


Abstract
A fundamental question in microarray analysis is the estimation of the number of expressed probes in different RNA samples. Negative control probes available in the latest microarray platforms, such as Illumina whole genome expression BeadChips, provide a unique opportunity to estimate the number of expressed probes without setting a threshold. A novel algorithm was proposed in this study to estimate the number of expressed probes in an RNA sample by utilizing these negative controls to measure background noise. The performance of the algorithm was demonstrated by comparing different generations of Illumina BeadChips, comparing the set of probes targeting well-characterized RefSeq NM transcripts with other probes on the array and comparing pure samples with heterogenous samples. Furthermore, hematopoietic stem cells were found to have a larger transcriptome than progenitor cells. Aire knockout medullary thymic epithelial cells were shown to have significantly less expressed probes than matched wild-type cells.
INTRODUCTION
Statistical analysis of microarray gene expression experiments has so far focused mostly on identifying genes which are differentially expressed between different conditions (1,2). However, there is an even more fundamental question which has so far been largely neglected, which is to detect which transcripts are actually expressed in each sample. Understanding how the size of the transcriptome varies with cell type and circumstance is of fundamental biological interest (3–5). For example, does the pluripotency of stem cells imply a greater number of distinct expressed transcripts than in committed cells (3). There are also technical implications, for example because most microarray normalization algorithms assume that different samples express similar numbers of transcripts (6).
Technologies that sequence randomly sampled transcripts from RNA samples provide possibilities to estimate statistically the size of the transcriptome (7,8). However, these statistical methods are heavily dependent on distributional assumptions about how expression levels vary between transcripts, and have not yet attracted widespread use. We provide instead a method for estimating the size of the transcriptome using inexpensive, readily available microarray data and making relatively few assumptions. Specifically, we propose an algorithm to estimate the proportion of probes on a whole-genome microarray that correspond to transcripts which are present in the RNA sample hybridized to a particular array. The only requirement is for a selection of good-quality negative control probes which are representative of the behavior of non-expressed probes. Throughout this article, we use the shorthand ‘expressed probe’ to mean a probe corresponding to a transcript which is expressed in the sample hybridised to that array.
Commercial microarray platforms often provide detection calls (present/absent) for each probe on an array (9). For example, Illumina BeadStudio software computes a detection P-value for each probe on an Illumina BeadChip, equal to the proportion of negative control probes which have intensities greater than that probe on the same array (10). These calls allow a subset of probes to be selected which are highly likely, based on their intensities, to be truly expressed. The situation is similar for Affymetrix arrays. Affymetrix MAS 5.0 software computes a present/absent call for each probe-set on an Affymetrix GeneChip. The present/absent call is made using a Wilcoxon test for each probe set after estimating a baseline from the intensities of mismatch probes on the same array (11). Present/absent calls can be refined using probe-sequence information (12) even without mismatch information (13).
A different approach is to judge the presence/absence for each probe relative to its range of expression in a large database of expression profiles (14). This approaches accounts for differences in probe performance, but makes calls only for probes which have a full range of expression in the database.
All detection call methods yield an estimate of the proportion of expressed probes, simply by counting the number of probes called as detected versus those that are not. However, the approach is skewed toward finding evidence in favor of expression. The rate of false negatives, probes which are expressed but are called absent, is not controlled or estimated. BeadStudio detection calls are typically relatively stringent, so the false negative rate is likely to be large (9). MAS 5.0 present calls are less stringent, but the false negative rate is still unknown. Detection call methods are not generally designed or intended to call probes that are expressed at low levels.
Our aim is different and more ambitious, to estimate the proportion of all probes which are expressed, regardless of how high or low that expression level might be. Rather than making present/absent calls for individual probes, we treat the size of the transcriptome as a phenotype in its own right. Our algorithm is designed to give a consistent and approximately unbiased estimate of the total number of expressed probes, without necessarily identifying individual probes.
