

Abstract
Transposable elements (TEs) can donate regulatory sequences that help to control the expression of human genes. The oncogene c-Myc is a promiscuous transcription factor that is thought to regulate the expression of hundreds of genes. We evaluated the contribution of TEs to the c-Myc regulatory network by searching for c-Myc binding sites derived from TEs and by analyzing the expression and function of target genes with nearby TE-derived c-Myc binding sites. There are thousands of TE sequences in the human genome that are bound by c-Myc. A conservative analysis indicated that 816–4564 of these TEs contain canonical c-Myc binding site motifs. c-Myc binding sites are over-represented among sequences derived from the ancient TE families L2 and MIR, consistent with their preservation by purifying selection. Genes associated with TE-derived c-Myc binding sites are co-expressed with each other and with c-Myc. A number of these putative TE-derived c-Myc target genes are differentially expressed between Burkitt’s lymphoma cells versus normal B cells and encode proteins with cancer-related functions. Despite several lines of evidence pointing to their regulation by c-Myc and relevance to cancer, the set of genes identified as TE-derived c-Myc targets does not significantly overlap with two previously characterized c-Myc target gene sets. These data point to a substantial contribution of TEs to the regulation of human genes by c-Myc. Genes that are regulated by TE-derived c-Myc binding sites appear to form a distinct c-Myc regulatory subnetwork.
Introduction
Almost half of the human genome sequence is made up of interspersed repeat sequences, which are remnants of formerly mobile transposable elements (TEs).1,2 These TE sequences have shaped the structure, function and evolution of their host genomes in a number of ways.3,4 For example, TEs are the source of a variety of regulatory sequences, including transcription factor binding sites (TFBS), alternative transcription start sites and small RNAs, that help to control the expression of host genes.5 The gene regulatory properties of TEs have received a great deal of attention in recent years, particularly since eukaryotic genome sequences and functional genomic datasets began accumulating over the last decade.
The ability of TEs to donate sequences that regulate nearby genes was first noticed in individual molecular genetic studies where regulatory elements were found to be located inside of repetitive sequence elements. In perhaps the first example of this kind of study, the sex-limited protein (Slp) encoding gene in mouse was shown to be regulated by androgen response elements located in the long terminal repeat sequence of an upstream endogenous retrovirus.6 An accumulation of such anecdotal cases was taken to support the possibility that TEs may have broad genome-scale effects on gene regulation.7,8 In the genomics era, three distinct classes of approaches have been taken to elucidate the regulatory contributions of TEs on the genome scale: (i) computational prediction of TE-derived regulatory sequences, (ii) identification of highly conserved TE sequences with comparative genomics and (iii) co-location of experimentally characterized regulatory sequences and TEs.
Computational analyses of TE sequences using position weight matrices that represent cis-regulatory sequence motifs have shown that TEs harbour numerous putative TFBS.9,10 These data, taken together with the genomic abundance of TEs, underscore their potential ability to regulate the expression of numerous host genes. A problem with this approach is that the ab initio prediction of cis-regulatory sequence motifs is prone to numerous false positives. To overcome this limitation, authors have used sequence shuffling, or simulation, to build null background sequence sets and then find TFBS that are over-represented among TE sequences relative to the background sets.10 Even with such a control for sequence composition in place, it is still difficult to know which of these TE-derived TFBS may actually be functionally relevant in terms of regulating the expression of host genes. It can also be difficult to distinguish between sequences that regulate the expression of the element itself versus those that regulate nearby host genes.
Comparative genomics studies have been used to help identify TE sequences that are likely to encode functions for their host genomes. The rationale behind this approach is that conserved TE sequences have been preserved by purifying selection because of their functional, presumably regulatory, importance to the host organism.11 The comparative genomics approach to the identification of TE-derived regulatory sequences was pioneered by Silva et al. who identified numerous ancient L2 and MIR intergenic TE sequences that were highly conserved among mammals and therefore likely to be functionally important.12 Since that time, a number of studies have turned up thousands of conserved non-coding sequences that are derived from TEs.13–17 These findings indicate that a substantial fraction of TE sequences in mammalian genomes have been conserved by virtue of their functional (regulatory) relevance.18 However, this evolutionary approach to the identification of TE-derived regulatory sequences is overly conservative in some cases because it will not detect regulatory sequences that are derived from relatively recently inserted, or lineage-specific, TEs. Indeed, TEs are the most dynamic and rapidly evolving sequences in eukaryotic genomes, and most TE insertions are not shared between evolutionary lineages.19 Accordingly, it has been shown that numerous experimentally characterized TE-derived regulatory sequences are not conserved between species.19–21
One of the most promising genome-scale approaches for the characterization of TE-derived regulatory sequences involves co-locating experimentally characterized regulatory elements and TE annotations on genomic sequences. This approach was first used on a relatively small scale by mapping the locations of hundreds of TFBS characterized in individual experiments to human TEs and then extrapolating to the entire human genome.22 This study suggested that thousands of human genes may be regulated by TE-derived regulatory sequences, but it was not possible to know whether this was actually the case. In order for the co-localization approach to really work on the genome-scale, high-throughput experimental data on the locations of regulatory sequences are needed. These data have become widely available in the last few years thanks to the invention of techniques like chromatin immunoprecipitation followed by microarray, ChIP-chip, or high-throughput sequencing, ChIP-Seq, analysis.23 There are now hundreds-of-thousands of experimentally characterized TFBS that have been mapped to the human genome using these techniques, and recent studies have shown that many of these sites are derived from TEs.24,25 Many of these TE-derived TFBS are lineage-specific and may define recently evolved regulatory subnetworks that elaborate on previously existing networks as is the case for p53 binding sites derived from human endogenous retroviruses.25
One particularly interesting transcription factor for which there is a human genome-wide map of binding sites is c-Myc.26 c-Myc has been reported to regulate a large set of genes,27–29 and it is considered an oncogene by virtue of its deregulation in a variety of cancers. For instance, c-Myc is markedly deregulated in lymphomas where it is over-expressed relative to normal B cells. A recent report evaluated the contribution of TEs to c-Myc binding sites on the human genome.24 These authors found that c-Myc bound regions were not statistically enriched for co-localization with any particular TE family, and based on this observation concluded that c-Myc TFBS do not reside on repeats. However, our own preliminary data revealed that numerous c-Myc bound regions were in fact derived from human TE sequences, and we wanted to further explore the relationship between c-Myc binding and TEs to address this discrepancy.
