


Abstract
The current arsenal of molecular tools for site-directed cleavage of single-stranded DNA (ssDNA) is limited. Here, we describe a method for targeted DNA cleavage that requires only the presence of an A nucleotide at the target position. The procedure involves hybridization of a complementary oligonucleotide probe to the target sequence. The probe is designed to create a deliberate G:A mismatch at the desired position of cleavage. The DNA repair enzyme MutY glycosylase recognizes the mismatch structure and selectively removes the mispaired A from the duplex to create an abasic site in the target strand. Addition of an AP-endonuclease, such as Endonuclease IV, subsequently cleaves the backbone dividing the DNA strand into two fragments. With an appropriate choice of an AP-cleaving enzyme, the 3′- and 5′-ends of the cleaved DNA are suitable to take part in subsequent enzymatic reactions such as priming for polymerization or joining by DNA ligation. We define suitable standard reaction conditions for glycosylase/AP-cleaving enzyme (G/AP) cleavage, and demonstrate the use of the method in an improved scheme for in situ detection using target-primed rolling-circle amplification of padlock probes.
INTRODUCTION
Many of our successes in genetic research stem from the ability to manipulate DNA molecules in a highly precise manner. Amidst the current molecular tools, only a small subset has been developed for targeted cleavage of single-stranded DNA (ssDNA). A common route towards cleavage of synthetic ssDNA as well as genomic targets that have been rendered single-stranded (1) is by hybridization of an oligonucleotide probe to the desired site of cleavage. The probe and target form a short double-stranded (ds) DNA segment that can be targeted by a larger assortment of traditional cleavage strategies. Restriction endonucleases (2,3), for example, can be directed to cleave at the duplex structure if the duplex contains an appropriate recognition sequence. Although there are over 600 of these enzymatic scissors available commercially (4), the dependence on a multi-nucleotide recognition sequence can hamper the choice of exactly where the cut should occur. This is an important concern when targeting specific genomic sequences/duplexes, as the desired site of cleavage may not coincide with the natural distribution of the recognition sequence within the genome. Also, each additional sequence to be cleaved requires a different restriction enzyme which can dramatically increase costs. There exist a few nicking enzymes (4) (http://www.neb.com/nebecomm/tech_reference/restriction_enzymes/feature_article_nicking.asp) that cleave just one strand of the duplex, however these enzymes suffer the same limitation of recognition sequence dependence as any other restriction enzyme.
To circumvent some of these problems, a cleavage strategy involving targeting oligonucleotides has been developed (5). In this approach, oligonucleotides are engineered to contain two parts; a sequence that base pairs with the target of interest, and a sequence that folds into a hairpin loop and forms the recognition sequence for FokI endonuclease. Hybridization of the probe targets the desired sequence, and cleavage of the probe-target hybrid is mediated by FokI bound in the hairpin loop (5). Optimal performance is observed when the probe-target duplex is formed first followed by addition of the restriction enzyme (5). Inherent to this method is the use of restriction enzymes that cleave outside of their recognition sequence. In general, these type IIS enzymes are thought to bind to DNA as monomers for the most part, but tend to involve dimerization of the cleavage domains of adjacent enzyme molecules for DNA cleavage (6,7). For this reason, many type IIS enzymes are much more active on DNA molecules that contain multiple recognition sites. We propose an alternative approach that combines the targeting accuracy of probe hybridization, while avoiding the potential limitations of type IIS restriction enzymes.
The technique described here maintains the use of oligonucleotide probes for targeting the location of the cleavage site. Rather than depending on hairpins or other extraneous structures, however, the probe incorporates a G residue that mispairs with an A in the target sequence.
Cleavage of the target strand can then be mediated by a pair of DNA repair enzymes (Figure 1) (8). Addition of the DNA repair enzyme MutY glycosylase (9,10), which specifically recognizes G–A mismatches, selectively removes the adenine from the G:A duplex (11) creating an apurinic (AP) site in the target DNA strand. Next, the abasic site can be severed using an enzyme with AP-cleaving activity such as Endonuclease IV. As there are several enzymes used here that recognize and cleave AP sites through different chemical mechanisms (12), we will refer to them collectively as AP-cleaving enzymes. The combined glycosylase/AP-cleaving (G/AP) reactions thus cleave the target strand while preserving the probe molecule intact; a critical feature in some probe-directed analyses as exemplified by the padlock probe application described here. Briefly, padlock probes are linear oligonucleotides which can be circularized by DNA ligase when correctly hybridized to target DNA sequences (13). If the padlock probe hybridizes close to the 3′-end of the target DNA fragment, the target itself can serve as the primer for the rolling circle amplification (RCA). This target-primed RCA enables intracellular localization of sequences for in situ analyses of cells and tissues (14). The approach is effective; however, RCA is inhibited in cases where the padlock probe hybridizes away from the 3′-end of the target (15). Here, we show that simple integration of a single nucleotide mismatch in the padlock probe is sufficient to direct G/AP cleavage of the target DNA, thus producing the primer for the RCA reaction. In addition, with an appropriate selection of AP-cleaving enzyme the cleaved fragments are suitable to participate in polymerization, ligation, or other further enzymatic reaction steps.
Figure 1.
