

Abstract
Both prostaglandin H synthase (PGHS) isoforms utilize a radical at Tyr385 to abstract a hydrogen atom from arachidonic acid, initializing prostaglandin synthesis. A Tyr348–Tyr385 hydrogen bond appears to be conserved in both isoforms; this hydrogen bonding has the potential to modulate the positioning and reactivity of the Tyr385 side chain. The EPR signal from the Tyr385 radical undergoes a time-dependent transition from a wide doublet to a wide singlet species in both isoforms. In PGHS-2, this transition results from radical migration from Tyr385 to Tyr504. Localization of the radical to Tyr385 in the recombinant human PGHS-2 Y504F mutant was exploited in examining the effects of blocking Tyr385 hydrogen bonding by introduction of a further Y348F mutation. Cyclooxygenase and peroxidase activities were found to be maintained in the Y348F/Y504F mutant, but the Tyr385 radical was formed more slowly and had greater rotational freedom, as evidenced by observation of a transition from an initial wide doublet species to a narrow singlet species, a transition not seen in the parent Y504F mutant. The effect of disrupting Tyr385 hydrogen bonding on the cyclooxygenase active site structure was probed by examination of cyclooxygenase inhibitor kinetics. Aspirin treatment eliminated all oxygenase activity in the Y348F/Y504F double mutant, with no indication of the lipoxygenase activity observed in aspirin-treated wild-type PGHS-2. Introduction of the Y348F mutation also strengthened the time-dependent inhibitory action of nimesulide. These results suggest that removal of Tyr348–Tyr385 hydrogen bonding in PGHS-2 allows greater conformational flexibility in the cyclooxygenase active site, resulting in altered interactions with inhibitors and altered Tyr385 radical behavior.
Prostaglandin H synthases (PGHSs) are membrane-bound hemoproteins that catalyze the first committed step in prostanoid biosynthesis, the conversion of arachidonic acid to PGH2 (1). There are two isoforms found in vertebrates that are ~60% identical in sequence: the constitutive or “housekeeping” enzyme (PGHS-1)1 and the inducible enzyme (PGHS-2) (2). Both isoforms contain a histidine-ligated heme group that reacts with peroxides to form a two-electron oxidized intermediate (compound I) (3–5). Compound I can then undergo an intramolecular electron transfer, oxidizing a nearby tyrosine residue, Tyr385 (6, 7). The Tyr385 radical connects the peroxidase and cyclooxygenase activities in PGHS-1 and -2, as it abstracts the 13-(S) hydrogen atom of arachidonic acid to initiate cyclooxygenase catalysis (8).
Tyr348 is positioned near Tyr385 in all crystal structures of PGHS-1 and -2 holoenzymes; the average distance between the phenolic oxygens is 2.8 ± 0.2 Å (9–20), which is ideal for formation of a hydrogen bond between the two tyrosine residues (Figure 1) (21). In addition, high-frequency EPR (HF-EPR) and electron–nuclear double resonance (ENDOR) studies of PGHS-1 suggest that the initial Tyr385 radical species is hydrogen-bonded (22, 23). Hydrogen bonding with Tyr348 has the potential to modulate the characteristics of the Tyr385 radical. The rate of Tyr385 radical formation is one factor controlling the efficiency of coupling between the PGHS peroxidase and cyclooxygenase reactions (5). Furthermore, hydrogen bonding between Tyr385 and Tyr348 has been shown to be an important structural factor in the interaction of PGHS-2 with aspirin that facilitates the acetylation reaction (24).
Figure 1.
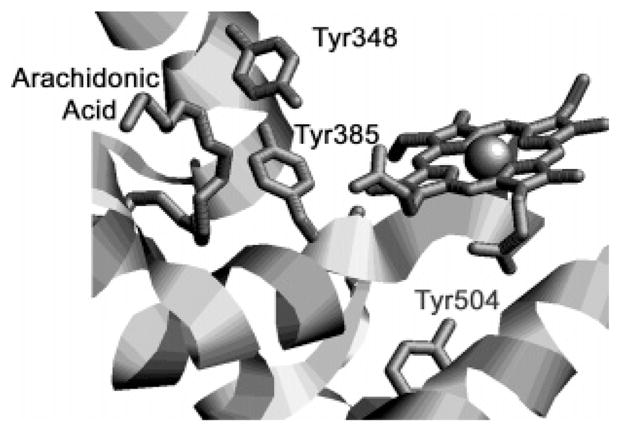
Crystal structure of oPGHS-1 complexed with arachidonic acid (PDB entry 1DIY), showing the relative positions of Tyr385, Tyr348, Tyr504, arachidonic acid, and CoPPIX (14).
Mutation of Tyr504 to phenylalanine in PGHS-2 prevents radical migration, yielding a single Tyr385 radical species, a 29 G wide doublet (WD) with 18 G center splitting (25). The Y504F mutant of PGHS-2 thus provides a useful model for evaluating the effects of Tyr348–Tyr385 hydrogen bonding on Tyr385 radical characteristics. We have compared the properties of the Y348F/Y504F double mutant with those of the Y504F mutant and found marked changes in Tyr385 radical structure and kinetics as well as in the kinetics of interaction with cyclooxygenase inhibitors. Loss of hydrogen bonding between Tyr385 and Tyr348 in PGHS-2 thus affects both the conformation of the Tyr385 side chain and inhibitor interactions in the cyclooxygenase active site.
EXPERIMENTAL PROCEDURES
Materials
AA was from NuChek Preps (Elysian, MN). EtOOH was purchased as a 5% aqueous solution from Polysciences (Warrington, PA) or obtained as a 6.25% solution from G. Barney Ellison (University of Colorado, Boulder, CO); the identity of the latter was confirmed by NMR and mass spectrometry. The polyclonal antibody against the PGHS-2 C-terminus, PPHP, and nimesulide were from Cayman Chemical (Ann Arbor, MI), and Tween-20 and n-octyl β-D-glucopyranoside were from Anatrace (Maumee, OH). All other reagents were obtained from Sigma-Aldrich.
Construction of a Plasmid for PGHS-2 with Y348F and Y504F Mutations
The QuikChange multisite mutagenesis kit (Stratagene, La Jolla, CA) was used for construction of the hPGHS-2 double mutant (Y348F/Y504F). The 5′-phosphorylated primer sequences containing the mutations (base changes underlined) were as follows: Y348F, 5′-GTGATTGAAGATTTTGTGCAACACTTGAGTGGC-3′; Y504F, 5′-GATGCTGTGGAGCTGTTTCCTGCCCTTCTGGTAG-3′. The mutagenesis reaction was carried out using equal amounts of the 5′-phosphorylated primers; the reaction product was used to transform Escherichia coli XL-10 competent cells. The cDNA containing the desired mutation was inserted into the pVL1393 vector, and the integrity of the resulting transfer vector construct was confirmed by restriction enzyme digestion and DNA sequencing.
Baculovirus Generation, Expression, and Purification of the Recombinant Protein
Procedures for generation, amplification, and titer determination of recombinant baculovirus containing cDNA encoding recombinant PGHS-2 protein and for recombinant protein expression have been described previously (25, 26). The detergent-solubilized preparations of the recombinant PGHS-2 proteins used for characterization of cyclooxygenase and peroxidase kinetics were prepared as described elsewhere (26). For RFQ-EPR and single-turnover experiments, the detergent-solubilized preparations were further purified by gel filtration chromatography on an AcA34 column (25). Apoenzymes were reconstituted with heme as previously described (27).
Protein Characterization
Expression of recombinant PGHS-2 was monitored by electrophoresis under denaturing conditions on 10% polyacrylamide gels, with the proteins visualized either by Coomassie blue staining or by immunoblotting using the antibody against PGHS-2. Both visualization techniques revealed a major band at ~73 kDa for all the recombinant PGHS-2 constructs, indicating that they were expressed in the baculovirus system as full-length, detergent-soluble proteins. The concentrations of recombinant PGHS-2 apoenzymes were determined by a dot-blot assay using homogeneous PGHS-2 as the standard (26). PGHS-2 holoenzyme concentrations were determined from their absorbance at 406 nm (165 mM−1 cm−1).
