

Abstract
In the mammalian brain, the specificity of excitatory synaptic transmission depends on rapid diffusion of glutamate away from active synapses and the powerful uptake capacity of glutamate transporters in astrocytes. The extent to which neuronal glutamate transporters influence the lifetime of glutamate in the extracellular space remains unclear. Here we show that EAAC1, the predominant neuronal glutamate transporter at excitatory synapses in hippocampal area CA1, buffers glutamate released during synaptic events and prolongs the time course of its clearance by astrocytes. EAAC1 does not significantly alter activation of receptors in the synaptic cleft. Instead, it reduces recruitment of perisynaptic/extrasynaptic NR2B-containing NMDARs, thereby facilitating induction of long-term potentiation by short bursts of high-frequency stimulation. We describe novel roles of EAAC1 in regulating glutamate diffusion and propose that NMDARs at different subsynaptic locations can make distinct contributions to the regulation of synaptic strength.
Introduction
Excitatory synaptic transmission in the hippocampus is mediated by a variety of glutamate receptors, differentially activated according to their location and the concentration time course of glutamate in the extracellular space (ECS) (Asztely et al., 1997; Scanziani et al., 1997; Diamond, 2001). This latter parameter depends solely on the morphology of the synaptic environment (which influences diffusion dynamics) (Rusakov and Kullmann, 1998) and the efficacy of glutamate uptake (Diamond, 2001; Arnth-Jensen et al., 2002; Scimemi et al., 2004). Glutamate transporters are abundantly expressed in glial cells: they take up synaptically released neurotransmitter and maintain low ambient extracellular glutamate levels. In the hippocampus, pyramidal cells in area CA1 express the glutamate transporter EAAC1 in the perisynaptic region of excitatory synapses (He et al., 2000), but EAAC1's density of expression and how much glutamate it binds and transports across the cell membrane remain unknown.
A thorough examination of EAAC1's role in synaptic transmission could be critical to understand how neurons convey spatially confined signals among each other, but has not been achieved, primarily because of lack of pharmacological compounds with a sufficiently high selectivity for EAAC1. A previous report provided indirect evidence that EAAC1 limits NMDAR activation by glutamate spillover between synapses (Diamond, 2001). That study concluded that EAAC1 protects receptors within quiescent synapses from glutamate released at neighboring ones. However, it did not address experimentally whether EAAC1 also limits glutamate escape from active synapses to nearby regions, and perhaps alters the relative activation of NMDAR subtypes differentially expressed in synaptic and extrasynaptic areas, like those containing the NR2A or NR2B subunit (Li et al., 1998; Tovar and Westbrook, 1999). In this case, EAAC1 may also regulate the magnitude and direction of forms of synaptic plasticity that rely on NMDAR activation (Liu et al., 2004; Massey et al., 2004; Morishita et al., 2007; von Engelhardt et al., 2008).
Here we explore the role of EAAC1 on the dynamics of glutamatergic signaling and plasticity with experiments on acute hippocampal slices from transgenic mice bearing a non-functional mutation in the EAAC1 gene (Peghini et al., 1997). Recording transporter-mediated currents (TCs) from CA1 astrocytes in stratum radiatum, we show that EAAC1 regulates the lifetime of glutamate in the ECS immediately surrounding active synapses. Patch-clamp experiments from CA1 pyramidal cells show that the NMDAR component of glutamatergic EPSCs is enhanced in the absence of EAAC1, suggesting that EAAC1 also restricts NMDAR activation. This effect is associated with an enhanced sensitivity of NMDAR EPSCs to NR2B-selective antagonists, suggesting that it is primarily due to differential recruitment of NR2B-containing NMDARs (NR2B-NMDARs). Enhancing the activation of NR2B-NMDARs does not affect the maximal range of long-term potentiation (LTP) or long-term depression (LTD) of Schaffer collateral synapses, but it alters the balance between potentiation and depression elicited by a physiologically relevant induction protocol, theta-burst stimulation (TBS). Unexpectedly, by limiting the recruitment of perisynaptic/extrasynaptic NR2B-NMDARs, EAAC1 facilitates the expression of TBS-induced LTP.
These results unveil novel contributions of neuronal glutamate transporters to hippocampal synaptic transmission and argue for potentially complimentary roles of synaptic and extrasynaptic NMDARs in regulating synaptic strength, depending on their extent of activation during synaptic events.
Materials and Methods
Electrophysiology.
EAAC1 knock-out (KO; Memorec Stoffel) and wild-type (WT) mice [from postnatal day (P) 14 to P21] were deeply anesthetized with isoflurane and decapitated, in accordance with the with National Institute of Neurological Disorders and Stroke Animal Care and Use Committee guidelines. Transverse hippocampal slices (250 μm thick) were prepared using a vibrating blade microtome (VT1000S, Leica). The slicing solution (4°C, continuously bubbled with a mixture of 95% O2-5% CO2) contained (in mm): 119 NaCl, 2.5 KCl, 0.5 CaCl2, 1.3 MgSO4 · 7H2O, 4 MgCl2, 26.2 NaHCO3, 1 NaH2PO4, 22 glucose, 320 mOsm, pH 7.4. After cutting, slices were kept in this solution in a submersion chamber at 34°C for 30 min and at RT for up to 5 h. The artificial CSF (ACSF) used for electrophysiological recordings contained (in mm): 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgSO4 · 7H2O, 26.2 NaHCO3, 1 NaHPO4, 22 glucose, 300 mOsm, pH 7.4, saturated with 95% O2-5% CO2. In experiments performed in the absence of external Mg2+, the CaCl2 concentration was increased to 3.8 mm. All recordings were performed at 34−36°C. A cut perpendicular to stratum pyramidale separated CA3 and CA1 areas, to minimize spreading of recurrent activity. Constant voltage electrical stimuli (50 μs) were delivered through bipolar stainless steel electrodes (115 μm pole spacing; Frederick Haer Company) located ∼100 μm away from the recorded cell. In synaptic plasticity experiments, two pathways were stimulated with monopolar electrodes positioned 100 μm apart (Frederick Haer Company). The following drugs were routinely added to the recording solution to block GABAA, GABAB, mGluRII, mGluRIII and A1 adenosine receptors, to avoid potential confounding factors due to the presence of EAAC1 at inhibitory terminals and potential differences in the recruitment of A1 adenosine and presynaptic glutamate metabotropic receptors in WT and KO mice: picrotoxin (100 μm), 3-[[(3,4-dichlorophenyl)methyl] amino]propyl] diethoxymethyl)phosphinic acid (CGP 52432; 5 μm), 2-[(1S,2S)-2-carboxycyclopropyl]-3-(9H-xanthen-9-yl)-d-alanine (LY 341495; 1 μm), (R,S)-α-methylserine-O-phosphate (MSOP; 100 μm), 8-cyclopentyl-1,3-dipropylxanthine (DPCPX; 1 μm). For astrocytic recordings, AMPAR and NMDARs were blocked with 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzoquinoxaline-7-sulfonamide disodium salt (NBQX; 10 μm), and (R,S)-3-(2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (CPP; 10 μm), respectively. Pyramidal cells and astrocytes were identified under infrared-differential interference contrast using an upright fixed-stage microscope (Axioskop 2FS; Zeiss). Recordings from pyramidal cells were made with patch pipettes containing (in mm): 120 CsCH3SO3, 10 EGTA, 20 HEPES, 2 MgATP, 0.2 NaGTP, 5 QX-314Br, 290 mOsm, pH 7.2. In those experiments in which we tested for presynaptic NR2B-NMDARs in 0 Mg2+ (Fig. S5D, available at www.jneurosci.org as supplemental material), an intracellular solution containing 5H-dibenzo[a,d]cyclohepten-5,10-imine (MK-801; 1 mm) was used to back-fill the patch electrodes. EPSC decay became progressively faster and reached a steady state ∼20 min after reaching the whole-cell configuration (data not shown), an indication of the time required for MK-801 to diffuse into the cell and block postsynaptic NMDARs. No developmental trend in the effects of ifenprodil was observed within the age range of the animals used for our experiments (data not shown). In other experiments we replaced EGTA with the faster Ca2+-buffer BAPTA (10 mm) (Fig. S5A, available at www.jneurosci.org as supplemental material). To block EAAC1 in individual neurons, we replaced CsCH3SO3 with equimolar NMDG+, a larger cation that cannot replace K+ in the relocation step of glutamate transport (Diamond, 2005). At positive potentials, NMDAR EPSCs were blocked ∼20 min after reaching the whole-cell configuration and electrical recordings at negative potentials were commenced afterward. Astrocytic recordings were obtained by replacing CsCH3SO3 in the internal solution with equimolar KCH3SO3 (QX-314 was omitted). For field recordings, pipettes were filled with ACSF. The stimulus intensity was set to ∼70% of that required to elicit a population spike. In each experiment, independence of the two pathways was tested with a crossed paired-pulse protocol: two stimuli delivered 100 ms apart to one pathway elicited paired-pulse facilitation. If the two stimuli were delivered to different pathways at the same interpulse interval, no facilitation in either pathway was observed (data not shown).
Field recordings were obtained with the same voltage-clamp amplifier (Axopatch 1D, Molecular Devices) used to record whole-cell EPSCs and TCs. In this amplifier, voltage is recorded using modified voltage-clamp circuitry (rather than a voltage follower), rendering field EPSP (fEPSP) amplitudes sensitive to changes in electrode resistance. To circumvent this problem, field responses were recorded under voltage clamp, and electrode resistance was monitored with a voltage step (0.5 mV) before each stimulus trial, allowing any changes in resistance to be detected. Under stable recording conditions fEPSCs, scaled to mV according to the amplitude of the current response to the test voltage step, matched closely the amplitude and time course of fEPSPs recorded in the “I = 0” configuration (data not shown) (Diamond et al., 1998).
TBS consisted of 5 bursts applied at 5 Hz, with each burst comprising 6 pulses at 100 Hz. During in-slice comparison experiments (Fig. S6, available at www.jneurosci.org as supplemental material), TBS was applied to one of two independent pathways and fEPSC slope was monitored for 30 min. Then we washed in ifenprodil for ∼15 min, adjusted the stimulus intensity to elicit fEPSCs similar to those recorded in baseline conditions, and delivered a TBS to the second pathway (Fig. S6, available at www.jneurosci.org as supplemental material). Tetani consisted of 100 pulses at 100 Hz, low-frequency stimulation (LFS) delivered to induce LTD consisted of 900 pulses at 1 Hz, and depotentiation was induced with 600 pulses at 1 Hz. The initial slope of fEPSCs was calculated from a linear fit of 10–90% of the rising phase.
For flash photolysis experiments, 4-methoxy-7-nitroindolinyl-caged-l-glutamate (MNI-l-glutamate; 100 μm) was perfused in the recording chamber and uncaged using a Flashmic Xe lamp (Rapp OptoElectronic) connected to the epi-illumination port. The lamp output was set to maximum and filled the entire back aperture of the 40× objective. Consequently, the entire field of view was illuminated with a collimated beam, corresponding to an uncaging area of ∼662.5 μm diameter [field number (26.5)/magnification of the objective (40×)]. Based on the manufacturer's test results, in the configuration used, the flash width at 50% of peak height was 330 μs and the flash was 99% complete in 680 μs.
Reagents were obtained from Sigma and Tocris Bioscience. Currents were filtered at 2 KHz, converted with an 18 bit 200 kHz A/D board (Instrutech) digitized at 5 KHz and collected through a custom made software (J.S.D.) written in IgorPro (Wavemetrics). Patch and field electrodes (#0010 glass; World Precision Instruments) had tip resistances of 2.5 and 1 MΩ, respectively. Series resistance (∼20 MΩ for neurons, ∼10 MΩ for astrocytes) was continuously monitored, not compensated, and experiments were discarded if this changed by >20%.
Data analysis.