We applied our algorithm to the increasingly popular Illumina whole-genome expression BeadChips, for which a set of good-quality negative control probes is available. We comprehensively tested the efficacy of the algorithm on a range of different experimental scenarios that could be expected to produce groups with different transcriptome sizes. Initial validation was performed by comparing the number of expressed probes between chip generations and for verified coding sequences versus predicted sequences. These measurements accurately portrayed the progress in chip design and sequence annotation. The algorithm also effectively tracked a controlled increase in transcriptome size, which was achieved by comparing chips generated from homogenous and heterogenous populations. Finally, we showed that our algorithm could identify changes in transcription at the physiological level by studying differentiation stages of hemapoietic cells and the regulation of RNA transcript numbers by the thymic transcription factor AIRE.
MATERIALS AND METHODS
Algorithmic approach
The intensity distribution of regular probes on any particular array is a mixture of the intensities of probes which are expressed and those which are not expressed. We can express this as the mathematical mixture
where freg is the overall probability density function of the intensities of regular probes, f0 the probability density for non-expressed regular probes, f1 the probability density for expressed regular probes and π0 the proportion of regular probes that are not expressed. The aim of this article is to estimate π0. The corresponding cumulative distribution function can be similarly written
| (1) |
If the array contains a large number of good-quality negative control probes, then the empirical distribution of intensities from these probes will give a good estimate of F0. Meanwhile, Freg can be readily estimated from the empirical distribution of regular probe intensities. If we could also estimate F1(y), for any particular y, then we could solve (1) for π0.
It is natural to assume that the intensities of expressed probes are made up of background intensities and signal intensities, i.e. if y is the intensity of a randomly chosen expressed probe, then
where b is the background intensity and s is the signal intensity (18). Here s is a measure of the expression level of the probe's; transcript while b represents measurement error arising from technical sources. It is also natural to assume that the background intensities follow the same distribution f0 as that of non-expressed probes. Therefore, the distributions of expressed and non-expressed probes are related through the convolution equation
or
| (2) |
where fs and Fs are the probability density and cumulative distribution functions of the signals of expressed probes.
Let b1, … , bm be the observed intensities of negative control probes for one array. Approximating f0 in (2) by the empirical distribution of the bi gives
| (3) |
Now we need an estimator for Fs.
Any plot of microarray intensities shows a very strongly right skew distribution. It is reasonable to assume that most transcripts have low levels of expression and that higher levels of expression are progressively less common. Therefore, we follow the previous practice of a number of several highly successful background correction and normalization methods (18–21) and assume that Fs can be adequately modelled by an exponential distribution. Let y1, … , yn be the observed intensities of regular probes for our array. The mean parameter E(s) = α of Fs is estimated by
where 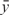 and
and 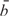 are the averages of observed intensities for regular probes and negative control probes, respectively. This yields our estimator for π0. For any y we estimate
are the averages of observed intensities for regular probes and negative control probes, respectively. This yields our estimator for π0. For any y we estimate
and
Finally we estimate F1(y) from (3), using the exponential form for Fs.
This yields an estimate of the proportion of non-expressed probes as
Any y yields an estimate. In practice, we use
where bmed is the median of the negative control intensities.
The estimated expression proportions were found to be stable around bmed when testing on all negative control probes (Supplementary Figure S1). All three distribution function estimators should be accurately estimated for y in this neighborhood.
Microarray data sets
The data sets used in this study are summarized in Table 1. Particular attention is given to data sets 2 and 4. For data set 2, CD45− Ly51− MHCIhi mTECs were isolated from C57BL/6 Aire+/+ and Aire−/− mice (22). For data set 4, C57BL/6 mouse hematopoietic stem cells are found in the Lineage- Sca1+ Kit+ (LSK) fraction of bone marrow tissue (23). Unless otherwise indicated in Table 1, all data is from in-house experiments conducted by the authors.
Table 1.
