

Abstract
Plaque rupture is the main cause of myocardial infarctions and strokes. Ruptured plaques have thin, highly inflamed, and collagen-poor fibrous caps that contain elevated levels of proteases, including metalloproteinases (MMPs), which might weaken plaque caps and promote rupture. On the other hand, MMPs facilitate migration and proliferation vascular smooth muscle cells, which should promote fibrous cap stability. Given the dual effects of MMPs, therapies should selectively target harmful MMPs or the processes that cause MMP activity to rise to destructive levels.
Introduction
There are at least 23 structurally related matrix metalloproteinases (MMPs) that share a similar Zn2+-based catalytic mechanism. The substrate specificity of MMPs includes many extracellular matrix (ECM) and cell surface proteins (Nagase et al. 2006). Although most MMPs are secreted, six membrane-type MMPs are located on the cell surface (Figure 1). The activity of MMPs is regulated by transcription and translation of pro-protease genes and by pro-form activation. Inactivation by binding to four tissue inhibitors of MMPs (TIMPs) normally prevents MMP activity from becoming excessive (Figure 1) (Nagase et al. 2006). It has been suggested that overactivity of MMPs leads to destruction rather than to controlled remodeling of the ECM (Newby 2005). If this happens in the fibrous cap of atherosclerotic plaques, it could precipitate plaque rupture and hence unstable coronary syndromes, including myocardial infarction. This review considers the recent evidence supporting this proposal and its value in suggesting new treatments.
Figure 1.

Metalloproteinase production and activity. Metalloproteinases are synthesized as proforms in the rough endoplasmic reticulum (RER) and mostly secreted via the endosomal pathway. Secreted MMPs are activated by removal of a propeptide in the extracellular compartment. Membrane-type MMPs (e.g., MMP-14) are activated by furins in the endosomes and expressed on the cell surface. A few active MMPs have the ability to make the initial one-fourth, three-fourths cleavage of fibrillar collagens, whereas other MMPs cooperate in the destruction of cleaved collagen and other matrix components. Tissue inhibitors of MMPs are secreted and inactivate MMPs by blocking their catalytic sites.
Metalloproteinase Activity and Localization in Atherosclerotic Plaques
As previously reviewed (Newby 2005), only pro-MMP-2, TIMP-1, and TIMP-2 are found in normal arteries, and no MMP activity is detected by in situ zymography. Highly inflamed atheromatous plaques show increased global MMP activity (Choudhary et al. 2006), and levels of MMP-1, MMP-3, MMP-8, and MMP-9 are significantly greater in atheromatous than in fibrous human plaques (Sukhova et al. 1999, Sluijter et al. 2006). Histologic sections of rabbit and mouse atherosclerotic plaques and the rupture-prone shoulder regions of human atherosclerotic plaques (reviewed in Newby 2005, Dollery and Libby 2006) show increased levels of MMP-1, MMP-3, and MMP-9. Rupture-prone regions of human plaques also have increased MMPs 8, 11, 14, and 16. Metalloproteinases 1, 2, 8, 13, and 14 can cleave strength-giving fibrillar type-I and type-III collagens (Nagase et al. 2006). Indeed, MMP-1 and MMP-13 colocalize with cleaved collagen in plaques (Sukhova et al. 1999). Metalloproteinases 9 and 12 fragment elastin, whereas MMP-3 and MMP-7 degrade cleaved collagens and proteoglycan core proteins (Nagase et al. 2006). Hence, a large complement of MMPs acting together could in theory completely degrade the arterial ECM. Extracellular matrix degradation by MMPs could cause reduced fibrous cap thickness and collagen content, which are typical features of plaques vulnerable to rupture. Another feature of vulnerable plaques is abundance of macrophagederived foam cells, which are a prominent source of several MMPs (see below). However, endothelial and vascular smooth muscle cells (VSMCs) also secrete several MMPs (see below). Indeed, not all MMPs are markers for plaque inflammation; levels of MMP-2 are increased in fibrous rather than atheromatous human carotid plaques (Sluijter et al. 2006).