Despite the lack of enrichment for c-Myc binding sites in a particular TE class or family observed previously, we found thousands of TE-derived c-Myc binding sites in the human genome using a conservative approach that integrated data from the experimental characterization of c-Myc bound regions with c-Myc binding site motif prediction. Gene expression and gene set enrichment analyses indicate that many of these TE-derived c-Myc binding sites are likely to be functionally relevant with respect to the regulation of human gene expression. However, most genes associated with TE-derived c-Myc binding sites do not correspond to genes previously characterized as targets of c-Myc regulation. This raises the possibility that TE-derived c-Myc targets define a distinct c-Myc regulatory subnetwork.
Results and discussion
TE-derived c-Myc binding sites
We integrated experimental data on c-Myc bound genomic sequences, probabilistic transcription factor binding site (TFBS) analysis and TE genome-annotations to identify TE-derived c-Myc binding sites in the human genome. The locations of c-Myc bound human genome sequences were previously determined using genome-wide chromatin immunoprecipitation (ChIP) and paired-end-ditag (PET) sequencing on P493 B cells.26 We co-located these c-Myc bound human genome sequences with TE sequences annotated by RepeatMasker (http://www.repeatmasker.org). This analysis resulted in a set of 259 294 TE sequences co-located with c-Myc bound regions. The precise locations of TE-derived c-Myc binding sites were then determined by running the program Clover30 on the c-Myc bound TE sequences. Clover was run using two c-Myc position–frequency matrices (Fig. S1, ESI‡) with P-value thresholds of 0.01 and 0.001. This analysis resulted in a total of 4564 TE-derived c-Myc binding sites for P ≤ 0.01 and 816 TE-derived c-Myc sites for P ≤ 0.001. Thus, there is a substantial potential for human TE sequences to contribute to c-Myc gene regulatory networks. Here it should be noted that the use of Clover for the identification of specific c-Myc binding sites represents a conservative approach that eliminates many c-Myc bound human TE sequences that do not contain canonical c-Myc binding site sequence motifs. In fact, running Clover resulted in a two orders-of-magnitude reduction in the number of TE-derived c-Myc bound regions identified in the human genome. While this approach may result in the loss of some bona fide TE-derived c-Myc binding sites, it also yields increased confidence in the functional relevance of the smaller set of TE-derived sites we identified.
In order to evaluate the contribution of distinct TEs to c-Myc binding sites, we divided human TEs into 8 classes/families based on the Repbase database31 classification system: L1, L2, LINE other, Alu, MIR, SINE other, DNA and LTR. The observed numbers of individual TE insertions with c-Myc binding sites for each class/family are shown in Table 1 (P ≤ 0.01) and Table S1, ESI‡ (P ≤ 0.001), and a comparison of the observed versus expected percentages for each TE class/family are shown in Fig. 1A (P ≤ 0.01) and Fig. S2, ESI‡ (P ≤ 0.001). Members of the abundant L1 and Alu element families have lower observed than expected percentages, while L2 and MIR elements have higher than expected percentages. The relative ages of these families can be estimated by calculating the sequence divergence between individual elements and subfamily consensus sequences; younger elements have lower divergence since they inserted in the genome more recently. L1s and Alus are younger element families, many of which are primates-specific, whereas L2 and MIR are more ancient families that radiated early in mammalian evolution (Fig. 1B). In other words, relatively older TE families contribute more c-Myc binding sites than expected based on their percentage in the genome, whereas younger families contribute fewer c-Myc binding sites than expected. A similar pattern was found in a recent study that analyzed experimentally characterized human TE-derived binding sites from numerous distinct transcription factors.21 The enrichment of c-Myc TFBS in more ancient TEs is consistent with the notion that these sequences have been conserved in the genome by purifying selection based on their functional relevance.12 Nevertheless, Alu elements show the highest number of c-Myc binding sites, since they are the most numerous elements in the genome. TFBS derived from relatively young, even polymorphic in some cases, elements like Alu are of interest since they may impart lineage- or condition-specific regulatory properties on nearby genes.18–21,25 We explore this possibility later in the manuscript.
Table 1.
Number of TEs that contain c-Myc binding sites for each TE class/family
| TE class/familya | Observed numberb | Observed percentc (%) | Expected percentd (%) |
|---|---|---|---|
| L1 | 940 | 20.60 | 21.9 |
| L2 | 546 | 11.96 | 9.7 |
| LINE other | 47 | 1.03 | 1.6 |
| Alu | 994 | 21.78 | 28.1 |
| MIR | 733 | 16.06 | 13.9 |
| SINE other | 27 | 0.59 | 0.1 |
| DNA | 411 | 9.01 | 9.3 |
| LTR | 866 | 18.97 | 15.5 |
| Total | 4564 | 100.00 | 100.0 |
Name of TE classes or families.