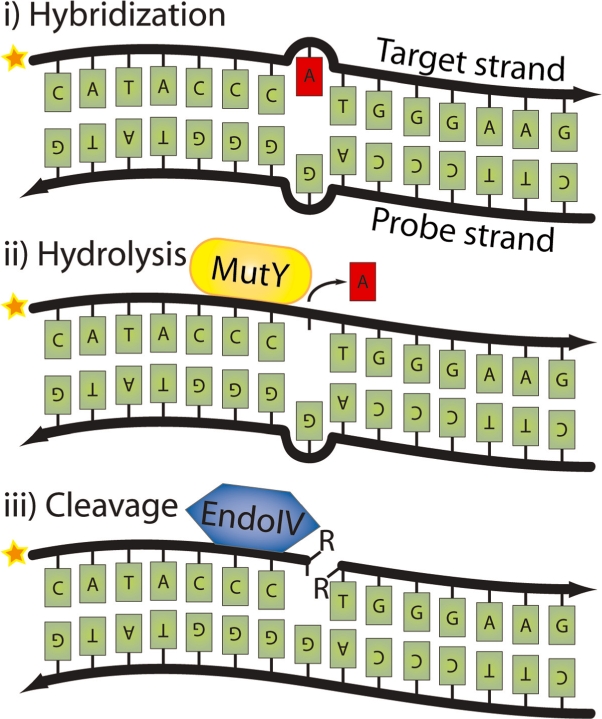
Strategy for strand-specific cleavage using MutY glycosylase and AP-cleaving enzymes. When oligonucleotides are hybridized to each other, a G:A mismatch is created (i) which is recognized by MutY glycosylase. (ii) MutY glycosylase removes the mispaired A and (iii) the abasic site is nicked by an AP-cleaving enzyme, for example Endonuclease IV. The star indicates the 32P-labeling of the target strand. The R’s in the figure indicate residual side groups.
Herein, we explore the enzymatic properties of MutY glycosylase, a series of AP-cleaving enzymes, and conditions for combining the two activities. We also apply the site-directed cleavage mechanism for in situ detection of mtDNA using padlock probes and target-primed RCA.
MATERIALS AND METHOD
32-P labeling of target strands
Oligonucleotide sequences are found in Table 1.
Table 1.
Oligonucleotide sequences
| Test | Oligo | Names | Sequences |
|---|---|---|---|
| MutY glycosylase test | Probe | probe_MutY | 5′-TAAGAAGAGGAATTGCCTTTCCTACGACCTCAATGCACATGTTTGGCTCCTCTTCCCAGGGGTATGTTGT-3′ |
| Target | target_MutY | 5′-*TCAGCTGACAACATACCCATGGGAAGAGGA-3′ | |
| AP test | Probe | probe_AP | 5′-TCAGCTGACAACGTACCCATGGGAAGAGGA-3′ |
| Target | target_AP | 5′-*TCCTCTTCCCAGGGGTAUGTTGTCAGCTGA-3′ | |
| Ligation after cleavage | Probe | probe_relig | 5′-ATCAGTCGACAAAGAGACCCTGGAAGGGTACCGTGCGCCTGGTAGCAAATAGTCCGTCATGTGTTAGATGC-3′ |
| Tracking probe 1 | track_relig1 | 5′-*CTACCAGGCGCACGGTACC-3′ | |
| Tracking probe 2 | track_relig2 | 5′-ATGACGGAATATTTG-3′ | |
| Ligation probe | lig_relig | 5′-TAACACATGACGGAC-3′ | |
| Cleavage of blocked end | Padlock probe | plp_cleave | 5′-TAAGAAGAGGAATTGCCTTTCCTACGACCTCAATGCACATGTTTGGCTCCTCTTCCCAGGGGTATGTTGT-3′ |
| Target blocked | target-blocked_cleave | 5′-GCAATTCCTCTTCTTAACAACATACCCATGGUUU-3′ | |
| Target non-blocked | target-nonblocked_cleave | 5′-CAATTCCTCTTCTTAACAACATACCCATG-3′ | |
| Detection probe | det_cleave | 5′-Cy3-CCTCAATGCACATGTTTGGCTCC-3′ | |
| In situ detection of mtDNA | Padlock probe | plp_insitu | 5′-TAAGAAGAGGAATTGCCTTTCCTACGACCTCAATGCACATGTTTGGCTCCTCTTCCCAGGGGTATGTTGT-3′ |
| Detection probe | det_insitu | 5′-Cy5-CCTCAATGCACATGTTTGGCTCC-3′ |
Bold letters indicate the site of the mismatch upon oligonucleotide annealing.
*Where the 32P-label is introduced.
Underlined sequences indicate the target complements of the padlock probes.
Italic letters in padlock probes indicate detection probe sequences.
32P-labeling of the target strands (target_MutY and target_AP (20 nM)) 5′-termini was performed using 60 nM [γ-32P]ATP (Amersham) and 0.2 U/μl T4 polynucleotide kinase (T4 PNK) (Fermentas) in the supplied Reaction buffer A (1×) for 15 min at 37°C, followed by inactivation of the T4 PNK for 5 min at 65°C. Eighty nanomolar target strand (track_relig1) was 32P-labeled with 80 nM [γ-32P]ATP and 0.2 U/μl OptiKinase (USB) in the supplied OptiKinase buffer (1×) for 30 min at 37°C followed by inactivation of the OptiKinase for 10 min at 65°C. After labeling, oligonucleotides were purified in illustra MicroSpin™ G-50 Columns (GE Healthcare) at 3000 r.p.m. for 2 min.
Hybridization of oligonucleotides and site-specific cleavage of mismatch with MutY glycosylase and AP-cleaving enzyme
Two nanomolar of labeled target (target_MutY for the MutY glycosylase test and target_AP for the AP test) and 4 nM of probe (probe_MutY for MutY glycosylase test and probe_AP for AP test) were hybridized in 1 × G/AP buffer (20 mM Tris–HCl, 30 mM NaCl, 1 mM EDTA, 100 mM KCl and 1 mM DTT) for 10 min at 65°C and then slowly cooled to room temperature. For the MutY glycosylase test, 640 nM MutY glycosylase (USB) was added to the hybridized duplex. The cleavage of the target strand was performed with 1 M NaOH incubated for 20 min at 95°C. For comparison of the AP-cleaving enzymes, 0.05 U/μl UDG (Fermentas) was combined pair wise with 0.05 U/μl of the following AP-cleaving enzymes: Ape 1 (New England Biolabs), Endonuclease III (New England Biolabs), Endonuclease IV (Fermentas), Endonuclease V (Fermentas), Exonuclease III (Fermentas) and Fpg (New England Biolabs). Each reaction also contained 0.1 μg/μl BSA (New England Biolabs) and 1 × G/AP buffer, mixed with 1 nM of the annealing product. Incubations were for 30 min at 37°C for total cleavage.