Effects of MMPs on Vascular Cells
Metalloproteinases can promote macrophage invasion (Shipley et al. 1996, Johnson et al. 2006a, 2006b), thereby increasing plaque inflammation. Metalloproteinases also promote angiogenesis (Pepper 2001), another feature associated with vulnerable plaques. Together with ECM destruction and fibrous cap thinning, these actions could precipitate plaque rupture. On the other hand, MMP-2, MMP-9, and MMP-14 promote VSMC migration and proliferation, which could increase fibrous cap thickness and promote plaque stability. As previously reviewed (Newby 2006), MMPs detach VSMC from their basement membranes before cell migration. Metalloproteinases also mediate shedding of VSMC surface cadherins; this enables translocation of the transcriptional regulator, β-catenin, to the cell nucleus and promotes VSMC proliferation. Extracellular matrix remodeling also enables signaling from cell surface integrin receptors to focal adhesions, which modulates key cell-cycle genes, including cyclin D1 and p21/p27 cyclin-dependent kinase inhibitors. In summary, MMPs have the ability to promote or prevent plaque rupture (Newby 2005). Which action predominates may depend on the spectrum of MMPs expressed, their levels of activity, and the stage of plaque development.
Insights from Experimental Animal Studies
Acute plaque ruptures are rare in mice, except perhaps in the brachiocephalic artery under extreme hypercholesterolemia (Johnson et al. 2005). Hence, increased lesion size, abundance of macrophages, and decreased content of collagen or other matrix proteins are generally used surrogate markers, by analogy with unstable human plaques.
The effects of MMP overactivity on plaque stability have been investigated by deleting TIMP genes or overexpressing individual MMPs in apoplipoprotein E knockout mice. Two studies agreed that TIMP-1 gene deletion increased vessel wall MMP activity and elastin degradation, implying that MMPs destabilize plaques (Silence et al. 2002, Lemaitre et al. 2003). On the other hand, transgenic macrophage-specific overexpression of the human collagenase, MMP-1, gene produced smaller, more stable plaques (Lemaitre et al. 2001). Similar overexpression of pro-MMP-9 had little effect on plaque stability, but overexpression of active MMP-9 induced plaque disruption, without significantly affecting lesion size or macrophage content (Gough et al. 2006). Local adenovirusmediated overexpression of pro-MMP-9 had no effect on size of early carotid lesions, but disrupted advanced lesions (de Nooijer et al. 2006). In summary, unphysiologically high levels of MMP-9 activity promoted plaque instability, especially in advanced plaques provoked by extreme hypercholesterolemia. Overexpression of active metalloelastease, MMP-12, promoted inflammation and reduced collagen in plaques in rabbits, but again only during extreme hypercholesterolemia (Liang et al. 2006).
Knocking out MMPs or modifying their substrates has also been used to define the roles of individual MMPs in the apoplipoprotein E null mouse. Expressing collagenase-resistant collagen-I or deleting the mouse collagenase, MMP-13, gene resulted in collagen accumulation giving more stable plaques (Fukumoto et al. 2004, Deguchi et al. 2005).
Knocking out the gelatinase, MMP-2, by contrast, produced less stable lesions with fewer smooth muscle cells compared with macrophages (Kuzuya et al. 2006), consistent with the ability of MMP-2 to aid migration of VSMCs. The effects of MMP-9 deficiency was less clear; one study showed reduced elastolysis in the aortic root implying greater stability (Luttun et al. 2004), but another (Johnson et al. 2005) showed increased plaque size in the brachiocephalic artery, with less collagen and more macrophages, implying less stable plaques. These conflicting results might be rationalized by the opposing effects on plaque stability of MMP-9-facilitated migration of VSMCs and macrophages.
Knockout of MMP-3 also led to discordant results; larger, more stable atherosclerotic plaques were found in the aorta (Silence et al. 2001) but larger, less stable plaques in the brachiocephalic artery (Johnson et al. 2005). Metalloproteinase 7 deletion had no effect (Johnson et al. 2005).
Metalloelastase, MMP-12, deletion caused smaller, more stable lesions in the brachiocephalic artery (Johnson et al. 2005) and reduced elastin degradation in the aortic arch (Luttun et al. 2004). Both studies suggested that MMP-12 destabilizes plaques.