Observed number of TEs in each class/family.
Observed percent: the observed number of TEs in each class/family divided by the total observed number (4564) of TEs containing c-Myc binding sites.
Expected percent: the total number of TEs in each class/family in human genome divided by the total number of all TEs in human genome.
Fig. 1.

Family origins and relative ages for human TEs bound by c-Myc. Observed versus expected percentages of c-Myc binding sites derived from different TE classes/families. (A) The observed percentages (blue) of TEs containing c-Myc binding sites in each TE class/family are plotted along with the expected percentages (maroon) of TEs in each class/family based on their background percentages in the human genome. (B) Percent divergence from subfamily consensus sequences for human TEs that are bound by c-Myc. The relative percentages of each of the six TE families are shown for each percent divergence bin. Younger elements have lower divergence from their consensus sequences, and older elements have higher divergence.
Regulatory effects of TE-derived c-Myc binding sites
In order to evaluate the potential regulatory effects of TE-derived c-Myc binding sites, we mapped the TE-derived sites to the vicinity of human genes and analyzed these genes’ tissue-specific expression patterns. Human genes with TE-derived c-Myc binding sites within ±10 kb were considered as potential c-Myc regulated target genes. This resulted in a total set of 1550 human genes with proximal TE-derived c-Myc binding sites. The expression patterns of these putative target genes over 79 human tissues and cell lines were compared to each other, and to the expression patterns of c-Myc, using the Novartis human gene expression atlas of Affymetrix microarray data.32
For each class/family of TEs, the expression patterns of all putative c-Myc target genes were compared using the Pearson correlation coefficient (PCC). Table 2 shows the number of target gene Affymetrix probes for each TE class/family along with the average PCCs, Z scores and P-values. All 8 TE classes/families have sets of putative c-Myc target genes that are positively and significantly co-expressed, on average, across human tissues. Target genes with Alu-derived c-Myc binding sites, the most numerous class, show the highest levels of average co-expression and the greatest statistical significance. It should be noted that while the average co-expression levels for the distinct TE class/family target gene sets are all positive and, for the most part, highly statistically significant, the average PCC values are still quite low (i.e. close to 0). This suggests that while there is certainly an enrichment for co-expressed gene pairs among the TE-derived c-Myc target genes, the total set of target genes for each class has a broad range of tissue-specific expression patterns. This is consistent with the fact that genes with proximal TE-derived c-Myc binding sites are also likely to be regulated by additional transcription factors as well as different classes of regulators such as epigenetic modifications and/or small RNAs.
Table 2.
Pearson correlation coefficients (PCC) of gene expression within each target gene class
| Target gene classa | Number of probesb | Average PCCc | Z scored | P-valuee |
|---|---|---|---|---|
| L1 | 357 | 0.027 | 31.17 | 3.85 × 10−213 |
| L2 | 297 | 0.031 | 29.63 | 7.96 × 10−193 |
| LINE other | 20 | 0.033 | 2.29 | 0.022 |
| Alu | 550 | 0.044 | 73.06 | 0 |
| MIR | 390 | 0.021 | 26.10 | 4.64 × 10−150 |
| SINE other | 17 | 0.055 | 2.32 | 0.021 |
| DNA | 205 | 0.028 | 17.57 | 4.04 × 10−69 |
| LTR | 257 | 0.021 | 16.63 | 4.13 × 10−62 |
Name of TE classes or families.
Number of Affymetrix probes corresponding to genes with c-Myc binding sites derived from TE of specific classes/families.
Average of Pearson correlation coefficients (PCC) of each pair of probes within specific TE classes/families.
Z-transformation of PCC values.
P-values indicate the significance levels of Z scores.
In order to further explore the relationship between human gene expression and the presence of TE-derived c-Myc binding sites, tissue-specific expression levels of putative target genes were compared to the expression of the regulator c-Myc. This allowed us to more directly investigate whether those target genes are actually regulated by c-Myc. To do this, we calculated the target genes’ average expression levels in each tissue and compared them with the c-Myc expression data by calculating pairwise PCCs across tissues between the TE classes/families and c-Myc. The results of the PCC analysis are shown in Table 3, and the average expression levels for TE classes/families and c-Myc across 79 tissues are shown in Fig. 2. 7 out of 8 TE class/family target gene sets show statistically significant co-expression with c-Myc. Furthermore, for these 7 TE classes/families, the average PCC values between the putative target genes with TE-derived binding sites and c-Myc are an order of magnitude greater (Table 3) than the average PCC values among all pairs of target genes (Table 2). This indicates that the target genes’ tissue-specific expression patterns are distributed around the expression pattern of c-Myc in such a way as to be more similar to c-Myc, on average, than they are to each other. This can be visually appreciated by comparing the average tissue-specific expression levels of the TE class/family target genes to the expression pattern of c-Myc (Fig. 2). Target genes with TE-derived c-Myc binding sites are clearly more highly expressed, on average, in the same tissues where c-Myc is also highly expressed. The most striking cases of c-Myc-to-target gene co-expression can be seen for both normal and cancerous T cells and B cells, including CD4+ and CD8+ T cells, CD19+ B cells and several lymphoma and leukemia cell lines (Fig. 2).
Table 3.