Cyclooxygenase Activity
Oxygen uptake was assayed polarographically at 30 °C (28); 1 unit of cyclooxygenase activity has an optimal velocity of 1 nmol of O2/min. Cyclooxygenase KM values were determined by measuring the activity with 1–100 μM AA and fitting the values to the Michaelis–Menten equation using SigmaPlot 8.0 (SPSS Inc.). The time-dependent inhibition kinetics with nimesulide and aspirin were determined as previously described (29). The kinetic parameters for time-dependent inhibition were estimated by fitting the values for surviving activity to eqs 1–3 of Callan et al. (30); these equations are derived from the mechanism in eq 1
 |
(1) |
where EI represents the initial inhibitor–enzyme complex that is in a slow, reversible equilibrium with a higher-affinity complex, EI*. These inhibitor binding steps are in competition with substrate binding (forming ES) and reaction (forming EP).
Lipoxygenase Activity
Measurements were performed at room temperature using a Bio-SEQUENTIAL DX-18MV stopped-flow instrument (Applied Photophysics, Leatherhead, U.K.). One syringe contained 120 μM AA in 0.1 M Tris-HCl (pH 8.0), 1 mM phenol, and 0.1% Tween-20, while the other contained 0.1–1 μM enzyme in the same buffer. Lipoxygenase activity was measured by monitoring the initial velocity of hydroperoxy fatty acid formation at 235 nm during the first few seconds of the reaction, using an extinction coefficient of 23.3 mM−1 cm−1 (31). Use of the initial velocity and short reaction times ensures that decomposition of prostaglandin endoperoxides to 12-hydroxyheptatrienoic acids (32) will be minimal, and thus, the absorbance changes are due primarily to lipoxygenase activity.
Peroxidase Activity
Peroxidase activity was measured by monitoring the initial velocity of TMPD oxidation at 611 nm during reaction with 1.6 mM H2O2 by stopped flow (25); 1 unit represents 1 nmol of peroxide reduced/min. Peroxidase KM values were determined by assessing cosubstrate oxidation in reactions with 2–150 μM PPHP and 10 mM guaiacol using an extinction coefficient at 436 nm of 3.33 × 103 M−1 cm−1 (26). Data were fitted to the Michaelis–Menten equation using SigmaPlot.
Peroxide-Induced Radicals
The initial EPR analysis of tyrosyl radicals was done by hand-mixing the detergent-solubilized PGHS-2 preparations [15 μM heme in 100 mM KPi (pH 7.2), 0.04% octyl glucoside, and 50 μM phenol] with 15 equiv of EtOOH on ice for 10 s and then freezing the sample in an ethanol/dry ice bath. For complexes of PGHS-2 with nimesulide and diclofenac, the enzymes were preincubated with a 5-fold molar excess of inhibitor at room temperature for 0.5–1 h before reacting with EtOOH. Aspirin-treated PGHS-2 was prepared by preincubation of the enzyme with 1.5–12 mM aspirin for 1 h at room temperature. In all cases, cyclooxygenase activity after inhibitor treatment was <20% of that of the control.
Detailed kinetic analyses of reactions with peroxide were performed in rapid freeze quench (RFQ) experiments using an Update Instruments (Madison, WI) System 1000 chemical/freeze quench apparatus with a model 1019 syringe ram, a model 715 ram controller, and a 0.008 in. diameter nozzle. The ram velocity was 2.0 cm/s, and the dead time was 4–5 ms. An isopentane bath at 125–130 K was used to chill the packing assembly for sample collection and packing (33).
Anaerobic Reactions with Peroxide and Arachidonate
Partially purified Y348F/Y504F PGHS-2 (15 μM heme) in 100 mM KPi (pH 7.2) and 0.1% Tween-20 was reacted sequentially with 15 equiv of EtOOH and then with 5 equiv of fatty acid (AA or d8-AA) on ice using the manual mixing apparatus described previously (8).
EPR spectra were recorded on a Bruker EMX spectrometer using a modulation amplitude of 2.0 G, a modulation frequency of 100 kHz, a time constant of 327 ms, a microwave power of 1 mW, and a temperature of 113 K. Radical concentrations were determined by double integration of the EPR signals with reference to a copper standard (34); calculations of radical concentrations for RFQ samples used a packing correction factor of 0.45 (33). Simulations of spectra used the Simfonia package provided by Bruker.
RESULTS
Cyclooxygenase and Peroxidase Activities
Substitution of phenylalanine for tyrosine at residue 348 had modest effects on cyclooxygenase reaction kinetics under standard conditions (Table 1). The Y348F substitution in Y504F PGHS-2 resulted in a 20% increase in the cyclooxygenase rate, whereas the same substitution in wild-type PGHS-2 produced a 25–50% decrease in cyclooxygenase activity (19, 24, 25). Similarly, introduction of a Y348F substitution had modest and variable effects on the cyclooxygenase KM value in wild-type PGHS-2 and the Y504F mutant, giving an 80% increase in the former and a 20% decrease in the latter (Table 1). Cyclooxygenase site interactions with fatty acid thus do not appear to involve hydrogen bonding with Tyr348, as expected from crystallographic results (14). A Y348F substitution had no significant effect on cyclooxygenase self-inactivation in wild-type PGHS-2 and slightly enhanced self-inactivation in the Y504F mutant. This indicates that the Tyr348–Tyr385 hydrogen bond does not have a major influence on the destructive side reactions of cyclooxygenase intermediates. Substitution of phenylalanine for tyrosine at residue 348 also had little effect on the steady-state peroxidase kinetics of wild-type PGHS-2, although introduction of the Y348F mutation into the Y504F parent mutant decreased peroxidase activity by 50% (Table 1).
Table 1.
Effects of Y348F Mutation on Cyclooxygenase and Peroxidase Kinetic Parametersa
| cyclooxygenase |
Peroxidase |
||||
|---|---|---|---|---|---|
| PGHS-2 construct | specific activityb (units/μg) | KM (μM AA) | kinactc (min−1) | specific activityb (units/μg) | KM (μM PPHP) |
| wild-typed | 5.8 ± 0.1 | 2.1 ± 0.4e | 2.3 ± 0.1 | 14.1 ± 0.3 | 138 ± 42 |
| Y348Fd | 2.7 ± 0.1 | 3.8 ± 0.7 | 2.7 ± 0.3 | 15.0 ± 0.6 | 72 ± 22 |
| Y504Fd | 3.9 ± 0.0 | 4.2 ± 0.4 | 2.2 ± 0.1 | 34.0 ± 0.9 | 437 ± 79 |
| Y348F/Y504F | 4.7 ± 0.3 | 3.4 ± 0.7 | 3.4 ± 0.3 | 15.6 ± 0.5 | 266 ± 36 |
Values represent averages ± the standard deviation of at least three assays of detergent-solubilized enzyme preparations.
Calculated by dividing the Vmax value by the amount of recombinant protein determined by the immunoblot assay or Soret absorbance as described in Experimental Procedures.
The cyclooxygenase self-inactivation rate constant was calculated by dividing the optimal velocity (nanomoles of O2 per minute) by the extent of reaction after self-inactivation was complete (nanomoles of O2 consumed) (49).
Values from ref 25.
Value from ref 26.
Effects of the Tyr348 Mutation on Tyrosyl Radical EPR Spectra
Reaction of Y348F/Y504F PGHS-2 with excess EtOOH resulted in the formation of a 30 G (peak to trough) doublet signal with 18 G center splitting that increased in intensity over the first 0.2 s, reaching a maximum of 0.23 spin/heme (Figure 2, A-1); the rate of formation of this radical was 15 s−1. Subsequently, the radical intensity slowly decayed at a rate of 0.08 s−1. The increase in radical intensity was not accompanied by any line shape change (Figure 2, A-2), but there was a loss of center splitting, converting the doublet to a 30 G wide singlet (WS) signal as the intensity decayed over the next 10 s (Figure 2, A-3). Power saturation studies indicated that the P1/2 value for the doublet was 1.8 mW at 113 K.