Electrophysiological recordings were analyzed within the Igor Pro environment using custom made software (A.S. and J.S.D.). Astrocytes from WT and KO mice displayed similar resting membrane potential (VR WT −83.7 ± 3.3 mV, n = 7, VR KO −87.5 ± 2.8 mV, n = 6, p = 0.052), input resistance (Rinput WT 14 ± 7 MΩ, n = 7, Rinput KO 14 ± 8, n = 6, p = 0.93) and membrane time constant (τ WT 3.3 ± 1.8 ms, n = 7; τ KO 4.2 ± 1.9, n = 6, p = 0.39) (data not shown). When recording from these cells, isolation of TCs elicited by wide-field flash photolysis (FTCs) was obtained by subtracting the stimulus artifact recorded with the light path blocked from the actual response (Diamond, 2005). In the presence of glutamate receptor antagonists, FTCs consisted of a fast-rising and fast-decaying component that completely recovered in <20 ms. FTCs displayed similar amplitude and kinetics in WT and KO slices (data not shown). Synaptically activated TCs (STCs) were isolated as described previously (Diamond, 2005). Briefly, single and pairs of stimuli, 100 ms apart, were delivered every 10 s. Single STCs were subtracted from paired STCs to isolate the current evoked by the second pulse. Single STCs were then shifted in time so that their onset matched that of paired STCs and subtracted from them. The resulting current represented the facilitated portion of the STC in response to paired stimuli. Because the sustained potassium current superimposed to STCs does not facilitate, this method allows for almost perfect isolation of the STC, similarly to what can be achieved by subtracting the sustained potassium current in high concentrations of threo-β-benzyloxyaspartate (TBOA) from astrocytic currents recorded in control conditions (Diamond, 2005). The following approach was adopted whenever a proportion of the sustained potassium current was still present after subtraction. In 4 astrocytes, we isolated the potassium current by adding TBOA (50–100 μm) to the recording solution. A monoexponential fit of the rising phase of the residual current with the following function,
 |
enabled estimating τonset = 4.9 ± 0.7 ms (A was set as the amplitude of the sustained current). This waveform was scaled and subtracted from STCs for optimal isolation of the facilitated component (Diamond, 2005). Throughout the text, unless otherwise stated, we will refer to the facilitated STCs isolated with this analytical approach as STCs. The peak of STCs was 6 times larger than the steady-state value of the potassium current in WT and KO mice (WT 6.4 ± 2.0, n = 7, KO 6.6 ± 2.1, n = 6, p = 0.91).
The steps of the deconvolution approach used to derive the time course of glutamate clearance are schematized in Fig. S7A (available at www.jneurosci.org as supplemental material). TCs were binomially smoothed (order = 50) and fitted with the following equation (Nielsen et al., 2004):
 |
Fitted TCs were used to derive the time course of glutamate clearance (Diamond, 2005). This fit allowed us to control for potential glitches in the TC recordings and to resolve small differences in the estimated clearance time courses. The residual components of the fits did not differ between WT and KO mice (an analysis of STC residuals is displayed in Fig. S8A,B, available at www.jneurosci.org as supplemental material). Small differences between the fit and the STC waveform were reproduced on an idealized STCs and used to derive a hypothetical clearance time course (Fig. S8C, available at www.jneurosci.org as supplemental material). For each of them, we calculated a χ2 value, to derive information on the error on the <t TC introduced by our fitting procedure. The analysis indicates that our fits lead to ≤5% error in derived clearance time (Fig. S8D, available at www.jneurosci.org as supplemental material) and therefore cannot account for the differences between WT and KO reported in Figure 1.
TC introduced by our fitting procedure. The analysis indicates that our fits lead to ≤5% error in derived clearance time (Fig. S8D, available at www.jneurosci.org as supplemental material) and therefore cannot account for the differences between WT and KO reported in Figure 1.
Figure 1.

Deleting EAAC1 does not alter the glial uptake capacity, but speeds the time course of synaptically released glutamate clearance. A, Examples of raw and peak normalized STCs in WT (black) and KO mice (red). Each trace represents the averages of 20 trials. B, The bar chart shows the summary of the kinetic properties of STCs in WT (white) and KO mice (red). STCs decay faster in the absence of EAAC1 (rise time, p = 0.17; t50 **p = 0.002; <t ***p = 0.0002; WT n = 8, KO n = 13). C, Left: schematic of the experimental design. Electrical stimuli were delivered through a bipolar electrode in stratum radiatum to evoke glutamate release from Schaffer collateral fibers. Right: Raw WT (black) and KO (red) STCs. D, Deconvolution-derived clearance of synaptically released glutamate is faster in KO mice (*p = 0.02). Mean ± SEM. E, The STC clearance derived from centroid analysis is also faster in KO mice (*p = 0.02). F, Left: Schematic of the experimental design. MNI-l-glutamate (100 μm) was uncaged over the entire field of view. Right: Raw WT and KO FTCs. G, Deconvolution-derived clearance of caged glutamate is similar in WT and KO mice (p = 0.61). H, The FTC clearance derived from centroid analysis is similar in WT and KO mice (p = 0.29). Data in C–H have been obtained from the same population of astrocytes (WT n = 7, KO n = 6). I, Relationship between the STC charge transfer and the amplitude of the fiber volley preceding the STC. No difference in the extent of glutamate transport per afferent fiber recruitment is observed between WT and KO mice (WT n = 5, KO n = 6; raw STC charge p = 0.22; raw STC FV amplitude p = 0.42). J, STCs have a similar sensitivity to the GLT-1 antagonist DHK (300 μm) in WT (n = 5) and KO mice (n = 4, amplitude p = 0.71, t50
p = 0.47, <t
p = 0.37). The inset shows representative examples of STCs recorded in control conditions and in the presence of DHK, in WT and KO mice.
TCs can be thought as functions of glutamate clearance and of a filter, which represents all the distortion factors that convert a fast process like glutamate clearance into a much slower TC (i.e., electrical properties of the astrocytes, transporters kinetics, asynchronicity of transmitter release, etc). In absolute terms, this would be a nonlinear filter because, for example, the transporters glutamate binding sites could become saturated. However, it has previously been shown that glutamate transporters in hippocampal astrocytes are not saturated by synaptically released glutamate (Diamond and Jahr, 2000) and that, under experimental conditions similar to those of our experiments, the filter behaves approximately linearly (Diamond, 2005). Therefore, under these experimental conditions, the filter operates within a linear range. Accordingly: (1) the TC time course remains constant across large changes in response amplitude such as those caused by paired-pulse facilitation (Fig. S7B, available at www.jneurosci.org as supplemental material); (2) the amplitude of simulated TCs (obtained using a diffusion model described in the following section) increases linearly with the amount of glutamate molecules released, whereas its <t remains constant (Fig. S9B, available at www.jneurosci.org as supplemental material); (3) the amplitude and <t of simulated TCs does not change when releasing 2000 glutamate molecules in brief succession from one another (Fig. S9C, available at www.jneurosci.org as supplemental material).
Since we are likely operating in a linear regime of the filter, we represent TCs as convolutions of the filter itself and the clearance time course. In TBOA (10 μm), when the uptake capacity of astrocytes is diminished, glutamate clearance is prolonged and accounts almost completely for the time course of the TC (the properties of the filter are assumed to be similar in control and in TBOA) (Diamond, 2005). Therefore, we approximate glutamate clearance in TBOA (10 μm) with a monoexponential function that rises instantaneously and decays with the same exponential decay as TCs. The error introduced by this fitting procedure is small and does not differ between WT and KO STCs and FTCs (data not shown). The instantaneous rise approximation is a simplification because, for example, the diffusion time of glutamate from active release sites to glial membranes is obviously not instantaneous. Accordingly, FTCs rise faster than STCs (WT: STC rise 1.73 ± 0.50 ms, FTC rise 1.13 ± 0.50, n = 7, ***p = 0.0002; KO: STC rise 1.40 ± 0.15 ms, FTC rise 1.02 ± 0.18 ms, n = 6, ***p = 7.4E-5). Any error introduced by this approach, however, is negligible as similar results can be obtained when a different analytical approach is used (see below). The filter is derived by deconvolving the approximated glutamate clearance in TBOA from TCs in TBOA. Then, deconvolution of this filter from TCs recorded in control conditions yields an estimate of the glutamate clearance time course with intact uptake. Successful deconvolution was performed in all recorded astrocytes. The STC and FTC filter displayed similar time course in WT and KO mice (Fig. S7D, available at www.jneurosci.org as supplemental material).
A second analytical approach (Fig. 1E,H) takes advantage of the linearity of the filter and of the additive property of centroids under convolution. The centroid,
 |
was calculated in a time window corresponding to 0.1 of the peak, before and after its onset. Since TC = filter * clearance, then <tTC = <tfilter + <tclearance. <tfilter and <tclearance were extrapolated by plotting <tTC versus the inverse of the transporter concentration ([T]) normalized by its value in control conditions [we used the initial slope of TCs as a measure of [T], since it is the least filtered portion of TCs and is indicative of relative transporter abundance (Diamond, 2005)]. A linear fit of the data obtained in control conditions and in TBOA provided information on <tfilter (the y-intercept, where [T] → ∞) and <tclearance (the slope). The deconvolution-derived <tTC clearance tended to be slower than the slope-derived <tTC clearance (Fig. S7C, available at www.jneurosci.org as supplemental material) (Diamond, 2005), presumably because of a slight overestimate of glutamate clearance in TBOA (in fact, the deconvolution-derived <tfilter tended to be faster than the slope-derived <tfilter in both WT and KO mice (Fig. S7E, available at www.jneurosci.org as supplemental material).
The more rapid STC glutamate clearance in KO mice was also observed in the absence of MNI-l-glutamate in the recording solution (data not shown), ruling out potential artifacts due to the presence of this compound in the extracellular solution.
Miniature events were detected using an optimally scaled template (Clements and Bekkers, 1997) adapted for Igor Pro (A.S.). Paired-pulse ratio (PPR) of EPSCs was calculated by subtracting single from paired EPSCs averaged across 20 traces, after first peak normalization.
Coefficient of variation (CV) was calculated according to the following equation:
 |
where σbaseline2 is the variance of the baseline noise and σsignal2 is the variance of the response calculated across an identical time window, and is the response mean.
The accuracy of slope and the charge transfer in a 50 ms time window between 30 and 80 ms after the stimulus artifact (Q30-80) of composite EPSCs as measures of AMPAR and NMDAR components was validated by analysis of AMPAR and NMDAR EPSCs kinetics (AMPAR EPSC rise time in ACSF and CPP (10 μm) WT 2.1 ± 1.8 ms, n = 14; NMDAR EPSC rise time in NBQX (10 μm) WT 5.3 ± 1.6 ms, n = 6, **p = 0.0017; AMPAR EPSCs t50 in ACSF WT 8.1 ± 3.9 ms, n = 13; KO 8.7 ± 2.4 ms, n = 11, p = 0.61) and by the distinct voltage dependence of each measure in the presence of extracellular magnesium (Fig. S3A,B, available at www.jneurosci.org as supplemental material).
Data averages are presented as mean ± SD, unless indicated otherwise in the text and figure legends. Statistical significance was determined by Student's paired or unpaired t test, as appropriate and differences were considered significant at p < 0.05 (*p < 0.05; **p < 0.01; ***p < 0.001). Post hoc power analysis was performed with G*Power 3.0.10 (Franz Faul, Universität Kiel, Germany) and allowed us to compute the achieved power given α = 0.05 and a sample size corresponding to that of individual experiments.
Modeling.
The geometry of the synaptic and extrasynaptic environment was created in silico with an open source program (Blender), imported in a Monte Carlo simulation environment (MCell) and rendered with a visualization software (DReAMM). The simulations were run within a spherical space (world) of radius r = 790 nm. The presynaptic and postsynaptic elements were placed at the center of the world, and simulated as hemispheres with radius r = 250 nm, separated from each other by a synaptic cleft of height h = 20 nm. The synapse was surrounded by 4 arcs, 300 nm thick and 20 nm far from each other. Two adjacent arcs were covered by glial transporters (density d = 10,800 μm−2) (Danbolt, 2001), whereas the two other ones expressed few NMDARs, with a surface density similar to that used in extrasynaptic areas (d = 60 μm−2). The distance between the arcs and the synapse was varied between 20 and 200 nm, to test the effects of different ECS widths on NMDAR activation. The postsynaptic density (PSD) had a radius r = 110 nm, and was populated with NMDARs (300 μm−2). Outside the PSD, the density of these receptors was set to 60 μm−2. NMDARs were also scattered on the outer surface of the postsynaptic element. The NMDARs kinetic rates were set in accordance to (Lester and Jahr, 1992), whereas glutamate transporters were modeled using the simplified GLT-1 scheme (Bergles et al., 2002) described previously (Diamond, 2005), with the exception that the reverse translocation rate was set to 200 s−1. This change was necessary to prevent sustained delayed activation of NMDARs due to glutamate unbinding from glial transporters. All rates were adjusted for Q10 = 3, to approximate the receptor and transporter kinetics at 35°C. The transporter rates were also adjusted for [Na]o = 146.2 mm, [Na]i = 5 mm, [Glut]i = 10 μm, pHi = 7.2. The STC waveforms were calculated as described previously (Diamond, 2005), from the forward and backward reactions of the translocation step.