Pearson correlation coefficients (PCC) between expression levels of TE-derived target genes and c-Myc
| Target gene classa | PCCb | tc | P-valued |
|---|---|---|---|
| L1 | 0.37 | 3.45 | 9.18 × 10−04 |
| L2 | 0.35 | 3.28 | 1.57 × 10−03 |
| LINE other | −0.10 | −0.87 | 0.39 |
| Alu | 0.48 | 4.79 | 7.95 × 10−06 |
| MIR | 0.41 | 3.93 | 1.86 × 10−04 |
| SINE other | 0.62 | 7.00 | 8.17 × 10−10 |
| DNA | 0.34 | 3.14 | 2.41 × 10−03 |
| LTR | 0.32 | 2.94 | 4.36 × 10−03 |
Name of TE classes or families.
Pearson correlation coefficients (PCC) between the average tissue-specific expression levels of all target genes with a TE class/family and c-Myc.
PCC transformed into t-values by t = PCC × sqrt(df/(1−PCC2)) where df = 77.
P-values indicate the significance levels of t scores (following Student’s t distribution).
Fig. 2.

Average expression levels of TE-derived c-Myc target genes, and c-Myc expression levels, across 79 tissues/cell lines. Average tissue-specific expression levels are shown for TE-derived c-Myc target genes from 8 TE classes/families, and tissue-specific gene expression levels are shown for c-Myc. High expression levels are shown in red and low expression levels are shown in blue.
We performed a permutation test to more precisely identify the specific tissues where both c-Myc and the target genes with TE-derived c-Myc binding sites are over-expressed. To do this, the average tissue-specific expression levels of all target genes were computed and compared to 1000 randomly permuted (over the same gene set) tissue-specific average expression level vectors. The same analysis was done using c-Myc tissue-specific expression levels as the test set. For each tissue, the observed test set average, or c-Myc, expression level was then compared to the distribution of permuted values. There are 22 significantly (P < 0.05) over-expressed tissues among the TE c-Myc binding site target genes including the aforementioned normal and cancerous T and B cells as well as several brain tissues (Table S2 and Fig. S3, ESI‡). When the c-Myc expression levels were similarly compared to the permuted expression levels, 6 tissues were identified as significantly over-expressed, all of which were over-expressed in the TE-derived c-Myc target gene set. Taken together, the data comparing the expression patterns of the target genes and c-Myc provide an additional, and more compelling, line of expression evidence in support of the functional relevance of TE-derived c-Myc binding sites.
A TE-specific c-Myc regulatory network
To further explore the functional relevance of the human genes with TE-derived c-Myc binding sites, we compared the set of putative TE-derived c-Myc target genes to two previously published sets of genes identified to be regulated by c-Myc. Basso et al. characterized a set of 2063 c-Myc target genes by reverse engineering a c-Myc regulatory network from high-throughput gene expression data.27 Zeller et al. used literature mining to create the Myc target gene database (http://www.myccancergene.org) reporting 1697 experimentally characterized c-Myc target genes.29 We computed the overlap of these two c-Myc target gene sets with each other and the overlap of each with our own TE-derived target gene set; the statistical significance levels of the c-Myc target gene set overlaps were assessed using the hypergeometric distribution (Fig. 3). The two previously available c-Myc target gene sets have a substantial, and highly statistically significant (P < 1.8 × 10−112), overlap of 434 genes. This indicates that the distinct expression and literature-based c-Myc target gene search protocols converge on a shared core of c-Myc regulated target genes. On the other hand, the 1550 TE-derived c-Myc target genes we identified have a low, and non-significant (0.81 < P < 0.99), overlap with the previously characterized sets of genes. This result can be interpreted in two ways. It could mean that the TE-derived c-Myc target genes we identified do not represent a functionally relevant set of genes that are in fact regulated by c-Myc. This interpretation is not consistent with the expression data we report here indicating that genes with TE-derived c-Myc binding sites are co-expressed with each other and with c-Myc. The low overlap between our TE-derived target gene set and the previously published sets could also be taken to indicate that TE sequences yield a distinct and specific c-Myc regulatory network. This interpretation is consistent with previously published results indicating that TEs can provide lineage-specific regulatory sequences.18–21,25
Fig. 3.
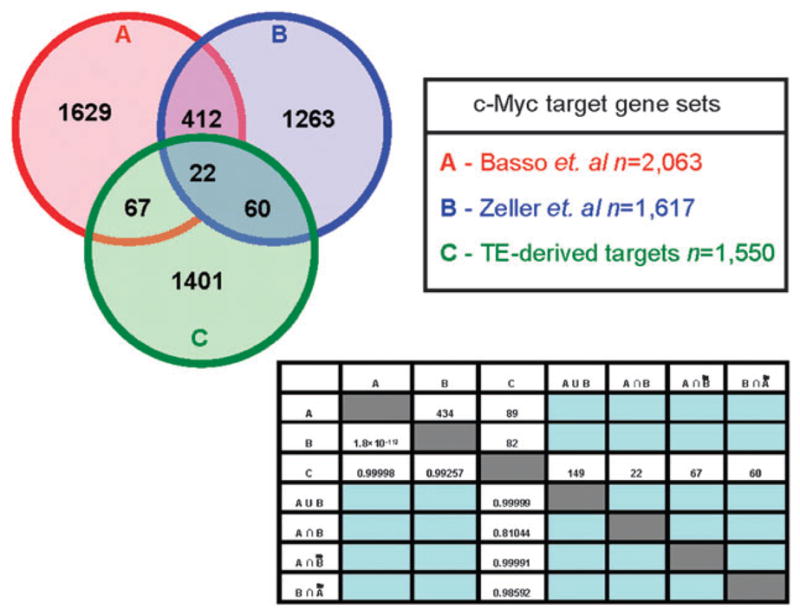
Overlap between TE-derived c-Myc target genes identified here and two previously characterized c-Myc target gene sets. Circle A represents the c-Myc target gene dataset from Basso et al.,27 circle B represents the c-Myc target gene dataset from Zeller et al.,29 and circle C represents the putative TE-derived c-Myc target genes identified here. The numbers above the diagonal of the matrix are the number of genes that overlap between two different datasets, and the numbers below the diagonal of the matrix are the significance levels (P-values) of the overlap calculated by the hypergeometric test.