Figure 2.

Tyrosyl radical kinetics during the reaction of Y348F/Y504F PGHS-2 and Y504F PGHS-2 with EtOOH. (A) The double mutant (68 μM heme) in 100 mM KPi (pH 7.2), 50 μM phenol, 0.04% octyl glucoside, and 10% glycerol was reacted at room temperature with 15 equiv of EtOOH. (A-1) Time course of Y348F/Y504F tyrosyl radical intensity as determined by double integration of the EPR signals (●) in experiments with two separate enzyme preparations and time course for the PGHS-2 Y148F/Y348F/Y404F/Y504F quadruple mutant (○) (25). (A-2 and A-3) EPR spectra from the double mutant reaction samples freeze trapped at the indicated times. (B) Y504F PGHS-2 (48 μM heme) was reacted with 15 equiv of EtOOH at room temperature (25). (B-1) Time course of radical intensity determined by double integration. (B-2 and B-3) EPR spectra from the Y504F mutant reaction samples freeze trapped at the indicated times.
This change in EPR line shape from a doublet to WS observed in the double mutant contrasts with the lack of line shape change observed for the Y504F single mutant, which remained a wide doublet at all aging times, and also showed much faster accumulation of radical (Figure 2, B-1–B-3). The conversion from a 30 G doublet to a 30 G WS in the double mutant but not in the Y504F parent suggests that disruption of the Tyr348–Tyr385 hydrogen bond alters the dynamics of the Tyr385 radical. In previous experiments using the PGHS-2 Y148F/Y348F/Y404F/Y504F quadruple mutant, in which the Tyr348–Tyr385 hydrogen bond also is disrupted, this doublet to WS conversion was not detected due to the much faster decay of radical intensity in the quadruple mutant (0.3 s−1) compared to the double mutant (0.08 s−1), as is apparent in Figure 2, A-1. At the 10 s time point, where the double mutant WS is fully formed (dotted vertical line in Figure 2, A-1), little radical intensity remained in the quadruple mutant.
Treatment of Y348F/Y504F with the cyclooxygenase inhibitor, nimesulide, prior to reaction with peroxide resulted in a 13 G (peak to trough) narrow singlet (NS) species (Figure 3A), which had a P1/2 value of 0.14 mW. The peak NS intensity was 0.1–0.14 spin/heme, approximately half of the radical intensity observed with the untreated enzyme. The same NS species also was formed upon reaction of EtOOH with the double mutant pretreated with diclofenac or aspirin (data not shown), showing that formation of the NS species is not dependent on cyclooxygenase inhibitor structure.
Figure 3.

(A) Experimental (—) and simulated (– – –) EPR spectra of the doublet, wide singlet (WS), and narrow singlet (NS) EPR signals from Y348F/Y504F PGHS-2 reacted with peroxide. The doublet was observed after reaction of the double mutant (38 μM heme) with 15 equiv of EtOOH for 104 ms. The simulated spectrum represents the sum of 80% of the Y504F WD simulated spectrum and 20% of the NS simulated spectrum. The WS was observed after reaction of the double mutant (15 μM heme) with 15 equiv of EtOOH on ice for ~10 s. The wide singlet simulation represents the sum of 48% simulated Y504F WD and 52% simulated NS. The NS spectrum was recorded after preincubation of the double mutant (15 μM heme) with 5 equiv of nimesulide and subsequent reaction with 15 equiv of EtOOH for 10 s on ice. Parameters for simulation of the Y504F WD and NS spectra are given in Table 3. (B) Structural diagrams of the proposed conformations of the β-methylene groups of tyrosyl radicals that give rise to the WD and NS spectra.
Analysis of the Narrow Singlet, Doublet, and Wide Singlet Tyrosyl Radicals Observed with the Y348F/Y504F Double Mutant
The Bruker Simfonia program was used to simulate EPR powder pattern line shapes to determine whether differences in the side chain conformation of the Tyr385 radical might account for the various signals that are observed. The modified McConnell relationship, in which Aiso = ρC1(β0 + β2 cos2 θ), where Aiso is the isotropic hydrogen hyperfine coupling arising from a β-hydrogen, ρC1 is the spin density at C1 of the phenyl ring [0.38, determined from previous studies (35)], and β0 and β2 are constants [0 and 162.4 MHz, respectively (36)], allows calculation of θ, the dihedral angle between the pz orbitals of C1 and the β-hydrogen (37) (Figure 3B).
The 13 G NS species observed for the double mutant pretreated with inhibitor could be closely simulated using the parameters in Table 3 (Figure 3A). Starting g-tensor and hydrogen hyperfine values were those previously reported for PGHS-1 (22, 35). The highly anisotropic g-tensor values in Table 3 were necessary to closely simulate the observed spectrum, as was the inclusion of two β-hydrogens. These parameters may not be unique, and other spectroscopic methods such as high-field EPR or ENDOR will be needed for precise determination. Analysis of the H1β and H2β coupling values derived from the NS species simulations indicates that the dihedral angle values are 50° and −70°, respectively (Figure 3B), giving a tyrosine radical with a low-energy, relaxed ring conformation almost identical with that in the PGHS-1 inhibitor complex NS (22, 35).
Table 3.
Parameters Used for Simulation of the WD EPR Spectrum from the Y504F Single Mutant and the NS EPR Spectrum from the Y348F/Y504F Double Mutanta
| Y504F PGHS-2 wide doublet (WD) | Y348F/Y504F narrow singlet (NS) | |
|---|---|---|
| gx | 2.003 | 2.0084 |
| gy | 2.003 | 2.0055 |
| gz | 2.003 | 2.0023 |
| H3,5 Ax | 16.8 | 20 |
| H3,5 Ay | 16.8 | 6 |
| H3,5 Az | 16.8 | 18.5 |
| Hβ1 Ax | 46.2 | 45 |
| Hβ1 Ay | 46.2 | 5 |
| Hβ1 Az | 46.2 | 28 |
| Hβ2 Ax | 8.4 | 5 |
| Hβ2 Ay | 8.4 | 5 |
| Hβ2 Az | 8.4 | 22 |
Absolute values for hyperfine couplings are given in megahertz.
The Y348F/Y504F doublet could not be adequately simulated as a single species, but rather as an arithmetic combination of the “pure” Y504F 30 G WD and the inhibitor-treated Y348F/Y504F 13 G NS, in a 4:1 ratio (Figure 3A). This combination reproduced the width and central splitting of the doublet. A similar examination of the Y148F/Y348F/Y404F/Y504F quadruple mutant doublet indicated that this signal also probably results from a 4:1 mixture of pure WD and 13 G NS spectra (data not shown). This suggests that the Tyr385 radical in both the double and quadruple mutants is present in two distinct rotational conformations even at the earliest time points in the reaction with EtOOH. One, a strained conformation with a dihedral angle of 11°, gives rise to the WD spectrum, whereas the 13 G NS spectrum results from another conformation with a more relaxed dihedral angle of 50°. Either the Tyr385 side chain adopts two conformations in these resting enzymes, or a fraction of the Tyr385 radical undergoes rapid side chain rotation after it forms. Similarly, the Y348F/Y504F WS is best simulated as a 1.1:0.9 mixture of the pure WD species and the Y348F/Y504F inhibitor complex NS species (Figure 3A).
Effect of the Tyr348 Mutation on Enzyme-Bound Substrate Radical Structure
Reaction of the Y348F/Y504F PGHS-2 WS species with arachidonic acid under anaerobic conditions generated a new EPR signal (Figure 4A). This is probably not a single species, but rather represents a combination of the 13 G NS tyrosyl radical (Figure 3A) with an enzyme-bound arachidonate pentadienyl radical, as the observed spectrum can be closely simulated as the sum of the NS simulated spectrum and a seven-line pentadienyl spectrum (38) (Figure 4A). When the experiment was repeated with d8-AA, the resulting spectrum could be accurately simulated by a combination of the NS spectrum and a five-line pentadienyl radical spectrum (38) (Figure 4B). Subsequent exposure of the AA and d8-AA reaction intermediates to air resulted in conversion of the pentadienyl/NS mixtures to exclusively NS species (data not shown), indicating that the enzyme-bound arachidonate radicals reacted with oxygen to re-form tyrosyl radical species in the less sterically hindered conformation.