Data sets used in this study
| ID | Platform | Number regular probes | Number negative controls | Experiment description |
|---|---|---|---|---|
| 1 | MouseWG-6 V1.1 | 46 657 | 1603 | Six cell types: hematopoietic stem cells, CMPs, GMPs, pro DC precursors, neutrophils and macrophages. Number of arrays per cell type: 4, 2, 1, 3, 1 and 3, respectively. |
| 2 | MouseWG-6 V1.1 | 46 657 | 1603 | Two cell types: wild type and Aire knockout MHCIIhi mTECs. Number of arrays per cell type: 3. |
| 3 | MouseWG-6 V2 | 45 281 | 936 | Three cell types: pro DC precursors, neutrophils and macrophages. Number of arrays per cell type: 9, 3 and 3, respectively. |
| 4 | MouseWG-6 V2 | 45 281 | 936 | Four cell types: hematopoietic stem cells, CMPs, GMPs and MEPs. Number of arrays per cell type: 3. |
| 5 | HumanWG-6 V1 | 47 312 | 1517 | Six conditions: MCF7 and Jurkat samples were mixed at six different proportions (see Figure 4a). Number of arrays per condition: 2. |
| 6 | HumanWG-6 V1 | 47 312 | 1517 | Four conditions: Universal Human Reference RNA(UHRR) and Human Brain Reference RNA(HBRR) were mixed at four different proportions (see Figure 4b). Number of arrays per condition: 5. Published in ref. (15) |
| 7 | HumanWG-6 V2 | 48 687 | 1374 | Six conditions: three subtypes of T lymphocytes taken from two patients infected with hepatitis C virus. Number of arrays per condition: 1. Published in ref. (16) |
| 8 | HumanHT-12 | 48 799 | 759 | Twelve samples. Bone marrow from seven malaria-infected and five uninfected donors. Number of arrays per sample: 1. |
| 9 | HumanWG-6 V3 | 48 803 | 759 | Four cell types: Lin−CD49fhiEpCAM−, Lin−CD49f−EpCAM−, Lin−CD49f−EpCAM+ and Lin−CD49f+EpCAM+ mammary subpopulations. Number of arrays per cell type: 3. Published in ref. (17) |
Data input and annotation
All microarray data was read and manipulated using the Bioconductor R software package limma (24). Probe annotation files were downloaded from Illumina web site (http://www.illumina.com).
RESULTS
Negative control intensities
Illumina BeadChips include a set of negative control probes (10). The negative control probes have randomly permutated sequences and appear in all our investigations to be a good representation of the behavior of non-expressed probes. The number of negative control probes ranges from 750 to 1600 for different types of BeadChips (different species and different versions). Each WG-6 BeadChip encompasses six arrays.
Figure 1 shows the intensity distributions for regular probes and negative control probes from a data set using Illumina MouseWG-6 version 2 BeadChips (Table 1, data set 3). There are 45 281 regular probes and 936 negative control probes in each array. On every array, the main body of negative control intensities is below the median and overlapping the lower quartile of the negative controls. The negative control probes consistently track the regular probes in the sense that an array having high regular probe intensities also has high negative control probe intensities. This pattern increases our confidence that the negative control probes provide an unbiased estimate of the background intensities. The similar pattern has been observed for other types of BeadChips.
Figure 1.

Intensity distributions for regular probes and negative control probes. Data from 15 arrays using Illumina MouseWG-6 version 2 was used for this plot. Numbers of regular probes and negative control probes are 45 281 and 936, respectively on each array. Intensities are on log2 scale.
Our algorithm estimates the proportion of expressed probes on each array, by comparing the empirical intensity distribution of the negative control probes with that of the regular probes. A mathematical mixture model is used to infer the intensity distribution of expressed probes, and hence to estimate the expressed proportion. In the following, we demonstrate the performance on this estimator on different data sets and on different BeadChip versions.
Expression proportions by platform
Figure 2 shows estimated expression proportions for all Illumina WG-6 BeadChip platforms. To make this plot, we used all arrays from all data sets described in Table 1 with a few exceptions. The thymic epithelial cells (data set 2), the reference RNA samples (data set 6) and the erythrocyte progenitors (from data set 4) were excluded, so as to make the cell types on the different platforms as similar as possible. There is a consistent trend to higher proportions of expressed probes in later versions of both mouse and human BeadChips, presumably because of improved probe design in the later platforms. In mouse, 30% v1.1 probes were replaced in v2. In human, 82% of v1 probes were replaced or removed in v2, and a further 23% of v2 probes were replaced in v3. Our v3 BeadChips had larger expression proportions than our HT-12 BeadChips, despite having exactly the same set of probes. This may be because the v3 samples are from adult stem cells and early progenitors, which have been found to express more genes than lineage restricted cells (25,26).