In order to try and discriminate between these two possible scenarios, (i) functional irrelevance of the TE-derived c-Myc binding sites versus (ii) a TE-specific c-Myc regulatory network, we evaluated the overlap of the TE-derived c-Myc target gene set with a series of gene set collections from the molecular signatures database (MSigDB) (http://www.broad.mit.edu/gsea/msigdb/index.jsp). The MSigDB gene sets represent groups of genes with similar features or properties such as co-regulated genes, genes with similar cis-regulatory motifs and genes with similar gene ontology (GO) functional annotations.33 Thus, gene set enrichment analysis with the MSigDB can be used to evaluate whether the TE-derived c-Myc target genes have similar biological functions or regulation. The TE-derived c-Myc target genes were broken down into class/family-specific sets and run against MSigDB. This analysis resulted in numerous statistically significant gene set enrichments (Table S3, ESI‡), the most relevant of which include a number of cancer related gene modules. These data suggest that many of the TE-derived c-Myc target genes are functionally related and associated with cancer. For instance, c-Myc target genes with L2-derived binding sites are enriched for a cluster of genes with expression patterns indicative of lymphoma and immune response, based on their tissue-specific expression levels. Both MIR and LTR elements donate c-Myc binding sites to genes classified as being involved in B cell lymphoma via so-called clinical annotations, which associate microarrays with known clinical attributes. In other words, TE-derived c-Myc target genes that are from different families, and are identified with different methodologies, converge on genes that function in B cells and in cancer. In addition, DNA element-derived c-Myc target genes are enriched for genes involved in the MAPK signalling pathway, which regulates cellular response to growth factors and mediates the action of many oncogenes.
Differential expression in Burkitt’s lymphoma versus normal B cell
c-Myc is a well known oncogene that is over-expressed in a number of different cancers, particularly lymphomas.28 In light of its role in cancer, we asked whether TE-derived c-Myc target genes showed differential expression between cancer and normal cells. To do this, we used a microarray gene expression dataset, from the Oncomine database, comparing Burkitt’s lymphoma (n = 31) versus normal B cell (n = 25).27 We identified 53 TE-derived c-Myc target genes that show statistically significant (P < 0.05) differential expression between normal and cancer (Fig. 4); 16 of the c-Myc binding sites that map to these genes are derived from Alu elements. c-Myc is also known to be over-expressed in Burkitt’s lymphoma cells, and we calculated the PCC across the 56 cancer and normal cell lines for these 53 genes’ expression data with c-Myc’s to see if the differentially expressed target genes are co-regulated with c-Myc (Table S4, ESI‡). There are 32 TE-derived c-Myc target genes that show positive correlations (0.23 ≤ PCC ≤ 0.80) with c-Myc and 21 target genes with negative correlations (−0.66 ≤ PCC ≤ −0.38); all PCC are statistically significant (P < 0.05). These data indicate that TE-derived c-Myc binding sites contribute to the cancer-related expression of c-Myc regulated genes. TE-derived c-Myc target genes are both up-regulated and down-regulated in cancer, while c-Myc is over-expressed in lymphoma relative to normal B cells. This finding may be attributed to the fact that c-Myc can both positively and negatively regulate the expression of its target genes.28 The fact that the majority of correlations are positive is consistent with our results showing the overall average positive correlation between TE-derived c-Myc target genes and c-Myc (Table 3 and Fig. 2).
Fig. 4.

Differential expression of TE-derived c-Myc target genes in Burkitt’s lymphoma versus normal B cells. Each row shows the expression levels of a gene in Burkitt’s lymphoma cells (n = 31 on the left) and normal cells (n = 25 on the right); over-expression is shown in red and under-expression in green.
In order to further evaluate the function of these differentially expressed genes, gene set enrichment analysis was performed on the set of 53 TE-derived c-Myc target genes that are deregulated in Burkitt’s lymphoma. To do this, the genes were sorted according to the TE class/family of their c-Myc binding sites and each set was evaluated against the MSigDB gene sets. A number of statistically significant enrichments for cancer-related gene sets were detected, particularly for MIR and L1 elements (Table S5, ESI‡). For instance, the genes encoding ITPR1 and AKT2 bear MIR-derived c-Myc binding sites and show up in several enriched gene sets including members of the B cell antigen receptor signalling pathway genes and the gene set related to the PIP3 signalling pathway in B lymphocytes. For instance, AKT genes encode serine–threonine protein kinases that promote cell proliferation by phosphorylating targets that lead to the activation of the anti-apoptotic transcription factor NF-kB. ITPR1 encodes an intracellular channel that mediates release of calcium from the endoplasmic reticulum, which can also lead to cell proliferation via stimulation of the CALML6 protein upstream in the calcium signalling pathway. In addition, the genes LRPPC and PRKCB1 both have L1-derived c-Myc binding sites and are known to be deregulated in B cell lymphoma.