5′-Ligation of oligonucleotide after MutY glycosylase and Endonuclease IV treatment
The tracking oligo was created by adding 15 nM of the labeled track_relig1 with 15 nM track_relig2 and hybridizing to 15 nM probe_relig in a mixture of 0.2 U/μl Ampligase (Epicentre), 20 mM Tris–HCl pH 8.3, 25 mM KCl, 10 mM MgCl2, 0.5 mM NAD, 0.01 % Triton X-100 and 0.1 μg/μl BSA for 2 min at 75°C, 1 min at 45°C, 2 min at 30°C and finally 2 min at 45°C. The duplex was cleaved at the mismatched site by the addition of 80 nM of MutY glycosylase and 0.1 U/μl Endonuclease IV in a mixture with 0.1 μg/μl BSA and 1 × G/AP buffer for 30 min at 37°C for total cleavage. Fourteen nanomolars of the ligation strand (lig_relig) was mixed with 3.5 nM cleaved product in a mixture of 0.2 U/μl Ampligase (Epicentre), 20 mM Tris–HCl pH 8.3, 25 mM KCl, 10 mM MgCl2, 0.5 mM NAD, 0.01 % Triton X-100 and 0.1 μg/μl BSA for 2 min at 75°C, 1 min at 45°C, 2 min at 30°C and finally 30 min at 45°C. For ligation with T4 DNA ligase, the duplex was mixed with 0.1 U/μl T4 DNA ligase (Fermentas), 10 mM Tris–Acetate pH 7.5, 10 mM MgAc2, 50 mM KAc, 1 mM ATP (Fermentas) and 0.1 μg/μl BSA for 2 min at 75°C, 1 min at 45°C, 2 min at 30°C, 30 min at 37°C and finally 10 min at 65°C (the T4 DNA ligase was added prior to the incubation at 37°C).
Polyacrylamide gel
The samples were mixed with loading buffer (95% formamide, 25 mM EDTA and 5 mg/ml blue dextran) and denatured for 2 min at 90°C. The samples were loaded onto a 12% polyacrylamide gel (25 × 25 × 0.03 cm) [20 ml 12% polyacrylamide/7 M urea with 100 μl 10% ammonium persulphate (Sigma Aldrich) and 10 μl 100% TEMED (Fluka)] and separated for 1 h at 20 W. The gels were exposed to a Phosphoimaging screen (Science Imaging Scandinavia IP-MS2325) for ∼1 h before analysis on a Phosphoimager using the Image Reader and Image Process software.
Site-specific cleavage of blocked 3′-end with MutY glycosylase/Endonuclease IV
One micromolar of the padlock probe (plp_cleave) was phosphorylated with 0.2 U/μl T4 PNK enzyme, the supplied reaction buffer A (1×) and 1 mM ATP for 15 min at 37°C following inactivation of the enzyme at 65°C for 10 min. The hybridization and ligation of the padlock probe (10 nM) to the 3′ blocked and non-blocked templates (target-blocked_cleave, target-nonblocked_cleave) (30 nM) was performed in mixtures of 25 mU/μl T4 DNA ligase, 1 × Φ29 DNA polymerase buffer (Fermentas), 1 mM ATP, 0.2 μg/μl BSA for 30 min at 37°C and 10 min at 65°C for inactivation of ligase. For cleavage of the blocked 3′-end, 1 nM of the oligonucleotide duplexes were mixed with 12 nM MutY glycosylase, 25 mU/μl Endonuclease IV in 1 × G/AP buffer for 45 min at 37°C. After removal of blocked 3′-end, RCA was performed by mixing 100 pM of the circularized oligonucleotides with 4 mU/μl Φ29 DNA polymerase (Fermentas), 1 × Φ29 DNA polymerase buffer, 100 μM dNTP (Fermentas) and 0.2 μg/μl BSA for 60 min at 37°C and 10 min at 65°C for inactivation of polymerase. Hybridization of 5 nM detection probe (det_cleave) to the rolling circle product (RCP) (50 pM) was performed in 0.1% Tween-20 (Sigma), 20 mM Tris–HCl, 500 mM NaCl and 20 mM EDTA at 65°C for 10 min, then at 37°C for 30 min. The final samples were pumped through a thermoplastic microchannel, mounted in a standard confocal fluorescence microscope (LSM 5 META, Zeiss) to detect individual RCPs. The samples were run in triplicates and the raw data files generated an output file that allowed the RCPs to be counted using Matlab. The technique is described in ref. (16).
Cells and pretreatments for in situ detection
143B cells were cultured in Dulbecco’s Modified Eagles medium (Gibco) without phenol red, with 10% fetal calf serum (Sigma), 50 μg/ml uridine, 0.1% glucose, 292 μg/ml l-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. The cells were grown on glass microscope slides and fixed in 70% ethanol for 20 min at room temperature. The cells were permeabilized using 0.01% pepsin (Sigma) in 0.1 M HCl for 90 s at 37°C, followed by washing in PBS. Subsequent incubations in the procedure for in situ detection were carried out in a volume of 20 μl under 22 × 22 mm coverslips, except for the RCA reaction, which was performed in a volume of 40 μl in a silicone chamber.
Target preparation with restriction enzyme and exonuclease
For hybridization of the padlock probe to the DNA, we first rendered the target sequence single-stranded by using a combination of restriction enzymes and exonucleases. The genomic DNA was initially restriction digested with 0.5 U/μl of MscI, BsmI or BtsI (New England Biolabs) for 30 min at 37°C in the NEB buffer 4 (MscI and BtsI) or NEB buffer 2 (BsmI) with 0.2 μg/μl BSA followed by a rinse in a wash buffer (0.1 M Tris–HCl pH 7.5, 0.15 M NaCl and 0.05% Tween-20). The digested ends were made single-stranded by exonucleolysis using 0.4 U/μl of the 5′-3′ T7 exonuclease (New England Biolabs) at 37°C for 15 min in the buffer supplied, supplemented with 0.2 μg/μl BSA and 5% glycerol. The slides were then rinsed in wash buffer.
