

Abstract
Rationale
Atherosclerotic lesions express matrix metalloproteinase-8 (MMP8) which possesses proteolytic activity on matrix proteins particularly fibrillar collagens and on non-matrix proteins such as angiotensin I (Ang I).
Objective
We studied whether MMP8 plays a role in atherogenesis.
Methods and Results
In atherosclerosis-prone apoE deficient mice, inactivating MMP8 resulted in a substantial reduction in atherosclerotic lesion formation. Immunohistochemical examinations showed that atherosclerotic lesions in MMP8 deficient mice had significantly fewer macrophages but increased collagen content. In line with results of in vitro assays showing Ang I cleavage by MMP8 generating angiotensin II (Ang II), MMP8 knockout mice had lower Ang II levels and lower blood pressure. In addition, we found that products of Ang I cleavage by MMP8 increased vascular cell adhesion molecule-1 (VCAM-1) expression and that MMP8 deficient mice had reduced VCAM-1 expression in atherosclerotic lesions. Intravital microscopy analysis showed that leukocyte rolling and adhesion on vascular endothelium was reduced in MMP8 knockout mice. Furthermore, we detected an association between MMP8 gene variation and extent of coronary atherosclerosis in patients with coronary artery disease. A relationship between MMP8 gene variation, plasma VCAM-1 level and atherosclerosis progression was also observed in a population-based, prospective study.
Conclusion
These results indicate that MMP8 is an important player in atherosclerosis.
Keywords: Atherosclerosis, matrix metalloproteinase, gene
Chronic inflammation plays an important role in the pathogenesis of atherosclerosis which in turn is the main underlying cause of the majority of coronary heart disease and stroke1. One of the earliest steps in atherogenesis is the attachment of circulating leukocytes to the vascular endothelium consequent to the expression of adhesion molecules. Following adhesion to endothelial cells, leukocytes penetrate the endothelium into the arterial intima, differentiate into macrophages, uptake modified lipoproteins and become lipid-laden foam cells which represent a major component of atherosclerotic lesions1. In addition, vascular smooth muscle cells and extracellular matrix proteins, particularly types I and III collagens and proteoglycans, are also major constituents of atherosclerotic plaques1.
The matrix metalloproteinases (MMPs) are zinc-dependent proteases that can degrade extracellular matrix proteins2. Some MMPs can also cleave non-matrix proteins such as cytokines and growth factors2. There is substantial evidence indicating that MMPs participate in a number of diseases with matrix remodeling and inflammation. Several MMPs have been demonstrated to be involved in atherosclerosis in which these different proteases appear to exert divergent effects3-14.
MMP8 (also known as collagenase-2) has also been suggested to be implicated in atherosclerosis15-17. MMP8 possesses proteolytic activity on several matrix proteins particularly type I collagen and on some non-matrix proteins such as angiotensin-I (Ang I)18. It has been shown that endothelial cells, macrophages and smooth muscle cells in atherosclerotic lesions express MMP815, that its expression is increased in rapidly progressing lesions16, and that elevated serum MMP8 concentrations are associated with the presence of atherosclerosis and with unfavorable cardiovascular outcome17. However, it has remained unknown whether MMP8 plays a causal role in atherogenesis or is simply a bystander marker. To address this issue, we studied whether inactivating the MMP8 gene would have an effect on atherogenesis in atherosclerosis-prone apoE deficient mice. We found that inactivating MMP8 resulted in a significant reduction in the extent of atherosclerosis, with a decrease in macrophages but an increase in collagen content in the atherosclerotic lesions. In addition, we found that MMP8 knockout mice had lower levels of angiotensin-II (Ang II), lower blood pressure, decreased vascular cell adhesion molecular-1 (VCAM-1) expression and reduced vascular leukocyte recruitment. Furthermore, we observed that in humans, MMP8 gene variation was associated with VCAM-1 level and severity and progression of atherosclerosis.
Methods
The methods are briefly described here, and further details are provided in the Online Supplements.
Animals
MMP8−/−/apoE+/+ mice19 were crossbred with MMP8+/+/apoE−/− mice to generate MMP8+/−/apoE+/− double heterozygous mice. MMP8+/−/apoE+/− double heterozygous mice were bred to produce MMP8−/−/apoE−/− double knockout mice and MMP8+/+/apoE−/− controls (littermates of MMP8−/−/apoE−/− double knockout mice). Six week old males of both types of mouse were fed a Western diet for 12 weeks.
Characterization of atherosclerotic lesions
The extent of aortic atherosclerotic lesions in MMP8−/−/apoE−/− double knockout mice and controls was analyzed by en face staining of aortas with oil red O, as previously described20. The contents of macrophages, smooth muscle cells, collagen type I, Ang II and VCAM-1 in aortic atherosclerotic lesions were examined by immunohistochemical analyses.
Real-time reverse-transcriptase-PCR (real-time RT-PCR)
MMP2, MMP9, MMP13 and MMP14 expression levels in MMP8−/−/ApoE−/− mice and control mice were quantified by real-time RT-PCR.
