

Abstract
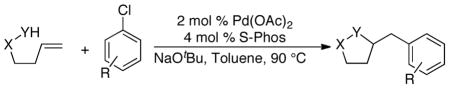
The development of conditions that allow use of inexpensive aryl chlorides as electrophiles in Pd-catalyzed alkene carboamination and carboetherification reactions is described. A catalyst composed of Pd(OAc)2 and S-Phos minimizes N-arylation of the substrate and prevents formation of mixtures of regioisomeric products. A number of heterocycles, including pyrrolidines, isoxazolidines, tetrahydrofurans, and pyrazolidines, are efficiently generated with this method.
Over the past several years, our group has developed a new type of cross-coupling reaction in which alkenes bearing pendant aminopropyl groups are transformed to substituted pyrrolidines via Pd-catalyzed carboamination reactions with aryl bromides. These alkene difunctionalization reactions provide a convergent and efficient means to access substituted N-aryl, N-acetyl, or N-Boc pyrrolidines with a high degree of stereocontrol.1,2 This strategy has also been employed for the generation of several other oxygen- or nitrogen-containing heterocycles.3,4,5
To further expand the scope and utility of these transformations, we sought to employ inexpensive aryl chlorides as electrophilic coupling partners in these reactions.6 In our prior studies we had found that chelating phosphine ligands with wide bite angles, such as Dpe-phos, Xantphos, or dppb, provided optimal results in many transformations of aryl bromides.2 However, Pd-catalysts supported by these ligands are not sufficiently active to facilitate transformations of aryl chlorides, which are considerably less reactive than the corresponding aryl bromides. Thus, to achieve our goal, we would need to discover catalysts that both activate aryl chlorides, and also promote the alkene carboamination process.
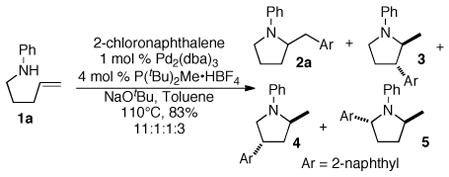
Due to the significant economic advantages associated with using aryl chlorides in place of aryl bromides, considerable research effort has been expended on the development of ligands for Pd-catalyzed cross-coupling reactions of these relatively unreactive electrophiles.7 Many of these ligands are highly effective in Suzuki couplings, N-arylations, and other carbon-carbon or carbon-heteroatom bond-forming processes.7,8,9 However, our initial efforts to employ these ligands in Pd-catalyzed carboamination reactions of γ–(N-arylamino)alkenes (e.g., 1a) provided unsatisfactory results. Use of Buchwald’s biphenyl(dialkyl)phosphines8 led to competing N-arylation of these substrates, and many other electron-rich ligands led to mixtures of regioisomeric products. For example, the Pd/P(tBu)2Me-catalyzed reaction of 1a with 2-chloronaphthalene afforded an 11:1:1:3 mixture of 2a:3:4:5 (eq 1).
 |
(1) |
After considerable optimization, we discovered that PCy2Ph provided acceptable results in many Pd-catalyzed carboamination reactions of aryl chlorides with γ-N-(arylamino)alkenes. As shown in Table 1, both electron-donating and electron-withdrawing groups are tolerated on the aryl chloride, and in all cases the major products were formed with ≥ 90% regioselectivity. Transformations involving acyclic internal alkene substrates were unsuccessful, affording complex mixtures of products.10 However, cyclopentene-derived substrate 1d was converted to bicyclic heterocycles 2f–2g in good yields and with high diastereoselectivities.11
TABLE 1.