Figure 4.

EPR spectra of radicals formed during anaerobic reaction of Y348F/Y504F PGHS-2 with AA or d8-AA. (A) Reaction at 0 °C of Y348F/Y504F PGHS-2 (15 μM heme) with 15 equiv of EtOOH for 26 s and then with 4 equiv of AA for 11 s (—). The simulated EPR spectrum (– – –) is an arithmetic combination of 59% Y348F/Y504F PGHS-2 NS and 41% AA-pentadienyl radical. (B) Reaction at 0 °C of Y348F/Y504F PGHS-2 (15 μM heme) with 15 equiv of EtOOH for 19 s and then with 4 equiv of d8-AA for 13 s (—). The simulated EPR spectrum (– – –) is an arithmetic combination of 54% Y348F/Y504F PGHS-2 NS and 46% d8-AA-pentadienyl radical. Simulation parameters are given in Table 3 and ref 38.
Effects of the Tyr348 Mutation on Cyclooxygenase Inhibition by Aspirin
Preincubation with aspirin produced a similar progressive inhibition of cyclooxygenase activity in wild-type PGHS-2 and the Y504F mutant (Figure 5A,B). In both cases, the activity loss leveled off at longer preincubation times, as expected from the shift from cyclooxygenase to lipoxygenase catalysis (39). Introduction of the Y348F substitution into the Y504F mutant markedly affected the inhibitory effects of aspirin, making it a stronger and more complete inhibitor (Figure 5C). At the highest aspirin level that was tested, inhibition of the double mutant continued as a first-order process to 5% surviving activity, indicating that there was no longer a shift from cyclooxygenase to lipoxygenase catalysis. This lack of increased lipoxygenase activity during time-dependent inhibition of the double mutant by aspirin was confirmed by direct lipoxygenase assays (data not shown). Analysis of the double mutant inactivation kinetics using eq 1 indicated a Ki value of 2 ± 1 mM and a k2 value of 0.4 ± 0.2 min−1; k−2 was set to zero as there appeared to be no residual activity and thus no reverse reaction.
Figure 5.
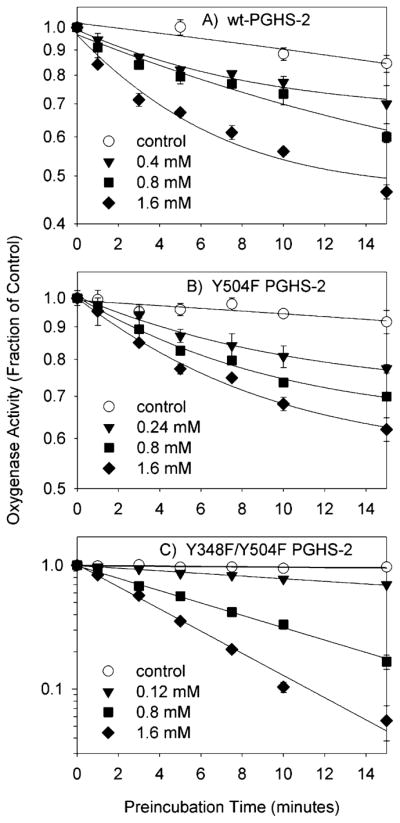
Kinetics of cyclooxygenase inhibition by aspirin in wild-type PGHS-2 and the Y504F and Y348F/Y504F mutants. The initial velocity of oxygen consumption was measured after preincubation of PGHS-2 (A), the Y504F mutant (B), or the Y348F/Y504F mutant (C) with the indicated levels of aspirin for the indicated times. Data shown are the averages of triplicate measurements, normalized to the uninhibited control; standard deviations are indicated by error bars. Lines represent exponential or linear fits to the data.
Effects of the Tyr348 Mutation on Cyclooxygenase Inhibition by Nimesulide
Initial screening experiments showed that the Y348F/Y504F double mutant had a considerably lower IC50 for nimesulide (0.14 μM) than did wild-type PGHS-2 (0.68 μM) (Figure 6). The inhibition kinetics were then probed more closely by preincubation experiments (Figure 7 and Table 2). The Y504F mutant and wild-type PGHS-2 had similar time-dependent inhibition kinetics, with essentially no difference in Ki or k2 values, although the higher k−2 value in Y504F indicates that the EI* complex was slightly less stable in this mutant. Introduction of the Y348F substitution, however, substantially changed the interaction with nimesulide compared to that in the Y504F parent, with a 3-fold increase in initial binding affinity (Ki of 0.27 μM compared to a Ki of 0.90 μM), and increased the stability of the EI* complex (increased k2 and decreased k−2 values). These changes combine to make nimesulide a much stronger cyclooxygenase inhibitor when the Tyr348 hydroxyl is absent.
Figure 6.

Cyclooxygenase inhibition by nimesulide in PGHS-2 (●) and the Y348F/Y504F double mutant (○). The enzyme was preincubated for 1 min at 30 °C with the indicated level of nimesulide before the reaction was initiated by addition of arachidonate (100 μM). Activities are normalized to uninhibited control values; data represent averages of triplicate measurements, and standard deviations are indicated by error bars.
Figure 7.
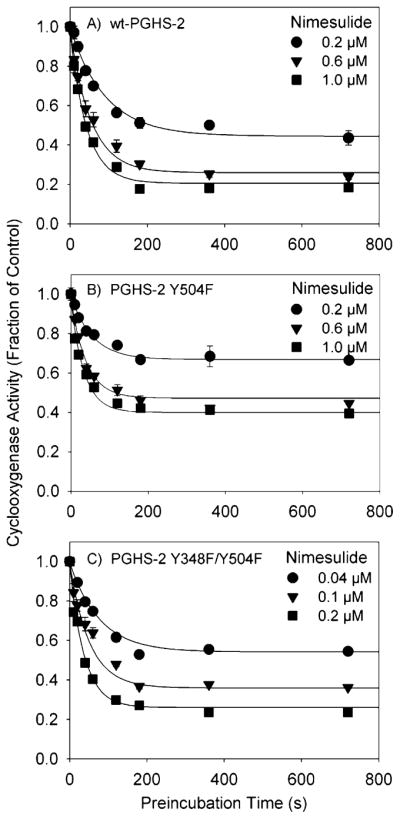
Kinetics of cyclooxygenase inhibition by nimesulide in PGHS-2 and the Y504F and Y348F/Y504F mutants. The initial velocity of oxygen consumption was measured after preincubation of PGHS-2 (A), the Y504F mutant (B), or the Y348F/Y504F double mutant (C) with the indicated levels of nimesulide for the indicated times. Data are the averages of triplicate measurements, normalized to the control; standard deviations are given by error bars. The curves represent theoretical fits to the data using the model described by Callan et al. (30).
Table 2.
Kinetic Parameters for Cyclooxygenase Inhibition by Nimesulidea
| PGHS-2 construct | Ki (μM) | k2 (min−1) | k−2 (min−1) |
|---|---|---|---|
| wild-type | 1.0 ± 0.2 | 2.2 ± 0.3 | 0.29 ± 0.02 |
| Y504F | 0.9 ± 0.2 | 2.0 ± 0.2 | 0.73 ± 0.05 |
| Y348F/Y504F | 0.27 ± 0.06 | 2.8 ± 0.5 | 0.43 ± 0.03 |
Data from the experiments shown in Figure 5 were analyzed as described in Experimental Procedures.
DISCUSSION
In these experiments, we focused our attention on one of the most remarkable aspects of PGHS cyclooxygenase site structure, the hydrogen bond between Tyr348 and Tyr385. Tyr385 and Tyr504 are the only tyrosine residues in PGHS-2 readily oxidized during reaction with peroxide, and a Y504F mutation localizes the peroxide-induced radical to Tyr385, whose side chain remains in a single conformation (25). Therefore, any differences in the EPR signals of the Y348F/Y504F mutant compared to those of the Y504F mutant can be ascribed to loss of the Tyr348–Tyr385 hydrogen bond.