Neuronal glutamate transporters were randomly scattered at different densities (d = 0–10,800 μm−2) in a perisynaptic ring (width w = 70 nm), 180 nm away from the center of the PSD, that extended to the outer perimeter of the apposition zone between presynaptic and postsynaptic hemispheres. At t = 0, 2000 glutamate molecules were released from a point source placed in the center of the PSD, close to the presynaptic element. Glutamate diffused away with a diffusion coefficient D* = 3.3 × 10−6 cm2/s (Nielsen et al., 2004). Each simulation consisted of 104 iterations with a time step Δt = 1 μs (therefore spanning a 10 ms time window), and was run 100 times. Glutamate was removed from the simulation environment once it hit the inner surface of the world. Glutamate hitting the surfaces of the arcs or of the presynaptic and postsynaptic elements bounced back in the ECS. The glutamate concentration was monitored at every Δt, in the ECS above the PSD (r = 110 nm, h = 20 nm), above the perisynaptic ring (w = 70 nm, h = 20 nm), or above a 100-nm-wide spherical surface, centered 425 nm away from the release site (h = 20–200 nm). The efficiency was calculated from the ratio of the net flow of glutamate transporters through the outward-facing glutamate-bound state and the inward-facing glutamate-bound state. It was decreased by increasing proportionately the binding/unbinding rate of glutamate for EAAC1. These changes altered only negligibly the EC50 of the peak simulated TC in response to 0.3–30 ms glutamate pulses (data not shown). Increasing the backward translocation rate also reduced efficiency, but this caused an increase in perisynaptic glutamate and enhanced NMDAR activation, contrary to our experimental observations (see Fig. 4). The glutamate waveforms were exported in ChanneLab (Synaptosoft) to simulate NMDARs responses.
Figure 4.
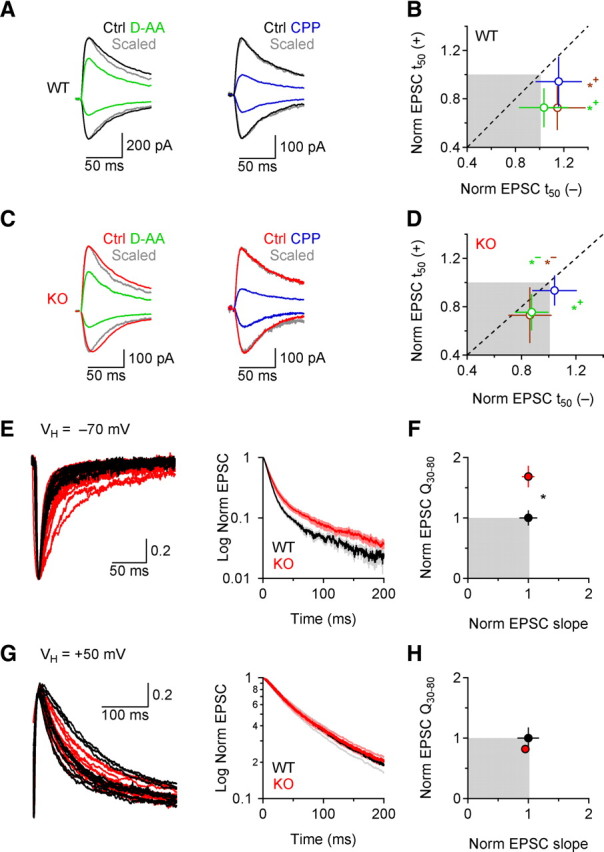
A larger population of NMDARs is activated when postsynaptic glutamate transport is impaired. A, NMDAR EPSCs in WT mice in control conditions (black) and in D-AA (100 μm; green) or CPP (1 μm; blue), at ±50 mV. Peak-scaled EPSCs are shown in gray. B, Summarized effects of D-AA (green; n = 11), l-AP5 (250 μm; brown; n = 9) and CPP (blue; n = 19) on the decay of NMDAR EPSCs at ±50 mV in WT mice (CPP+50 mV-D-AA+50 mV *p = 0.014; CPP+50 mV-l-AP5+50 mV *p = 0.036; CPP−50 mV-D-AA−50 mV p = 0.21; CPP−50 mV-l-AP5−50 mV p = 0.92). C, As in A, in KO mice. D, As in B, with summarized effects of D-AA (n = 8), l-AP5 (n = 10), and CPP (n = 10) in KO mice (CPP+50 mV-D-AA+50 mV *p = 0.021; CPP+50 mV-l-AP5+50 mV p = 0.107, CPP−50 mV-D-AA−50 mV *p = 0.023, CPP−50 mV-l-AP5−50 mV *p = 0.040; n = 6). Drug application in A–D was performed in separate groups of cells. EPSC analysis was performed 20 min after drug application, after confirming that the effects on the EPSC profile were stable. E, Left: Raw traces of composite peak-normalized EPSCs recorded at −70 mV, in 0 Mg2+. Right: the average peak-normalized decay is plotted on a semilog scale (WT n = 13, KO n = 27). Mean ± SEM F, Summary of the NMDAR and AMPAR contribution to composite EPSCs. Initial slope and Q30-80 were normalized by the corresponding mean values in WT mice. The NMDAR component is larger in KO than in WT mice (*p = 0.016). Mean ± SEM. G, Left: Raw traces of peak-normalized composite EPSCs recorded at +50 mV. Right: The average peak-normalized decay is plotted on a semilog scale (WT n = 11, KO n = 11). Mean ± SEM. H, As in F, at +50 mV (p = 0.34). Mean ± SEM.
Results
EAAC1 delays the arrival of synaptically released glutamate at astrocytic membranes
The rate of glutamate clearance from the ECS is reflected in the time course of TCs recorded from astrocytes (Bergles and Jahr, 1997; Diamond, 2005). We examined whether EAAC1 influences glutamate diffusion in the hippocampal neuropil by recording TCs from astrocytes in stratum radiatum of acute mouse hippocampal slices (Bergles and Jahr, 1997; Diamond and Jahr, 1997; Diamond, 2005). EAAC1, shown by immunogold electron microscopy (EM) to be present in the perisynaptic region of excitatory synapses (He et al., 2000), could affect STCs by binding glutamate released during synaptic events before the transmitter reaches glial transporters. The net effect on glutamate diffusion (and therefore STCs) would depend on whether EAAC1 released glutamate back in the ECS (acting as a buffer), or carried it across the membrane (acting as a pure transporter). In the first case EAAC1 would delay the arrival of glutamate at astrocytic membranes, thereby slowing glial uptake without affecting the total amount taken up. Alternatively, if EAAC1 transported all of the bound glutamate, it would reduce the amount to be taken up by glial transporters without changing the time course of the STCs (Diamond, 2005). To distinguish between these two possibilities, we elicited STCs in astrocytes by stimulating Schaffer collaterals in the presence of glutamate, GABA and A1 adenosine receptor blockers (Materials and Methods). STCs exhibited faster decay kinetics in EAAC1 KO mice (Fig. 1A,B). The time course of glutamate clearance reflected in the STCs, however, is partially obscured (filtered) by the electrotonic properties of astrocytes (Diamond, 2005). Accordingly, the effect of deleting EAAC1 was much more evident when, in separate experiments, we accounted for the filtering and used the STCs to derive the actual time course of glutamate clearance (Materials and Methods). Glial glutamate clearance was faster in KO than in WT mice (WT STC <tclearance 3.8 ± 0.8 ms, n = 7; KO STC <tclearance 2.8 ± 0.2 ms, n = 6, *p = 0.02; Fig. 1C,D). Similar results were also obtained when estimating glutamate clearance using a different analytical approach (Materials and Methods), ruling out potential biases when deriving the extent of filtering (WT STC <tclearance 3.5 ± 1.3 ms, n = 7; KO STC <tclearance 1.8 ± 0.7 ms, n = 6, *p = 0.02; Fig. 1E).
These results suggest that EAAC1 acts as a glutamate buffer, provided that the uptake capacity of astrocytes is not impaired in KO mice. To test this, we estimated glutamate clearance from FTCs MNI-l-glutamate (100 μm), in the same astrocytes from which we recorded STCs. Flash-uncaged glutamate was taken up at a similar rate by glial transporters in WT and KO slices, as shown by both deconvolution (WT FTC <tclearance 3.2 ± 1.3, n = 7; KO FTC <tclearance 2.8 ± 0.1, n = 6, p = 0.50; Fig. 1F,G) and centroid analysis (WT FTC <tclearance 2.3 ± 1.2, n = 7; KO FTC <tclearance 1.7 ± 0.5, n = 6, p = 0.29; Fig. 1H). This indicated that removing EAAC1 does not affect the glial glutamate uptake capacity. If this were true, one would also expect the STC charge transfer, which varies with the amount of glutamate transported into astrocytes, to be similar, on average, in WT and KO slices. Although significant cell-to-cell variability may preclude detection of small changes, the charge transfer per active fiber, estimated by measuring the STC charge transfer relative to the amplitude of the fiber volley (FV) that precedes the STCs in whole-cell astrocyte recordings (Bergles and Jahr, 1997), appeared distributed across a similar range in WT and KO mice (Fig. 1I).
Do the expression of glial transporters and the properties of the extracellular diffusion environment change with deletion of EAAC1? A series of control experiments tested for several potentially confounding differences between WT and KO tissue. Western blot and immunofluorescence analyses revealed no detectable differences in GLAST or GLT-1 expression, or in that of the neuronal glutamate transporters EAAT4 and EAAT5 (Fig. S1A–D). In addition, STCs displayed a similar sensitivity to the GLT-1 antagonist dihydrokainic acid (DHK) (300 μm) in WT and KO slices (Fig. 1J). These findings argue against altered expression of glial transporters in KO mice, or changes in the contribution of glial transporter subtypes to STCs.
The effects of EAAC1 deletion on TCs described in Figure 1 were also not confounded by changes in the structure of the neuropil. Several approaches were taken to examine this possibility. The density and size of dendritic spines on CA1 pyramidal neurons were not altered in KO mice (Fig. 2A–C), and no difference in ECS fraction was detected by EM analysis of stratum radiatum in acute slices, even after prolonged in vitro storage, or in perfused animals (Fig. 2D,E). To evaluate whether the properties of extracellular diffusion differed between WT and KO mice, we first examined the fluorescence intensity distribution (in space and time) of Texas red-conjugated dextrans and quantum dots injected into the neuropil (Thorne and Nicholson, 2006). The estimated diffusion coefficient, hindrance and hydrodynamic diameter of each probe were similar in WT and KO mice (Fig. 3A–C, Table 1). In addition, no difference was observed in the diffusion of Alexa Fluor 350, a fluorescent probe with a molecular weight (349.29 Da) similar to that of glutamate (147.13 Da), using high-frequency line scans in a thin focal plane in WT and KO slices (Fig. 3D,E) (Savtchenko and Rusakov, 2005; Zheng et al., 2008). Therefore, the structural and diffusion properties of the hippocampal neuropil are not altered by genetic deletion of EAAC1.
Figure 2.