Taken together, the MMP knockout experiments support the dual role of MMPs. Several MMPs had clear deleterious effects on plaque stability, consistent with the associative evidence from human plaques. The effect of MMP-12 was particularly striking. On the other hand, plaque-stabilizing effects were also noted, consistent with the known effects of MMPs on migration and proliferation of VSMCs.
Effects of Reducing MMP Activity
Adenovirus-mediated TIMP-1 overexpression decreased plaque size and macrophage content at the aortic root (Rouis et al. 1999), whereas TIMP-2 decreased plaque size and markers of plaque rupture in brachiocephalic arteries (Johnson et al. 2006a, 2006b). These encouraging data suggest that MMP inhibition can stabilize plaques. On the other hand, oral administration of synthetic MMP inhibitors with a broad spectrum of activity similar to TIMP-2 had no effect on lesion size or stability in mice (Prescott et al. 1999, Johnson et al. 2006a, 2006b). Tetracyclines, which inhibit MMP activity and expression, also had no effect (Manning et al. 2003). These broad-spectrum MMP inhibitors may prevent both the harmful and beneficial effects of MMPs and therefore have no overall effect; a narrow therapeutic window also hinders clinical application (Peterson 2006). More selective inhibitors have been generated (reviewed in Peterson 2006), including a series of compounds that are highly selective for MMP-12 (Devel et al. 2006). It will be important to test whether such inhibitors replicate the benefits seen in the mouse MMP knockout studies.
Targeting MMP Production
Given the limitations of direct MMP inhibitors, preventing MMP up-regulation and activation of pro-MMPs could be more fruitful.
Endothelial and VSMCs
As reviewed in detail elsewhere (Newby 2005), production of pro-MMP-2, MMP-14, TIMP-1, and TIMP-2 is constitutive. Inflammatory cytokines (e.g., interleukin [IL]-1 or tumor necrosis factor α [TNF-α]), growth factors (e.g., platelet derived growth factor or fibroblast growth factor-2), and CD40L (a component of activated T-lymphocyte membranes) induce MMP-1, MMP-3, and MMP-9 secretion and increase surface MMP-14. Activation of several protein kinase C isoforms (Hussain et al. 2002), mitogen-activated protein kinases (Cho et al. 2000), and the nuclear factor κB (NF-κB) pathway (Bond et al. 2001) appears to combine in mediating MMP up-regulation. Targeting specific cytokines or signaling pathways could therefore reduce MMP activity in plaques. For example, inhibition of CD40L stabilizes plaques in mice (Lutgens et al. 1999), which could in part be due to effects on MMPs. Compared with other MMPs and TIMPs, TIMP-3 shows an unusual pattern of up-regulation by transforming growth factor (TFG-β) and platelet derived growth factor in VSMCs (Fabunmi et al. 1996), perhaps contributing to the atheroprotective effects of TGF-β.
Monocyte/Macrophages
Peripheral blood monocytes do not express any MMP at high levels. However, attachment to tissue culture plastic or a variety of ECM components triggers up-regulation of MMP-9, MMP-10, MMP-14, and MMP-19 (Bar-Or et al. 2003). Monocytes probably up-regulate these mechanisms during their initial recruitment into inflammatory foci. Engagement of cell surface lectins and integrins causes rapid synthesis of prostaglandin E2, which turns on MMP transcription via the EP4 prostaglandin receptor and cyclic adenosine monophosphate accumulation (Cipollone et al. 2005, Pavlovic et al. 2006) (Figure 2). As a consequence, antagonists at the EP4 receptor that strongly suppress MMP production (Cipollone et al. 2005, Pavlovic et al. 2006) could be useful in stabilizing plaques. Up-regulation of MMP-9 and MMP-14 also occurs during transformation of monocytes to macrophages and could promote macrophage invasion through the intima.
Figure 2.