Tyr348 and Tyr385 Radical Kinetics
The tyrosyl radical accumulated ~10 times more slowly in the Y348F/Y504F double mutant than in the Y504F single mutant (Figure 2). Closer consideration of earlier data from the Y148F/Y348F/Y404F/Y504F quadruple mutant shows that its tyrosyl radical kinetics are consistent with those of the Y348F/Y504F double mutant (Figure 2). Tyr348 does not lie on the optimal pathway predicted for electron transfer from Tyr385 to the heme (25), and so a Y348F mutation by itself would not be expected to alter the electron path or electron transfer kinetics. However, if the Tyr348–Tyr385 hydrogen bond is important for positioning Tyr385 for optimal electron transfer to the oxidized heme, then mutation of Tyr348 might well alter the rate of Tyr385 radical formation. Simulations of the initially formed doublet spectra (Figure 3) suggest that there are two conformer populations present during tyrosyl radical formation in the double mutant. The major species is a strained 30 G doublet analogous to the WD seen in the Y504F mutant, and a minor proportion of a 13 G NS species is also present. It seems unlikely, however, that this small population of a NS species is responsible for the 10-fold slower rate of radical accumulation observed in the double mutant. Perhaps Tyr348 accepts a proton from Tyr385, and removal of this acceptor decreases the rate of electron transfer from Tyr385 to the heme. Tyr385 is located in a highly hydrophobic environment, which might accentuate the importance of a nearby proton acceptor. Despite their proximity, differences in the surrounding protein matrix appear to influence the reactivity of these two residues. For example, Tyr385 is preferentially oxidized during reaction with peroxide, whereas Tyr348 is not, despite their similar predicted electron transfer coupling (25).
Another possible role for Tyr348 might be to “tune” the distribution of the radical between Tyr385 and Tyr504. The cyclooxygenase specific activity of the Y348F single mutant is 50% of that of wild-type PGHS-2 (Table 1). The peroxide-induced EPR signal in the Y348F mutant is noticeably narrower than that in wild-type PGHS-2 (25), consistent with a higher proportion of radical on Tyr504 (an NS signal) than on Tyr385 (a WD signal). Addition of a Y504F mutation (to form the double mutant) partially restores cyclooxygenase activity, to 80% of the wild-type level, presumably by forcing the radical to remain on Tyr385. Similarly, the peroxidase activity of the Y504F single mutant is twice that of the wild-type enzyme, and introduction of the Y348F mutation results in a decrease in peroxidase activity to wild-type levels. This suggests that the relative amounts of radical at Tyr385 and Tyr504 determine the relative cyclooxygenase and peroxidase activities and that Tyr348 helps control this distribution of radical.
Tyr348 and Tyr385 Conformation
Simulations indicate that the doublet to WS transition observed in the double mutant but not the parent Y504F single mutant (Figure 2) is due to a growing population of the NS species over time (Figure 3). The phenyl ring of the Tyr385 radical thus appears free to slowly rotate into a lower-energy conformation in the absence of the Tyr348–Tyr385 hydrogen bond. Radical migration to another tyrosine residue is very unlikely to contribute to the doublet to singlet transition in the double mutant, as this mutant lacks Tyr504, the only other tyrosine residue besides Tyr385 that is oxidized in peroxide-treated PGHS-2 (25). It is notable that the conversion observed in the double mutant is considerably slower than the radical migration to Tyr504 observed in wild-type PGHS-2, which is complete within 50 ms (Figure 8) (40). This increased conformational flexibility of the Tyr385 radical side chain does not grossly affect cyclooxygenase activity, as the specific activity of the double mutant and the KM for arachidonate were only slightly different from the values for the Y504F mutant (Table 1). In addition, the introduction of the Y348F mutation had little effect on the enzyme-bound arachidonyl radical (Figure 4). All of this suggests that the increased movement of the Tyr385 radical in the absence of hydrogen bonding with Tyr348 is not large enough to perturb substrate binding in the cyclooxygenase site.
Figure 8.

EPR spectra of tyrosyl radicals formed during reaction of PGHS-2, Y348F/Y504F PGHS-2, and PGHS-1 with EtOOH. Individual EPR spectra are scaled to the same amplitude and overlaid to facilitate line shape comparisons. (A) PGHS-2 (11.2 μM heme) in 100 mM KPi (pH 7.2), 0.1% Tween-20, and 10% glycerol was reacted at room temperature with 5 equiv of EtOOH for the indicated times. Data are from ref 27. (B) The Y348F/Y504F PGHS-2 double mutant (68 μM heme) in 100 mM KPi (pH 7.2), 50 μM phenol, 0.04% octyl glucoside, and 10% glycerol was reacted at room temperature with 15 equiv of EtOOH for the indicated times. (C) PGHS-1 (8.5 μM heme) in 100 mM KPi (pH 7.2), 0.1% Tween-20, and 10% glycerol was reacted at room temperature with 5 equiv of EtOOH for the indicated times. Data are from ref 27.
Tyr348 and the Effects of Cyclooxygenase Inhibitors on Tyr385
The effects of the Y348F mutation on radical formation in inhibitor-bound enzyme provide another indication of the importance of the Tyr348–Tyr385 hydrogen bond in regulating Tyr385 radical formation. The inhibitor complex of the Y504F PGHS-2 single mutant is practically EPR silent when reacted with peroxide (25), indicating that inhibitor binding alters the environment of Tyr385 and prevents radical formation on that residue. In the Y348F/Y504F double mutant complexed with nimesulide, however, reaction with peroxide produced a pronounced Tyr385 NS radical (Figure 3), indicating that Tyr385 is in a more reactive position when not hydrogen-bonded to Tyr348. Pretreatment of the double mutant with diclofenac, which hydrogen bonds to Tyr385 and Ser530 without disrupting the Tyr348–Tyr385 hydrogen bond in wild-type PGHS-2 (19), also results in NS radical formation (data not shown). The similarity of the EPR signals seen after peroxide treatment of the nimesulide and diclofenac complexes of the PGHS-2 double mutant, and in the aspirin-treated double mutant, indicates that the new conformation adopted by Tyr385 in the absence of the Tyr348 hydrogen bond is not dictated by the inhibitor structure.
The NS spectrum seen with the inhibitor-pretreated double mutant enzyme has a significantly narrower line shape (13 G) than that from the inhibitor-pretreated wild-type PGHS-2 inhibitor complex (21 G). However, analysis of hyperfine constants suggests that very similar conformations of the tyrosyl radical, with one β-hydrogen at an angle of 50° to the pz orbital axis (Figure 3), are responsible for the NS signals in the wild-type and double mutant PGHS-2 enzymes. The differences in signal line shapes therefore arise largely from anisotropic gx values, which are dependent upon the local electrostatic environment. Some degree of anisotropy in inhibitor-treated PGHS-2 might be expected on the basis of the previous high-field EPR studies on inhibitor-treated PGHS-1 (22). The high gx value for the inhibitor complex of the PGHS-2 double mutant could arise from several factors. The most likely is a loss of hydrogen bonding of the Tyr385 radical. Another potential influence is a change in dipolar and exchange couplings between the heme and the Tyr385 radical caused by tyrosine reorientation, although this change is expected to be small given the distance between the heme and Tyr385 (22, 41). Overall, a reasonable interpretation is that the phenyl rings of the Tyr385 radicals in inhibitor complexes of the wild type and the double mutant adopt the same rotational conformation of the phenyl ring (θ in Figure 3), but lack a crucial H-bond with Tyr348 and perhaps also differ in other side chain bond angles of Tyr385 or other active site residues, leading to different local electronic environments at Tyr385. The high gx value is also expected for the doublet EPR of the double mutant; however, the simulation seems to show low constraints on the g values, and an isotropic g factor gives a reasonable simulation of our data (Table 1 and Figure 3). A different Tyr385 side chain conformation in the double mutant is further supported by the observation of a NS signal after aspirin pretreatment, whereas a wide singlet signal is seen in aspirin-treated wild-type PGHS-2 (42).