Figure 2.

Proportions of expressed probes estimated for different BeadChip types. All data described in Table 1 are included except for datasets 2 and 6 and the erythrocyte progenitors from data set 4. For each BeadChip type, the boxes show the minimum, first quartile, median, second quartile and maximum of estimated expression proportions across all its arrays.
Regardless of platform, far fewer probes were detected when using BeadStudio's; detection P-values instead of our estimate (Supplementary Section 2). The BeadStudio detection calls are presumably less able to detect lowly expressed probes. The increasing pattern of expression proportions along the BeadChips versions was also lost (Supplementary Figure S2).
RefSeq versus non-RefSeq probes
RefSeq NM transcripts from the RefSeq database are curated mature messenger RNA transcripts that have verified coding sequences. For each BeadChip type, we divided the regular probes on the array into RefSeq NM probes and other probes, using annotation provided by Illumina. Probes designed to interrogate these transcripts are naturally more likely to be truly expressed in most samples, compared with probes designed to interrogate predicted transcripts, and this was confirmed by our data for every BeadChip type (Figure 3).
Figure 3.

Proportions of expressed probes by RefSeq annotation and BeadChip type. Data are as for Figure 2.
Interestingly, the RefSeq expression proportions were higher for human than for mouse, regardless of BeadChips version. The difference remained when estimating the expression proportion at the gene or transcript level (Supplementary Section 3). At the gene level estimation, the median numbers of expressed genes in HumanWG-6 version 3 and MouseWG-6 version 2 are 14 597 and 9 467, respectively.
Mixture experiments
A microarray experiment in which pure samples are mixed at different proportions is called a mixture experiment in this study. The mixed sample, which is a mixture of the two pure samples, should have a larger proportion of expressed probes than either of the pure samples because it includes distinct transcripts from both samples. Two mixture experiments were examined here: an in-house mixture experiment and the MAQC experiment (15). In the in-house mixture experiment, MCF7 and Jurkat samples were mixed at six different proportions: 100% versus 0%, 94% versus 6%, 88% versus 12%, 76% versus 24%, 50% versus 50% and 0% versus 100% (Data set 5 in Table 1). In the MAQC experiment, UHRR and HBRR samples were mixed at four different proportions: 100% versus 0%, 75% versus 25%, 25% versus 75% and 0% versus 75% (Data set 6 in Table 1). Estimation of the expression proportion was performed on RefSeq NM probes.
As expected, almost all the mixed samples have higher proportions of expressed probes than pure samples in both our in-house mixture experiment and the MAQC experiment (Figure 4a and b). It is interesting to see that MAQC arrays have larger proportions of expressed probes than arrays in our in-house mixture experiment and other arrays (RefSeq NM groups) in this study. This is not surprising because the UHRR sample consists of RNAs from 10 human cancer cell lines and therefore includes many more expressed distinct mRNA transcripts than samples in the ‘usual' experiments.
Figure 4.

Expression proportion estimation for samples from two mixture experiments. (a) Estimated expression proportions for samples from the in-house mixture experiment. Jurkat and MCF7 samples were mixed at the proportions of 100:0, 94:6, 88:12, 76:24, 50:50 and 0:100. Error bars are standard errors (n = 2). (b) Estimated expression proportions for samples used in the MAQC project. Universal Human Reference RNA(UHRR) and Human Brain Reference RNA(HBRR) samples were mixed at the proportions of 100:0, 75:25, 25:75 and 0:100. Error bars are standard errors (n = 5). (c) RefSeq NM probes commonly and exclusively expressed in MCF7 and Jurkat samples. (d) RefSeq NM probes commonly and exclusively expressed in UHRR and HBRR samples.
The HBRR sample is also found to have a large proportion of expressed probes (73.8%). It was reported that the proportion of expressed genes in mouse brain was 80% (27). The expression proportion estimation at gene level reveals that the average proportion of expressed genes in the HBRR sample was 79.6%, which was very close to the reported proportion.