Carboamination of γ-(N-Arylamino)alkenes with Aryl Chloridesa
| entry | amine | product | regioselectivityb | yieldc |
|---|---|---|---|---|
| 1 |
 1a |
 2a |
43:3:0:1 | 79% |
| 2 | 1a |
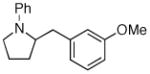 2b |
38:3:0:1 | 74% |
| 3 | 1a |
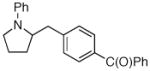 2c |
27:2:0:1 | 79% |
| 4 |
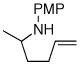 1b |
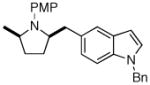 2d |
10:1:0:0 | 66% (>20:1 dr) |
| 5 |
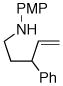 1c |
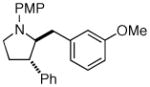 2e |
>20:1 | 65% (>20:1 dr)d |
| 6 |
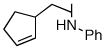 1d |
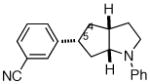 2f |
11:1e | 70% (>20:1 dr)f |
| 7 | 1d |
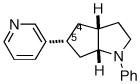 2g |
12:1e | 69% (>20:1 dr)f |
Conditions: amine (1.0 equiv), aryl chloride (1.1–1.4 equiv), NaOtBu (1.2 equiv), Pd2(dba)3 (1 mol %), PCy2Ph (4 mol %), toluene (0.25 M), 110 °C.
Determined by GC and GC/MS analysis. The minor regioisomers formed are analogous to 3–5 shown in eq 1.
Isolated yield (average of two or more experiments).
PCy3•HBF4 was used in place of PCy2Ph.
The minor regioisomer was arylated at C4 rather than C5.
P(tBu)2Me•HBF4 was used in place of PCy2Ph.
The mechanism of the carboamination reactions involves the syn-aminopalladation of intermediate I, followed by C–C bond-forming reductive elimination from intermediate II to afford the desired products (Scheme 1).1,2 Diarylamine side products (III) result from competing C–N bond forming reductive elimination of intermediate I.12,13 Undesired regioisomers 3–5 are generated through β-hydride elimination of II, followed by a series of hydridopalladation/β-hydride elimination steps.1,2 In light of this mechanism, the difficulties we encountered during our studies on carboamination reactions between aryl chlorides and γ-(N-arylamino)alkenes can be ascribed to two factors: (a) use of electron-rich ligands slows reductive elimination from II, leading to increased amounts of regioisomers; and (b) use of bulky, electron-rich ligands that facilitate C–C bond forming reductive elimination leads to competing N-arylation via C–N bond-forming reductive elimination from I.
SCHEME 1.

Mechanism and Side Reactions
This mechanistic analysis suggests that transformations of the analogous N-Boc-protected substrates may be less problematic. The electron-withdrawing Boc-group is known to slow the rate of C–N bond-forming reductive elimination that leads to N-arylation.1b,12 Thus, bulky electron-rich ligands could be used to facilitate the C–C bond-forming reductive elimination from intermediates analogous to II with less concern about competing N-arylation. In addition, the electron-withdrawing Boc-group also disfavors β–hydride elimination pathways that provide regioisomers,1b which should further aid in the selective formation of a single product.
We have recently illustrated that the electron-rich ligand S-Phos14,15 provides excellent results in Pd-catalyzed carboetherification reactions between unsaturated alcohols and aryl bromides,16 and this ligand appeared to be a good candidate for use in alkene difunctionalization reactions between aryl chlorides and substrates containing relatively non-nucleophilic heteroatoms. As such, we examined the Pd-catalyzed coupling of 6a with 4-chlorotoluene and were gratified to find this transformation afforded the desired product 7a in 74% yield (eq 2).
