques 2005, 38, 257–264. [DOI] [PubMed] [Google Scholar]
- Brakmann S.; Lobermann S. Angew. Chem., Int. Ed. Engl. 2001, 40, 1427–1429. [DOI] [PubMed] [Google Scholar]
- Augustin M. A.; Ankenbauer W.; Angerer B. J. Biotechnol. 2001, 86, 289–301. [DOI] [PubMed] [Google Scholar]
- Ramanathan A.; Pape L.; Schwartz D. C. Anal. Biochem. 2005, 337, 1–11. [DOI] [PubMed] [Google Scholar]
- Yu H.; Chao J.; Patek D.; Mujumdar R.; Mujumdar S.; Waggoner A. S. Nucleic Acids Res. 1994, 22, 3226–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z.; Chao J.; Yu H.; Waggoner A. S. Nucleic Acids Res. 1994, 22, 3418–3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z.; Waggoner A. S. Cytometry 1997, 28, 206–211. [PubMed] [Google Scholar]
- Lacenere C.; Garg M. K.; Stoltz B. M.; Quake S. R. Nucleosides Nucleotides Nucleic Acids 2006, 25, 9–15. [DOI] [PubMed] [Google Scholar]
- Gardner A. F.; Jack W. E. Nucleic Acids Res. 1999, 27, 2545–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner A. F.; Jack W. E. Nucleic Acids Res. 2002, 30, 605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Mitaxov V.; Waksman G. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 9491–9496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astatke M.; Ng K.; Grindley N. D.; Joyce C. M. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 3402–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M.; Baskin D.; Hood L.; Loeb L. A. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 9670–9675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick E.; Vigna K. L.; Loeb L. A. EMBO J. 2001, 20, 7303–7312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summerer D.; Marx A. Angew. Chem., Int. Ed. Engl. 2002, 41, 3620–36223516. [DOI] [PubMed] [Google Scholar]
- Summerer D.; Rudinger N. Z.; Detmer I.; Marx A. Angew. Chem., Int. Ed. Engl. 2005, 44, 4712–4715. [DOI] [PubMed] [Google Scholar]
- Jestin J. L.; Kristensen P.; Winter G. Angew. Chem., Int. Ed. 1999, 38, 1124–1127. [DOI] [PubMed] [Google Scholar]
- Xia G.; Chen L.; Sera T.; Fa M.; Schultz P. G.; Romesberg F. E. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 6597–6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghadessy F. J.; Ong J. L.; Holliger P. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 4552–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawfik D. S.; Griffiths A. D. Nat. Biotechnol. 1998, 16, 652–656. [DOI] [PubMed] [Google Scholar]
- Ghadessy F. J.; Ramsay N.; Boudsocq F.; Loakes D.; Brown A.; Iwai S.; Vaisman A.; Woodgate R.; Holliger P. Nat. Biotechnol. 2004, 22, 755–759. [DOI] [PubMed] [Google Scholar]
- d’Abbadie M.; Hofreiter M.; Vaisman A.; Loakes D.; Gasparutto D.; Cadet J.; Woodgate R.; Paabo S.; Holliger P. Nat. Biotechnol. 2007, 25, 939–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loakes D.; Gallego J.; Pinheiro V. B.; Kool E. T.; Holliger P. J. Am. Chem. Soc. 2009, 131, 14827–14837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong J. L.; Loakes D.; Jaroslawski S.; Too K.; Holliger P. J. Mol. Biol. 2006, 361, 537–550. [DOI] [PubMed] [Google Scholar]
- Jager S.; Famulok M. Angew. Chem., Int. Ed. Engl. 2004, 43, 3337–3340. [DOI] [PubMed] [Google Scholar]
- Tasara T.; Angerer B.; Damond M.; Winter H.; Dorhofer S.; Hubscher U.; Amacker M. Nucleic Acids Res. 2003, 31, 2636–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemori T.; Ishino Y.; Toh H.; Asada K.; Kato I. Nucleic Acids Res. 1993, 21, 259–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghadessy F. J.; Holliger P. Protein Eng. Des. Sel. 2004, 17, 201–204. [DOI] [PubMed] [Google Scholar]