Measurements of cholesterol and triglycerides
Plasma cholesterol and triglyceride levels in the MMP8−/−/apoE−/− double knockout mice and controls were determined with the use of commercially available assay kits.
Angiotensin I cleavage by MMP8 and mass spectrometry
Ang I was incubated with purified human MMP8. An uncleaved Ang I control was generated by incubating Ang I in the assay buffer without MMP8. Ang I and cleavage products in the above solutions (from the cleavage reaction and the control, respectively) were extracted and analysed by liquid chromatography-electrospray tandem mass spectrometry. The mass spectral data were processed into peak lists and searched against the Swiss Prot database. The relative intensity of the angiotensin peptides was calculated using the total ion count of each peptide.
Enzyme-linked immuno-sorbent assay (ELISA)
Commercially available ELISA kits were used to measure plasma Ang I and Ang II. Concentrations of VCAM-1 in plasma samples were also measured by ELISA kits.
Blood pressure measurement
Blood pressure was measured in conscious mice using a volume pressure recording sensor and occlusion tail cuff.
Analyses of other angiotensin II forming enzymes
Tissue extracts were prepared from aortas from MMP8+/+/ApoE−/− or MMP8−/−/ApoE−/− mice and assayed for other angiotensin II forming enzymes including angiotensin-converting enzyme (ACE), chymase and cathespins.
Flow Cytometry
Mouse endothelial cells (C166) were incubated with Ang I that had been subjected to cleavage by MMP8 or uncleaved Ang I, in the absence or presence of the Ang II type 1 receptor (AT1R) antagonist losartan and the Ang II type 2 receptor (AT2R) antagonist PD123319, and then subjected to flow cytometric analysis of VCAM-1 expression.
Intravital microscopy
Mesenteries of anesthetized mice were superfused with bicarbonate-buffered solution at 37°C at a rate of 2ml/min. Cell rolling was determined by counting the number of leukocytes rolling per minute in each vessel. Cell adhesion was quantified by counting, for each vessel, the number of adherent leukocytes in a 100μm length.
Human subjects
We studied MMP8 gene variation in relation to extent of atherosclerosis in a group (n=2000) of patients with coronary artery disease (CAD) documented angiographically as having >50% diameter stenosis in at least one coronary artery21. We also examined MMP8 gene variation in relation to progression of atherosclerosis in subjects of the Bruneck Study, a population-based, prospective study of atherosclerosis22-25.
Functional analysis of MMP8 gene rs1940475 SNP
MMP8 zymogens with Glu87 or Lys87 were synthesized and subjected to activation by 4-aminophenylmercuric acetate, followed by Western blot analysis.
Statistical analyses
The statistical analyses performed in this study are described in Online Data Supplements.
Results
Reduced atherosclerosis in MMP8 deficient mouse
Extensive aortic atherosclerotic lesions were observed in MMP8+/+/apoE−/− mice (controls) fed a Western diet (Figure 1A). In contrast, MMP8−/−/apoE−/− double knockout mice had substantially less aortic atherosclerosis (36% reduction compared with controls, p=0.007, Figure 1A). Compared with the lesions in the controls, those in the double knockout mice had significantly fewer macrophages (70% reduction, p=0.002, Figure 1B). In addition, there was a trend towards lower smooth muscle cell content in the lesions in the double knockout mice than lesions in the controls (p=0.057, Figure 1C). On the other hand, collagen content was higher in the lesions in the double knockouts than lesions in the controls (differing by 27%, p=0.034, Figure 1D). There was no significant difference in plasma cholesterol and triglyceride levels between the two groups (p=0.887 for cholesterol and p=0.117 for triglyceride). The levels of expression of the other mouse collagenases including MMP2, MMP3, MMP13 and MMP14 did not differ between the two groups (p=0.108, p=0.611, p=0.509, and p=0.318, respectively, Online Supplement Figure 2).
Figure 1. Reduced atherosclerosis in MMP8−/−/apoE−/− mice.

Panel A: representative images of Oil Red O stained en face preparations of aortas of MMP8+/+/apoE−/− and MMP8−/−/apoE−/− mice, and chart showing percentages of aortic surface area with atherosclerosis in the two groups of mice.
Panels B, C and D: representative images of immunostaining of macrophages (B), smooth muscle cells (C) and collagen (D) in aortic root atherosclerotic lesions of MMP8+/+/apoE−/− and MMP8−/−/apoE−/− mice, and charts showing percentages of mean macrophage, smooth muscle cell, and collagen contents in the two groups of mice.
Data presented are mean and standard error of mean.
Reduced Ang II level and VCAM-1 expression in MMP8 knockout mouse
MMP8 has been shown to cleave Ang I producing Ang II26, a well established atherogenic factor27-29, and angiotensin1-7 (Ang1-7). To verify this finding, we incubated Ang I with MMP8 and then performed liquid chromatography-electrospray tandem mass spectrometry analysis. The analysis showed cleavage of Ang I by MMP8, generating Ang II and Ang1-7, with the former being the major product (Figure 2 and Online Supplement Table 5).
Figure 2. Cleavage of Ang I by MMP8, producing Ang II and Ang1-7.