Padlock probe hybridization and ligation
Two micromolar of the padlock probe (plp_insitu) was phosphorylated in 0.2 U/μl T4 PNK enzyme, the supplied reaction buffer A (1×) and 1 mM ATP for 15 min at 37°C followed by inactivation of the enzyme at 65°C for 10 min. Hybridization and ligation were performed in a single reaction containing 100 nM of the padlock probe in a solution of 10 mM Tris–Acetate pH 7.5, 10 mM MgAc2, 50 mM KAc, 250 mM NaCl, 1 mM ATP, 0.2 μg/μl BSA and 0.1 U/μl T4 DNA ligase for 15 min at 37°C. The slides were first washed in 2 × SSC, 0.05% Tween-20 for 5 min at 37°C then rinsed in wash buffer. Cleavage of the target strand at the mismatch site was performed by 1.54 μM MutY glycosylase and 0.1 U/μl Endonuclease IV in 1 × G/AP buffer containing 1 μg/μl BSA for 30 min at 37°C. This was followed by rinsing the slides in wash buffer and dehydration in a series of 70%, 85% and 100% ethanol.
The RCA reactions were performed in rubber silicone reaction chambers in G/AP buffer, 0.25 mM dNTP, 0.2 μg/μl BSA, 5% glycerol and 1 U/μl Φ29 DNA polymerase at 37°C for 30 min on a shaking platform. The silicone chambers were removed from the slides before rinsing them in wash buffer. Following polymerization, 250 nM of the detection probe (det_insitu) was hybridized to the RCPs in 2xSSC and 20% formamide at 37°C for 15 min. After rinsing in wash buffer, the slides were dehydrated before being mounted for microscopy in Vectashield (Vector Laboratories) containing 10 ng/ml DAPI.
Image analysis
For visualization of the cells and RCPs, an epifluorescence microscope (Axioplan II, Zeiss) equipped with a 100 W mercury lamp and excitation and emission filters for DAPI and Cy5, was used together with a CCD camera (C4742-95, Hamamatsu) for acquiring images. A 40× objective (Plan-neofluar, Zeiss) was used and images were collected using the AxioVision software 4.4 (Zeiss). The RCPs were quantified using the BlobFinder software (17) (version 3.0_beta) and images were thresholded for visualization in Adobe Photoshop CS3 (Adobe Systems Inc).
RESULTS
MutY glycosylase activities
The primary role of glycosylases in DNA repair is the recognition and removal of modified, damaged or mismatched nucleotide bases (18). The removal mechanism involves hydrolysis of the N-glycosidic bond that connects the nucleobases to the sugar backbone of the DNA molecule (Figure 1). This hydrolysis leaves the DNA backbone intact, producing an apurinic (G and A) or apyrimidinic (C, T and U) (AP) lesion at the position of the incorrect nucleotide. In normal DNA repair, base excision is followed by successive enzymatic steps of cleavage, polymerization and ligation which serve to fully repair the DNA molecule (19).
The MutY adenine glycosylase employed here is a member of the base excision repair (BER) pathway in Escherichia coli. The enzyme recognizes non-cognate basepairing between A and G (10,20), A and oxidized guanine (7,8-dihydro-8-oxo-2’-deoxy-guanosine or OG) (21–23), and to a lesser extent A to C (24) (Figure 2A). In the presence of duplex DNA structures containing G:A or OG:A mismatched base pairs, MutY glycosylase selectively removes the undamaged A from the structure. Although seemingly counter-intuitive, the mechanism compensates for the cell’s replication machinery which frequently mis-incorporates A residues across from oxidized G (25). A two-phase repair process is thus necessary to reconstitute the original DNA molecule; first, the replacement of the A residue with a C in one strand, followed by replacement of the OG with an unmodified G in the complementary DNA strand.
Figure 2.

Overview of the glycosylases and AP-cleaving enzymes presented herein. (A) (i) The hydrolysis mechanism creating abasic sites, (ii) the different glycosylases MutY, TDG (thymine DNA glycosylase) and UDG, the corresponding substrates they are recognizing and possible AP-activity, (iii) hydrolysis of target strand by MutY and cleavage of abasic sites by NaOH and/or 95°C heat. (B) The different AP-cleaving enzymes and their effects and efficiencies. (i) β- or δ-elimination of the DNA phosphodiester backbone depending on the AP-cleaving enzyme, (ii) the different AP-cleaving enzymes that have been compared are Ape 1, Endonuclease III, Endonuclease IV, Endonuclease V, Exonuclease III and Fpg. The way of elimination (β or δ) of abasic sites are presented as well as what phosphodiester bond they have as cleavage site and what terminus they leave at the 5′- and 3′-end, (iii) efficiencies of AP-cleaving enzymes on abasic sites. The abasic sites are created through removal of uracils by UDG and cleaved by addition of different AP-cleaving enzymes. Positive control (pos) represents NaOH and 95°C heat, while negative control (neg) is without addition of AP-cleaving enzyme.
The enzymatic activity of MutY glycosylase was initially examined in a model system composed of a pair of base-paired oligonucleotides. Hybridization resulted in a DNA duplex containing a centrally located G:A mismatch. The A containing strand was end labeled with 32P. MutY glycosylase enzyme was then added in 50-fold excess over substrate and after incubation, base (NaOH) and/or heat was used to break the oligonucleotide at the abasic sites created by MutY glycosylase. Cleaved and uncleaved DNA molecules were separated by denaturing PAGE and results were recorded on a Phosphoimager.
As shown in Figure 2A, the presence of MutY glycosylase alone was sufficient to generate the cleaved fragment with a ratio of cleaved to uncleaved of ∼50%. This observation indicates that MutY glycosylase from this commercial vendor appears to harbor some AP-cleaving activity, allowing the enzyme preparation to exert both G and AP activities. Upon addition of base and high temperature incubation, the fuzzy band disappears and the ratio between cleaved and non-cleaved band intensities reaches a maximum of ∼85% for this model system. The cleavage efficiency from the literature and from other model systems in our lab indicate that the MutY glycosylase efficiency ranges between 50% and 99% (23).