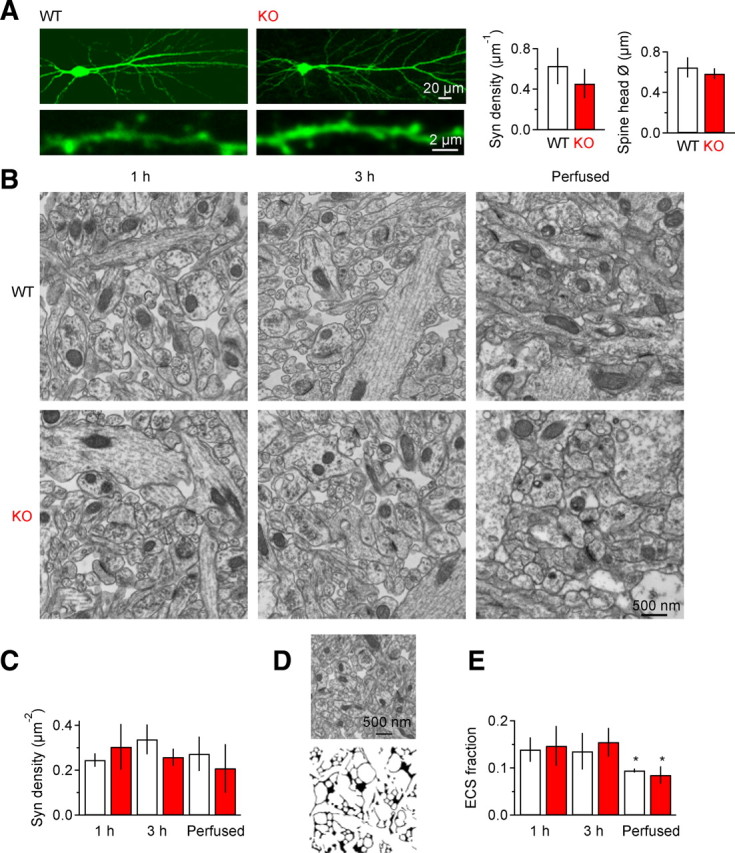
The structure of the neuropil does not change between WT and EAAC1 KO mice. A, Biocytin-filled CA1 pyramidal cell in WT (left) and KO mice (middle). Bottom panels are close-up views of dendritic regions in stratum radiatum. Right bar charts: Summary of spine density and size analysis in WT (n = 9) and KO mice (n = 6, density WT-KO p = 0.07, Power = 0.61; spine head diameter WT-KO p = 0.18, Power = 0.41). B, Electron micrographs showing the structure of the hippocampal neuropil in stratum radiatum 1 and 3 h after acute slice preparation and in perfused animals (right panel) in a WT and KO mouse. C, The total density of synapses does not change in vitro and is similar to that of perfused tissue in WT (n = 3) and KO mice (n = 3; 1 h WT-KO p = 0.39, Power = 0.20; 3 h WT-KO p = 0.14, Power = 0.44; perfused WT-KO p = 0.44, Power = 0.17). D, Examples of the analytical approach used to calculate the ECS fraction from EM data. An EM picture is shown together with its corresponding binary image. The ECS is drawn in black. E, Estimates of the ECS. No change is detected between WT and KO mice (1 h WT-KO p = 0.80, Power = 0.08; 3 h WT-KO p = 0.53, Power = 0.14; perfused WT-KO p = 0.41, Power = 0.19), but the ECS tends to be smaller after perfusion (WT 1 h-perfused *p = 0.04, 3 h-perfused p = 0.14; KO 1 h-perfused 0.08, 3 h-perfused *p = 0.03).
Figure 3.

Diffusion in the hippocampal neuropil is similar in WT and KO mice. A, Representative images of TRdx3 diffusion in stratum radiatum of WT (top) and KO mice (bottom). Middle: Fluorescence intensity profiles and theoretical fits for these images. Right: A linear regression of γ2/4 over time yields an estimate of D* for TRdx3. B, Summary of the apparent diffusion coefficient (D*), tortuosity (λ), and hydrodynamic diameter (dH) for various fluorescent probes measured at 34°C in WT and KO neuropil. (D* TRdx3 WT n = 11, KO n = 9, p = 0.36, Power = 0.11; TRdx10 WT n = 8, KO n = 8, p = 0.18, Power = 0.38; TRdx40 WT n = 10, KO n = 8, p = 0.89, Power = 0.06; TRdx70 WT n = 14, KO n = 12, p = 0.82, Power = 0.10; QD655 WT n = 5, KO n = 3 p = 0.57, Power = 0.12) (λ TRdx3 p = 0.94, Power = 0.15; TRdx10 p = 0.20, Power = 0.36; TRdx40 p = 0.84, Power = 0.09; TRdx70 p = 0.46, Power = 0.40) (dH TRdx3 p = 0.60, Power = 0.13; TRdx10 p = 0.21, Power = 0.35; TRdx40 p = 0.81, Power = 0.08; TRdx70 p = 0.12, Power = 0.49). The numerical values are presented in Table 1. C, By using a model of parallel plane geometry for the hippocampal neuropil (Thorne and Nicholson, 2006), we show a similar diffusion behavior of the Texas red probes in WT and KO slices. The predicted values of λθ = 0 (the estimated tortuosity for a vanishingly small molecule) are λ = 2.18 ± 0.13 (WT) and λ = 2.06 ± 0.13 (KO). The estimated ECS width is 155.1 ± 9.5 nm (WT) and 120.4 ± 8.4 nm (KO). D, Fluorescence intensity profiles for Alexa Fluor 350 pressure applied in free medium (left), WT (middle) or KO slices (right). Each image represents the average of 10 consecutive trials, and the intensity profiles are measured 200 ms and 1 s after the puff. E, Summary of the diffusion coefficient and tortuosity measured in each condition at room temperature (Top: Dfree 0.64 ± 0.16 μm2/ms, n = 6; D*WT 0.32 ± 0.08 μm2/ms, n = 8; D*KO 0.30 ± 0.07 μm2/ms, n = 9, Dfree-D*WT ***p = 0.0002, Dfree-D*KO ***p = 6.1E-5, D*WT-D*KO p = 0.68, Power = 0.13). Bottom: λWT 1.45 ± 0.17; λKO 1.48 ± 0.17, p = 0.71, Power = 0.09). The estimated values of the diffusion coefficients at 34°C, obtained by multiplying the previous values by Q10 = 1.3 are: Dfree 0.84 ± 0.21 μm2/ms; D*WT 0.41 ± 0.10 μm2/ms; D*KO 0.39 ± 0.09 μm2/ms.
Table 1.
Summary of the diffusion parameters estimated through the integrative optical imaging approach in free solution, in WT and KO slices
| Probe | Dfree (μm2/ms) |
D* (μm2/ms) |
λ |
dH (nm) |
|||
|---|---|---|---|---|---|---|---|
| WT | KO | WT | KO | WT | KO | ||
| TRdx3 | 0.33 ± 0.11 (12) | 0.074 ± 0.025 (11) | 0.080 ± 0.039 (9) | 2.19 ± 0.36 | 2.07 ± 0.47 | 9.13 ± 3.02 | 8.32 ± 3.80 |
| TRdx10 | 0.37 ± 0.13 (8) | 0.080 ± 0.006 (8) | 0.085 ± 0.010 (8) | 2.16 ± 0.08 | 2.09 ± 0.12 | 7.71 ± 0.56 | 7.23 ± 0.86 |
| TRdx40 | 0.18 ± 0.13 (9) | 0.021 ± 0.004 (10) | 0.020 ± 0.004 (8) | 2.99 ± 0.30 | 3.02 ± 0.37 | 30.58 ± 6.42 | 31.39 ± 8.10 |
| TRdx70 | 0.19 ± 0.09 (7) | 0.008 ± 0.005 (14) | 0.009 ± 0.002 (12) | 5.48 ± 1.51 | 4.84 ± 0.59 | 103.47 ± 55.47 | 76.27 ± 18.28 |
| QD655 | 0.075 ± 0.048 (7) | 0.0004 ± 0.0004 (5) | 0.0008 ± 0.0015 (3) | N.A. | N.A. | N.A. | N.A. |
N.A., not applicable. The numbers in parentheses represent the number of experiments done.
Recent reports indicate that lack of EAAC1 results in reduced glutathione synthesis and increased susceptibility to oxidative stress in 6- to 8-week-old mice (Aoyama et al., 2006). To test this in the mice studied here, we monitored the immunoreactivity for 4-hydroxy-2-nonenal (HNE), an indicator of oxidative stress formed by oxidant reactions with proteins and lipids, at different times of in vitro incubation and in the presence of H2O2 (200 μm), in P14–P21 and 8-week-old animals (Fig. S1G, available at www.jneurosci.org as supplemental material). No difference between WT and KO slices was observed, arguing against major changes in the vulnerability to oxidants depending on the presence of EAAC1.
Together, these results indicate that, at the ages studied here, EAAC1 buffers a significant amount of glutamate released during synaptic events and that this effect is not due to changes in the morphological characteristics, diffusion properties and resilience to oxidative damage of the WT and KO hippocampus.
EAAC1 limits activation of NMDARs located outside the synaptic cleft
Given the impact of EAAC1 on glutamate diffusion in the ECS (Fig. 1), we examined whether it affects excitatory signaling between neurons. Previous work suggested that, in the rat hippocampus, EAAC1 limits spillover of synaptically released glutamate from active synapses onto quiescent, neighboring ones, sensed by high-affinity, slowly desensitizing NMDARs (Diamond, 2001). These receptors, however, are present not only within the PSD but, at lower levels, also in perisynaptic/extrasynaptic membranes (Takumi et al., 1999). Here we tested whether EAAC1 limits spillover activation of NMDARs in mouse hippocampus, and whether this results in a differential recruitment of NMDARs that reside in synaptic or extrasynaptic regions.
Because the recruitment of distant NMDARs is delayed relative to NMDARs at directly activated synapses, they may contribute a slower component of the EPSC (Diamond, 2001; Arnth-Jensen et al., 2002; Nielsen et al., 2004). If a sufficiently large proportion of these receptors encounters diluted glutamate concentrations, low-affinity competitive NMDAR antagonists could speed the decay of the EPSC (Diamond, 2001). At +50 mV (i.e., when postsynaptic glutamate transport is impaired (Wadiche et al., 1995), the low-affinity competitive NMDAR antagonists d-aminoadipate (D-AA; 100 μm) or l-2-amino-5-phosphonopentanoic acid (l-AP5; 250 μm) sped the EPSC decay in WT and KO mice (Fig. 4A–D). At −50 mV, a potential that is permissive for glutamate transport, D-AA and l-AP5 sped the EPSC decay in KO but not WT mice (Fig. 4A–D) (WT-KO: D-AA *p = 0.025, l-AP5 **p = 0.0063). CPP (1 μm), a high-affinity NMDAR antagonist, reduced the EPSC amplitude as much as D-AA and l-AP5 (to ∼40% of control), but did not affect the decay at either potential (Fig. 4A–D), ruling out potential bias due to voltage-clamp errors. The apparently subtle effects of low-affinity antagonists on the EPSC kinetics, which nevertheless reflects a substantially different glutamate transient at NMDARs in WT and KO slices (Diamond, 2001), is likely due to the fact that many of the NMDARs mediating EPSCs are located in the synaptic cleft, where the glutamate concentration is relatively unaffected by EAAC1 (Fig. S2, available at www.jneurosci.org as supplemental material; see Fig. 9). In fact, postsynaptic receptor activation by single transmitter quanta was not influenced by EAAC1, as the amplitude and decay of miniature EPSCs (mEPSCs) were comparable in WT and KO mice in the presence (Fig. S2A, available at www.jneurosci.org as supplemental material) and absence of external Mg2+ (Fig. S2B, available at www.jneurosci.org as supplemental material) and DGG, a low-affinity competitive AMPAR antagonist whose efficacy at reducing the AMPAR EPSC depends on the time course of glutamate in the synaptic cleft (Christie and Jahr, 2006), exerted similar effects on AMPAR EPSC amplitude and PPR in WT and KO mice (Fig. S2C–E, available at www.jneurosci.org as supplemental material).
Figure 9.