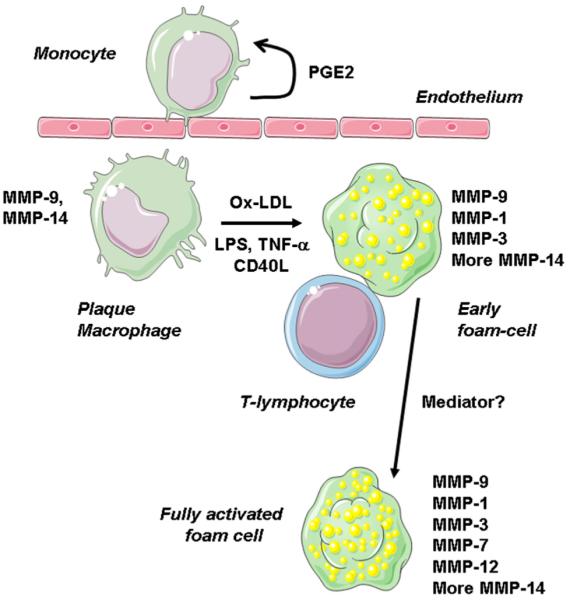
Staged up-regulation of MMPs in plaque macrophages. Blood monocytes express low levels of MMPs but up-regulate MMP-9 and MMP-14 on contact with cells and surfaces, probably via postaglandin E2 and cyclic adenosine monophosphate. Differentiation to macrophages also up-regulates MMP-9 and MMP-14, which together with pro-MMP-2 from smooth muscle cells, facilitate movement into the intima. Lipopolysaccharide, inflammatory cytokines, CD40L, and oxidized LDL up-regulate MMP-1 and further increase MMP-14. Metalloproteinase 3 is also found in early fatty streaks. Transcription factors NF-κB and serum amyloid A activating factor 1 are implicated. Metalloproteinases 7 and 12 are up-regulated in deep macrophages of advanced plaques; the pathways responsible are unclear.
Exposure of human monocyte-derived macrophages to lipopolysaccharide, TNF-α, CD40L, or oxidized low-density lipoprotein (LDL) up-regulates MMP-1 and MMP-14 via NF-κB (Chase et al. 2002) and serum amyloid A activating factor 1 (Ray et al. 2004) transcription factors, respectively (Figure 2). Metalloproteinase 3 is also expressed in a NF-κB-dependent way (Chase et al. 2002), although the mediators responsible are unclear. These MMPs probably mediate intimal remodeling in early atherosclerosis because they appear in early fatty streak lesions (Aikawa et al. 1998, Aikawa et al. 2001). In addition, MMP-1 and MMP-3 are selectively up-regulated in foam cells compared with those in nonfoamy macrophages (Galis et al. 1995, Chase et al. 2002). Metalloproteinase 12 is also up-regulated in foam cells but, unlike MMP-1, it is not up-regulated by treating macrophages with oxidized LDL alone (Thomas et al. 2007). Moreover, MMP-12 seems more selectively localized to the core of advanced plaques, in macrophages that have decreased arginase-I expression, a marker of additional activation (Thomas et al. 2007). In human plaques, MMP-12 (and MMP-7) localizes selectively to macrophages at the borders of the lipid core (Halpert et al. 1996). These data suggest that expression of MMP-12 marks a late stage of foam cell maturation (see Figure 2).
Other Mechanisms for Suppressing MMP Activity
As previously reviewed (Newby 2005), physiologic regulators that prevent MMP overexpression include nitric oxide, heparin and heparan sulfate proteoglycans, TGF-β, interferon-γ, IL-4, and IL-10. The established atheroprotective roles of TGF-β and IL-10 could be partly explained by effects on MMPs. On the other hand, interferon-γ (a cytokine secreted by T helper 1 lymphocytes) and IL-4 (a cytokine secreted by T helper 2 lymphocytes) and are on balance both proatherogenic despite their inhibitory effects on MMPs.
Statins are potent lipid-lowering drugs that prevent atherosclerosis progression and coronary events. Statins reduce expression and secretion of MMP-1, MMP-2, MMP-3, and MMP-9 from macrophages and smooth muscle cells in vitro and in rabbit and human atheroma (Bellosta et al. 1998, Aikawa et al. 2001, Crisby et al. 2001, Luan et al. 2003), but they increase TIMP-1 expression (Crisby et al. 2001). Hence, statin treatment may stabilize plaques in part by inhibiting MMP secretion. peroxisomal proliferator-activated receptor α and γ ligands, which inhibit MMP-9 secretion from smooth muscle cells (Marx et al. 1998a, 1998b) and macrophages (Marx et al. 1998a, 1998b, Shu et al. 2000) also have established therapeutic potential against atherosclerosis to which inhibition of MMP expression might contribute.