AP-cleaving activity
AP-cleaving enzymes are responsible for cleaving abasic sites during DNA repair. AP-endonucleases such as Ape 1, Endonuclease IV and Endonuclease V recognize AP lesions and cleave the phosphodiester backbone by a hydrolytic mechanism (26). Fpg and other AP-lyases sever the sugar–phosphate backbone through elimination reactions (12). Depending on whether the cleavage occurs 3′ or 5′ of the ribose, the reactions occur via β- or δ-elimination reaction, respectively (Figure 2B). Endonuclease V is a notable exception as it cleaves at the 2nd phosphodiester bond 3′ of the abasic lesion (27).
We created an experimental set-up to evaluate the activity of AP-cleaving enzymes on abasic substrates in a manner that was independent of MutY glycosylase activity. The substrate consisted of hybridized, complementary oligonucleotides with one strand containing a centrally located 2′-deoxyuridine (dU). The U strand was labeled with 32P. The U was removed by treatment with uracil DNA glycosylase (UDG), generating an abasic site in the oligonucleotide duplex. Six different enzymes were tested (present in at least a 50-fold molar excess of enzyme to substrate) along with base treatment and heat as a chemical control (Figure 2B). Endonuclease III, Endonuclease IV, Exonuclease III and MutM [formamidopyrimidine–DNA glycosylase (Fpg)] showed near 100% conversion of the 30 base fragment to the 17 base fragment as expected upon cleavage at the abasic site. Ape 1 showed a ∼65% cleavage in these experiments, and Endonuclease V appeared not to cleave the substrate to any significant extent. A possible explanation for the poor performance of Ape 1 and Endonuclease V is that buffer and assay conditions necessary for the present protocol differ greatly from the manufacturers’ recommended buffers and assay conditions for these two enzymes.
DNA target cleavage and ligation
Cleavage of DNA molecules by the combined action of glycosylases and AP-endonucleases or AP-lyases has been observed during the exploration of the enzymes roles in DNA repair (26). Several methods in molecular biology have further adapted the use of G/AP-cleaving enzymes for the modification of synthetic and amplified DNA molecules (28). For example, the USER™ cloning kit (New England Biolabs) uses a combination of UDG and Endo VIII for digestion of uracil containing PCR products for subsequent cloning (29). The procedure does not require in vitro ligation as the bacterial host repair mechanism is employed to join the nick sites upon transformation. In mutation detection or allele discrimination, the presence of a mutation results in cleavage of a mismatched probe molecule (30). The cleavage event is subsequently detected as a reduction in probe length or loss of energy transfer between donor and acceptor fluorophores located on opposing sides of the cleavage site. The purpose of this work is to expand the use of these DNA repair enzymes, beyond applications on synthetic or amplified DNA molecules, for modification of non-amplified DNA for subsequent manipulation of the generated DNA fragments. However, the combinations of enzymes as well as the cleavage conditions defined herein are also useful for manipulation of synthetic oligonucleotides.
A series of experiments were designed to investigate the efficiency of ligation using DNA fragments generated by G/AP reactions (Figure 3A). As an initial step, an internally labeled tracking oligo was created by ligating a 5′-32P labeled oligonucleotide with an adjacently hybridized oligonucleotide. The resultant 34 nt tracking oligo formed an imperfect duplex with the ligation template, resulting in a G:A mismatch. Following MutY glycosylase treatment, the abasic sites were cleaved with AP-cleaving enzyme Endonuclease IV which reduced the length of the tracking probe to 17 nt. Then, a 23 nt oligonucleotide was added to anneal immediately 5′ to the nick site. Successful ligation by T4 ligase thus resulted in increasing the length of the tracking oligo to 40 nt. Since the tracking oligo was radio-labeled, changes in the oligonucleotides length were detectable by PAGE (Figure 3B). Well over 75% of the oligonucleotides underwent both cleavage in the G/AP reaction and ligation at the cleaved site. Interestingly, as can be seen in Figure 2B, Endonuclease IV leaves a sugar at the 5′ side of the lesion after lysis of abasic sites. The presence of the deoxyribose-5′-phosphate (dR5P) should inhibit ligation, however, Endonuclease IV proved to be one of the better AP-cleaving enzymes to use in this configuration. This is probably due to the spontaneous loss of extremely labile 5′-terminal dR5P (31) in aqueous solutions.
Figure 3.
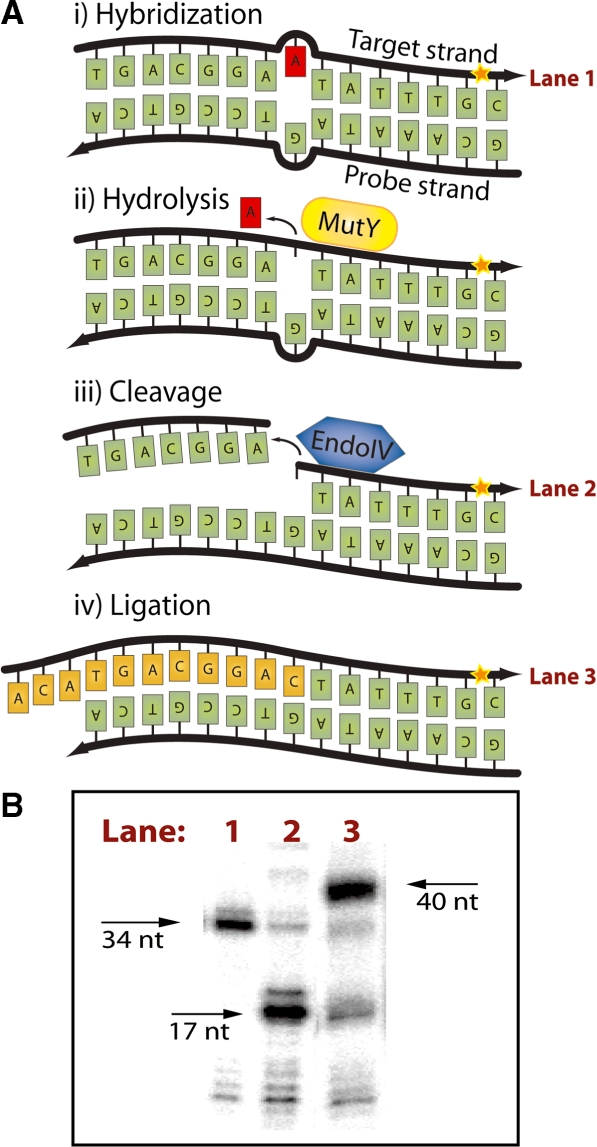
Ligation after MutY/Endo IV strand-specific nicking. (A) Mechanism for 5′-end ligation after nicking with MutY/Endo IV. Endo IV leaves a ligatable 5′-end which can be joined to the 3′-OH of a probe hybridized in juxtaposition through ligation. (i) Probe hybridization creating G:A mismatch. (ii) Recognition of mismatch and removal of an A residue by MutY. (iii) AP-site recognition by Endo IV and cleavage of 5′ DNA backbone. (iv) Ligation of probe to the 5′-end after cleavage. (B) MutY/Endo IV cleavage efficiency and 5′ ligation efficiency after MutY/Endo IV. Lane 1, labeled oligonucleotide (34 nt). Lane 2, cleaved product after MutY/Endo IV (17 nt). Lane 3, the ligation probe joined by ligation to the 5′-end terminus created by Endo IV (40 nt product).