Panel A: Chromatogram of mass spectrometry analysis of uncleaved Ang I, showing the detection of Ang I peptide ion (m/z value 432.84 3+). Panel B: Chromatogram of mass spectrometry analysis of products of Ang I cleavage by MMP8, showing the presence of Ang II (m/z value 523.79 2+) and Ang1-7 (450.25 2+) peptide ions, in addition to Ang I (m/z value 432.91 3+). The presence of Ang II, Ang1-7 and Ang I in the respective peaks were confirmed by peptide fragmentation sequencing.
We then examined whether there was a difference in Ang II level between MMP8−/−/apoE−/− double knockout mice and controls. Indeed, we found that Ang II levels in atherosclerotic lesions and blood were lower in double knockout mice than in controls (p=0.002 and p=0.026, respectively, Figure 3A and 3B). In contrast, plasma levels of Ang I, the processor of Ang II, were higher in the double knockout group than in the control group (p=0.018, Figure 3C), which might be explained by reduced Ang I cleavage in double knockout mice. Consistent with the finding of reduced Ang II levels in double knockout mice, we found that blood pressure was lower in double knockout mice than in controls (p=0.017 for systolic and p=0.023 for diastolic blood pressure, Figure 4). We found no difference between MMP8−/−/apoE−/− double knockout mice and controls in the levels of the other Ang II forming enzymes ACE, chymases and cathepsins (Online Supplement Figure 3).
Figure 3. Reduced Ang II levels in MMP8−/−/apoE−/− mice.

Panel A: representative images of Ang II staining in atherosclerotic lesions in MMP8−/−/apoE−/− and MMP8+/+/apoE−/− mice respectively, and column chart showing lower relative intensity of Ang II staining in lesions in MMP8−/−/apoE−/− mice than in MMP8+/+/apoE−/− mice. Panels B and C: charts showing lower plasma Ang II level, but higher plasma Ang I level, in MMP8−/−/apoE−/− mice than in MMP8+/+/apoE−/− mice. Data presented as mean and standard error of mean.
Figure 4. Reduced blood pressure in MMP8−/−/apoE−/− mice.
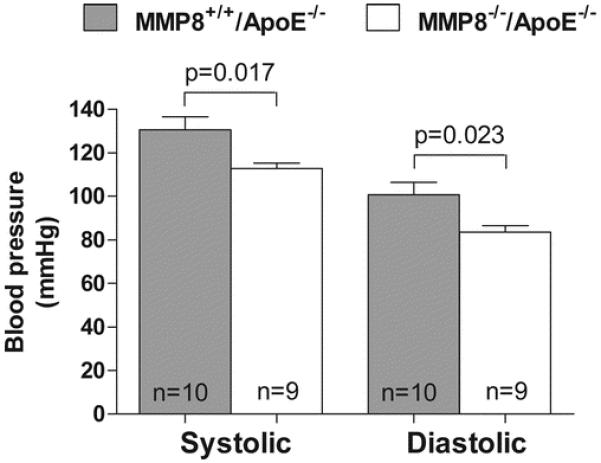
Chart showing lower systolic and diastolic blood pressure in MMP8−/−/ApoE−/− mice than in MMP8+/+/ApoE−/− mice. Data shown are mean ± standard error of mean.
Previous studies demonstrated that Ang II induced the expression of adhesion molecules28, 29 such as VCAM-130-34 which plays an important role in atherogenesis35. In agreement, our experiments showed that Ang II in a wide range of concentration increased VCAM-1 expression in endothelial cells (Online Supplement Figure 4). In addition, we found that the products of Ang I cleavage by MMP8 increased VCAM-1 expression. As shown in Figure 5A and 5B, flow cytometric analysis showed that cultured endothelial cells treated with products of Ang I cleavage by MMP8 expressed more VCAM-1 than endothelial cells treated with uncleaved Ang I (p=0.002). This effect was abolished in the presence of the AT1R antagonist losartan and the AT2R antagonist PD123319 (Figure 5C and 5D).
Figure 5. Products of Ang I cleavage by MMP8 increase VCAM-1 expression in endothelial cells.
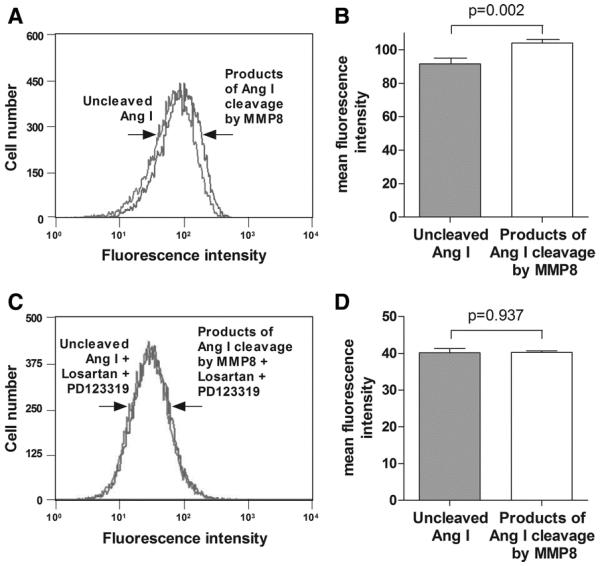
Panels A and B: representative plot (A) and mean fluorescence intensity (B) in flow cytometric analyses of VCAM-1 expression on endothelial cells incubated with uncleaved Ang I or with products of Ang I cleavage by MMP8. Panels C and D: representative plot (C) and mean fluorescence intensity (D) in flow cytometric analyses of VCAM-1 expression on endothelial cells incubated with uncleaved Ang I or with products of Ang I cleavage by MMP8, in the presence of the AT1R antagonist losartan and the AT2R antagonist PD123319. Error bars in the column charts are standard error of mean.