Conclusions
Histologic studies provide strong associative evidence that some MMPs promote plaque vulnerability in man. Experiments in genetically modified mice provide important proof of principle that high levels of MMP activity can promote instability of advanced plaques formed in extreme models of hypercholesterolemia. Other studies show plaque-stabilizing effects of some MMPs, consistent with their known effects to promote migration and proliferation of VSMC. The end result, plaque growth or rupture, may depend on the spectrum of MMPs expressed, their level of activity, and the stage of plaque development. The observation that macrophages express a wider spectrum of MMPs, including MMP-7 and MMP-12, in advanced human and rabbit plaques suggests the intriguing possibility that this pattern marks a distinct macrophage phenotype. Currently available broad-spectrum MMP inhibitors have little net effect on plaque progression or vulnerability in animal models at clinically tolerable concentrations. Hence, interventions selectively targeting individual MMPs seem worthwhile. Given the location of MMP-12 in advanced plaques and the protective effects of MMP-12 knockout in mice, selective inhibitors of this MMP seem particularly interesting. Targeting the mainly inflammatory pathways that lead to overproduction MMPs is another attractive approach.
Footnotes

This study is supported by grants from the British Heart Foundation and the European Vascular Genomics Network.
References
- Aikawa M, Rabkin E, Okada Y, et al. Lipid lowering by diet reduces matrix metalloproteinase activity and increases collagen content of rabbit atheroma. A potential mechanism of lesion stabilisation. Circulation. 1998;97:2433–2444. doi: 10.1161/01.cir.97.24.2433. [DOI] [PubMed] [Google Scholar]
- Aikawa M, Rabkin E, Sugiyama S, et al. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation. 2001;103:276–283. doi: 10.1161/01.cir.103.2.276. [DOI] [PubMed] [Google Scholar]
- Bar-Or A, Nuttall RK, Duddy M, et al. Analyses of all matrix metalloproteinase members in leukocytes emphasize monocytes as major inflammatory mediators in multiple sclerosis. Brain. 2003;126:2738–2749. doi: 10.1093/brain/awg285. [DOI] [PubMed] [Google Scholar]
- Bellosta S, Via D, Canavesi M, et al. HMG-CoA reductase inhibitors reduce MMP-9 secretion by macrophages. Arterioscler Thromb Vasc Biol. 1998;18:1671–1678. doi: 10.1161/01.atv.18.11.1671. [DOI] [PubMed] [Google Scholar]
- Bond M, Chase AJ, Baker AH, Newby AC. Inhibition of transcription factor NF-κB reduces matrix metalloproteinase-1, -3 and -9 production by vascular smooth muscle cells. Cardiovasc Res. 2001;50:556–565. doi: 10.1016/s0008-6363(01)00220-6. [DOI] [PubMed] [Google Scholar]
- Chase A, Bond M, Crook MF, Newby AC. Role of nuclear factor-κB activation in metalloproteinase-1, -3 and -9 secretion by human macrophages in vitro and rabbit foam cells produced in vivo. Arterioscler Thromb Vasc Biol. 2002;22:765–771. doi: 10.1161/01.atv.0000015078.09208.92. [DOI] [PubMed] [Google Scholar]
- Cho A, Graves J, Reidy MA. Mitogen-activated protein kinases mediate matrix metalloproteinase-9 expression in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2000;20:2527–2532. doi: 10.1161/01.atv.20.12.2527. [DOI] [PubMed] [Google Scholar]
- Choudhary S, Higgins CL, Chen IY, et al. Quantitation and localization of matrix metalloproteinases and their inhibitors in human carotid endarterectomy tissues. Arterioscler Thromb Vasc Biol. 2006;26:2351–2358. doi: 10.1161/01.ATV.0000239461.87113.0b. [DOI] [PubMed] [Google Scholar]
- Cipollone F, Fazia ML, Iezzi A, et al. Association between prostaglandin E receptor subtype EP4 overexpression and unstable phenotype in atherosclerotic plaques in human. Arterioscler Thromb Vasc Biol. 2005;25:1925–1931. doi: 10.1161/01.ATV.0000177814.41505.41. [DOI] [PubMed] [Google Scholar]