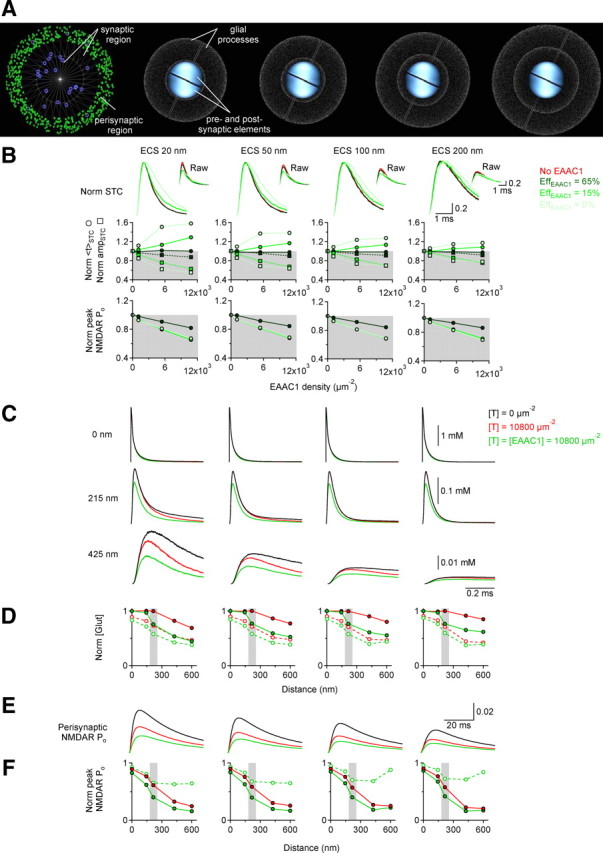
Three-dimensional Monte Carlo model of the synaptic and extrasynaptic environment. A, The first diagram illustrates the arrangement of NMDARs (blue symbols) and EAAC1 molecules (green symbols) in the postsynaptic area directly facing the presynaptic terminal (r = 250 nm). The PSD is the inner white circle (r = 110 nm) and the perisynaptic area the 70-nm-wide ring at the edge of the apposition zone. NMDARs are randomly scattered in the PSD at d = 300 μm−2, and at d = 60 μm−2 beyond the PSD. In this example, for EAAC1, d = 1080 μm−2. The next images show the arrangement of the presynaptic and postsynaptic elements and of neighboring astrocytic processes (wedges). The ECS width between the synapse and the wedges is 20 nm, 50 nm, 100 nm, 200 nm, respectively. The white dots represent glial glutamate transporters, present at d = 10,800 μm−2. At t = 0, 2000 glutamate molecules were released from a point source 20 nm above the center of the PSD (see Materials and Methods for details). B, Top: The four panels show the simulated astrocytic STCs obtained at the different values of ECS width described in A. The red traces represent simulated STCs obtained in the absence of EAAC1. The green traces represent simulated STCs obtained in the presence of perisynaptic EAAC1 (d = 10,800 μm−2), for different levels of efficiency of glutamate transport (dark green, EAAC1 efficiency = 65%, like that of glial transporters; green, EAAC1 efficiency = 15%; light green, EAAC1 efficiency = 0%). The insets display STC profiles, normalized by that observed in 20 nm ECS width, with no glial or neuronal transporters. Middle: Summary results of the influence on <tSTC (filled circles and continuous lines) and ampSTC (filled squares and dotted lines) of varying the density of expression of EAAC1, its transport efficiency, and the width of ECS around an active synapse. Bottom: Effect of varying EAAC1's transport efficiency on perisynaptic NMDAR activation. C, Effect of glial transporters and EAAC1 (efficiency = 15%) on the glutamate concentration profile monitored in the extracellular volume above the PSD (0 nm), the perisynaptic ring (215 nm), and a 100-nm-wide extrasynaptic region centered 425 nm away from the center of the PSD (425 nm). The concentration of glial transporters ([T]) is set to 10,800 μm−2. The concentration of EAAC1 ([EAAC1]) is set to 0 μm−2 in the black and red traces, whereas it matches the one of glial transporters (10,800 μm−2) in the green traces (both glial and neuronal transporters are present in these simulations). Glial transporters curtail the decay of the glutamate transient within the synapse, and also decrease the peak glutamate concentration extrasynaptically. EAAC1 decreases the peak but, due to its low transport efficiency, prolongs the decay of the glutamate transient in perisynaptic and extrasynaptic regions. D, Summary of the effects of glial transporters (red) and EAAC1 (green) on the peak (solid lines) and the integral (dashed lines) of the glutamate concentration at different distances from the release site. The gray shaded area represents the region in which EAAC1 is expressed. E, Example traces showing the effect of glial and glutamate transporters on the activation of perisynaptic NMDARs. F, Summary of the effects of glial transporters (red) and EAAC1 (green) on the peak open probability of NMDARs at increasing distances from the release site, for different values of ECS width. The dashed line represents the relative effect of EAAC1 compared with what measured in the presence of glial transporters only. EAAC1 limits NMDAR activation particularly in the perisynaptic region. The level to which this effect extends in the extrasynaptic compartment depends on the ECS width.
The enhanced NMDAR activation in the EAAC1 KO was also apparent when monitoring composite AMPAR/NMDAR EPSCs in 0 Mg2+, to permit NMDAR activation at all potentials (Fig. 4E–H). Composite EPSCs decayed more slowly in KO mice at −70 mV (Fig. 4E,F) but not at +50 mV (Fig. 4G,H). The AMPAR and NMDAR components were quantified by measuring the initial slope (∼10–90%) and Q30-80, respectively (Fig. S3A,B, available at www.jneurosci.org as supplemental material) (Materials and Methods). At −70 mV, the NMDAR but not the AMPAR component of composite EPSCs was larger in KO compared with WT mice (NMDAR: KO 1.68 ± 0.18 of WT, *p = 0.016, n = 27; AMPAR: KO 1.05 ± 0.10 of WT, p = 0.77, mean ± SEM; Fig. 4F), whereas at +50 mV no difference between WT and KO mice was observed (NMDAR: KO 0.82 ± 0.04 of WT, p = 0.34, n = 11; AMPAR: KO 0.95 ± 0.07 of WT, p = 0.78, mean ± SEM; Fig. 4H). A number of experimental findings confirmed that these effects on NMDAR EPSCs were not due to changes in release probability in the EAAC1 KO: AMPAR mEPSC frequency was similar in WT and KO slices (Fig. S4A, available at www.jneurosci.org as supplemental material), as were the amplitude, PPR and CV of AMPAR and NMDAR EPSCs (Fig. S4B–F, available at www.jneurosci.org as supplemental material) (Materials and Methods).
These results confirm and extend those previously observed for rats (Diamond, 2001) and suggest that the glutamate transient detected by NMDARs at a distance from active release sites can vary considerably depending on the presence of EAAC1. Together with the STC results (Fig. 1), they indicate that the NMDARs mostly affected by EAAC1 are those located in perisynaptic/extrasynaptic regions, between the sites of expression of EAAC1 and the nearest neighboring glial processes.
If glutamate escaping active synapses reached neighboring quiescent synapses, it would lead to an increase in the apparent quantal content—and, therefore, a reduction in the CV—of NMDAR EPSCs (CVNMDAR) versus AMPAR EPSCs (CVAMPAR) (Kullmann, 1994) (Fig. S3E, available at www.jneurosci.org as supplemental material) given the similar release probabilities at WT and KO synapses (Fig. S4A,D, available at www.jneurosci.org as supplemental material). CVNMDAR was smaller than CVAMPAR (Kullmann, 1994), but neither measure differed between WT and KO mice at positive or negative membrane potentials (Fig. S3C–F, available at www.jneurosci.org as supplemental material), arguing against this possibility. Therefore, unexpectedly, EAAC1 prevents primarily glutamate escape from active synapses and activation of nearby perisynaptic/extrasynaptic NMDARs, rather than receptors at neighboring synaptic contacts (Diamond, 2001).
EAAC1 protects NR2B-NMDARs
CA1 pyramidal cells express NMDARs containing NR2A (NR2A-NMDARs) and/or NR2B subunits (Monyer et al., 1994). In culture, most extrasynaptic NMDARs contain the NR2B subunit (Li et al., 1998; Tovar and Westbrook, 1999), while the NR2A subunit is preferentially targeted to the PSD (Li et al., 1998). The previous findings raise the possibility that EAAC1, by restricting receptor activation at some distance from active release sites, may influence the recruitment of particular NMDAR subtypes that prevail in extrasynaptic regions. To determine the subunit composition of the NMDARs protected by EAAC1, we examined the effects of ifenprodil and Ro-25,6981, antagonists with higher affinity for NR2B-NMDARs than for NR2A-NMDARs (Williams, 1993; Priestley et al., 1995; Kew et al., 1996). At +50 mV [i.e., with neuronal transporters impaired (Wadiche et al., 1995)], ifenprodil (5 μm) reduced NMDAR EPSC amplitude to a similar extent in WT and KO slices (WT: 0.74 ± 0.17 of control, **p = 0.002, n = 9; KO: 0.64 ± 0.27 of control, **p = 0.002, n = 10; WT-KO p = 0.38; Fig. 5A,C). In contrast, at −50 mV, ifenprodil reduced the EPSC amplitude more in KO than in WT mice (WT: 0.95 ± 0.19 of control, p = 0.43, n = 9; KO: 0.71 ± 0.27 of control, **p = 0.007, n = 10; WT-KO *p = 0.040; Fig. 5A,C). Although ifenprodil is not a voltage-dependent drug, its ability to block NR2B-NMDARs does vary with a voltage-dependent phenomenon like Ca2+-dependent inactivation of NMDARs and also with occupancy of the NMDAR glycine-binding site (Legendre and Westbrook, 1991). However, we obtained similar results when applying ifenprodil in the presence of d-Serine (100 μm) and intracellular BAPTA (10 mm) (Fig. S5A, available at www.jneurosci.org as supplemental material), ruling out any bias due to a possible voltage dependence of ifenprodil's effect. Similar results were also obtained when using the more potent NR2B-NMDAR antagonist Ro-25,6981 (0.5 μm, a concentration that in culture blocks NR2B-NMDARs as much as ifenprodil (5 μm) (Fischer et al., 1997) (Fig. S5B, available at www.jneurosci.org as supplemental material), supporting the hypothesis that a larger population of NR2B-NMDARs is recruited during synaptic events when the activity of EAAC1 is impaired. We ruled out presynaptic actions of ifenprodil at Schaffer collateral synapses, because it did not change AMPAR EPSC amplitude or PPR, even when using a 0 Mg2+ external solution and in the presence of MK-801 (1 mm) in the recording pipette (Fig. S5C–F, available at www.jneurosci.org as supplemental material) (Materials and Methods).
Figure 5.

EAAC1 limits activation of NR2B-containing NMDARs. A, NMDAR EPSCs in WT and KO mice in control conditions (black, WT; red, KO) and after application of ifenprodil (5 μm), at ±50 mV. Gray, peak-scaled EPSCs in ifenprodil. B, NMDAR EPSCs at −50 with internal NMDG+, in WT mice. C, Summarized effects of NR2B antagonists on EPSC amplitude at ±50 mV [ifenprodil WT+50 mV **p = 0.002, WT−50 mV p = 0.43, n = 9 (white circle), KO+50 mV **p = 0.002, KO−50 mV **p = 0.007, n = 10 (red circle), WT-KO+50 mV p = 0.38, WT-KO−50 mV *p = 0.040)] The hollow white square represents the mean reduction of EPSC amplitude in ifenprodil at −50 mV in WT mice, with internal NMDG+ (ifenprodil+NMDG+ WT−50 mV **p = 0.003, n = 7). D, NMDAR EPSCs in WT and KO mice in ifenprodil, in control conditions (black, WT; red, KO) and after D-AA (100 μm, green). Gray: peak-scaled EPSCs. E, Summary of D-AA effects on EPSC t50 in ifenprodil at ±50 mV, in WT (white circle) and KO mice (red circle) (WT D-AA+50 mV p = 0.24, WT D-AA−50 mV **p = 0.006, n = 8; KO D-AA+50 mV p = 0.37, KO D-AA−50 mV *p = 0.043, n = 5, WT-KO+50 mV p = 0.13, WT-KO−50 mV p = 0.13).
If the reduced NR2B-NMDAR activation in WT mice were due to EAAC1, blocking neuronal transporters in the recorded neuron should rescue an effect of ifenprodil on NMDAR EPSCs at −50 mV. Even without selective EAAC1 antagonists, this can be achieved in individual cells by replacing intracellular Cs+ with NMDG+, a larger cation that does not support countertransport and effectively locks transporters in the inward-facing configuration (Diamond, 2005). Under these conditions, ifenprodil reduced NMDAR EPSC amplitude at −50 mV in WT (0.77 ± 0.13 of control, **p = 0.003, n = 7; Fig. 5B,C) to a similar extent as in KO neurons filled with Cs+-based solution (Fig. 5C) (WTNMDG+-KOCs+ p = 0.61). Although NR2B-NMDARs exhibit slower kinetics than NR2A-NMDARs (Flint et al., 1997; Chen et al., 1999), neither ifenprodil nor Ro-25,6981 sped the NMDAR EPSC decay (Fig. 5A,B), likely because of the small NR2B-NMDAR component in these EPSCs: the reduction of EPSC decay by NR2B antagonists correlated with the reduction of EPSC amplitude, an effect that was not observed with CPP (Fig. S5G,H, available at www.jneurosci.org as supplemental material).
These results indicate that EAAC1 protects primarily NR2B-NMDARs. If these receptors are mainly located outside the synaptic cleft, ifenprodil should occlude the effects of low-affinity antagonists that we observed (Fig. 4A–D). Consistent with this prediction, when NR2B-NMDARs were blocked by ifenprodil, D-AA no longer sped the NMDAR EPSC decay at either potential in WT or KO mice (Fig. 5D,E). Taken together, the results suggest that EAAC1 prevents glutamate spillover from active synapses onto perisynaptic/extrasynaptic NR2B-NMDARs.