Tyr348 and Interactions with Cyclooxygenase Inhibitors
Crystallographic structures show that Tyr348 and Tyr385 lie directly across the substrate channel from Ser530 in both PGHS isoforms (9, 12). In bromoacetyl-derivatized PGHS-1, the Tyr385 phenolic oxygen is equidistant between Tyr348 OH and the closest oxygen of the bromoacetyl group on Ser530, ~3.0 Å from each (Figure 9) (10). This positioning of Tyr385 and the derivatized Ser530 side chain blocks access by arachidonate and accounts for the complete loss of oxygenase activity in aspirin-treated PGHS-1. Crystallographic data are not available for aspirin-treated PGHS-2; however, the observation of lipoxygenase activity (39, 43) indicates that a hydrogen bonding network between Tyr385 and acetylated Ser530 does not exist in aspirin-treated PGHS-2. Consistent with this, aspirin pretreatment perturbs the tyrosyl radical in PGHS-1 but not in PGHS-2 (42, 44). Thus, it was surprising to find that aspirin pretreatment produced complete inhibition of oxygenase activity in the PGHS-2 double mutant (Figure 5). Our interpretation is that Tyr385 in PGHS-2 has greater conformational freedom when not hydrogen bonded to Tyr348 and can swing out into the substrate channel. Once thus displaced, Tyr385 hydrogen bonds instead to the acetyl group on Ser530, “locking” the Tyr385 side chain in the channel and eliminating all oxygenase activity. It is worth noting that the observation of slower Ser530 acetylation in a Y348F single mutant compared to wild-type PGHS-2 was interpreted to mean that a Tyr348–Tyr385–aspirin hydrogen bond network speeds the acetylation reaction (24). The Tyr348–Tyr385 hydrogen bond thus appears to influence both the kinetics of Ser530 acetylation by aspirin and the consequences of that acetylation for oxygenase catalysis.
Figure 9.
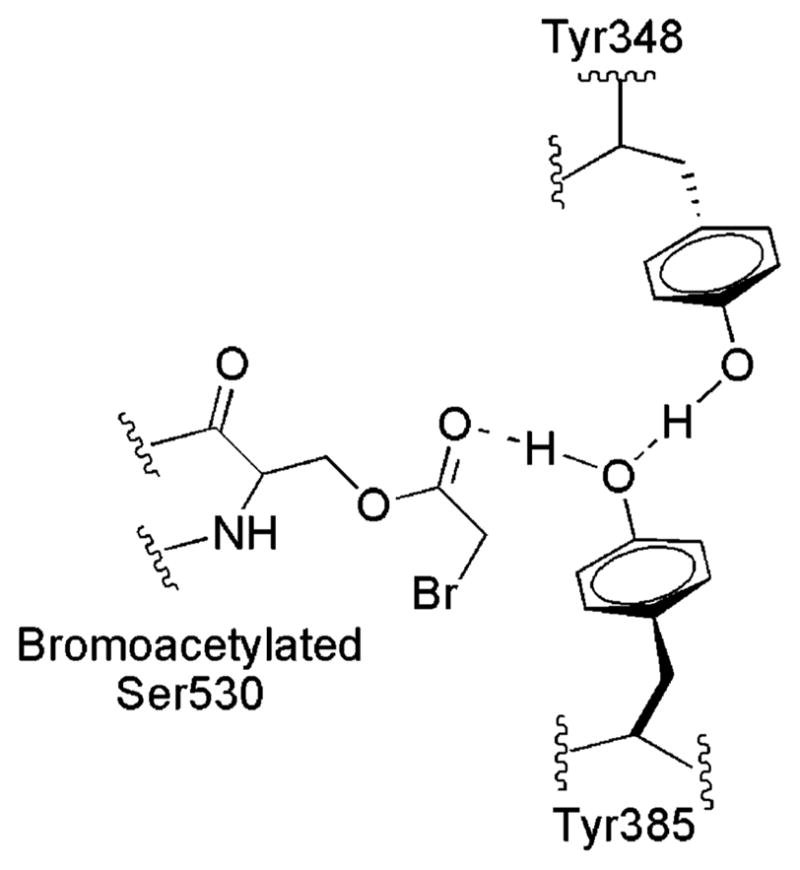
Schematic structure of bromo-acetylated oPGHS-1 (PDB entry 1PTH) depicting the interactions among Tyr385, Tyr348, Ser530, and the bromoacetyl group based on crystallographic data from ref 10.
Almost all cyclooxygenase inhibitors, including nimesulide and diclofenac, bind reversibly to the cyclooxygenase site of PGHS, rather than chemically modifying an active site residue as aspirin does. The inhibition by nimesulide and diclofenac has been analyzed as two distinct phases, a rapid, reversible binding of the inhibitor (E + I ⇔ EI) and a second, slower transition (EI ⇔ EI*) that is poorly reversible (30, 45); cyclooxygenase site mutations can conceivably effect either or both of these phases of inhibitor action. The Y348F mutation was found to decrease the Ki of the initial fast substrate binding phase for nimesulide, with smaller effects on the second, slower transition (Table 2). Therefore, the k2 and k−2 parameters do not reflect the structural change, seen in the acetylated double mutant, that leads to the Tyr385 NS EPR signal. The lack of a major effect of Tyr348 mutation on the EI ⇔ EI* transition in PGHS-2 contrasts with the major consequence of mutating Tyr355, which is considered to be part of a hydrogen bonding network in the substrate channel entrance: a 7-fold increase in the value of k2 for indomethacin (46). It is evident that the EI ⇔ EI* transition underlying the time-dependent inhibition of PGHS is a complex process, with Tyr348 and Tyr355 participating in distinct aspects of inhibitor binding and conformational changes.
PGHS-2 Y348F Mutants as PGHS-1 Mimics
One surprising aspect of these results is the similarity between the Y348F/Y504F PGHS-2 mutant and PGHS-1 in terms of their tyrosyl radical characteristics and their interactions with inhibitors. Introduction of the Y348F mutation into the Y504F PGHS-2 mutant results in a WD → WS transition that more closely resembles that of PGHS-1 than that of PGHS-2 (Figure 8). In wild-type PGHS-2, this transition is due to radical migration to Tyr504 and is complete within 50 ms, whereas the WD → WS conversions are considerably slower in the PGHS-2 double mutant (~10 s) and in PGHS-1 (~50 s) (Figure 8) (40). This suggests either that the radical migration from Tyr385 to Tyr504 is 1000-fold slower in PGHS-1 than in PGHS-2 or that the WD → WS conversion in PGHS-1 is caused by a slow Tyr385 conformational change. Mutational studies in PGHS-1 will be needed to resolve this issue.
Another hallmark difference between the PGHS isoforms is that PGHS-2 converts from cyclooxygenase to lipoxyge-nase catalysis upon acetylation by aspirin (39, 43), whereas PGHS-1 is completely inhibited (47, 48). This difference has been attributed to the slightly larger substrate binding channel in PGHS-2, permitting substrate binding and abstraction of the C13 hydrogen even when Ser530 is acetylated (13). Introduction of the Y348F mutation into Y504F PGHS-2 results in complete oxygenase inhibition upon acetylation, just as observed in PGHS-1.
CONCLUSIONS
The Tyr348–Tyr385 hydrogen bond in PGHS-2 appears to play an important role in regulating Tyr385 radical characteristics and in the positioning of the Tyr385 side chain in the cyclooxygenase pocket. These roles are reflected by changes in tyrosyl radical kinetics and conformation, and in cyclooxygenase inhibitor kinetics when the interactions between Tyr348 and Tyr385 are disrupted.
Acknowledgments
We thank Dr. G. Barney Ellison for the generous gift of EtOOH.