- Crisby M, Nordin-Fredriksson G, Shah PK, et al. Pravastatin treatment increases collagen content and decreases lipid content, inflammation, metalloproteinases, and cell death in human carotid plaques: implications for plaque stabilization. Circulation. 2001;103:926–933. doi: 10.1161/01.cir.103.7.926. [DOI] [PubMed] [Google Scholar]
- De Nooijer R, Verkleij CJN, Von der Thusen JH, et al. Lesional overexpression of matrix metalloproteinase-9 promotes intraplaque hemorrhage in advanced lesions but not at earlier stages of atherogenesis. Arterioscler Thromb Vasc Biol. 2006;26:340–346. doi: 10.1161/01.ATV.0000197795.56960.64. [DOI] [PubMed] [Google Scholar]
- Deguchi J-O, Aikawa E, Libby P, et al. Matrix metalloproteinase-13/collagenase-3 deletion promotes collagen accumulation and organization in mouse atherosclerotic plaques. Circulation. 2005;112:2708–2715. doi: 10.1161/CIRCULATIONAHA.105.562041. [DOI] [PubMed] [Google Scholar]
- Devel L, Rogakos V, et al. Development of selective inhibitors and substrate of matrix metalloproteinase-12. J Biol Chem. 2006;281:11152–11160. doi: 10.1074/jbc.M600222200. [DOI] [PubMed] [Google Scholar]
- Dollery CM, Libby P. Atherosclerosis and proteinase activation. Cardiovasc Res. 2006;69:625–635. doi: 10.1016/j.cardiores.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Fabunmi RP, Baker AH, Murray EJ, Booth RFG, Newby AC. Divergent regulation by growth factors and cytokines of 95-kDa and 72-kDa gelatinases and tissue inhibitors of metalloproteinases-1, -2 and -3 in rabbit aortic smooth muscle cells. Biochem J. 1996;315:335–342. doi: 10.1042/bj3150335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto Y, Deguchi J-O, Libby P, et al. Genetically determined resistance to collagenase action augments interstitial collagen accumulation in atherosclerotic plaques. Circulation. 2004;110:1953–1959. doi: 10.1161/01.CIR.0000143174.41810.10. [DOI] [PubMed] [Google Scholar]
- Galis ZS, Sukhova GK, Kranzhöfer R, et al. Macrophage foam cells from experimental atheroma constitutively express matrix-degrading proteases. Proc Natl Acad Sci. 1995;92:402–406. doi: 10.1073/pnas.92.2.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough PJ, Gomez IG, Wille PT, Raines EW. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J Clin Invest. 2006;116:59–69. doi: 10.1172/JCI25074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpert I, Sires UI, Roby JD, et al. Matrilysin is expressed by lipid-laden macrophages at sites of potential rupture in atherosclerotic lesions and localizes to areas of versican deposition, a proteoglycan substrate for the enzyme. Proc Natl Acad Sci U S A. 1996;93:9748–9753. doi: 10.1073/pnas.93.18.9748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain S, Assender JW, Bond M, et al. Activation of protein kinase Czeta is essential for cytokine-induced metalloproteinase-1, -3, and -9 secretion from rabbit smooth muscle cells and inhibits proliferation. J Biol Chem. 2002;277:27345–27352. doi: 10.1074/jbc.M111890200. [DOI] [PubMed] [Google Scholar]
- Johnson JL, George SJ, Newby AC, Jackson CL. Divergent effects of matrix metallo-proteinases 3, 7, 9, and 12 on atherosclerotic plaque stability in mouse brachiocephalic arteries. Proc Natl Acad Sci U S A. 2005;102:15575–15580. doi: 10.1073/pnas.0506201102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JL, Baker AH, Oka K, et al. Suppression of atherosclerotic plaque progression and instability by tissue inhibitor of metalloproteinase-2: involvement of macrophage migration and apoptosis. Circulation. 2006a;113:2435–2444. doi: 10.1161/CIRCULATIONAHA.106.613281. [DOI] [PubMed] [Google Scholar]