We also examined whether there was a difference in VCAM-1 expression level between MMP8−/−/apoE−/− double knockout mice and controls. We observed that VCAM-1 expression in endothelial and other cells in atherosclerotic lesions was substantially reduced in the double knockout mice compared with controls (Figure 6A, 6B and 6C). Plasma levels of soluble VCAM-1 were also lower in double knockout mice than in controls (Figure 6D).
Figure 6. Reduced VCAM-1 expression in MMP8−/−/apoE−/− mice compared with MMP8+/+/apoE−/− mice.

Panels A, B and C show results of immunohistochemical analyses of VCAM-1 in aortic atherosclerotic lesions, (A), representative images; (B), mean VCMA-1 staining in lesions; (C), mean VCAM-1 staining in the endothelial layer. Panel D shows mean levels of soluble VCAM-1 in plasma. Error bars in the column charts are standard error of mean.
Reduced leukocyte recruitment in MMP8 knockout mice
Intravital microscopy examination showed that leukocyte rolling and adhesion on vascular endothelium was attenuated (by approximately 50%) in double knockout mice compared with controls (p=0.049 and p=0.009 respectively, Figure 7), which might be in part explained by the reduction of Ang II and VCAM-1 levels in the double knockout mice.
Figure 7. Reduced leukocyte recruitment in MMP8−/−/apoE−/− mice.

Panel A: Intravital microscopic images showing leukocyte rolling (arrow point 1) and adhesion (arrow point 2) on mesenteric postcapillary venules in vivo. Panel B: Diagrams showing that leukocyte rolling and adhesion is reduced in MMP8−/−/apoE−/− mice compared with MMP8+/+/apoE−/− mice. Data presented as the mean and standard error of mean.
Association of MMP8 gene variation with VCAM-1 level and atherosclerosis in humans
Further to the animal and in vitro studies described above, we investigated whether in humans, there was a relationship between MMP8 gene variation and inter-individual differences in atherosclerosis. To address this question, we ascertained whether MMP8 gene variation was associated with extent of atherosclerosis in patients (n=2000) with CAD. In addition, we examined whether MMP8 gene variation was associated with atherosclerosis progression in a population-based, prospective study (the Bruneck study, n=782).
In the study of CAD patients, we initially genotyped 1000 subjects for a panel of 16 single nucleotide polymorphisms (SNPs) consisting of all common SNPs (with minor allele frequency ≥0.05) identified by re-sequencing the MMP8 gene proximal promoter (2kb) and coding regions and tagging SNPs selected from the HapMap database to capture (r2≥0.8) common SNPs (with minor allele frequency ≥0.05) in the introns, 5′ upstream sequence and 3′-untranslated region. An association was detected between the extent of coronary atherosclerosis and SNP rs1940475 (dbSNP database identification number) (p=0.01). We then genotyped 1000 additional CAD patients for this SNP and analyzed the data for this SNP in the entire sample (n=2000). The analysis showed a highly significant association between the SNP and extent of coronary atherosclerosis (p=0.0008), with the T allele having a lower frequency in patients who had >50% stenosis in two or three coronary arteries than in patients with >50% stenosis in one coronary artery (odds ratio=0.80, 95% confidence interval=0.70-0.91, Table 1). The association remained significant after taking into account the number of SNPs tested in this study, as the p-value (0.0008) for the association observed was smaller than the significance threshold (0.005) calculated using the SNPSpD method36, 37 to take into consideration of the number of SNPs tested and the degree of linkage disequilibrium between the SNPs.
Table 1. Association between MMP8 gene variation and extent of coronary atherosclerosis in patients with coronary artery disease (n=2000).
| rs1940475 SNP | Subjects with significant atherosclerosis* in one coronary artery |
Subjects with significant atherosclerosis* in two or three coronary arteries |
Odds ratio (95% confidence interval) † for significant atherosclerosis* in two or three coronary arteries |
|---|---|---|---|
| Genotype | n (%) | n (%) | |
| C/C | 170 (22.2) | 342 (27.7) | reference |
| C/T | 398 (52.0) | 632 (51.2) | 0.77 (0.62-0.97), p=0.028 |
| T/T | 198 (25.8) | 260 (21.1) | 0.63 (0.49-0.83), p=0.0008 |
| Total | 766 (100.0) | 1234 (100.0) | |
| Allele | frequency | frequency | |
| C | 0.48 | 0.53 | reference |
| T | 0.52 | 0.47 | 0.80 (0.70-0.91), p=0.0008 |
Signifcant atherosclerosis was defined by >50% stenosis.
calculated by logistic regression analysis with adjustment for age, gender, smoking, hypercholesterolemia, hypertension and diabetes.