The estimated expression proportions for the mixed samples and pure samples can be used to infer the numbers of genes expressed commonly and uniquely in the two samples (see Supplementary Section 4 for details). This showed 56% of RefSeq NM probes to be expressed in both MCF7 and Jurkat, with 2.4% uniquely expressed in Jurkat and 3.2% in MCF7 (Figure 4c). For the MAQC data, 70% of RefSeq NM probes were expressed in both UHRR and HBRR, with 3–4% uniquely expressed in each individual source (Figure 4d).
Hematopoietic stem and progenitor cells
Stem cells are unique in their ability to self renew and differentiate into mature cells. Recent work suggests that embryonic stem cells maintain their differentiation potential through a unique chromatin state, that keeps lineage-specific genes poised for activation, yet is able to be permanently shut down as cells were lineage restricted and the genes would not be required (25,26). This chromatin structure, termed ‘bivalent domains’, results in expression for many lineage specific genes at a low level. Accessibility is lost during lineage restriction, correlating with a decreased number of expressed genes. Whether this is true for tissue-specific stem cells is unknown.
Hematopoietic stem cells (LSKs) are thought to differentiate into lineage restricted progenitors including common myeloid progenitors (CMPs) and common lymphoid progenitors (CLPs) (28). CMPs in turn produce more restricted progenitors including granulocyte macrophage progenitors (GMPs) and megakaryocyte erythrocyte progenitors (MEPs) (29) (Figure 5a). It has been hypothesized that hematopoietic stem cells may express a wider variety of transcripts than restricted progenitors, although many of these transcripts may be expressed at low levels (3). Our algorithm shows that LSK cells do indeed have a higher expression proportion than the three types of progenitor cells. More generally, increasing lineage restriction and decreasing pluripotency is associated with lower expression proportions in cells further down the family tree (Figure 5b).
Figure 5.

Correspondence between hematopoietic stem cell differentiation tree and estimated expression proportions for different cell types. (a) Hematopoietic stem cells differentiate into different progenitor cells. (b) Estimated proportions of expressed probes for four different cell types. Error bars are standard errors (n = 3).
Promiscuous expression in the thymus
Effective deletion of autoreactive T cells is essential for establishing immunological tolerance and preventing autoimmune disease. Medullary thymic epithelial cells (mTECs) play a unique role in this process due to their ability to ‘promiscuously’ express a range of autoantigens that are normally restricted to peripheral tissues (30,31). The intrathymic expression of these antigens exposes thymocytes to the peripheral environment during their development and facilitates the negative selection of those cells displaying autoreactive receptors: a mechanism that has proved important in preventing autoimmunity against tissue-specific antigens (32,33,34).
The autoimmune regulator, Aire, is a transcription factor that promotes promiscuous expression in mTECs and its absence results in a reduction in the intrathymic expression of many tissue-restricted antigen genes (4,5,35). At the phenotypic level, AIRE mutations in humans are responsible for the multi-organ autoimmune syndrome APS-1 (36,37), which is mimicked in part by Aire-deficient mouse models (4,22,38).
The estimated proportion of expressed probes for our wild-type mTEC samples was 0.52 (standard error 0.009, n = 3). As expected, this was greater than for other cell types using the same platform (Figure 2). In our Aire−/− mTEC samples, the proportion of expressed probes was markedly reduced to 0.44 (standard error 0.016, n = 3).
The number of genes whose expression is activated by Aire has been reported to be in the range 200–1200 (4). This appears to be an underestimate. Our estimation at the gene level shows that there are 2006 more genes expressed in the wild type compared with the Aire−/− cells.
DISCUSSION
We have validated our algorithm by showing that it can track improvements in probe design and annotation. Newer BeadChips show steadily increasing expression proportions for the same cell types as probe design is improved.
Our estimator of proportion expressed has a variety of potential applications. By examining mixed samples, we have shown that our estimator can distinguish heterogeneous cell samples from pure samples. We were further able to determine the number of distinct transcripts uniquely expressed in each of the pure samples. We have also demonstrated that the estimator can detect multi-potential gene expression in stem cells, and can describe promiscuous expression associated with T-cell deletion in the thymus. The ability to quantify these effects in terms of numbers of probes, and numbers of genes, is a marked step forward in understanding these processes. We give the first quantitative demonstration that hematopoietic stem cells have a larger expressed transcriptome than more committed progenitors. In the thymus we show that twice as many genes are affected by the regulator Aire as previously reported. In the future, we plan to apply this technique across a extensive collection of hematopoietic cell lineages, to describe the process of differentiation and commitment. Comparisons across cells in different activated states, such as naive, memory and effector T cells, is also likely to throw light on the nature of the molecular response.