Footnotes
Abbreviations: PGHS-1, prostaglandin H synthase-1; PGHS-2, prostaglandin H synthase-2; EtOOH, ethyl hydrogen peroxide; WD, Y504F 29–30 G wide doublet tyrosyl radical; WS, Y348F/Y504F 30 G wide singlet tyrosyl radical; NS, Y348F/Y504F 13 G narrow singlet tyrosyl radical; EPR, electron paramagnetic resonance spectroscopy; ENDOR, electron nuclear double resonance spectroscopy; AA, arachidonic acid; d8-AA, 5,6,8,9,11,12,14,15-d8-arachidonic acid; PPHP, trans-5-phenyl-4-pentenyl-1-hydroperoxide; TMPD, N,N,N′,N′-tetramethyl-p-phenylenediamine; RFQ, rapid freeze quench; PPIX, protoporphyrin IX.
This work was supported by U.S. Public Health Service Grants GM52170 (to R.J.K.) and GM44911 (to A.-L.T.) and Postdoctoral Fellowship DK61929 (to C.E.R.).
References
- 1.Smith WL, Dewitt DL. Prostaglandin endoperoxide H synthases-1 and -2. Adv Immunol. 1996;62:167–215. doi: 10.1016/s0065-2776(08)60430-7. [DOI] [PubMed] [Google Scholar]
- 2.Herschman HR. Prostaglandin synthase 2. Biochim Biophys Acta. 1996;1299:125–140. doi: 10.1016/0005-2760(95)00194-8. [DOI] [PubMed] [Google Scholar]
- 3.Lambeir AM, Markey CM, Dunford HB, Marnett LJ. Spectral properties of the higher oxidation states of prostaglandin H synthase. J Biol Chem. 1985;260:14894–14896. [PubMed] [Google Scholar]
- 4.Tang MS, Copeland RA, Penning TM. Detection of an Fe2+-protoporphyrin-IX intermediate during aspirin-treated prostaglandin H2 synthase II catalysis of arachidonic acid to 15-HETE. Biochemistry. 1997;36:7527–7534. doi: 10.1021/bi962750k. [DOI] [PubMed] [Google Scholar]
- 5.Lu G, Tsai AL, Van Wart HE, Kulmacz RJ. Comparison of the peroxidase reaction kinetics of prostaglandin H synthase-1 and -2. J Biol Chem. 1999;274:16162–16167. doi: 10.1074/jbc.274.23.16162. [DOI] [PubMed] [Google Scholar]
- 6.Dietz R, Nastainczyk W, Ruf HH. Higher oxidation states of prostaglandin H synthase. Rapid electronic spectroscopy detected two spectral intermediates during the peroxidase reaction with prostaglandin G2. Eur J Biochem. 1988;171:321–328. doi: 10.1111/j.1432-1033.1988.tb13793.x. [DOI] [PubMed] [Google Scholar]
- 7.Shimokawa T, Kulmacz RJ, DeWitt DL, Smith WL. Tyrosine 385 of prostaglandin endoperoxide synthase is required for cyclooxygenase catalysis. J Biol Chem. 1990;265:20073–20076. [PubMed] [Google Scholar]
- 8.Tsai AL, Kulmacz RJ, Palmer G. Spectroscopic evidence for reaction of prostaglandin H synthase-1 tyrosyl radical with arachidonic acid. J Biol Chem. 1995;270:10503–10508. doi: 10.1074/jbc.270.18.10503. [DOI] [PubMed] [Google Scholar]
- 9.Picot D, Loll PJ, Garavito RM. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature. 1994;367:243–249. doi: 10.1038/367243a0. [DOI] [PubMed] [Google Scholar]
- 10.Loll PJ, Picot D, Garavito RM. The structural basis of aspirin activity inferred from the crystal structure of inactivated prostaglandin H2 synthase. Nat Struct Biol. 1995;2:637–643. doi: 10.1038/nsb0895-637. [DOI] [PubMed] [Google Scholar]
- 11.Loll PJ, Picot D, Ekabo O, Garavito RM. Synthesis and use of iodinated nonsteroidal antiinflammatory drug analogs as crystallographic probes of the prostaglandin H2 synthase cyclooxygenase active site. Biochemistry. 1996;35:7330–7340. doi: 10.1021/bi952776w. [DOI] [PubMed] [Google Scholar]
- 12.Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY, Gildehaus D, Miyashiro JM, Penning TD, Seibert K, Isakson PC, Stallings WC. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 1996;384:644–648. doi: 10.1038/384644a0. [DOI] [PubMed] [Google Scholar]
- 13.Luong C, Miller A, Barnett J, Chow J, Ramesha C, Browner MF. Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nat Struct Biol. 1996;3:927–933. doi: 10.1038/nsb1196-927. [DOI] [PubMed] [Google Scholar]
- 14.Malkowski MG, Ginell SL, Smith WL, Garavito RM. The productive conformation of arachidonic acid bound to prostaglandin synthase. Science. 2000;289:1933–1937. doi: 10.1126/science.289.5486.1933. [DOI] [PubMed] [Google Scholar]
- 15.Thuresson ED, Malkowski MG, Lakkides KM, Rieke CJ, Mulichak AM, Ginell SL, Garavito RM, Smith WL. Mutational and X-ray crystallographic analysis of the interaction of dihomo-γ-linolenic acid with prostaglandin endoperoxide H synthases. J Biol Chem. 2001;276:10358–10365. doi: 10.1074/jbc.M009378200. [DOI] [PubMed] [Google Scholar]
- 16.Malkowski MG, Thuresson ED, Lakkides KM, Rieke CJ, Micielli R, Smith WL, Garavito RM. Structure of eicosapentaenoic and linoleic acids in the cyclooxygenase site of prostaglandin endoperoxide H synthase-1. J Biol Chem. 2001;276:37547–37555. doi: 10.1074/jbc.M105982200. [DOI] [PubMed] [Google Scholar]
- 17.Loll PJ, Sharkey CT, O’Connor SJ, Dooley CM, O’Brien E, Devocelle M, Nolan KB, Selinsky BS, Fitzgerald DJ. O-Acetylsalicylhydroxamic acid, a novel acetylating inhibitor of prostaglandin H2 synthase: Structural and functional characterization of enzyme–inhibitor interactions. Mol Pharm. 2001;60:1407–1413. doi: 10.1124/mol.60.6.1407. [DOI] [PubMed] [Google Scholar]
- 18.Selinsky BS, Gupta K, Sharkey CT, Loll PJ. Structural analysis of NSAID binding by prostaglandin H2 synthase: Time-dependent and time-independent inhibitors elicit identical enzyme conformations. Biochemistry. 2001;40:5172–5180. doi: 10.1021/bi010045s. [DOI] [PubMed] [Google Scholar]
- 19.Rowlinson SW, Kiefer JR, Prusakiewicz JJ, Pawlitz JL, Kozak KR, Kalgutkar AS, Stallings WC, Kurumbail RG, Marnett LJ. A novel mechanism of cyclooxygenase-2 inhibition involving interactions with Ser-530 and Tyr-385. J Biol Chem. 2003;278:45763–45769. doi: 10.1074/jbc.M305481200. [DOI] [PubMed] [Google Scholar]
- 20.Gupta K, Selinsky BS, Kaub CJ, Katz AK, Loll PJ. The 2.0 Å resolution crystal structure of prostaglandin H2 synthase-1: Structural insights into an unusual peroxidase. J Mol Biol. 2004;335:503–518. doi: 10.1016/j.jmb.2003.10.073. [DOI] [PubMed] [Google Scholar]
- 21.Jeffrey GA. An Introduction to Hydrogen Bonding. Oxford University Press; New York: 1997. [Google Scholar]
- 22.Dorlet P, Seibold SA, Babcock GT, Gerfen GJ, Smith WL, Tsai A-L, Un S. High-field EPR study of tyrosyl radicals in prostaglandin H(2) synthase-1. Biochemistry. 2002;41:6107–6114. doi: 10.1021/bi015871f. [DOI] [PubMed] [Google Scholar]
- 23.Wilson JC, Wu G, Tsai AL, Gerfen GJ. Determination of the structural environment of the tyrosyl radical in prostaglandin H-2 synthase-1: A high-frequency ENDOR/EPR study. J Am Chem Soc. 2005;127:1618–1619. doi: 10.1021/ja043853q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hochgesang GP, Rowlinson SW, Marnett LJ. Tyrosine-385 is critical for acetylation of cyclooxygenase-2 by aspirin. J Am Chem Soc. 2000;122:6514–6515. [Google Scholar]