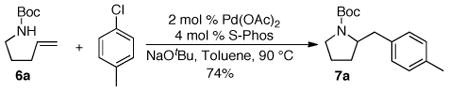 |
(2) |
In order to explore the scope of this method, we examined the coupling of a range of N-Boc-protected γ-aminoalkene derivatives. As shown in Table 2, the transformations are effective with a variety of aryl chlorides, including electron-rich, electron-poor, and ortho-substituted compounds. In addition, satisfactory results were also obtained with the heteroaromatic electrophiles N-benzyl-5-chloroindole, 2-chloropyridine, and 2-chloropyrazine (entries 2, 6, and 8). The synthesis of cis-2,5-disubstituted products was achieved with excellent stereocontrol (entries 12–13), and good to excellent selectivity was obtained in the synthesis of trans-2,3-disubstituted products. In all cases the products were generated with complete regioselectivity.17 Although substitution at the allylic position of the γ-aminoalkene derivative was tolerated (entries 9–11), efforts to employ substrates bearing internal alkenes were unsuccessful due to competing substrate decomposition.17
TABLE 2.
Carboamination of γ-(N-Boc)Aminoalkenes with Aryl Chloridesa
| entry | amine | product | yieldb |
|---|---|---|---|
| 1 | 6a |
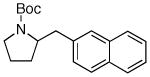 7b 7b
|
74% |
| 2 | 6a |
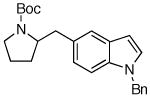 7c 7c
|
66% |
| 3 | 6a |
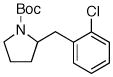 7d 7d
|
65% |
| 4 |
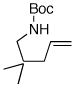 6b |
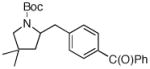 7e 7e
|
73% |
| 5 | 6b |
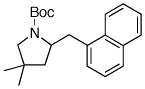 7f 7f
|
81% |
| 6 | 6b |
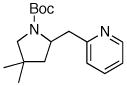 7g 7g
|
72% |
| 7 |
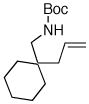 6c |
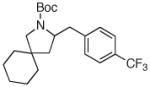 7h 7h
|
63% |
| 8 |
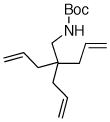 6d |
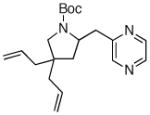 7i 7i
|
56% |
| 9 |
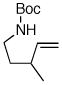 6e |
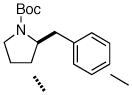 7j 7j
|
61% (4:1 dr) |
| 10 |
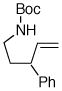 6f |
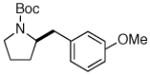 7k 7k
|
64% (> 20:1 dr) |
| 11 | 6f |
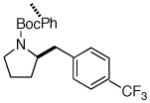 7l 7l
|
63% (> 20:1 dr) |
| 12 |
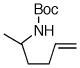 6g |
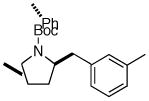 7m 7m
|
71% (> 20:1 dr) |
| 13 | 6g |
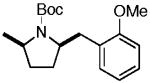 7n 7n
|
69% (> 20:1 dr) |
Conditions: amine (1 equiv), aryl chloride (1.2 equiv), NaOtBu (1.2 equiv), Pd(OAc)2 (2 mol %), S-Phos (4 mol %), toluene (0.25 M), 90 °C.
Isolated yield (average of two or more experiments).
Following our success with N-Boc-aminopropyl alkenes, we proceeded to examine the utility of the Pd/S-Phos catalyst in carboamination and carboetherification reactions of aryl chlorides that generate other heterocycles. As shown in Table 3, the conversion of urea 8, hydroxylamine 9, and hydrazine 10 to the corresponding imidazolidin-2-one 14, isoxazolidine 15, and pyrazolidine 16 proceeded smoothly. The heterocyclic products were obtained in good chemical yield, and 15 was formed as a single diastereomer. The coupling of tertiary alcohol 11 with 1-chloronaphthalene afforded tetrahydrofuran 17 in 89% yield, although a 13:1 mixture of regioisomers was generated. However, attempts to effect a similar transformation between secondary alcohol 12 and 4-chloroanisole failed to yield the desired tetrahydrofuran product.18 Instead, oxidation of the alcohol was observed, which suggests that alkene oxypalladation from an intermediate analogous to I is relatively slow with S-Phos as ligand. As a result, β-hydride elimination from this intermediate is the predominant reaction pathway with substrate 12. The conversion of amine 13 to morpholine 19 was also unsuccessful due to competing N-arylation of the substrate.18,19
TABLE 3.