The estimator can be applied to subclasses of probes. The expression proportion computed from the RefSeq annotated probes alone provides an estimate of the number of well-characterized messenger RNA transcripts that are expressed. The expression proportion computed from the unannotated probes could suggest the existence of novel messenger RNA transcripts.
The human BeadChips showed higher numbers of RefSeq genes expressed than mouse BeadChips. This is not sufficient to conclude that the human transcriptome is larger than that of mouse, because there may be differences in RefSeq annotation or probe performance between the species, and the cell types profiled for the two species were not identical. Indeed the mouse results in Figure 3 exclude the thymic epithelium cells, which had the highest expression proportions of any mouse samples. However, the difference was preserved across all versions of the BeadChips, and the mouse cell types include hematopoitetic stem cells which were expected to have larger than average transcriptomes. Apart from the universal reference RNA samples, the human samples with highest expression proportions were mammary stem cells.
Our expression proportions tend to be much higher than the proportion of probes called as detected by Illumina BeadStudio detection calls. This was expected because detection calls cannot estimate probes with low-level expression. Even more importantly, our measure is more stable and predictable across replicate arrays, cell types and BeadChip versions. This may be because the detection call P-values rely on an upper tail statistic of the negative controls, a type of extreme statistic subject to relatively high variability, whereas our method uses the entire distribution of the negative controls, with greatest weight near the median.
The proportion of probes called as expressed by Illumina detection calls can be varied by choosing the cutoff P-value higher or lower. The same is true of Affymetrix present/absent calls. A cutoff P-value of 0.01 underestimates the expression proportion, whereas Illumina detection calls with P=0.5 give expression proportions which are much too high (data not shown). In general, there is no P-value cutoff for the detection call that gives a consistent estimate of the propotion expressed across all BeadStudio platforms and biological samples, because the detection call approach does not attempt to estimate the expression distribution of expressed probes.
Our results have a number of technical implications relating to microarray normalization and pre-processing. Most microarray normalization strategies assume that all the samples have transcriptomes of similar size. For example, quantile normalization is a well accepted method which assumes that the overall expression distribution is identical for every sample (6). These normalization methods may give unexpected and undesirable results when applied to samples with markedly different transcriptomes. We found that, for MouseWG-6 version 2 BeadChips, expression proportions for different cell types and samples varied from a minimum of 0.38 to a maximum of 0.49, meaning that one sample could have up to 5000 more expressed probes than another (Figure 2). Knowing the proportion of expressed probes will be useful for customizing normalization strategies for different microarray experiments.
Certain popular background correction algorithms for microarray data require an estimate of the mean intensity of expressed probes (18,19,20,21). An estimate of the expression proportion could refine this estimate.
Filtering out probes which do not express in any condition in a microarray experiment has been demonstrated to increase the power to detect differentially expressed genes (39,40). However, lowly expressed probes, including possibly important genes such as transcription factors, may be lost if the threshold is set too high. Knowing the expression proportion for each array gives valuable guidance regarding the number of probes to filter.
Our algorithm can be readily applied to microarray platforms other than Illumina, provided that negative control probes are included that provide a good estimate of the background intensities. Affymetrix and Agilent have both included negative control probes into their latest expression platforms including Affymetrix Mouse Gene 1.0 ST Array, Agilent Whole Mouse Genome Oligo 4 × 44k Microarray etc.
Our algorithm, utilizing the negative control probes on the array, adds another string to the bow of microarray expression analysis. The algorithm is implemented in the freely available Bioconductor R package limma (24).
SUPPLEMENTARY DATA
Supplementry Data are available at NAR Online.
FUNDING
Funding for open access charge: National Health and Medical Research Council (Program grant 490037).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Shuo Li for providing data prior to publication, Matthew Ritchie and Mette Langaas for valuable discussions, Andrew Holloway for preparing the mixture data, Francois-Xavier Hubert for his assistance in mTEC sample preparation, Charity Law, Belinda Phipson and Di Wu for the raw data retrieval and Leming Shi for providing the complete raw Illumina data generated from MAQC-I project.