- 25.Rogge CE, Liu W, Wu G, Wang LH, Kulmacz RJ, Tsai AL. Identification of Tyr504 as an alternative tyrosyl radical site in human prostaglandin H synthase-2. Biochemistry. 2004;43:1560–1568. doi: 10.1021/bi035717o. [DOI] [PubMed] [Google Scholar]
- 26.Bambai B, Rogge CE, Stec B, Kulmacz RJ. Role of Asn-382 and Thr-383 in activation and inactivation of human prostaglandin H synthase cyclooxygenase catalysis. J Biol Chem. 2004;279:4084–4092. doi: 10.1074/jbc.M304762200. [DOI] [PubMed] [Google Scholar]
- 27.Tsai AL, Wu G, Palmer G, Bambai B, Koehn JA, Marshall PJ, Kulmacz RJ. Rapid kinetics of tyrosyl radical formation and heme redox state changes in prostaglandin H synthase-1 and -2. J Biol Chem. 1999;274:21695–21700. doi: 10.1074/jbc.274.31.21695. [DOI] [PubMed] [Google Scholar]
- 28.Kulmacz RJ, Lands W. In: Prostaglandins and Related Substances: A Practical Approach. Benedetto C, McDonald-Gibson RG, Nigam S, Slater TF, editors. IRL Press; Washington, DC: 1987. [Google Scholar]
- 29.Guo Q, Wang LH, Ruan KH, Kulmacz RJ. Role of Val509 in time-dependent inhibition of human prostaglandin H synthase-2 cyclooxygenase activity by isoform-selective agents. J Biol Chem. 1996;271:19134–19139. doi: 10.1074/jbc.271.32.19134. [DOI] [PubMed] [Google Scholar]
- 30.Callan OH, So OY, Swinney DC. The kinetic factors that determine the affinity and selectivity for slow binding inhibition of human prostaglandin H synthase-1 and -2 by indomethacin and flurbiprofen. J Biol Chem. 1996;271:3548–3554. doi: 10.1074/jbc.271.7.3548. [DOI] [PubMed] [Google Scholar]
- 31.Graff G, Anderson LA, Jaques LW. Preparation and purification of soybean lipoxygenase-derived unsaturated hydroperoxy and hydroxy fatty-acids and determination of molar absorptivities of hydroxy fatty-acids. Anal Biochem. 1990;188:38–47. doi: 10.1016/0003-2697(90)90525-e. [DOI] [PubMed] [Google Scholar]
- 32.Capdevila JH, Morrow JD, Belosludtsev YY, Beauchamp DR, DuBois RN, Falck JR. The catalytic outcomes of the constitutive and the mitogen inducible isoforms of prostaglandin H2 synthase are markedly affected by glutathione and glutathione peroxidase(s) Biochemistry. 1995;34:3325–3337. doi: 10.1021/bi00010a023. [DOI] [PubMed] [Google Scholar]
- 33.Tsai AL, Berka V, Kulmacz RJ, Wu G, Palmer G. An improved sample packing device for rapid freeze-trap electron paramagnetic resonance spectroscopy kinetic measurements. Anal Biochem. 1998;264:165–171. doi: 10.1006/abio.1998.2774. [DOI] [PubMed] [Google Scholar]
- 34.Tsai AL, Palmer G, Kulmacz RJ. Prostaglandin H synthase. Kinetics of tyrosyl radical formation and of cyclooxygenase catalysis. J Biol Chem. 1992;267:17753–17759. [PubMed] [Google Scholar]
- 35.Shi W, Hoganson CW, Espe M, Bender CJ, Babcock GT, Palmer G, Kulmacz RJ, Tsai A-L. Electron paramagnetic resonance and electron nuclear double resonance spectroscopic identification and characterization of the tyrosyl radicals in prostaglandin H synthase-1. Biochemistry. 2000;39:4112–4121. doi: 10.1021/bi992561c. [DOI] [PubMed] [Google Scholar]
- 36.Bender CJ, Sahlin M, Babcock GT, Barry BA, Chandrashekar TK, Salowe SP, Stubbe J, Lindstrom B, Petersson L, Ehrenberg A, Sjoberg BM. An ENDOR study of the tyrosyl free-radical in ribonucleotide reductase from Escherichia coli. J Am Chem Soc. 1989;111:8076–8083. [Google Scholar]
- 37.Fessenden RW, Schuler RH. Electron spin resonance studies of transient alkyl radicals. J Chem Phys. 1963;39:2147–2195. [Google Scholar]
- 38.Tsai AL, Palmer G, Xiao G, Swinney DC, Kulmacz RJ. Structural characterization of arachidonyl radicals formed by prostaglandin H synthase-2 and prostaglandin H synthase-1 reconstituted with mangano protoporphyrin IX. J Biol Chem. 1998;273:3888–3894. doi: 10.1074/jbc.273.7.3888. [DOI] [PubMed] [Google Scholar]
- 39.Holtzman MJ, Turk J, Shornick LP. Identification of a pharmacologically distinct prostaglandin H synthase in cultured epithelial cells. J Biol Chem. 1992;267:21438–21445. [PubMed] [Google Scholar]
- 40.Tsai A-L, Kulmacz RJ. Tyrosyl radicals in prostaglandin H synthase-1 and -2. Prostaglandins & Other Lipid Mediators. 2000;62:231–254. doi: 10.1016/s0090-6980(00)00083-6. [DOI] [PubMed] [Google Scholar]
- 41.Un S, Atta M, Fontecave M, Rutherford AW. G-Values as a probe of the local protein environment: High-field EPR of tyrosyl radicals in ribonucleotide reductase and photosystem-II. J Am Chem Soc. 1995;117:10713–10719. [Google Scholar]
- 42.Xiao G, Tsai A-L, Palmer G, Boyar WC, Marshall PJ, Kulmacz RJ. Analysis of hydroperoxide-induced tyrosyl radicals and lipoxygenase activity in aspirin-treated human prostaglandin H synthase-2. Biochemistry. 1997;36:1836–1845. doi: 10.1021/bi962476u. [DOI] [PubMed] [Google Scholar]
- 43.Meade EA, Smith WL, DeWitt DL. Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other non-steroidal anti-inflammatory drugs. J Biol Chem. 1993;268:6610–6614. [PubMed] [Google Scholar]
- 44.Kulmacz RJ, Palmer G, Tsai AL. Prostaglandin H synthase: Perturbation of the tyrosyl radical as a probe of anticyclooxygenase agents. Mol Pharm. 1991;40:833–837. [PubMed] [Google Scholar]
- 45.Lanzo CA, Sutin J, Rowlinson S, Talley J, Marnett LJ. Fluorescence quenching analysis of the association and dissociation of a diarylheterocycle to cyclooxygenase-1 and cyclooxygenase-2: Dynamic basis of cyclooxygenase-2 selectivity. Biochemistry. 2000;39:6228–6234. doi: 10.1021/bi992761o. [DOI] [PubMed] [Google Scholar]
- 46.So OY, Scarafia LE, Mak AY, Callan OH, Swinney DC. The dynamics of prostaglandin H synthases. Studies with prostaglandin H synthase-2 Y355F unmask mechanisms of time-dependent inhibition and allosteric activation. J Biol Chem. 1998;273:5801–5807. doi: 10.1074/jbc.273.10.5801. [DOI] [PubMed] [Google Scholar]
- 47.Ku EC, Wasvary JM. Inhibition of prostaglandin synthetase by SU-21524. Fed Proc. 1973;32:3302. [Google Scholar]
- 48.Rome LH, Lands WE. Structural requirements for time-dependent inhibition of prostaglandin biosynthesis by anti-inflammatory drugs. Proc Natl Acad Sci USA. 1975;72:4863–4865. doi: 10.1073/pnas.72.12.4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith WL, Lands WE. Oxygenation of unsaturated fatty acids by soybean lipoxygenase. J Biol Chem. 1972;247:1038–1047. [PubMed] [Google Scholar]