In line with the finding in the above study of CAD patients, the population-based, prospective study (the Bruneck study) showed that the T allele of SNP rs1940475 was associated with a protective effect against carotid atherosclerosis progression in a 10 year follow-up [odds ratio 0.77 (95% confidence interval 0.60-0.98) for atherosclerosis progression between 1990 and 1995, and odds ratio 0.74 (95% confidence interval 0.56-0.99) for atherosclerosis progression between 1995 and 2000] (Online Supplement Table 6). In addition, we found that plasma levels of soluble VCAM-1 were lower in individuals who carried the T allele (p=0.014, Online Supplement Table 7).
MMP8 is secreted from cells as a latent form, and activation is achieved by removing the propeptide domain of the zymogen18. The rs1940475 is a non-synonymous substitution resulting in a change from glutamic acid (Glu) to lysine (Lys) at amino acid residue 87 in the propeptide domain of MMP8. We carried out in vitro analysis to investigate whether this SNP could have an effect on MMP8 activation. The analysis indicated that MMP8 zymogen with Lys87 (produced by the T allele) is less amenable to activation than the zymogen with Glu87 (produced by the C allele) (Online Supplement Figure 5).
Discussion
Our study shows that inactivating MMP8 causes a substantial reduction in the extent of atherosclerosis in apoE deficient mice fed a Western diet. The atherosclerotic lesions in the MMP8 knockout mice have fewer macrophages but higher collagen content. Compared with controls, MMP8 deficient mice have higher Ang I and lower Ang II levels, likely reflecting the ability of MMP8 to cleave Ang I and generate Ang II. Consistently, we found that blood pressure was lower in MMP8 knockout mice than in controls. In agreement with the finding that Ang II induces adhesion molecule expression28-34, we found that products of Ang I cleavage by MMP8 increases VCAM-1 expression in cultured endothelial cells. In addition, we observed reduced expression of VCAM-1 in MMP8 knockout mice than in controls. Further to the findings from the mouse study, we detected an association between MMP8 gene variation and extent of coronary atherosclerosis in CAD patients, and an association of the MMP8 gene variation with plasma VCAM-1 level and progression of atherosclerosis in a population-based, prospective cohort study (the Bruneck study). Functional analysis indicated that the MMP8 genetic variant associated with reduced atherosclerosis had an effect on MMP8 zymogen activation. Taken together, these results indicate that MMP8 is an important player in atherogenesis.
Classically MMPs are implicated in matrix remodeling through their matrix protein degrading activity. However, relatively recent studies have revealed that MMPs have other important biological functions arising from their proteolytic activity on non-matrix proteins2. In particular, an increasing number of cytokines, chemokines and growth factors have been shown to be processed by MMPs, and there is growing evidence indicating that the actions of MMPs on these non-matrix proteins play important roles in tumorigenesis and a number of inflammatory diseases2, 38. Although currently the most appreciated roles of MMPs in atherosclerosis are mainly related to matrix protein degradation that facilitates cell migration and undermines atherosclerotic plaque stability, it is conceivable that actions of MMPs on non-matrix proteins also play a part in the development and progression of atherosclerosis. The findings that MMP8 can cleave Ang I generating Ang II and that MMP8 knockout mice have reduced Ang II levels seem to support this notion. It is plausible that the role of MMP8 in atherogenesis is partly related to its proteolytic activity on matrix proteins particularly type I collagen as well as its ability to cleave Ang I generating Ang II. Thus, the reduction in atherosclerotic lesion formation in MMP8 knockout mice could be partly due to reduced blood pressure and reduced VCAM-1 expression, as a result of lower Ang II levels.
The best known Ang I convertor is ACE, but other enzymes (such as chymases, cathepsins and MMP8) have also been shown to be able to cleave Ang I to generate Ang II26, 28. ACE is a zinc-dependent metalloproteinase and belongs to the metalloproteinase super-family that also includes MMPs39, 40. The catalytic sites of ACE and collagenases share similarities in amino acid sequence and structure41, 42. Interestingly, it has been shown that the ACE inhibitors captopril, lisinopril, quinapril and enalapril also inhibit MMP activity43-47 through interacting with the MMP active site, analogous to their interaction with the active site of ACE42, 47. Although ACE is likely to be the main converter of Ang I, it is probable that other enzymes such as MMP8, chymases and cathepsins also contribute to Ang I conversion, particularly at locations where ACE levels are low.