Synthesis of Other Heterocycles Using Aryl Chlorides as Electrophilesa
| entry | substrate | product | yieldb |
|---|---|---|---|
| 1 |
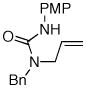 8 |
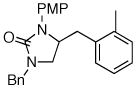 14 |
77% |
| 2 |
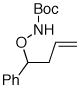 9 |
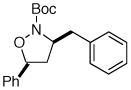 15 |
81% (20:1 dr) |
| 3 |
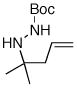 10 |
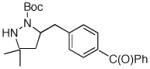 16 |
73% |
| 4 |
11 |
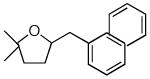 17 |
89%c |
| 5 |
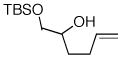 12 |
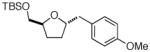 Not observed 18 |
0% |
| 6 |
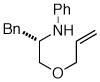 13 |
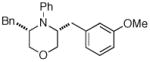 19 Not observed |
0% |
Conditions: substrate (1 equiv), aryl chloride (1.2 equiv), NaOtBu (1.2 equiv), Pd(OAc)2 (2 mol %), S-Phos (4 mol %), toluene (0.25 M), 90 °C or 110 °C.
Isolated yield (average of two or more experiments).
This material was obtained as a 13:1 mixture of regioisomers.
In conclusion, we have developed conditions that allow use of inexpensive and readily available aryl chloride electrophiles in many Pd-catalyzed carboamination and carboetherification reactions. These studies significantly expand the scope and utility of this method for heterocycle synthesis and also illustrate several remaining challenges for catalyst development in the field.
Experimental Section
Representative Procedure for Pd-Catalyzed Carboamination Reactions of Aryl Chlorides
(±)-tert-Butyl 2-(4-methylbenzyl)pyrrolidine-1-carboxylate (7a)
A Schlenk tube was evacuated, flame-dried, and backfilled with nitrogen. The tube was charged with Pd(OAc)2 (2.3 mg, 0.01 mmol), 2-dicyclohexylphosphino-2′,6′-dimethoxy-1,1′-biphenyl (S-Phos, 8.2 mg, 0.02 mmol), and NaOtBu (57.7 mg, 0.60 mmol). The tube was evacuated and backfilled with nitrogen three times. A solution of tert-butyl pent-4-en-1-ylcarbamate (93 mg, 0.50 mmol) and 4-chlorotoluene (71 μL, 0.60 mmol) in toluene (2 mL) was added to the Schlenk tube via syringe. The mixture was heated in a 90 °C oil bath with stirring until the starting material had been consumed as judged by GC analysis (7 h). The reaction mixture was cooled to room temperature, quenched with saturated aqueous NH4Cl (2 mL), and diluted with EtOAc (2 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (3 × 5 mL). The organic layers were concentrated in vacuo, and the crude product was purified by flash chromatography on silica gel to afford 95 mg (69%) of the title compound as a pale yellow oil. 1H NMR (400 MHz, C6D5CD3, 100 °C) δ 7.03–6.89 (m, 4 H), 4.03–3.91 (m, 1 H), 3.33–3.23 (m, 1 H), 3.20–3.01 (m, 2 H), 2.51 (dd, J = 8.9, 13.0 Hz, 1 H), 2.13 (s, 3 H), 1.55–1.28 (m, 13 H); 13C NMR (100 MHz, C6D5CD3, 100 °C) δ 154.6, 137.1, 135.9, 130.0, 129.6, 78.9, 59.5, 59.4, 47.2, 30.3, 29.1, 23.7, 21.2; IR (film) 1693, 1394, 1172 cm−1; MS (ESI): 298.1779 (298.1783 calcd for C17H25NO2, M + Na+).