Alu elements are the single most abundant class/family of TEs that provide c-Myc binding sites to human genes, and Alu-derived c-Myc binding sites are also over-represented among the set of target genes differentially expressed between Burkitt’s lymphoma and cancer. As alluded to previously, we were particularly interested in Alu elements since they have inserted relatively recently in the human genome, are potentially polymorphic, and have a known role in several cancers.34 We investigated the Alu-derived c-Myc target genes shown to be differentially regulated between Burkitt’s lymphoma versus normal B cells and found a small set of Alu c-Myc target genes that were tightly coherent with respect to several different characteristics (Fig. 5). These genes all have Alu-derived c-Myc binding sites that are located around the 5′ transcription start site, three of which are located within the proximal ±2 kb promoter region (Fig. 5A). All of these genes are up-regulated in Burkitt’s lymphoma and positively correlated with c-Myc expression (Table 4 and Fig. 5B). The specific c-Myc binding sites in these Alu sequences are all derived from one particular location in the element suggesting that the c-Myc TFBS evolved in an ancestral sequence and was distributed by transposition, as opposed to evolving in situ after the elements inserted (Fig. 5C and Fig. S4, ESI‡). Two of the five genes have c-Myc binding sites derived from AluSg subfamily sequences and the other three have c-Myc binding sites derived from the AluSx subfamily. AluSg and AluSx are particularly young Alu subfamilies that are polymorphic (i.e. show insertion site differences) among human populations.35 It is possible that polymorphic Alu elements change the regulatory network of c-Myc between individual humans and/or between cell types. Furthermore, if a gene is brought under the control of c-Myc by an Alu insertion it could lead to changes in expression of that gene associated with oncogenesis. These recently evolved Alu-derived c-Myc binding sites exemplify TE contributions to a specific c-Myc subnetwork, consistent with our characterization of numerous novel c-Myc target genes that are associated with TE-derived binding sites.
Fig. 5.

Target genes with Alu-derived c-Myc binding sites. (A) Approximate illustration of relative positions of Alu-derived c-Myc binding sites compared with target genes’ transcriptional start sites (TSS). (B) Differential expression of Alu-derived c-Myc target genes, and c-Myc, in Burkitt’s lymphoma versus normal B cells. (C) Multiple sequence alignment of the Alu element insertions with c-Myc binding site locations indicated.
Table 4.
Differential expression of Alu-derived c-Myc target genes
| Gene symbol | Differential expression (t-value)a | Differential expression (P-value)b | Correlation with c-Mycc | P-value of correlationd |
|---|---|---|---|---|
| SLC29A1 | 11.98 | 1.4 × 10−16 | 0.77 | 1.92 × 10−12 |
| LSM1 | 6.54 | 2.3 × 10−8 | 0.50 | 4.70 × 10−5 |
| OIP5 | 5.78 | 1.9 × 10−6 | 0.48 | 9.39 × 10−5 |
| AKAP1 | 11.21 | 1 × 10−14 | 0.79 | 1.42 × 10−13 |
| DKC1 | 5.45 | 1.3 × 10−6 | 0.64 | 5.27 × 10−8 |
Gene’s differential expression in Burkitt’s lymphoma cells versus normal B cells. T-values computed by the Student’s t-test.
Significance levels (P-values) of the differential expression.
Pearson correlation coefficients between cancer versus normal expression of Alu-derived c-Myc target genes and c-Myc.
Significance levels (P-values) of the correlation.
Materials and methods
Identification of TE-derived c-Myc binding sites
The locations of experimentally characterized c-Myc bound regions, characterized previously by genome-wide chromatin immunoprecipitation (ChIP) and paired-end-tag (PET) sequencing on P493 B cells,26 were taken from the GIS ChIP-PET track in UCSC genome browser (http://www.genome.ucsc.edu/).36 The positions of TEs were taken from the RepeatMasker track in UCSC genome browser. TE and c-Myc bound regions were co-localized using the UCSC table browser tool.37 TE-derived c-Myc bound regions were analyzed with the program Clover,30 using two c-Myc binding site motif position–frequency matrices from the TRANSFAC database38 (V$MYC_01 and V$MYC_02 see Fig. S1, ESI‡) to precisely locate c-Myc binding sites. Clover uses non-parametric approach with 1000 randomizations of the search sequence to generate a score and associated P-value. Clover was run using a conservative score threshold of 6 with two P-value thresholds P < 0.01 and P < 0.001.
Human TE sequences were divided into 8 classes/families using the Repbase classification system31 implemented with RepeatMasker: L1, L2, LINE-other (LINE elements excluding L1 and L2), Alu, MIR, SINE-other (SINE elements excluding Alu and MIR), DNA and LTR. Alu elements were further divided into subfamilies and members of individual subfamilies bound by c-Myc were aligned using ClustalW39 to identify the relative locations of c-Myc binding sites.
Analysis of TE-derived c-Myc target genes
Human Refseq40 genes were identified as putative TE-derived c-Myc regulatory targets if they had TE-derived c-Myc binding sites within 10 kb of the gene boundaries. Microarray gene expression data were taken from the Novartis mammalian gene expression atlas version 2 (GNF2),32 and Affymetrix probes from GNF2 were mapped to TE-derived c-Myc target genes using the UCSC genome browser annotations. Co-expression among TE-derived c-Myc target genes, and between target genes and c-Myc, was evaluated by calculating Pearson correlation coefficients (PCC) between pairs of genes across 79 different tissues or cell lines. Statistical significance levels (P-values) of PCC values, and averages, were computed using the Z transformation. A permutation test was used to identify sets of tissues that are over-expressed for c-Myc and among all TE-derived c-Myc target genes. To do this, tissue-specific gene expression vectors were randomly shuffled for each gene and average tissue-specific expression values were calculated for all randomly shuffled genes. 1000 sets of average tissue-specific expression values were used to compute null background expression level distributions for each tissue against which the observed values were compared. All P-values were corrected for multiple tests using the Benjamini–Hochberg false discovery rate.