- Derbyshire V.; Pinsonneault J. K.; Joyce C. M. Methods Enzymol. 1995, 262, 363–385. [DOI] [PubMed] [Google Scholar]
- Fogg M. J.; Pearl L. H.; Connolly B. A. Nat. Struct. Biol. 2002, 9, 922–927. [DOI] [PubMed] [Google Scholar]
- Patel P. H.; Suzuki M.; Adman E.; Shinkai A.; Loeb L. A. J. Mol. Biol. 2001, 18, 823–837. [DOI] [PubMed] [Google Scholar]
- Ness J. E.; Welch M.; Giver L.; Bueno M.; Cherry J. R.; Borchert T. V.; Stemmer W. P.; Minshull J. Nat. Biotechnol. 1999, 17, 893–896. [DOI] [PubMed] [Google Scholar]
- Franklin M. C.; Wang J.; Steitz T. A. Cell 2001, 105, 657–667. [DOI] [PubMed] [Google Scholar]
- Yan S. F.; Wu M.; Geacintov N. E.; Broyde S. Biochemistry 2004, 43, 7750–7765. [DOI] [PubMed] [Google Scholar]
- Cline J.; Braman J. C.; Hogrefe H. H. Nucleic Acids Res. 1996, 24, 3546–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eigen M. Naturwissenschaften 1971, 58, 465–523. [DOI] [PubMed] [Google Scholar]
- Vogelstein B.; Gillespie D. Proc. Natl. Acad. Sci. U.S.A. 1979, 76, 615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shouche Y. S.; Ramesh N.; Brahmachari S. K. Nucleic Acids Res. 1990, 18, 267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randolph J. B.; Waggoner A. S. Nucleic Acids Res. 1997, 25, 2923–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriaucionis S.; Heintz N. Science 2009, 324, 929–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gommers-Ampt J. H.; Borst P. FASEB J. 1995, 9, 1034–1042. [DOI] [PubMed] [Google Scholar]
- Kropinski A. M.; Bose R. J.; Warren R. A. Biochemistry 1973, 12, 151–157. [DOI] [PubMed] [Google Scholar]
- Hashimoto H.; Nelson M. G.; Switzer C. J. Am. Chem. Soc. 1993, 115, 7128–7134. [Google Scholar]
- Sakthivel K.; Barbas III C. F. Angew. Chem., Int. Ed. 1998, 110, 2998–3001. [Google Scholar]
- Gourlain T.; Sidorov A.; Mignet N.; Thorpe S. J.; Lee S. E.; Grasby J. A.; Williams D. M. Nucleic Acids Res. 2001, 29, 1898–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. E.; Sidorov A.; Gourlain T.; Mignet N.; Thorpe S. J.; Brazier J. A.; Dickman M. J.; Hornby D. P.; Grasby J. A.; Williams D. M. Nucleic Acids Res. 2001, 29, 1565–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Held H. A.; Benner S. A. Nucleic Acids Res. 2002, 30, 3857–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masud M. M.; Ozaki-Nakamura A.; Satou F.; Ohbayashi T.; Ozaki H.; Sawai H. Nucleic Acids Res. Suppl. 2001, 21–22. [DOI] [PubMed] [Google Scholar]
- Jager S.; Rasched G.; Kornreich-Leshem H.; Engeser M.; Thum O.; Famulok M. J. Am. Chem. Soc. 2005, 127, 15071–15082. [DOI] [PubMed] [Google Scholar]
- Varghese R.; Wagenknecht H. A. Chem. Commun. 2009, 2615–2624. [DOI] [PubMed] [Google Scholar]
- Gao J.; Strassler C.; Tahmassebi D.; Kool E. T. J. Am. Chem. Soc. 2002, 124, 11590–11591. [DOI] [PubMed] [Google Scholar]
- Wilson J. N.; Gao J.; Kool E. T. Tetrahedron 2007, 63, 3427–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong J.Directed evolution of DNA polymerases with altered substrate specificities; Cambridge University: Cambridge, U.K., 2004. [Google Scholar]
- Voigt C. A.; Kauffman S.; Wang Z. G. Adv. Protein Chem. 2000, 55, 79–160. [DOI] [PubMed] [Google Scholar]
- Romero P. A.; Arnold F. H. Nat. Rev. Mol. Cell. Biol. 2009, 10, 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom J. D.; Labthavikul S. T.; Otey C. R.; Arnold F. H. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 5869–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camps M.; Herman A.; Loh E.; Loeb L. A. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.