Supplementary Material
Acknowledgments
The authors thank the NIH-NIGMS (GM071650) for financial support of this work. Additional support was provided by the Camille and Henry Dreyfus Foundation (Camille Dreyfus Teacher Scholar Award), GlaxoSmithKline, Eli Lilly, and Amgen.
Footnotes
Supporting Information Available. Experimental procedures, spectroscopic data, and copies of 1H and 13C NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Ney JE, Wolfe JP. Angew Chem, Int Ed. 2004;43:3605. doi: 10.1002/anie.200460060. [DOI] [PubMed] [Google Scholar]; (b) Bertrand MB, Wolfe JP. Tetrahedron. 2005;61:6447. [Google Scholar]; (c) Ney JE, Wolfe JP. J Am Chem Soc. 2005;127:8644. doi: 10.1021/ja0430346. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Bertrand MB, Wolfe JP. Org Lett. 2006;8:2353. doi: 10.1021/ol0606435. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Bertrand MB, Neukom JD, Wolfe JP. J Org Chem. 2008;73:8851. doi: 10.1021/jo801631v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Rossiter LM, Slater ML, Giessert RE, Sakwa SA, Herr RJ. J Org Chem. 2009;74:9554. doi: 10.1021/jo902114u. [DOI] [PubMed] [Google Scholar]
- 2.For reviews on Pd-catalyzed carboamination reactions, see: Wolfe JP. Eur J Org Chem. 2007:571.Wolfe JP. Synlett. 2008:2913.
- 3.For related syntheses of imidazolidin-2-ones, isoxazolidines, pyrazolidines, piperazines, and morpholines via Pd-catalyzed carboamination reactions, see: Fritz JA, Wolfe JP. Tetrahedron. 2008;64:6838. doi: 10.1016/j.tet.2008.04.015.Lemen GS, Giampietro NC, Hay MB, Wolfe JP. J Org Chem. 2009;74:2533. doi: 10.1021/jo8027399.Giampietro NC, Wolfe JP. J Am Chem Soc. 2008;130:12907. doi: 10.1021/ja8050487.Nakhla JS, Schultz DM, Wolfe JP. Tetrahedron. 2009;65:6549. doi: 10.1016/j.tet.2009.04.017.Leathen ML, Rosen BR, Wolfe JP. J Org Chem. 2009;74:5107. doi: 10.1021/jo9007223.Peng J, Jiang D, Lin W, Chen Y. Org Biomol Chem. 2007;5:1391. doi: 10.1039/b701509g.Peng J, Lin W, Yuan S, Chen Y. J Org Chem. 2007;72:3145. doi: 10.1021/jo0625958.
- 4.For Cu- or Au-catalyzed carboamination reactions, see: Fuller PH, Chemler SR. Org Lett. 2007;9:5477. doi: 10.1021/ol702401w.Zeng W, Chemler SR. J Am Chem Soc. 2007;129:12948. doi: 10.1021/ja0762240. and references cited therein.Zhang G, Cui L, Wang Y, Zhang L. J Am Chem Soc. 2010;132:1474. doi: 10.1021/ja909555d.
- 5.For alkene carboamination reactions involving solvent C–H bond functionalization, see: Rosewall CF, Sibbald PA, Liskin DV, Michael FE. J Am Chem Soc. 2009;131:9488. doi: 10.1021/ja9031659.Sibbald PA, Rosewall CF, Swartz RD, Michael FE. J Am Chem Soc. 2009;131:15945. doi: 10.1021/ja906915w.
- 6.The use of aryl chlorides as electrophiles in Pd-catalyzed carboamination reactions that afford aziridines or pyrrolizidin-2-ones has recently been reported. See: Hayashi S, Yorimitsu H, Oshima K. Angew Chem, Int Ed. 2009;48:7224. doi: 10.1002/anie.200903178.Bagnoli L, Cacchi S, Fabrizi G, Goggiamani A, Scarponi C, Tiecco M. J Org Chem. 2010;75:2134. doi: 10.1021/jo1002032.