EAAC1 regulates long-term plasticity of Schaffer collateral synapses
If the extent of NMDAR activation dictates the direction of changes in synaptic strength at Schaffer collateral synapses (Cummings et al., 1996), how might EAAC1, by limiting perisynaptic/extrasynaptic NR2B-NMDAR activation, influence long-term synaptic plasticity? To address this question, we delivered different stimulus patterns to Schaffer collaterals and recorded fEPSCs with an extracellular electrode in stratum radiatum (Materials and Methods). Two stimulating electrodes were placed on opposite sides of the field electrode (along the axis of the Schaffer collaterals) to provide independent test and control pathways (established through a crossed paired-pulse protocol; Materials and Methods). TBS of the test pathway induced an input-specific increase in fEPSC slope in WT slices (1.52 ± 0.11 of baseline, n = 9, **p = 0.0013; mean ± SEM. Fig. 6A,B). In contrast, TBS induced no significant potentiation in KO slices (1.13 ± 0.07 of baseline, n = 7; p = 0.12, mean ± SEM; WT-KO Test *p = 0.013 Fig. 6C,D). This difference was caused by additional recruitment of NR2B-NMDARs in the KO, as TBS-induced LTP was rescued in the KO by the NR2B antagonist ifenprodil (1.41 ± 0.09 of baseline, n = 6, **p = 0.0058, mean ± SEM; KO Test-KO Test ifenprodil *p = 0.029; WT Test-KO Test ifenprodil p = 0.50; Fig. 6E,F; Fig. S6, available at www.jneurosci.org as supplemental material). TBS-induced LTP was not eliminated in WT mice by partially blocking GLT-1 with a low concentration of DHK (15 μm; Fig. 6G,H) [the IC50 of DHK for GLT-1 is 23 μm (Arriza et al., 1994)] (WT Test DHK 1.25 ± 0.08 of baseline, n = 13, *p = 0.016, mean ± SEM). In the presence of DHK, TBS-LTP was not significantly less than that observed in WT mice in control conditions (WT Test-WT Test DHK p = 0.059). This pharmacological manipulation is likely to enhance NMDAR activation throughout the neuropil and therefore not only in the perisynaptic/extrasynaptic regions as it happens when altering the activity of EAAC1, but also at neighboring, quiescent synapses.
Figure 6.
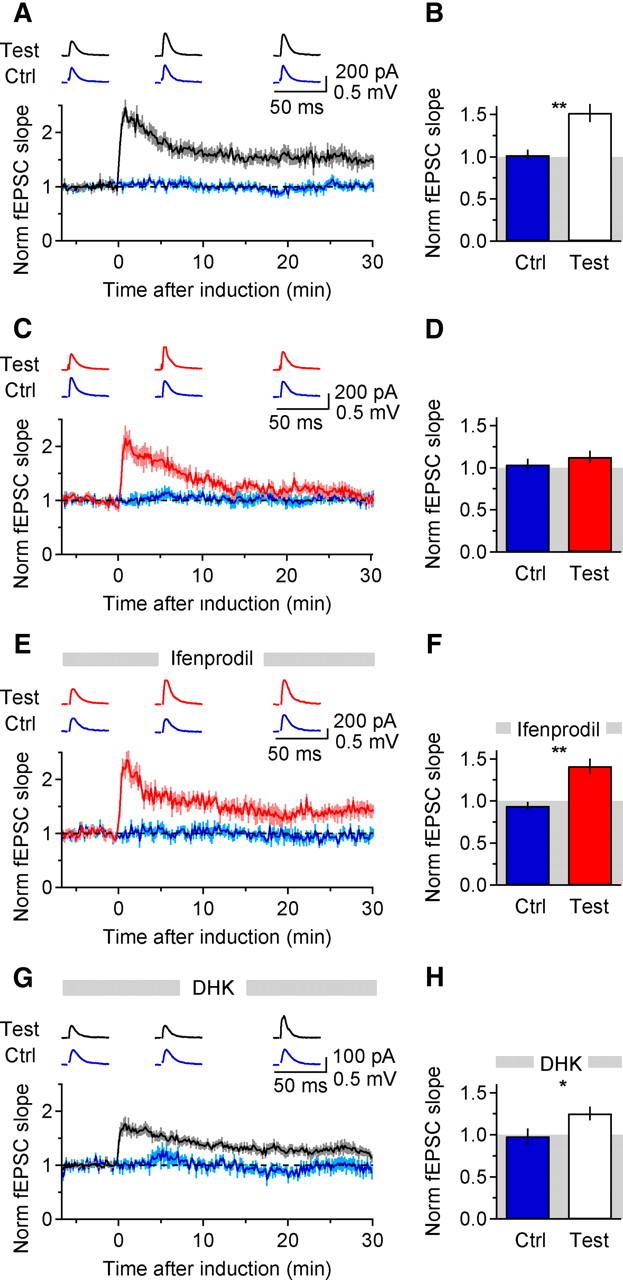
TBS-induced LTP is impaired in KO mice, but is rescued by ifenprodil. A, Time course of baseline-normalized fEPSC slope in WT slices (n = 9). Traces at the top are average responses in test and control pathways 5–0 min before, and 5–10 min and 25–30 min after TBS. The induction protocol consisted of 5 bursts at 5 Hz (100 Hz and 0.06 s each). B, Summarized effect of TBS on fEPSC slope in control and test pathways (WT Ctrl-Test **p = 0.001). C, Same as in A, in KO mice (n = 7). D, As in B, in KO mice (KO Ctrl-Test p = 0.45; WT Test-KO Test *p = 0.013). E, As in A, in KO mice and in the presence of ifenprodil (5 μm; n = 6). F, As in B, in KO mice and in the presence of ifenprodil (KO Ctrl-Test **p = 0.003; KO Test-KO Test ifenprodil *p = 0.029; WT Test-KO Test ifenprodil p = 0.50). All data in this figure represent mean ± SEM. G, Time course of baseline-normalized fEPSC slope in WT slices (n = 13) in the presence of DHK (15 μm). H, Summarized effect of TBS on fEPSC slope in control and test pathways (WT DHK Ctrl-Test *p = 0.016).
Is this difference in TBS-induced LTP simply due to a change in the absolute capacity of CA1 synapses for synaptic plasticity? LTD, elicited by LFS (1 Hz, 15 min) was similar in WT and KO mice (Fig. 7A,C). In both groups, LTD was insensitive to ifenprodil (Fig. 7B,D) (Morishita et al., 2007) (but see Liu et al., 2004; Massey et al., 2004). Maximal LTP, induced by three successive bursts of tetanic stimulation (T: 100 Hz, 1 s) was also comparable in WT and KO mice (Fig. 7E,F), as was depotentiation induced by LFS (1 Hz, 10 min) following maximal LTP (Fig. 7E,F). Therefore, deletion of EAAC1 does not affect the maximal range of synaptic plasticity at Schaffer collateral synapses.
Figure 7.
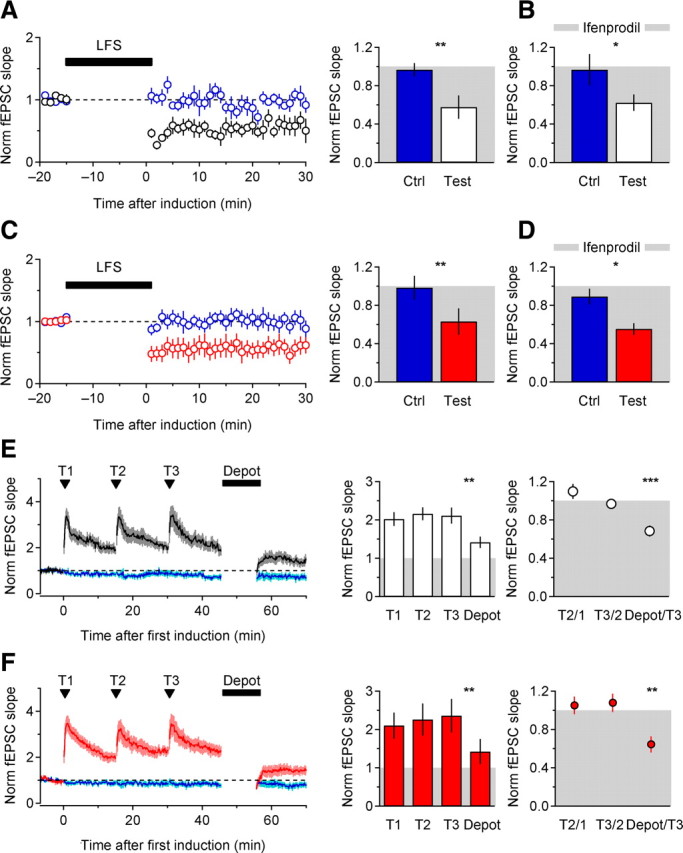
LTD, maximal LTP, and depotentiation are not affected by EAAC1. A, Left: Time course of baseline-normalized fEPSC slope before and after a LFS protocol (1 Hz, 15 min) in WT mice (n = 6). Right: summarized effects (WT Ctrl-Test **p = 0.002). B, Summary of the effect of ifenprodil (5 μm) on LFS-induced LTD, in WT slices (n = 8, WT Ctrl ifenprodil-Test ifenprodil *p = 0.048; WT Test-WT Test ifenprodil p = 0.75). C, As in A in KO mice (n = 7, KO Ctrl-Test **p = 0.006; WT Test-KO Test p = 0.77). D, As in B, in KO mice (n = 8, KO Ctrl ifenprodil-Test ifenprodil *p = 0.02, KO Test-KO Test ifenprodil p = 0.60; WT Test ifenprodil-KO Test ifenprodil p = 0.53). E, Left: Time course of baseline-normalized fEPSC slope following three tetani (100 Hz, 1 s) applied every 15 min and one LFS depotentiating protocol (1 Hz, 10 min) in WT mice (n = 8). Center: Summarized effect of tetanic stimulations and subsequent LFS on fEPSC slope, in WT mice (WT T3-Depot **p = 0.003). Right: fEPSC slope expressed relative to a 5 min baseline beginning 10 min after each tetanic stimulation (WT Depot/T3 ***p = 0.0008). F, As in E, in KO mice (KO T3-Depot **p = 0.007; KO Depot/T3 **p = 0.002). All data are displayed as mean ± SEM.
In KO mice, however, TBS could activate a subset of the perisynaptic/extrasynaptic NR2B-NMDARs activated with tetanic stimulations. This could result in a depressing signal arising in the perisynaptic/extrasynaptic region, counteracting the potentiation induced by the more abundant synaptic NMDARs. This requires TBS to be able to elicit both LTP and LTD and the latter form of plasticity to be more prominent in KO than in WT mice. We hypothesized that, if present, the TBS-induced depression would be revealed after occluding potentiation. Therefore, we monitored the effects of TBS in WT and KO mice after inducing maximal LTP with three tetanic stimulations (Fig. 8A,B). After LTP was maximized, TBS induced no further change in synaptic strength in WT slices (0.95 ± 0.08 of pre-TBS baseline, n = 13 p = 0.23, mean ± SEM; Fig. 8A), but consistently induced a small depression in KO mice (0.77 ± 0.08 of pre-TBS baseline, n = 11, *p = 0.038, mean ± SEM; Fig. 8B). This effect required NR2B-NMDARs because it was no longer observed in KO mice if TBS was delivered in the presence of ifenprodil (0.93 ± 0.03 of pre-TBS baseline, n = 17, p = 0.079, mean ± SEM; Fig. 8C). Therefore, the TBS-induced depression is enhanced in KO mice, and is mediated by NR2B-NMDARs. A similar rationale was adopted to isolate the TBS-induced potentiation: we maximized LTD with two rounds of LFS and then applied TBS. The potentiation induced by TBS was similar in WT and KO mice (WT: 1.73 ± 0.16 times of pre-TBS baseline, n = 14, **p = 0.002, mean ± SEM; KO: 1.51 ± 0.11 times of pre-TBS baseline, n = 12, **p = 0.003, mean ± SEM; TBS/LFS2 WT-KO p = 0.30; Fig. 8D,E), suggesting that instead the TBS-induced potentiation component is similar in WT and KO mice.
Figure 8.