Differential expression of target genes in cancer versus normal cells
TE-derived c-Myc target genes were mapped to the Burkitt’s lymphoma and normal B cell microarray dataset compiled by Basso et al.27 The Oncomine database41 was used to select genes from this dataset that were determined to be differentially expressed between cancer (Burkitt’s lymphoma n = 31) versus normal B cells (n = 25) using the Student’s t-test. Co-expression values between these differentially expressed TE-derived c-Myc target genes and c-Myc, across the 56 cancer and normal B cell lines, were computed using the PCC as described previously.
Gene set enrichment and c-Myc target gene analyses
Sets of TE-derived c-Myc target genes for each TE class/family were searched against a series of gene set collections from the molecular signatures database (MSigDB)33 to evaluate their shared functional and/or regulatory features. The extent and significance of the overlaps between the set of TE-derived c-Myc target genes identified here and two previously characterized c-Myc target gene sets were evaluated using the hypergeometric distribution:
where k = number of overlapping target genes, N = number of TE-derived c-Myc target genes, n = number of previously characterized c-Myc target genes, and m = human genes not previously characterized as c-Myc targets.
Conclusions
Recently, Bourque et al. analyzed the ability of human TEs to provide transcription factor binding sites genome-wide.24 They considered high-throughput binding site data for seven transcription factors, including c-Myc analyzed here, and concluded that five of these transcription factors bind to distinct families of human TEs. However, c-Myc was not one of the families identified in their study to bind to human TEs. This can be attributed to the enrichment criteria used to characterize transcription factors as binding human TEs. Specifically, they only considered transcription factors that bind to families of TEs with higher than expected frequency based on the abundance of the TE in the genome. This approach makes sense from a quantitative perspective, but it may be overly conservative if it misses bona fide functional transcription factor binding sites derived from TEs. We found that hundreds-of-thousands of human TEs have experimental evidence of being bound by c-Myc. Furthermore, many of these TE sequences harbor canonical c-Myc binding site sequence motifs, suggesting that the binding of c-Myc to the elements is not spurious. In addition, our own functional analysis of human genes with proximal TE-derived c-Myc binding sites suggests that many of these sites may indeed be functional with respect to mediating gene regulation by c-Myc. However, definitive proof of such function will have to await experimental characterization. Hopefully, the list of gene targets and TE-derived c-Myc binding sites uncovered by our analysis can be used to stimulate investigation of the regulatory properties of human TEs.
TE sequences in the human genome provide thousands of c-Myc binding sites, and genes that bear nearby TE-derived sites show evidence for regulation by c-Myc. TE-mediated regulation of human genes by c-Myc includes changes in expression that are characteristic of the difference between cancer versus normal B cells, and TE-derived target genes encode proteins with cancer-related functions. Nevertheless, the TE-derived c-Myc target genes identified in this study do not overlap, for the most part, with previously characterized c-Myc target genes. This suggests that expansion of TE sequences may provide a mechanism for the emergence of distinct lineage-specific regulatory subnetworks.25
Supplementary Material
Acknowledgments
I. K. J. and J. W. were supported by the School of Biology, Georgia Institute of Technology. I. K. J. was supported by an Alfred P. Sloan Research Fellowship in Computational and Evolutionary Molecular Biology (BR-4839). N. J. B. was supported by the Ovarian Cancer Institute Laboratory, School of Biology, Georgia Institute of Technology. This research was supported in part by the Intramural Research Program of the NIH, NLM, NCBI. LMR was supported by Corporación Colombiana de Investigatión Agropecuaria—CORPOICA.
Footnotes
This article is part of a Molecular BioSystems themed issue on Computational and Systems Biology.
Electronic supplementary information (ESI) available: Supplementary figures, tables and human genome coordinates for TE-derived binding sites and co-located genes. See DOI: 10.1039/b908494k
Contributor Information
Jianrong Wang, Email: jwang64@gatech.edu.
Nathan J. Bowen, Email: bowen@gatech.edu.
Leonardo Mariño-Ramírez, Email: lmarino@corpoica.org.co.