Previous studies have examined the effects of several MMPs on atherogenesis in apoE deficient mice or LDL receptor deficient mice4-11, 13, 14. These studies have suggested divergent effects of these enzymes in atherosclerotic lesion formation and stability48. It was shown that expressing human MMP1 in macrophages in apoE deficient mice resulted in smaller aortic atherosclerotic lesions with reduced fibrillar collagen content4. Similarly, inactivating MMP3 increased aortic atherosclerotic lesion sizes and collagen content6. Inactivating MMP13 also increased, whilst overexpressing MMP14 decreased, collagen accumulation in aortic atherosclerotic lesions without an apparent effect on lesion size13, 14. In contrast, inactivating MMP2 resulted in decreased aortic atherosclerotic lesions with reduced numbers of smooth muscle cells5. Inactivating MMP9 also led to smaller aortic atherosclerotic lesions with reduced numbers of smooth muscle cells but also with fewer macrophages and lower collagen content8-10. Moreover, it was shown that overexpressing active MMP9 in macrophages in apoE deficient mice induced aortic atherosclerotic plaque disruption11. MMP12 has also been shown to exert an effect on atherosclerotic lesion stability7. In contrast, inactivating MMP7 had no effect in these characteristics7. Thus the findings from these studies suggest that these MMPs exert divergent effects, some impacting on lesion size, some affecting collagen content, and some being required for macrophage and/or smooth muscle cell accumulation. As described earlier, our study showed that MMP8 knockout resulted in reduced atherosclerotic lesions with fewer macrophages and increased collagen content. Thus, in apoE deficient mice, MMP8 has a different profile of activities from the other MMPs mentioned above. However, similar to MMP-1, -3, -13 and -144, 6, 13, 14, MMP8 reduced collagen accumulation in atherosclerotic lesions, which could potentially have implications on atherosclerotic lesion stability.
The present study has some limitations. The study only assessed atherosclerosis in the aorta following a Western diet for 12 weeks. Further work is warranted to study the effect of MMP8 deficiency on atherosclerosis in other vascular sites, in shorter and longer durations of Western diet, and in chow diet. For the CAD patients and the Bruneck cohort, no Ang II level data were available and therefore we were unable to determine whether there was an association between Ang II level and MMP8 genotype. This would also warrant investigating in future work. It would also be interesting to investigate the relative contributions of the reduction in Ang II generation and the increase in collagen content to atherosclerotic lesion development in MMP8 knockout mice in future studies
In summary, our study indicates that MMP8 is an important player in atherogenesis. Taken together with the aforementioned studies of other MMPs, this work provides further evidence of involvement of MMPs in atherosclerosis and further supports the notion that different MMPs have divergent effects in the atherogenic process.
Supplementary Material
Acknowledgments
Sources of funding This work was supported by grants from the British Heart Foundation (PG/08/051/25141 and PG/98183) and the European Union (LSHC-CT-2003-503297). JD was supported by a fellowship from the Barts and The London Charity. RSS is funded by a Wellcome Trust Career Development Fellowship
Non-standard Abbreviations and Acronyms
- MMPs
matrix metalloproteinases
- MMP8
matrix metalloproteinase-8
- Ang I
angiotensin I
- Ang II
angiotensin II
- ACE
angiotensin converting enzyme
- AT1R
Ang II type 1 receptor
- AT2R
Ang II type 2 receptor
- Ang1-7
angiotensin1-7
- VCAM-1
vascular cell adhesion molecule-1
- CAD
coronary artery disease
- SNPs
single nucleotide polymorphisms
- RT-PCR
reverse-transcriptase polymerase chain reaction
- ELISA
Enzyme-linked immuno-sorbent assay
Footnotes
Disclosures None
References
- 1.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 2.Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8:221–233. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94:2493–2503. doi: 10.1172/JCI117619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lemaitre V, O’Byrne TK, Borczuk AC, Okada Y, Tall AR, D’Armiento J. ApoE knockout mice expressing human matrix metalloproteinase-1 in macrophages have less advanced atherosclerosis. J Clin Invest. 2001;107:1227–1234. doi: 10.1172/JCI9626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuzuya M, Nakamura K, Sasaki T, Cheng XW, Itohara S, Iguchi A. Effect of MMP-2 deficiency on atherosclerotic lesion formation in apoE-deficient mice. Arterioscler Thromb Vasc Biol. 2006;26:1120–1125. doi: 10.1161/01.ATV.0000218496.60097.e0. [DOI] [PubMed] [Google Scholar]