REFERENCES
- 1.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarray applied to the ionizing radiation response. Proc. Natl Acad. Sci. USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Gen. Mol. Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article 3. [DOI] [PubMed] [Google Scholar]
- 3.Akashi K, He X, Chen J, Iwasaki H, Niu C, Steenhard B, Zhang J, Haug J, Li L. Transcriptional accessibility for genes of multiple tissues and hematopoietic lineages is hierarchically controlled during early hematopoiesis. Blood. 2003;101:383–389. doi: 10.1182/blood-2002-06-1780. [DOI] [PubMed] [Google Scholar]
- 4.Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, von Boehmer H, Bronson R, Dierich A, Benoist C, et al. Projection of an immunological self shadow within the thymus by the Aire protein. Science. 2002;298:1395–1401. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- 5.Venanzi ES, Melamed R, Mathis D, Benoist C. The variable immunological self: genetic variation and nongenetic noise in aire-regulated transcription. Proc. Natl Acad. Sci. USA. 2008;105:15860–15865. doi: 10.1073/pnas.0808070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on bias and variance. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 7.Stern MD, Anisimov SV, Boheler KR. Can transcriptome size be estimated from sage catalogs? Bioinformatics. 2003;19:443–448. doi: 10.1093/bioinformatics/btg018. [DOI] [PubMed] [Google Scholar]
- 8.Zhu J, He F, Wang J, Yu J. Modeling transcriptome based on transcript-sampling data. PLoS ONE. 2008;3:e1659. doi: 10.1371/journal.pone.0001659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Archer KJ, Reese SE. Detection call algorithms for high-throughput gene expression microarray data. Brief Bioinform. 2010;11:244–252. doi: 10.1093/bib/bbp055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Illumina. 2008. [16 November, 2009, date last accessed]. BeadStudio Gene Expression Module User Guide. www.illumina.com. [Google Scholar]
- 11.Affymetrix. 2002. [16 November, 2009, date last accessed]. Statistical Algorithms Description Document, www.affymetrix.com/support/technical/whitepapers/sadd_whitepaper.pdf. [Google Scholar]
- 12.Schuster EF, Blanc E, Partridge L, Thornton JM. Correcting for sequence biases in present/absent calls. Genome Biol. 2007;8:R125. doi: 10.1186/gb-2007-8-6-r125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu Z, Irizarry RA. A statistical framework for the analysis of mciroarray probe-level data. Ann. Appl. Stat. 2007;1:333–357. [Google Scholar]
- 14.Zilliox MJ, Irizarry RA. A gene expression bar code for microarray data. Nat. Methods. 2007;4:911–913. doi: 10.1038/nmeth1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.MAQC Consortium. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat. Biotechnol. 2006;24:1151–1161. doi: 10.1038/nbt1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li S, Floess S, Hamann A, Gaudieri S, Lucas A, Hellard M, Roberts S, Paukovic G, Plebanski M, Loveland BE, et al. Analysis of FOXP3+ regulatory T cells that display apparent viral antigen specificity during chronic hepatitis C virus infection. PLoS Pathog. 2009;5:e1000707. doi: 10.1371/journal.ppat.1000707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, Asselin-Labat ML, Gyorki DE, Ward T, Partanen A, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in brca1 mutation carriers. Nat. Med. 2009;15:907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 18.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 19.McGee M, Chen Z. Parameter estimation for the exponential-normal convolution model for background correction of Affymetrix GeneChip data. Stat. Appl. Genet. Mol. Biol. 2006;5:Article 24. doi: 10.2202/1544-6115.1237. [DOI] [PubMed] [Google Scholar]
- 20.Silver J, Ritchie ME, Smyth GK. Microarray background correction: maximum likelihood estimation for the normal-exponential convolution model. Biostatistics. 2009;10:352–363. doi: 10.1093/biostatistics/kxn042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ritchie ME, Silver J, shlack AO, Holmes M, Diyagama D, Holloway A, Smyth GK. A comparison of background correction methods for two-colour microarrays. Bioinformatics. 2007;23:2700–2707. doi: 10.1093/bioinformatics/btm412. [DOI] [PubMed] [Google Scholar]