DNA target cleavage and polymerization
A model system was used to test if DNA fragments generated by G/AP cleavage could serve as primers for RCA reactions. In this setup, the sequence of a padlock probe was designed to create a G:A mismatch when hybridized to a target sequence (Figure 4A). Additionally, the target oligonucleotide was synthesized with a series of 2′-O-methyl-RNA residues to block degradation of the 3′-end (32). These modified nucleotides were non-complementary to the padlock probe, ensuring that the Φ29 DNA polymerase could not prime the RCA directly. Cleavage of the target by G/AP activity removes the blocked end generating a 3′-end that should be suitable to prime an RCA reaction.
Figure 4.

Removal of blocked 3′-end with MutY/Endo IV for RCA. (A) The mechanism of removal of the 3′ blocking end with MutY/Endo IV. (i) An oligonucleotide with a blocked 3′-end serves as a target for padlock hybridization and ligation. The duplex formed between the probe/target creates a G:A mismatch. (ii) Recognition and cleavage of mismatch by MutY glycosylase/Endo IV resulting in removal of the blocked 3′-end. (iii) RCA and detection of RCPs. (B) Results from quantification of RCPs with the digital RCA technique. Briefly the procedure involves hybridization of fluorescently labeled detection oligonucleotides to the RCPs followed by pumping the solution through a microfluidic channel that is mounted on a fluorescent microscope. The imaged RCPs show up as distinct fluorescent spots that are quantified using imaging software. The resulting number of RCPs was counted in the different blocked (B) or non-blocked (NB) samples: negative control (no MutY/Endo IV added) (B), MutY (B), Endo IV (B), MutY/Endo IV (B) and positive control (NB). Error bars indicate standard deviations.
Padlock probes were hybridized to 3′ blocked target oligonucleotides and ligated. Following exposure to either MutY glycosylase or Endonuclease IV, individually or in combination, RCA was performed and resulting RCPs were quantified in a microfluidic device (16). As can be seen in Figure 4B, exposure to glycosylase or AP-cleaving enzyme individually was insufficient to allow priming of the RCA reaction. However, with both enzymes, nearly 100% of the targets were converted to primers as estimated by comparing with targets that lacked the 3′ modification.
In situ detection of mtDNA
Padlock probes have been successfully implemented to detect mitochondrial sequences in situ (14). The design of such experiments involves cleavage of the mitochondrial genomes with restriction enzymes, followed by 5′ exonucleolytic degradation of one of the genome strands. The 5′ exonuclease serves to expose a single-stranded target sequence for hybridization of the padlock probe. Padlock probes are hybridized, ligated and amplified by RCA, where the cleaved genomic fragment serves as the primer for the Φ29 DNA polymerase. In cases where the DNA target extends a few nucleotides beyond the hybridization site of the padlock probe, the inherent 3′ exonucleolytic activity of Φ29 effectively digests the extension to initiate RCA. RCPs are subsequently detected using fluorescence microscopy.
One consistent observation has been that the reaction is more efficient when the restriction site is closer to the padlock probe hybridization site. This could be attributed to either inefficiencies in the genome preparation step (14), inhibition of Φ29’s exonucleolytic activity, or a combination thereof. A plausible explanation for the reduction in single-stranded exonucleolytic activity may be that longer fragments are more likely to form secondary structures. Additionally, 3′-ends involved in hairpins could act as a primer for ‘fold-back’ synthesis using the target strand as a template instead of the circularized padlock probe.
Figure 5A outlines the strategy taken to test if G/AP-mediated cleavage of mismatched sequences could increase the efficiency of the target-primed in situ RCA of padlock probes. Similar to previous experiments, restriction enzymes were first used to cut the genome, and an exonuclease was applied to prepare the single-stranded target for probe hybridization. Following ligation, samples were digested with G/AP enzymes, amplified by RCA, and exposed to fluorescence labeled detection probes. The cells were finally studied using an epifluorescence microscope.
Figure 5.

In situ detection of mitochondrial DNA using padlock probes and RCA. (A) (i) Genomic DNA is made single-stranded by restriction enzyme digestion and exonucleolytic digestion of one of the genomic DNA strands. This step generates a single-stranded DNA segment at the desired target site. (ii) Hybridization and ligation of a padlock probe. The resulting probe/target duplex contains a G:A mispair, respectively. The target strand is then cleaved at the mismatch site by MutY/Endo IV. (iii) The padlock probe is amplified by RCA followed by detection probe hybridization to the RCPs. (B) The effect of MutY/Endo IV when applied in situ on fixed cells detecting mitochondrial DNA. For comparison, the mitochondrial genome was cleaved at two different distances from hybridization site of the padlock probe. (i) Cleavage using restriction enzyme BsmI with a restriction site ∼40 bp 3′ from padlock target site without and (ii) with an addition of MutY/Endo IV. (iii) Cleavage using restriction enzyme BtsI ∼400 bp 3′ from padlock probe site without and (iv) with addition of MutY/Endo IV. The average number of RCPs per cell, as determined by the BlobFinder program, is displayed in the upper right corner of each picture. Cell nuclei were stained blue using DAPI and RCPs were stained using fluorescence labeled oligonucleotides and are shown in purple. Scale bar, 20 µm.