- Johnson JL, Fritsche-Danielson R, Behrendt M, et al. Effect of broad-spectrum matrix metalloproteinase inhibition on atherosclerotic plaque stability. Cardiovasc Res. 2006b;71:586–595. doi: 10.1016/j.cardiores.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Kuzuya M, Nakamura K, Sasaki T, et al. Effect of MMP-2 deficiency on atherosclerotic lesion formation in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2006;26:1120–1125. doi: 10.1161/01.ATV.0000218496.60097.e0. [DOI] [PubMed] [Google Scholar]
- Lemaitre V, O’Byrne TK, Borczuk AC, et al. ApoE knockout mice expressing human matrix metalloproteinase-1 in macrophages have less advanced atherosclerosis. J Clin Invest. 2001;107:1227–1234. doi: 10.1172/JCI9626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaitre V, Soloway PD, D’Armiento J. Increased medial degradation with pseudoaneurysm formation in apolipoprotein E-knockout mice deficient in tissue inhibitor of metalloproteinases-1. Circulation. 2003;107:333–338. doi: 10.1161/01.cir.0000044915.37074.5c. [DOI] [PubMed] [Google Scholar]
- Liang J, Liu E, Yu Y. Macrophage metalloelastase accelerates the progression of atherosclerosis in transgenic rabbits. Circulation. 2006;113:1993–2001. doi: 10.1161/CIRCULATIONAHA.105.596031. [DOI] [PubMed] [Google Scholar]
- Luan Z, Chase AJ, Newby AC. Statins inhibit secretion of metalloproteinases-1, -2, -3, and -9 from vascular smooth muscle cells and macrophages. Arterioscler Thromb Vasc Biol. 2003;23:769–775. doi: 10.1161/01.ATV.0000068646.76823.AE. [DOI] [PubMed] [Google Scholar]
- Lutgens E, Gorelik L, Daemen MJ, et al. Requirement for CD154 in the progression of atherosclerosis. Nat Med. 1999;5:1313–1316. doi: 10.1038/15271. [DOI] [PubMed] [Google Scholar]
- Luttun A, Lutgens E, Manderveld A, et al. Loss of matrix metalloproteinase-9 or matrix metalloproteinase-12 protects apolipoprotein E-deficient mice against atherosclerotic media destruction but differentially affects plaque growth. Circulation. 2004;109:1408–1414. doi: 10.1161/01.CIR.0000121728.14930.DE. [DOI] [PubMed] [Google Scholar]
- Manning MW, Cassis LA, Daugherty A. Differential effects of doxycycline, a broad-spectrum matrix metalloproteinase inhibitor, on angiotensin II-induced atherosclerosis and abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2003;23:483–488. doi: 10.1161/01.ATV.0000058404.92759.32. [DOI] [PubMed] [Google Scholar]
- Marx N, Schonbeck U, Lazar MA, et al. Peroxisome proliferator-activated receptor gamma activators inhibit gene expression and migration in human vascular smooth muscle cells. Circ Res. 1998a;83:1097–1103. doi: 10.1161/01.res.83.11.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx N, Sukhova G, Murphy C, et al. Macrophages in human atheroma contain PPAR gamma. Differentiation-dependent peroxisomal proliferator-activated receptor gamma expression and reduction of MMP activity through PPARgamma activation in mononuclear phagocytes in vitro. Am J Pathol. 1998b;153:17–23. doi: 10.1016/s0002-9440(10)65540-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69:562–573. doi: 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Newby AC. Dual role of matrix metalloproteinases (matrixins) in intimal thickening and atherosclerotic plaque rupture. Physiol Rev. 2005;85:1–31. doi: 10.1152/physrev.00048.2003. [DOI] [PubMed] [Google Scholar]
- Newby AC. Matrix metalloproteinases regulate migration, proliferation, and death of vascular smooth muscle cells by degrading matrix and non-matrix substrates. Cardiovasc Res. 2006;69:614–624. doi: 10.1016/j.cardiores.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Pavlovic S, Du B, Sakamoto K, et al. Targeting prostaglandin E2 receptors as an alternative strategy to block cyclooxygenase-2-dependent extracellular matrix-induced matrix metalloproteinase-9 expression by macrophages. J Biol Chem. 2006;281:3321–3328. doi: 10.1074/jbc.M506846200. [DOI] [PubMed] [Google Scholar]