- 22.Hubert FX, Kinkel SA, Crewther PE, Cannon PZ, Webster KE, Link M, Uibo R, O'B;ryan MK, Meager A, Forehan SP, et al. Aire-deficient C57BL/6 mice mimicking the common human 13-base pair deletion mutation present with only a mild autoimmune phenotype. J. Immunol. 2009;182:3902–3918. doi: 10.4049/jimmunol.0802124. [DOI] [PubMed] [Google Scholar]
- 23.Spangrude GJ, Heimfeld S, Weissman IL. Purification and characterization of mouse hematopoietic stem cells. Science. 1988;241:58–62. doi: 10.1126/science.2898810. [DOI] [PubMed] [Google Scholar]
- 24.Smyth GK. Limma: linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. Bioinformatics and Computational Biology Solutions using R and Bioconductor. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- 25.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 27.Lein ES, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 28.Kondo M, Weissman IL, Akashi K. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell. 1997;91:661–672. doi: 10.1016/s0092-8674(00)80453-5. [DOI] [PubMed] [Google Scholar]
- 29.Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–197. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- 30.Derbinski J, Schulte A, Kyewski B, Klein L. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat. Immunol. 2001;2:1032–1039. doi: 10.1038/ni723. [DOI] [PubMed] [Google Scholar]
- 31.Gotter J, Brors B, Hergenhahn M, Kyewski B. Medullary epithelial cells of the human thymus express a highly diverse selection of tissue-specific genes colocalized in chromosomal clusters. J. Exp. Med. 2004;199:155–166. doi: 10.1084/jem.20031677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liston A, Lesage S, Wilson J, Peltonen L, Goodnow CC. Aire regulates negative selection of organ-specific t cells. Nat. Immunol. 2003;4:350–354. doi: 10.1038/ni906. [DOI] [PubMed] [Google Scholar]
- 33.Liston A, Gray DH, Lesage S, Fletcher AL, Wilson J, Webster KE, Scott HS, Boyd RL, Peltonen L, Goodnow CC. Gene dosage–limiting role of Aire in thymic expression, clonal deletion, and organ-specific autoimmunity. J. Exp. Med. 2004;200:1015–1026. doi: 10.1084/jem.20040581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeVoss J, Hou Y, Johannes K, Lu W, Liou GI, Rinn J, Chang H, Caspi RR, Fong L, Anderson MS. Spontaneous autoimmunity prevented by thymic expression of a single self-antigen. J. Exp. Med. 2006;203:2727–2735. doi: 10.1084/jem.20061864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Derbinski J, Gbler J, Brors B, Tierling S, Jonnakuty S, Hergenhahn M, Peltonen L, Walter J, Kyewski B. Promiscuous gene expression in thymic epithelial cells is regulated at multiple levels. J. Exp. Med. 2005;202:33–45. doi: 10.1084/jem.20050471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.The Finnish-German APECED Consortium. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two phd-type zinc-finger domains. Nature. 1997;17:399–403. doi: 10.1038/ng1297-399. [DOI] [PubMed] [Google Scholar]
- 37.Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, Krohn KJ, Lalioti MD, Mullis PE, Antonarakis SE, et al. Positional cloning of the APECED gene. Nat. Genet. 1997;17:393–398. doi: 10.1038/ng1297-393. [DOI] [PubMed] [Google Scholar]
- 38.Ramsey C, Winqvist O, Puhakka L, Halonen M, Moro A, Kmpe O, Eskelin P, Pelto-Huikko M, Peltonen L. Aire deficient mice develop multiple features of APECED phenotype and show altered immune response. Hum. Mol. Genet. 2002;11:397–409. doi: 10.1093/hmg/11.4.397. [DOI] [PubMed] [Google Scholar]
- 39.Hackstadt AJ, Hess AM. Filtering for increased power for microarray data analysis. BMC Bioinformatics. 2009;10:11. doi: 10.1186/1471-2105-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi W, Smyth GK. Illumina WG-6 BeadChip strips should be normalized separately. BMC Bioinformatics. 2009;10:372. doi: 10.1186/1471-2105-10-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.