Two series of experiments were the initial restriction digestion of the genome occurred at different nucleotide distances from the padlock hybridization position. One set of experiments used the restriction enzyme BsmI which cleaved the target roughly 40 nt from the padlock probe hybridization site, and a second set of experiments used BtsI which cleaved 400 nt from the padlock site. As Figure 5B demonstrates, addition of the G/AP step results in a notable increase in the number of RCPs detected compared to reactions where the G/AP step was omitted. For both the 40 and 400 nucleotide overhangs, roughly 2.5–3.5 times more RCPs were observed compared to negative controls (no G/AP). Thus, addition of the G/AP treatment step in the target-primed RCA method enables some relaxation of assay design criteria.
DISCUSSION
Along with serving vital functions in maintaining healthy genomes, glycosylases and AP-cleaving enzymes are effective and versatile biotechnology tools for in vitro manipulation of nucleic acids. Cleavage of DNA fragments basically follows a two-step mechanism. First, MutY glycosylase disunites the base from the sugar-phosphate backbone. Scission of the target DNA strand is the consequence of subsequent hydrolysis of the apurinic site with one of several enzymes exhibiting AP-cleaving activity. The purpose of this work has been to optimize and demonstrate the use of G/AP as a general tool for probe-directed cleavage of single-stranded DNA sequences. For the in situ padlock probe application described here, G/AP-mediated cleavage effectively prepares the target to serve as a primer for the subsequent rolling circle reaction. Resulting RCPs remain localized and can reveal the cellular location of the target sequences.
Some key advantages of this probe-directed cleavage approach include the simplicity of design, which only requires substitution of a T for a G in the probe sequence. Standard nucleotides are used in the probes, avoiding the need for expensive synthesis of oligonucleotides with exotic modifications such as OG. With proper oligonucleotide design, multiple sequences could be targeted simultaneously with a common pair of G/AP enzymes and reaction conditions. It is furthermore of particular importance in the padlock probe application that the probe that directs the cleavage reaction remains intact. Thus, in our application, the site-directed cleavage can be directed by the padlock probe sequence without destroying the padlock probe itself. The approach is directly applicable to synthetic and single-stranded DNA species such as cDNA, however as demonstrated herein, the approach can also be used for cleavage of genomes if the target sequence is first rendered single-stranded.
Despite an extensive scientific literature regarding the characterization of G/AP, the protocol that we arrived at herein could only be discovered by empirical work. For example, in our hands the best combination for cleavage of single-stranded U-containing fragments is UDG and Fpg (Supplementary Figure 1). In addition, many enzymes have more than one catalytic activity and DNA repair enzymes are no exception. We noticed that quite a few of the AP-cleaving enzymes tested herein also show weak to moderate exonuclease activity (data not shown). The exonuclease activity is dependent on Mg++ concentration (33), so for in vitro use, simply omitting MgCl2 from buffer systems helped to reduce undesired exonuclease activity while leaving the AP-cleaving activity intact. Regarding the extensive debate in the scientific literature about the classification of MutY glycosylases (11,24), the enzyme as provided by the two commercial sources and used here appears to harbor both glycosylase and partial lyase activity. In our hands, MutY glycosylase thus exhibits bifunctional properties rather than behaving strictly as a monofunctional glycosylase. That being said, many of the AP-cleaving enzymes described here showed much higher cleaving efficiencies than MutY glycosylase on its own.
We conclude that in a growing repertoire of molecular tools, members of DNA repair pathways can serve as valuable companions to restriction enzymes for the catalytic manipulation of nucleic acids.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Foundations of Knut and Alice Wallenberg, Göran Gustafsson, and Erik K. Fernström; Swedish Research Council and the European Community's; Seventh Framework Program (FP7/2007-2013) under grant agreement [HEALTH-F4-2008-201418] entitled READNA; the Sixth Framework Program under the grant agreement entitled COMICS. Funding for open access charge: M.N.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to Dr Tomas Lindahl for his insightful discussions regarding glycosylases and AP-cleaving enzymes, and also to the reviewers for their constructive input into the article.