- Pepper MS. Role of the matrix metallo-proteinase and plasminogen activator-plasmin systems in angiogenesis. Arterioscler Thromb Vasc Biol. 2001;21:1104–1117. doi: 10.1161/hq0701.093685. [DOI] [PubMed] [Google Scholar]
- Peterson JT. The importance of estimating the therapeutic index in the development of matrix metalloproteinase inhibitors. Cardiovasc Res. 2006;69:677–687. doi: 10.1016/j.cardiores.2005.11.032. [DOI] [PubMed] [Google Scholar]
- Prescott MF, Sawyer WK, Von Linden-Reed J, et al. Effect of matrix metalloproteinase inhibition on progression of atherosclerosis and aneurysm in LDL receptor-deficient mice overexpressing MMP-3, MMP-12, and MMP-13 and on restenosis in rats after balloon injury. Ann NY Acad Sci. 1999;878:179–190. doi: 10.1111/j.1749-6632.1999.tb07683.x. [DOI] [PubMed] [Google Scholar]
- Ray BK, Shakya A, Turk JR, et al. Induction of the MMP-14 gene in macrophages of the atherosclerotic plaque: role of SAF-1 in the induction process. Circ Res. 2004;95:1082–1090. doi: 10.1161/01.RES.0000150046.48115.80. [DOI] [PubMed] [Google Scholar]
- Rouis M, Adamy C, Duverger N, et al. Adenovirus-mediate overexpression of tissue inhibitor of metalloproteinase-1 reduces atherosclerotic lesions in apolipoprotein E deficient mice. Circulation. 1999;100:533–540. doi: 10.1161/01.cir.100.5.533. [DOI] [PubMed] [Google Scholar]
- Shipley JM, Wesselschmidt RL, Kobayashi DK, et al. Metalloelastase is required for macrophage-mediated proteolysis and matrix invasion in mice. Proc Natl Acad Sci U S A. 1996;93:3942–3946. doi: 10.1073/pnas.93.9.3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu H, Wong BM, Zhou GC, et al. Activation of PPAR alpha or gamma reduces secretion of matrix metalloproteinase 9 but not interleukin 8 from human monocytic THP-1 cells. Biochem Biophys Res Commun. 2000;267:345–349. doi: 10.1006/bbrc.1999.1968. [DOI] [PubMed] [Google Scholar]
- Silence J, Lupu F, Collen D, Lijnen HR. Persistence of atherosclerotic plaque but reduced aneurysm formation in mice with stromelysin-1 (MMP-3) gene inactivation. Arterioscler Thromb Vasc Biol. 2001;21:1440–1445. doi: 10.1161/hq0901.097004. [DOI] [PubMed] [Google Scholar]
- Silence J, Collen D, Lijnen HR. Reduced atherosclerotic plaque but enhanced aneurysm formation in mice with inactivation of the tissue inhibitor of metalloproteinase-1 (TIMP-1) gene. Circ Res. 2002;90:897–903. doi: 10.1161/01.res.0000016501.56641.83. [DOI] [PubMed] [Google Scholar]
- Sluijter JPG, Pulskens WPC, Schoneveld AH, et al. Matrix metalloproteinase 2 is associated with stable and matrix metalloproteinases 8 and 9 with vulnerable carotid atherosclerotic lesions—a study in human endarterectomy specimen pointing to a role for different extracellular matrix metalloproteinase inducer glycosylation forms. Stroke. 2006;37:235–239. doi: 10.1161/01.STR.0000196986.50059.e0. [DOI] [PubMed] [Google Scholar]
- Sukhova GK, Schonbeck U, Rabkin E, et al. Evidence for increased collagenolysis by interstitial collagenases-1 and -3 in vulnerable human atheromatous plaques. Circulation. 1999;99:2503–2509. doi: 10.1161/01.cir.99.19.2503. [DOI] [PubMed] [Google Scholar]
- Thomas AC, Sala-Newby GB, Ismail Y, et al. Genomics of foam cells and nonfoamy macrophages from rabbits identifies arginase-I as a differential regulator of nitric oxide production. Arterioscler Thromb Vasc Biol. 2007;27:571–577. doi: 10.1161/01.ATV.0000256470.23842.94. [DOI] [PubMed] [Google Scholar]