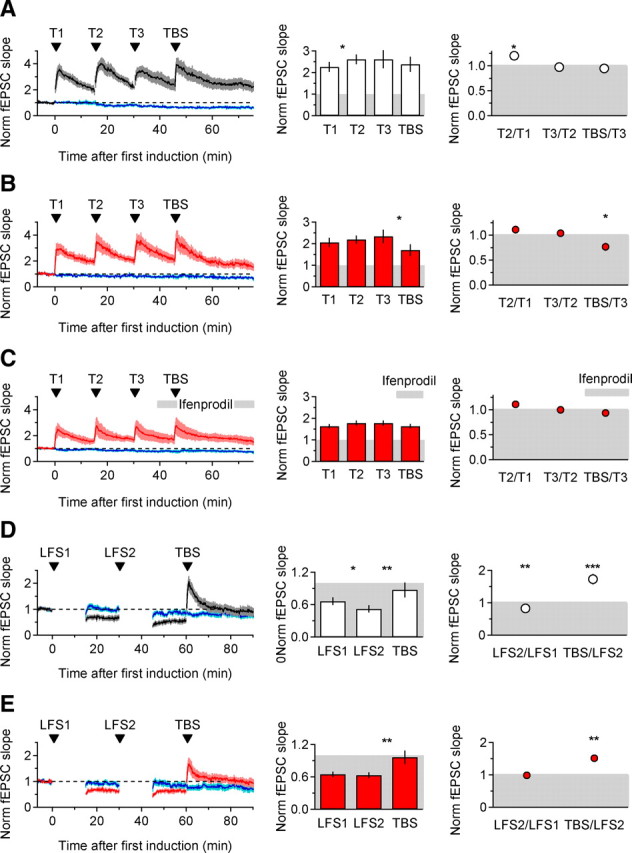
EAAC1 tips the balance between TBS-induced potentiation and depression. A, Average time course of baseline-normalized fEPSC slope following three tetanic stimulations (100 Hz, 1 s) and one TBS (5 bursts at 5 Hz, 100 Hz and 0.06 s each) in WT mice (n = 13; left). The bar chart summarizes the average magnitude of fEPSC slope, normalized to the initial baseline value, 10–15 min after each tetanus and 25–30 min after TBS (center: WT T1–T2 *p = 0.02, WT T3-TBS p = 0.23). Data in the right panel represent the fEPSC slope measured 10–15 min after T2, T3, and TBS relative to a baseline fEPSC slope measured 10–15 min after T1, T2, and T3, respectively (WT T2/T1 *p = 0.02; WT TBS/T3 p = 0.50). B, As in A, in KO mice (n = 11; KO T3-TBS *p = 0.038; KO TBS/T3 *p = 0.017). C, As in B, but ifenprodil was applied 8.3 min after T3 (n = 17; KO T3-TBS ifenprodil p = 0.058; KO TBS/T3 ifenprodil p = 0.079). In KO mice, maximal LTP was not significantly different in the absence (B) and presence (C) of ifenprodil (KO T1–T1 ifenprodil p = 0.051; KO T2–T2 ifenprodil p = 0.057; KO T3–T3 ifenprodil p = 0.062). D, Average time course of baseline-normalized fEPSC slope following two LFS stimulations (1 Hz, 15 min) and one TBS (5 bursts at 5 Hz, 100 Hz and 0.06 s each), in WT mice (n = 14; left). The bar chart summarizes the extent of change in fEPSC slope at the end of each test period (center: WT LFS1-LFS2 *p = 0.02, WT LFS2-TBS **p = 0.003). The plot on the right shows changes in fEPSC slope in respect to baseline measured before each stimulation protocol (WT LFS2/LFS1 **p = 0.002, WT TBS/LFS2 ***p = 0.0006). E, As in D, in KO mice (n = 12; center: KO LFS1-LFS2 p = 0.59, KO LFS2-TBS **p = 0.003; right: KO LFS2/LFS1 p = 0.80, KO TBS/LFS2 ***p = 0.0008). All data are displayed as mean ± SEM.
Based on these results we conclude that, by limiting activation of perisynaptic/extrasynaptic NR2B-NMDARs, EAAC1 shifts the balance between TBS-induced potentiation and depression, thereby tuning the plasticity of Schaffer collateral synapses.
Discussion
The neuronal transporter EAAC1 has been the subject of intense biochemical and biophysical investigations (Zerangue and Kavanaugh, 1996; Grewer et al., 2000; Watzke et al., 2001), but understanding its functional role in synaptic transmission has been hampered by insufficiently selective EAAC1 antagonists and lack of detectable currents associated with EAAC1 transport in hippocampal neurons (Bergles and Jahr, 1998) (our unpublished observation). Although a quantitative measure of EAAC1 expression in CA1 has not been reported, it is likely to be significantly lower than that of glial transporters (Danbolt, 2001), leading to the suggestion that EAAC1's primary function may be to take up cysteine and contribute to glutathione synthesis and oxidant metabolisms (Aoyama et al., 2006). Here we present several lines of evidence that strongly support novel and unsuspected physiological roles for EAAC1 in CA1 pyramidal cells: EAAC1 buffers glutamate released from active synapses, delaying its arrival at glial membranes (Fig. 1) and limiting the activation of perisynaptic/extrasynaptic NR2B-NMDARs (Fig. 5), thereby altering TBS-induced synaptic plasticity (Fig. 6).
Deletion of EAAC1 alters the time course of glial glutamate uptake without significantly diminishing the total amount of glutamate taken up by astrocytes (Fig. 1), suggesting that EAAC1 acts primarily as a glutamate buffer, rapidly binding glutamate and then releasing it back in the ECS. EAAC1's efficacy as a buffer depends on the density of its expression in the perisynaptic membrane and its transport efficiency (i.e., the probability that EAAC1 transports bound glutamate instead of releasing it back in the ECS). Both of these parameters are poorly constrained by experimental data (Grewer et al., 2000; Danbolt, 2001). A buffer that prolongs the time course of free glutamate in the perisynaptic space would not enhance NMDAR activation, as suggested by (Barbour, 2001). In fact, this scenario would be inconsistent with our experimental observations showing that EAAC1 reduces NMDAR activation (Fig. 4). This is presumably because glutamate unbinding from EAAC1 is more likely to be bound by glial glutamate transporters, expressed at high densities around the synapse (Danbolt, 2001), than by nearby NMDARs. To explore how different characteristics of EAAC1 (density of expression and transport efficiency) and parameters of the ECS (i.e., ECS width) might influence glutamate diffusion and NMDAR activation, we constructed a three-dimensional Monte Carlo diffusion model of the synaptic and extrasynaptic environment (Fig. 9A) (Materials and Methods). Initially, individual neuronal and glial transporters were represented by the same Markov model, developed to reflect the kinetic properties of GLT-1 (Bergles et al., 2002). Under these conditions, EAAC1 exhibited the same, relatively high (∼65%) transport efficiency as glial transporters: it reduced NMDAR activation and STC amplitude, but did not alter the STC time course (Fig. 9B; dark green sets). This suggests that EAAC1's effects cannot simply be attributed to its perisynaptic location. Increasing proportionately the extracellular glutamate binding and unbinding rates decreased EAAC1 efficiency without affecting equilibrium affinity for transmitter, the recycling rate, or the apparent EC50 in response to brief and long pulses of glutamate (data not shown). Consistent with our prediction, the less efficient EAAC1 not only reduced NMDAR recruitment, but also slowed the time course of the simulated STC (Fig. 9B). This effect varied in magnitude depending on EAAC1 efficiency, density of EAAC1 expression and the width of the ECS around the synapse (i.e., it was more pronounced in a narrow ECS, i.e., when there is less neurotransmitter dilution). Reducing the efficiency of EAAC1 led to a decreased peak and slower decay of the glutamate concentration in the perisynaptic region, with the overall effect of reducing activation of perisynaptic/extrasynaptic NMDARs (Fig. 9C,D). Notably, perisynaptic EAAC1 was much more effective than glial transporters at regulating the glutamate concentration and NMDAR activation in a spatially confined region that extended only a few hundred nanometers beyond the PSD (Fig. 9E,F). In agreement with our experimental results (Fig. S2, available at www.jneurosci.org as supplemental material) and previous reports (Barbour, 2001; Zheng et al., 2008), the model also indicates that NMDAR activation within the PSD is only marginally affected by either glial or neuronal glutamate transporters (Fig. 9F), as they all encounter glutamate after it diffuses away from the PSD. Lowering the efficiency of glial transporters also reduced NMDAR activation, but did not slow the STC time course (Fig. S9A, available at www.jneurosci.org as supplemental material), pointing to a possible unique role of EAAC1 in regulating the diffusion of synaptically released glutamate.
Does EAAC1 limit glutamate spillover onto nearby, quiescent synapses? The lack of change in CVNMDAR versus CVAMPAR at positive and negative membrane potentials in WT and KO mice (Fig. S4, available at www.jneurosci.org as supplemental material) and our model suggests that this is an unlikely scenario. The lack of differences in the decay of composite EPSCs and in the sensitivity of NMDAR EPSCs to low-affinity antagonists at +50 mV (Fig. 4) also indicate that heterocellular glutamate spillover is not significantly affected by EAAC1. Although this finding may seem at odds with the fact that the estimate for average glutamate diffusion distance in juvenile rat hippocampus (1.7 μm) (Diamond, 2005) exceeds the average distance among neighboring excitatory synapses (∼465 nm) (Rusakov and Kullmann, 1998), the peak glutamate concentration decreases steeply with distance and may be too low to recruit a significant portion of NMDARs at neighboring synapses (Barbour, 2001). Instead, EAAC1 influences primarily activation of NMDARs in the perisynaptic/extrasynaptic region of active synapses, between the PSD and the surrounding astrocytic processes. The preferential recruitment of NR2B-NMDARs in KO mice (Fig. 5; Fig. S5A,B, available at www.jneurosci.org as supplemental material) is consistent with their larger abundance at extrasynaptic sites (Tovar and Westbrook, 1999), higher affinity for glutamate (Priestley et al., 1995) and proposed role as the primary detectors of glutamate spillover (Lozovaya et al., 2004; Scimemi et al., 2004). Therefore, by varying the activity of EAAC1, one can alter specifically the extent of NR2B-NMDAR activation in a spatially restricted subcellular region, and analyze the contribution of this particular group of NMDARs to synaptic function.
The extent of NMDAR recruitment determines the direction of synaptic plasticity at Schaffer collateral synapses, as varying amounts of Ca2+-influx through these receptors can trigger intracellular signaling cascades leading to LTP or LTD (Cummings et al., 1996). Our results suggest that altering the extent of perisynaptic/extrasynaptic NMDAR activation can influence the direction of changes in synaptic strength. Accordingly, in the EAAC1 KO, activation of perisynaptic/extrasynaptic NR2B-NMDARs during TBS triggers a depressing signal that counteracts the induced potentiation (Fig. 6). These opposing signals are likely to originate from the cleft and perisynaptic/extrasynaptic regions of individual synapses, because deletion of EAAC1 does not appear to change the number of synapses activated (see CV analysis above) and impairing glutamate transport via GLT-1 does not eliminate TBS-LTP in WT. These results differ from those obtained in GLT-1 KO mice (Katagiri et al., 2001), but a direct comparison between our results (where we partially block GLT-1) and those of (Katagiri et al., 2001) (which uses slices from GLT-1 KO mice) is hard to make, primarily because in our hands complete block of GLT-1 with DHK (300 m) severely compromised the slice viability (data not shown). The fact that the depressing, NR2B-dependent signal induced by TBS in the KO is occluded by LFS-LTD (Fig. 8D,E), which does not require NR2B activation (Morishita et al., 2007) (Fig. 5B,D), also indicates that the two components of depression share common intracellular signaling pathways, despite being activated by spatially distinct NMDARs.
What insights can we gain from these findings on any presumed subunit-specificity of LTP/LTD? Recent reports argue for (Liu et al., 2004; Massey et al., 2004; von Engelhardt et al., 2008) and against (Berberich et al., 2005; Morishita et al., 2007) a specific role for NR2B-NMDARs in LTP and LTD. Reconciling these opposing findings with our results is complicated by the necessary considerations of animal species and age, as well as experimental conditions (particularly temperature, which strongly affects receptor and transporter kinetics and, to a lesser extent, neurotransmitter diffusion) and varied induction protocols. Our findings are inconsistent with a strict requirement for NR2B-NMDARs in LTP/LTD because, in our hands, the NR2B-selective antagonist ifenprodil impairs the induction of neither type of plasticity (Fig. 5; Fig. S9, available at www.jneurosci.org as supplemental material). One possibility is that NMDAR location and relative extent of activation, rather than specific subunit composition, may dictate the direction of plasticity. For example, LFS may elicit moderate calcium influx through NMDARs in the PSD, favoring depression, while tetanic stimulation may elicit relatively strong activation of both synaptic and extrasynaptic receptors, favoring potentiation. TBS may robustly activate synaptic receptors but, depending on the actions of EAAC1, sparsely activate only a few perisynaptic/extrasynaptic NMDARs, thereby triggering potentiating and depressing signals that counteract one another (Fig. 4). The ifenprodil-sensitivity of this effect may reflect the relative prevalence of NR2B-NMDARs in extrasynaptic regions (Tovar and Westbrook, 1999), but NR2B-NMDARs do not need to possess an intrinsic capacity to induce LTD. These effects may be more easily detected with moderate induction protocols (von Engelhardt et al., 2008), like TBS, than with more extreme protocols, like LFS and tetanic stimuli, that induce near-maximal LTD and LTP, respectively (Fig. 6).