- 6.Silence J, Lupu F, Collen D, Lijnen HR. Persistence of atherosclerotic plaque but reduced aneurysm formation in mice with stromelysin-1 (MMP-3) gene inactivation. Arterioscler Thromb Vasc Biol. 2001;21:1440–1445. doi: 10.1161/hq0901.097004. [DOI] [PubMed] [Google Scholar]
- 7.Johnson JL, George SJ, Newby AC, Jackson CL. Divergent effects of matrix metalloproteinases 3, 7, 9, and 12 on atherosclerotic plaque stability in mouse brachiocephalic arteries. Proc Natl Acad Sci U S A. 2005;102:15575–15580. doi: 10.1073/pnas.0506201102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galis ZS, Johnson C, Godin D, Magid R, Shipley JM, Senior RM, Ivan E. Targeted disruption of the matrix metalloproteinase-9 gene impairs smooth muscle cell migration arterial remodeling and geometrical. Circulation Research. 2002;91:852–859. doi: 10.1161/01.res.0000041036.86977.14. [DOI] [PubMed] [Google Scholar]
- 9.Luttun A, Lutgens E, Manderveld A, Maris K, Collen D, Carmeliet P, Moons L. Loss of matrix metalloproteinase-9 or matrix metalloproteinase-12 protects apolipoprotein E-deficient mice against atherosclerotic media destruction but differentially affects plaque growth. Circulation. 2004;109:1408–1414. doi: 10.1161/01.CIR.0000121728.14930.DE. [DOI] [PubMed] [Google Scholar]
- 10.Johnson C, Galis ZS. Matrix metalloproteinase-2 and -9 differentially regulate smooth muscle cell migration and cell-mediated collagen organization. Arterioscler Thromb Vasc Biol. 2004;24:54–60. doi: 10.1161/01.ATV.0000100402.69997.C3. [DOI] [PubMed] [Google Scholar]
- 11.Gough PJ, Gomez IG, Wille PT, Raines EW. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J Clin Invest. 2006;116:59–69. doi: 10.1172/JCI25074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liang J, Liu E, Yu Y, Kitajima S, Koike T, Jin Y, Morimoto M, Hatakeyama K, Asada Y, Watanabe T, Sasaguri Y, Watanabe S, Fan J. Macrophage metalloelastase accelerates the progression of atherosclerosis in transgenic rabbits. Circulation. 2006;113:1993–2001. doi: 10.1161/CIRCULATIONAHA.105.596031. [DOI] [PubMed] [Google Scholar]
- 13.Deguchi JO, Aikawa E, Libby P, Vachon JR, Inada M, Krane SM, Whittaker P, Aikawa M. Matrix metalloproteinase-13/collagenase-3 deletion promotes collagen accumulation and organization in mouse atherosclerotic plaques. Circulation. 2005;112:2708–2715. doi: 10.1161/CIRCULATIONAHA.105.562041. [DOI] [PubMed] [Google Scholar]
- 14.Schneider F, Sukhova GK, Aikawa M, Canner J, Gerdes N, Tang SM, Shi GP, Apte SS, Libby P. Matrix-metalloproteinase-14 deficiency in bone-marrow-derived cells promotes collagen accumulation in mouse atherosclerotic plaques. Circulation. 2008;117:931–939. doi: 10.1161/CIRCULATIONAHA.107.707448. [DOI] [PubMed] [Google Scholar]
- 15.Herman MP, Sukhova GK, Libby P, Gerdes N, Tang N, Horton DB, Kilbride M, Breitbart RE, Chun M, Schonbeck U. Expression of neutrophil collagenase (matrix metalloproteinase-8) in human atheroma: a novel collagenolytic pathway suggested by transcriptional profiling. Circulation. 2001;104:1899–1904. doi: 10.1161/hc4101.097419. [DOI] [PubMed] [Google Scholar]
- 16.Turu MM, Krupinski J, Catena E, Rosell A, Montaner J, Rubio F, varez-Sabin J, Cairols M, Badimon L. Intraplaque MMP-8 levels are increased in asymptomatic patients with carotid plaque progression on ultrasound. Atherosclerosis. 2006;187:161–169. doi: 10.1016/j.atherosclerosis.2005.08.039. [DOI] [PubMed] [Google Scholar]
- 17.Tuomainen AM, Nyyssonen K, Laukkanen JA, Tervahartiala T, Tuomainen TP, Salonen JT, Sorsa T, Pussinen PJ. Serum matrix metalloproteinase-8 concentrations are associated with cardiovascular outcome in men. Arterioscler Thromb Vasc Biol. 2007;27:2722–2728. doi: 10.1161/ATVBAHA.107.154831. [DOI] [PubMed] [Google Scholar]
- 18.Van Lint P, Libert C. Matrix metalloproteinase-8: cleavage can be decisive. Cytokine Growth Factor Rev. 2006;17:217–223. doi: 10.1016/j.cytogfr.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 19.Balbin M, Fueyo A, Tester AM, Pendas AM, Pitiot AS, Astudillo A, Overall CM, Shapiro SD, Lopez-Otin C. Loss of collagenase-2 confers increased skin tumor susceptibility to male mice. Nat Genet. 2003;35:252–257. doi: 10.1038/ng1249. [DOI] [PubMed] [Google Scholar]
- 20.Foteinos G, Hu Y, Xiao Q, Metzler B, Xu Q. Rapid endothelial turnover in atherosclerosis-prone areas coincides with stem cell repair in apolipoprotein E-deficient mice. Circulation. 2008;117:1856–1863. doi: 10.1161/CIRCULATIONAHA.107.746008. [DOI] [PubMed] [Google Scholar]