- 7.Reviews: Littke A, Ackermann L, editors. Modern Arylation Methods. Wiley-VCH; Weinheim, Germany: 2009. p. 25.Bedford RB, Cazin CSJ, Holder D. Coord Chem Rev. 2004;248:2283.Littke AF, Fu GC. Angew Chem Int Ed. 2002;41:4176. doi: 10.1002/1521-3773(20021115)41:22<4176::AID-ANIE4176>3.0.CO;2-U.
- 8.Surry DS, Buchwald SL. Angew Chem, Int Ed. 2008;47:6338. doi: 10.1002/anie.200800497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Stambuli JP, Kuwano R, Hartwig JF. Angew Chem, Int Ed. 2002;41:4746. doi: 10.1002/anie.200290036. [DOI] [PubMed] [Google Scholar]; (b) Navarro O, Kelly RA, III, Nolan SP. J Am Chem Soc. 2003;125:16194. doi: 10.1021/ja038631r. [DOI] [PubMed] [Google Scholar]; (c) Reddy CV, Kingston JV, Verkade JG. J Org Chem. 2008;73:3047. doi: 10.1021/jo702367k. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 10.Analysis by 1H NMR suggests that mixtures of pyrrolidine regioisomers are formed, and competing substrate N-arylation also occurs. This outcome may be due to a combination of relatively slow aminopalladation of the more hindered internal alkene combined with relatively slow reductive elimination from a secondary alkylpalladium intermediate analogous to II.
- 11.The installation of the aryl group at C5 rather than C6 in 2f–2g results from β-hydride elimination/hydridopalladation processes similar to those illustrated in Scheme I (II→IV→3–5). Selective formation of the 6-aryl isomers has been ascribed to the relative stabilities of intermediates along this pathway. For a detailed discussion of reaction mechanism and the origin of these products, see reference 1c.
- 12.(a) Muci AR, Buchwald SL. Top Curr Chem. 2002;219:131. [Google Scholar]; (b) Hartwig JF. Inorg Chem. 2007;46:1936. doi: 10.1021/ic061926w. [DOI] [PubMed] [Google Scholar]
- 13.The reactivity of intermediates I and II is highly dependent on ligand structure. For further discussion see references 1c and 2a.
- 14.S-Phos = 2-dicyclohexylphosphino-2′,6′-dimethoxy-1,1′-biphenyl.
- 15.Walker SD, Barder TE, Martinelli JR, Buchwald SL. Angew Chem, Int Ed. 2004;43:1871. doi: 10.1002/anie.200353615. [DOI] [PubMed] [Google Scholar]
- 16.Ward AF, Wolfe JP. Org Lett. 2010;12:1268. doi: 10.1021/ol1001472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Analysis of crude reaction mixtures by 1H NMR showed predominantly the desired product, with little or no evidence of side product formation. Modest yields obtained in some transformations may be due to base-mediated substrate decomposition. See: Tom NJ, Simon WM, Frost HN, Ewing M. Tetrahedron Lett. 2004;45:905.
- 18.Both of these transformations are effective with the corresponding aryl bromides when an appropriate phosphine ligand is used. For transformations that afford cis-3,5-disubstituted morpholines using P(2-furyl)3 as ligand, see reference 3e. For transformations that generate trans-2,5-disubstituted tetrahydrofurans using bis(2-diphenylphosphinophenyl)ether (dpe-phos) as ligand, see: Hay MB, Hardin AR, Wolfe JP. J Org Chem. 2005;70:3099. doi: 10.1021/jo050022+.
- 19.Efforts to employ a Boc-protected analog of 13 also failed to provide a substituted morpholine product. Base-mediated decomposition of the substrate was observed.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.