I. King Jordan, Email: king.jordan@biology.gatech.edu.
References
- 1.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, et al. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 2.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, et al. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 3.Biemont C, Vieira C. Nature. 2006;443:521–524. doi: 10.1038/443521a. [DOI] [PubMed] [Google Scholar]
- 4.Kazazian HH., Jr Science. 2004;303:1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- 5.Feschotte C. Nat Rev Genet. 2008;9:397–405. doi: 10.1038/nrg2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stavenhagen JB, Robins DM. Cell. 1988;55:247–254. doi: 10.1016/0092-8674(88)90047-5. [DOI] [PubMed] [Google Scholar]
- 7.Britten RJ. Proc Natl Acad Sci U S A. 1996;93:9374–9377. doi: 10.1073/pnas.93.18.9374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Britten RJ. Gene. 1997;205:177–182. doi: 10.1016/s0378-1119(97)00399-5. [DOI] [PubMed] [Google Scholar]
- 9.Shankar R, Grover D, Brahmachari SK, Mukerji M. BMC Evol Biol. 2004;4:37. doi: 10.1186/1471-2148-4-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thornburg BG, Gotea V, Makalowski W. Gene. 2006;365:104–110. doi: 10.1016/j.gene.2005.09.036. [DOI] [PubMed] [Google Scholar]
- 11.Witherspoon DJ, Doak TG, Williams KR, Seegmiller A, Seger J, Herrick G. Mol Biol Evol. 1997;14:696–706. doi: 10.1093/oxfordjournals.molbev.a025809. [DOI] [PubMed] [Google Scholar]
- 12.Silva JC, Shabalina SA, Harris DG, Spouge JL, Kondrashovi AS. Genet Res. 2003;82:1–18. doi: 10.1017/s0016672303006268. [DOI] [PubMed] [Google Scholar]
- 13.Bejerano G, Lowe CB, Ahituv N, King B, Siepel A, Salama SR, Rubin EM, Kent WJ, Haussler D. Nature. 2006;441:87–90. doi: 10.1038/nature04696. [DOI] [PubMed] [Google Scholar]
- 14.Kamal M, Xie X, Lander ES. Proc Natl Acad Sci U S A. 2006;103:2740–2745. doi: 10.1073/pnas.0511238103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishihara H, Smit AF, Okada N. Genome Res. 2006;16:864–874. doi: 10.1101/gr.5255506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santangelo AM, de Souza FS, Franchini LF, Bumaschny VF, Low MJ, Rubinstein M. PLoS Genet. 2007;3:1813–1826. doi: 10.1371/journal.pgen.0030166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie X, Kamal M, Lander ES. Proc Natl Acad Sci U S A. 2006;103:11659–11664. doi: 10.1073/pnas.0604768103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mikkelsen TS, Wakefield MJ, Aken B, Amemiya CT, Chang JL, Duke S, Garber M, Gentles AJ, Goodstadt L, Heger A, Jurka J, Kamal M, Mauceli E, Searle SM, Sharpe T, et al. Nature. 2007;447:167–177. doi: 10.1038/nature05805. [DOI] [PubMed] [Google Scholar]
- 19.Marino-Ramirez L, Lewis KC, Landsman D, Jordan IK. Cytogenet Genome Res. 2005;110:333–341. doi: 10.1159/000084965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marino-Ramirez L, Jordan IK. Biol Direct. 2006;1:20. doi: 10.1186/1745-6150-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Polavarapu N, Marino-Ramirez L, Landsman D, McDonald JF, Jordan IK. BMC Genomics. 2008;9:226. doi: 10.1186/1471-2164-9-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jordan IK, Rogozin IB, Glazko GV, Koonin EV. Trends Genet. 2003;19:68–72. doi: 10.1016/s0168-9525(02)00006-9. [DOI] [PubMed] [Google Scholar]
- 23.Euskirchen GM, Rozowsky JS, Wei CL, Lee WH, Zhang ZD, Hartman S, Emanuelsson O, Stolc V, Weissman S, Gerstein MB, Ruan Y, Snyder M. Genome Res. 2007;17:898–909. doi: 10.1101/gr.5583007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bourque G, Leong B, Vega VB, Chen X, Lee YL, Srinivasan KG, Chew JL, Ruan Y, Wei CL, Ng HH, Liu ET. Genome Res. 2008;18:1752–1762. doi: 10.1101/gr.080663.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang T, Zeng J, Lowe CB, Sellers RG, Salama SR, Yang M, Burgess SM, Brachmann RK, Haussler D. Proc Natl Acad Sci U S A. 2007;104:18613–18618. doi: 10.1073/pnas.0703637104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeller KI, Zhao X, Lee CW, Chiu KP, Yao F, Yustein JT, Ooi HS, Orlov YL, Shahab A, Yong HC, Fu Y, Weng Z, Kuznetsov VA, Sung WK, Ruan Y, et al. Proc Natl Acad Sci U S A. 2006;103:17834–17839. doi: 10.1073/pnas.0604129103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, Califano A. Nat Genet. 2005;37:382–390. doi: 10.1038/ng1532. [DOI] [PubMed] [Google Scholar]
- 28.Pelengaris S, Khan M, Evan G. Nat Rev Cancer. 2002;2:764–776. doi: 10.1038/nrc904. [DOI] [PubMed] [Google Scholar]
- 29.Zeller KI, Jegga AG, Aronow BJ, O’Donnell KA, Dang CV. Genome Biology. 2003;4:R69. doi: 10.1186/gb-2003-4-10-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frith MC, Fu Y, Yu L, Chen JF, Hansen U, Weng Z. Nucleic Acids Res. 2004;32:1372–1381. doi: 10.1093/nar/gkh299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kohany O, Gentles AJ, Hankus L, Jurka J. BMC Bioinformatics. 2006;7:474. doi: 10.1186/1471-2105-7-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB. Proc Natl Acad Sci U S A. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deininger PL, Batzer MA. Mol Genet Metab. 1999;67:183–193. doi: 10.1006/mgme.1999.2864. [DOI] [PubMed] [Google Scholar]
- 35.Wang J, Song L, Gonder MK, Azrak S, Ray DA, Batzer MA, Tishkoff SA, Liang P. Gene. 2006;365:11–20. doi: 10.1016/j.gene.2005.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ. Nucleic Acids Res. 2004;32:D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, Voss N, Stegmaier P, Lewicki-Potapov B, Saxel H, Kel AE, et al. Nucleic Acids Res. 2006;34:D108–D110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thompson JD, Higgins DG, Gibson TJ. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maglott DR, Katz KS, Sicotte H, Pruitt KD. Nucleic Acids Res. 2000;28:126–128. doi: 10.1093/nar/28.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.