What specific role, then, might EAAC1 play in this system? As mentioned above, our diffusion simulations suggest that EAAC1 may affect perisynaptic NMDAR activation more so than glial transporters (Fig. 9C–F), due to their more targeted localization and potentially reduced efficiency; moreover, modulation of EAAC1 (e.g., by postsynaptic membrane depolarization) could regulate perisynaptic NMDAR activation without significantly compromising the overall uptake capacity of the tissue.
In summary, the data presented here indicate that EAAC1 is capable of altering the dynamics of glutamate in the ECS and tunes the propensity of CA1 excitatory synapses to potentiate or depress by regulating the activation of NR2B-NMDARs in regions surrounding active synapses. Neuronal glutamate transporters may therefore be unsuspected crucial elements in the elaborate cellular basis of memory formation and seizure susceptibility in the hippocampus (Rothstein et al., 1996).
Footnotes
This work was supported by the National Institute of Neurological Disorders and Stroke (NINDS) Intramural Research Program and Human Frontier Science Program (RGP50/2006). We thank N. C. Danbolt for the EAAC1 antibody, the NINDS EM facility for technical assistance, and D. M. Kullmann and C. J. McBain for comments on this manuscript. Thanks to M. Dittrich for help with the MCell software and M. Beato, K. Paradiso, and A. V. Tzingounis for valuable discussions. A.S. performed electrophysiological recordings, EM analysis, 2P-LSM imaging, diffusion measurements, and modeling. H.T. performed molecular biology experiments. A.S. and J.S.D. designed the research and wrote this manuscript.
References
- Aoyama K, Suh SW, Hamby AM, Liu J, Chan WY, Chen Y, Swanson RA. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat Neurosci. 2006;9:119–126. doi: 10.1038/nn1609. [DOI] [PubMed] [Google Scholar]
- Arnth-Jensen N, Jabaudon D, Scanziani M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci. 2002;5:325–331. doi: 10.1038/nn825. [DOI] [PubMed] [Google Scholar]
- Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci. 1994;14:5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asztely F, Erdemli G, Kullmann DM. Extrasynaptic glutamate spillover in the hippocampus: dependence on temperature and the role of active glutamate uptake. Neuron. 1997;18:281–293. doi: 10.1016/s0896-6273(00)80268-8. [DOI] [PubMed] [Google Scholar]
- Barbour B. An evaluation of synapse independence. J Neurosci. 2001;21:7969–7984. doi: 10.1523/JNEUROSCI.21-20-07969.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berberich S, Punnakkal P, Jensen V, Pawlak V, Seeburg PH, Hvalby Ø, Köhr G. Lack of NMDA receptor subtype selectivity for hippocampal long-term potentiation. J Neurosci. 2005;25:6907–6910. doi: 10.1523/JNEUROSCI.1905-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE. Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron. 1997;19:1297–1308. doi: 10.1016/s0896-6273(00)80420-1. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE. Glial contribution to glutamate uptake at Schaffer collateral-commissural synapses in the hippocampus. J Neurosci. 1998;18:7709–7716. doi: 10.1523/JNEUROSCI.18-19-07709.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Tzingounis AV, Jahr CE. Comparison of coupled and uncoupled currents during glutamate uptake by GLT-1 transporters. J Neurosci. 2002;22:10153–10162. doi: 10.1523/JNEUROSCI.22-23-10153.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Luo T, Raymond LA. Subtype-dependence of NMDA receptor channel open probability. J Neurosci. 1999;19:6844–6854. doi: 10.1523/JNEUROSCI.19-16-06844.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie JM, Jahr CE. Multivesicular release at Schaffer collateral-CA1 hippocampal synapses. J Neurosci. 2006;26:210–216. doi: 10.1523/JNEUROSCI.4307-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, Bekkers JM. Detection of spontaneous synaptic events with an optimally scaled template. Biophys J. 1997;73:220–229. doi: 10.1016/S0006-3495(97)78062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings JA, Mulkey RM, Nicoll RA, Malenka RC. Ca2+ signaling requirements for long-term depression in the hippocampus. Neuron. 1996;16:825–833. doi: 10.1016/s0896-6273(00)80102-6. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Diamond JS. Neuronal glutamate transporters limit activation of NMDA receptors by neurotransmitter spillover on CA1 pyramidal cells. J Neurosci. 2001;21:8328–8338. doi: 10.1523/JNEUROSCI.21-21-08328.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS. Deriving the glutamate clearance time course from transporter currents in CA1 hippocampal astrocytes: transmitter uptake gets faster during development. J Neurosci. 2005;25:2906–2916. doi: 10.1523/JNEUROSCI.5125-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE. Transporters buffer synaptically released glutamate on a submillisecond time scale. J Neurosci. 1997;17:4672–4687. doi: 10.1523/JNEUROSCI.17-12-04672.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE. Synaptically released glutamate does not overwhelm transporters on hippocampal astrocytes during high-frequency stimulation. J Neurophysiol. 2000;83:2835–2843. doi: 10.1152/jn.2000.83.5.2835. [DOI] [PubMed] [Google Scholar]
- Diamond JS, Bergles DE, Jahr CE. Glutamate release monitored with astrocyte transporter currents during LTP. Neuron. 1998;21:425–433. doi: 10.1016/s0896-6273(00)80551-6. [DOI] [PubMed] [Google Scholar]
- Fischer G, Mutel V, Trube G, Malherbe P, Kew JN, Mohacsi E, Heitz MP, Kemp JA. Ro 25–6981, a highly potent and selective blocker of N-methyl-d-aspartate receptors containing the NR2B subunit. Characterization in vitro. J Pharmacol Exp Ther. 1997;283:1285–1292. [PubMed] [Google Scholar]
- Flint AC, Maisch US, Weishaupt JH, Kriegstein AR, Monyer H. NR2A subunit expression shortens NMDA receptor synaptic currents in developing neocortex. J Neurosci. 1997;17:2469–2476. doi: 10.1523/JNEUROSCI.17-07-02469.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewer C, Watzke N, Wiessner M, Rauen T. Glutamate translocation of the neuronal glutamate transporter EAAC1 occurs within milliseconds. Proc Natl Acad Sci U S A. 2000;97:9706–9711. doi: 10.1073/pnas.160170397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Janssen WG, Rothstein JD, Morrison JH. Differential synaptic localization of the glutamate transporter EAAC1 and glutamate receptor subunit GluR2 in the rat hippocampus. J Comp Neurol. 2000;418:255–269. [PubMed] [Google Scholar]
- Katagiri H, Tanaka K, Manabe T. Requirement of appropriate glutamate concentrations in the synaptic cleft for hippocampal LTP induction. Eur J Neurosci. 2001;14:547–553. doi: 10.1046/j.0953-816x.2001.01664.x. [DOI] [PubMed] [Google Scholar]
- Kew JN, Trube G, Kemp JA. A novel mechanism of activity-dependent NMDA receptor antagonism describes the effect of ifenprodil in rat cultured cortical neurones. J Physiol. 1996;497:761–772. doi: 10.1113/jphysiol.1996.sp021807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM. Amplitude fluctuations of dual-component EPSCs in hippocampal pyramidal cells: implications for long-term potentiation. Neuron. 1994;12:1111–1120. doi: 10.1016/0896-6273(94)90318-2. [DOI] [PubMed] [Google Scholar]
- Legendre P, Westbrook GL. Ifenprodil blocks N-methyl-d-aspartate receptors by a two-component mechanism. Mol Pharmacol. 1991;40:289–298. [PubMed] [Google Scholar]
- Lester RA, Jahr CE. NMDA channel behavior depends on agonist affinity. J Neurosci. 1992;12:635–643. doi: 10.1523/JNEUROSCI.12-02-00635.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JH, Wang YH, Wolfe BB, Krueger KE, Corsi L, Stocca G, Vicini S. Developmental changes in localization of NMDA receptor subunits in primary cultures of cortical neurons. Eur J Neurosci. 1998;10:1704–1715. doi: 10.1046/j.1460-9568.1998.00169.x. [DOI] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Lozovaya NA, Grebenyuk SE, Tsintsadze TSh, Feng B, Monaghan DT, Krishtal OA. Extrasynaptic NR2B and NR2D subunits of NMDA receptors shape ‘superslow’ afterburst EPSC in rat hippocampus. J Physiol. 2004;558:451–463. doi: 10.1113/jphysiol.2004.063792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir ZI. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004;24:7821–7828. doi: 10.1523/JNEUROSCI.1697-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Morishita W, Lu W, Smith GB, Nicoll RA, Bear MF, Malenka RC. Activation of NR2B-containing NMDA receptors is not required for NMDA receptor-dependent long-term depression. Neuropharmacology. 2007;52:71–76. doi: 10.1016/j.neuropharm.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Nielsen TA, DiGregorio DA, Silver RA. Modulation of glutamate mobility reveals the mechanism underlying slow-rising AMPAR EPSCs and the diffusion coefficient in the synaptic cleft. Neuron. 2004;42:757–771. doi: 10.1016/j.neuron.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Peghini P, Janzen J, Stoffel W. Glutamate transporter EAAC-1-deficient mice develop dicarboxylic aminoaciduria and behavioral abnormalities but no neurodegeneration. EMBO J. 1997;16:3822–3832. doi: 10.1093/emboj/16.13.3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priestley T, Laughton P, Myers J, Le Bourdellés B, Kerby J, Whiting PJ. Pharmacological properties of recombinant human N-methyl-d-aspartate receptors comprising NR1a/NR2A and NR1a/NR2B subunit assemblies expressed in permanently transfected mouse fibroblast cells. Mol Pharmacol. 1995;48:841–848. [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Rusakov DA, Kullmann DM. Extrasynaptic glutamate diffusion in the hippocampus: ultrastructural constraints, uptake, and receptor activation. J Neurosci. 1998;18:3158–3170. doi: 10.1523/JNEUROSCI.18-09-03158.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savtchenko LP, Rusakov DA. Extracellular diffusivity determines contribution of high-versus low-affinity receptors to neural signaling. Neuroimage. 2005;25:101–111. doi: 10.1016/j.neuroimage.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Salin PA, Vogt KE, Malenka RC, Nicoll RA. Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature. 1997;385:630–634. doi: 10.1038/385630a0. [DOI] [PubMed] [Google Scholar]
- Scimemi A, Fine A, Kullmann DM, Rusakov DA. NR2B-containing receptors mediate cross talk among hippocampal synapses. J Neurosci. 2004;24:4767–4777. doi: 10.1523/JNEUROSCI.0364-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takumi Y, Matsubara A, Rinvik E, Ottersen OP. The arrangement of glutamate receptors in excitatory synapses. Ann N Y Acad Sci. 1999;868:474–482. doi: 10.1111/j.1749-6632.1999.tb11316.x. [DOI] [PubMed] [Google Scholar]
- Thorne RG, Nicholson C. In vivo diffusion analysis with quantum dots and dextrans predicts the width of brain extracellular space. Proc Natl Acad Sci U S A. 2006;103:5567–5572. doi: 10.1073/pnas.0509425103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Engelhardt J, Doganci B, Jensen V, Hvalby Ø, Göngrich C, Taylor A, Barkus C, Sanderson DJ, Rawlins JN, Seeburg PH, Bannerman DM, Monyer H. Contribution of hippocampal and extra-hippocampal NR2B-containing NMDA receptors to performance on spatial learning tasks. Neuron. 2008;60:846–860. doi: 10.1016/j.neuron.2008.09.039. [DOI] [PubMed] [Google Scholar]
- Wadiche JI, Arriza JL, Amara SG, Kavanaugh MP. Kinetics of a human glutamate transporter. Neuron. 1995;14:1019–1027. doi: 10.1016/0896-6273(95)90340-2. [DOI] [PubMed] [Google Scholar]
- Watzke N, Bamberg E, Grewer C. Early intermediates in the transport cycle of the neuronal excitatory amino acid carrier EAAC1. J Gen Physiol. 2001;117:547–562. doi: 10.1085/jgp.117.6.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K. Ifenprodil discriminates subtypes of the N-methyl-d-aspartate receptor: selectivity and mechanisms at recombinant heteromeric receptors. Mol Pharmacol. 1993;44:851–859. [PubMed] [Google Scholar]
- Zerangue N, Kavanaugh MP. Flux coupling in a neuronal glutamate transporter. Nature. 1996;383:634–637. doi: 10.1038/383634a0. [DOI] [PubMed] [Google Scholar]
- Zheng K, Scimemi A, Rusakov DA. Receptor actions of synaptically released glutamate: the role of transporters on the scale from nanometers to microns. Biophys J. 2008;95:4584–4596. doi: 10.1529/biophysj.108.129874. [DOI] [PMC free article] [PubMed] [Google Scholar]