REFERENCES
- 1.Szybalski W. Universal restriction endonucleases: designing novel cleavage specificities by combining adapter oligodeoxynucleotide and enzyme moieties. Gene. 1985;40:169–173. doi: 10.1016/0378-1119(85)90039-3. [DOI] [PubMed] [Google Scholar]
- 2.Danna K, Nathans D. Specific cleavage of simian virus 40 DNA by restriction endonuclease of Hemophilus influenzae. Proc. Natl Acad. Sci. 1971;68:2913–2917. doi: 10.1073/pnas.68.12.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith HO, Wilcox KW. A restriction enzyme from Hemophilus influenzae. I. Purification and general properties. J. Mol. Biol. 1970;51:379–391. doi: 10.1016/0022-2836(70)90149-x. [DOI] [PubMed] [Google Scholar]
- 4.Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE–enzymes and genes for DNA restriction and modification. Nucleic Acids Res. 2007;35:D269–D270. doi: 10.1093/nar/gkl891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Podhajska AJ, Szybalski W. Conversion of the FokI endonuclease to a universal restriction enzyme: cleavage of phage M13mp7 DNA at predetermined sites. Gene. 1985;40:175–182. doi: 10.1016/0378-1119(85)90040-x. [DOI] [PubMed] [Google Scholar]
- 6.Bath AJ, Milsom SE, Gormley NA, Halford SE. Many type IIs restriction endonucleases interact with two recognition sites before cleaving DNA. J. Biol. Chem. 2002;277:4024–4033. doi: 10.1074/jbc.M108441200. [DOI] [PubMed] [Google Scholar]
- 7.Vanamee ES, Santagata S, Aggarwal AK. FokI requires two specific DNA sites for cleavage. J. Mol. Biol. 2001;309:69–78. doi: 10.1006/jmbi.2001.4635. [DOI] [PubMed] [Google Scholar]
- 8.Krokan HE, Standal R, Slupphaug G. DNA glycosylases in the base excision repair of DNA. Biochem. J. 1997;325(Pt 1):1–16. doi: 10.1042/bj3250001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nghiem Y, Cabrera M, Cupples CG, Miller JH. The mutY gene: a mutator locus in Escherichia coli that generates G.C—T.A transversions. Proc. Natl Acad. Sci. USA. 1988;85:2709–2713. doi: 10.1073/pnas.85.8.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Radicella JP, Clark EA, Fox MS. Some mismatch repair activities in Escherichia coli. Proc. Natl Acad. Sci. USA. 1988;85:9674–9678. doi: 10.1073/pnas.85.24.9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Williams SD, David SS. Evidence that MutY is a monofunctional glycosylase capable of forming a covalent Schiff base intermediate with substrate DNA. Nucleic Acids Res. 1998;26:5123–5133. doi: 10.1093/nar/26.22.5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bailly V, Verly WG. AP endonucleases and AP lyases. Nucleic Acids Res. 1989;17:3617–3618. doi: 10.1093/nar/17.9.3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nilsson M, Malmgren H, Samiotaki M, Kwiatkowski M, Chowdhary BP, Landegren U. Padlock probes: circularizing oligonucleotides for localized DNA detection. Science. 1994;265:2085–2088. doi: 10.1126/science.7522346. [DOI] [PubMed] [Google Scholar]
- 14.Larsson C, Koch J, Nygren A, Janssen G, Raap AK, Landegren U, Nilsson M. In situ genotyping individual DNA molecules by target-primed rolling-circle amplification of padlock probes. Nat. Methods. 2004;1:227–232. doi: 10.1038/nmeth723. [DOI] [PubMed] [Google Scholar]
- 15.Baner J, Nilsson M, Mendel-Hartvig M, Landegren U. Signal amplification of padlock probes by rolling circle replication. Nucleic Acids Res. 1998;26:5073–5078. doi: 10.1093/nar/26.22.5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jarvius J, Melin J, Goransson J, Stenberg J, Fredriksson S, Gonzalez-Rey C, Bertilsson S, Nilsson M. Digital quantification using amplified single-molecule detection. Nat. Methods. 2006;3:725–727. doi: 10.1038/nmeth916. [DOI] [PubMed] [Google Scholar]
- 17.Allalou A, Wahlby C. BlobFinder, a tool for fluorescence microscopy image cytometry. Comput. Methods Programs Biomed. 2009;94:58–65. doi: 10.1016/j.cmpb.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 18.Modrich P. Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 2006;281:30305–30309. doi: 10.1074/jbc.R600022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 20.Au KG, Clark S, Miller JH, Modrich P. Escherichia coli mutY gene encodes an adenine glycosylase active on G-A mispairs. Proc. Natl Acad. Sci. USA. 1989;86:8877–8881. doi: 10.1073/pnas.86.22.8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michaels ML, Cruz C, Grollman AP, Miller JH. Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc. Natl Acad. Sci. USA. 1992;89:7022–7025. doi: 10.1073/pnas.89.15.7022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Michaels ML, Tchou J, Grollman AP, Miller JH. A repair system for 8-oxo-7,8-dihydrodeoxyguanine. Biochemistry. 1992;31:10964–10968. doi: 10.1021/bi00160a004. [DOI] [PubMed] [Google Scholar]
- 23.Porello SL, Leyes AE, David SS. Single-turnover and pre-steady-state kinetics of the reaction of the adenine glycosylase MutY with mismatch-containing DNA substrates. Biochemistry. 1998;37:14756–14764. doi: 10.1021/bi981594+. [DOI] [PubMed] [Google Scholar]
- 24.Tsai-Wu JJ, Liu HF, Lu AL. Escherichia coli MutY protein has both N-glycosylase and apurinic/apyrimidinic endonuclease activities on A.C and A.G mispairs. Proc. Natl Acad. Sci. USA. 1992;89:8779–8783. doi: 10.1073/pnas.89.18.8779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michaels ML, Miller JH. The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine) J. Bacteriol. 1992;174:6321–6325. doi: 10.1128/jb.174.20.6321-6325.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Demple B, Harrison L. Repair of oxidative damage to DNA: enzymology and biology. Annu. Rev. Biochem. 1994;63:915–948. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- 27.Yao M, Hatahet Z, Melamede RJ, Kow YW. Purification and characterization of a novel deoxyinosine-specific enzyme, deoxyinosine 3′ endonuclease, from Escherichia coli. J. Biol. Chem. 1994;269:16260–16268. [PubMed] [Google Scholar]
- 28.Taylor GR. Enzymatic and chemical cleavage methods. Electrophoresis. 1999;20:1125–1130. doi: 10.1002/(SICI)1522-2683(19990101)20:6<1125::AID-ELPS1125>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 29.Bitinaite J, Rubino M, Varma KH, Schildkraut I, Vaisvila R, Vaiskunaite R. USER friendly DNA engineering and cloning method by uracil excision. Nucleic Acids Res. 2007;35:1992–2002. doi: 10.1093/nar/gkm041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Colbert T, Till BJ, Tompa R, Reynolds S, Steine MN, Yeung AT, McCallum CM, Comai L, Henikoff S. High-throughput screening for induced point mutations. Plant Physiol. 2001;126:480–484. doi: 10.1104/pp.126.2.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bailly V, Verly WG. The multiple activities of Escherichia coli endonuclease IV and the extreme lability of 5′-terminal base-free deoxyribose 5-phosphates. Biochem. J. 1989;259:761–768. doi: 10.1042/bj2590761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nilsson M, Gullberg M, Dahl F, Szuhai K, Raap AK. Real-time monitoring of rolling-circle amplification using a modified molecular beacon design. Nucleic Acids Res. 2002;30:e66. doi: 10.1093/nar/gnf065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kerins SM, Collins R, McCarthy TV. Characterization of an endonuclease IV 3′-5′ exonuclease activity. J. Biol. Chem. 2003;278:3048–3054. doi: 10.1074/jbc.M210750200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.