- 21.Ye S, Dunleavey L, Bannister W, Day LB, Tapper W, Collins AR, Day IN, Simpson I. Independent effects of the -219 G>T and epsilon 2/ epsilon 3/ epsilon 4 polymorphisms in the apolipoprotein E gene on coronary artery disease: the Southampton Atherosclerosis Study. Eur J Hum Genet. 2003;11:437–443. doi: 10.1038/sj.ejhg.5200983. [DOI] [PubMed] [Google Scholar]
- 22.Kiechl S, Willeit J. The natural course of atherosclerosis. Part I: incidence and progression. Arterioscler Thromb Vasc Biol. 1999;19:1484–1490. doi: 10.1161/01.atv.19.6.1484. [DOI] [PubMed] [Google Scholar]
- 23.Kiechl S, Willeit J. The natural course of atherosclerosis. Part II: vascular remodeling. Bruneck Study Group. Arterioscler Thromb Vasc Biol. 1999;19:1491–1498. doi: 10.1161/01.atv.19.6.1491. [DOI] [PubMed] [Google Scholar]
- 24.Kiechl S, Lorenz E, Reindl M, Wiedermann CJ, Oberhollenzer F, Bonora E, Willeit J, Schwartz DA. Toll-like receptor 4 polymorphisms and atherogenesis. N Engl J Med. 2002;347:185–192. doi: 10.1056/NEJMoa012673. [DOI] [PubMed] [Google Scholar]
- 25.Ye S, Willeit J, Kronenberg F, Xu Q, Kiechl S. Association of genetic variation on chromosome 9p21 with susceptibility and progression of atherosclerosis: a population-based, prospective study. J Am Coll Cardiol. 2008;52:378–384. doi: 10.1016/j.jacc.2007.11.087. [DOI] [PubMed] [Google Scholar]
- 26.Diekmann O, Tschesche H. Degradation of kinins, angiotensins and substance P by polymorphonuclear matrix metalloproteinases MMP 8 and MMP 9. Braz J Med Biol Res. 1994;27:1865–1876. [PubMed] [Google Scholar]
- 27.Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105:1605–1612. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmieder RE, Hilgers KF, Schlaich MP, Schmidt BM. Renin-angiotensin system and cardiovascular risk. Lancet. 2007;369:1208–1219. doi: 10.1016/S0140-6736(07)60242-6. [DOI] [PubMed] [Google Scholar]
- 29.Brasier AR, Recinos A, III, Eledrisi MS. Vascular inflammation and the renin-angiotensin system. Arterioscler Thromb Vasc Biol. 2002;22:1257–1266. doi: 10.1161/01.atv.0000021412.56621.a2. [DOI] [PubMed] [Google Scholar]
- 30.Pueyo ME, Gonzalez W, Nicoletti A, Savoie F, Arnal JF, Michel JB. Angiotensin II stimulates endothelial vascular cell adhesion molecule-1 via nuclear factor-kappaB activation induced by intracellular oxidative stress. Arterioscler Thromb Vasc Biol. 2000;20:645–651. doi: 10.1161/01.atv.20.3.645. [DOI] [PubMed] [Google Scholar]
- 31.Tummala PE, Chen XL, Sundell CL, Laursen JB, Hammes CP, Alexander RW, Harrison DG, Medford RM. Angiotensin II induces vascular cell adhesion molecule-1 expression in rat vasculature: A potential link between the renin-angiotensin system and atherosclerosis. Circulation. 1999;100:1223–1229. doi: 10.1161/01.cir.100.11.1223. [DOI] [PubMed] [Google Scholar]
- 32.Zhang L, Cheng J, Ma Y, Thomas W, Zhang J, Du J. Dual pathways for nuclear factor kappaB activation by angiotensin II in vascular smooth muscle: phosphorylation of p65 by IkappaB kinase and ribosomal kinase. Circ Res. 2005;97:975–982. doi: 10.1161/01.RES.0000190589.52286.41. [DOI] [PubMed] [Google Scholar]
- 33.Ramos CL, Huo Y, Jung U, Ghosh S, Manka DR, Sarembock IJ, Ley K. Direct demonstration of P-selectin- and VCAM-1-dependent mononuclear cell rolling in early atherosclerotic lesions of apolipoprotein E-deficient mice. Circ Res. 1999;84:1237–1244. doi: 10.1161/01.res.84.11.1237. [DOI] [PubMed] [Google Scholar]
- 34.Huo Y, Hafezi-Moghadam A, Ley K. Role of vascular cell adhesion molecule-1 and fibronectin connecting segment-1 in monocyte rolling and adhesion on early atherosclerotic lesions. Circ Res. 2000;87:153–159. doi: 10.1161/01.res.87.2.153. [DOI] [PubMed] [Google Scholar]
- 35.Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, Davis V, Gutierrez-Ramos JC, Connelly PW, Milstone DS. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest. 2001;107:1255–1262. doi: 10.1172/JCI11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nyholt DR. A simple correction for multiple testing for single-nucleotide polymorphisms in linkage disequilibrium with each other. Am J Hum Genet. 2004;74:765–769. doi: 10.1086/383251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li J, Ji L. Adjusting multiple testing in multilocus analyses using the eigenvalues of a correlation matrix. Heredity. 2005;95:221–227. doi: 10.1038/sj.hdy.6800717. [DOI] [PubMed] [Google Scholar]
- 38.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 39.Corvol P, Williams TA, Soubrier F. Peptidyl dipeptidase A: angiotensin I-converting enzyme. Methods Enzymol. 1995;248:283–305. doi: 10.1016/0076-6879(95)48020-x. [DOI] [PubMed] [Google Scholar]
- 40.Sturrock ED, Natesh R, van Rooyen JM, Acharya KR. Structure of angiotensin I-converting enzyme. Cell Mol Life Sci. 2004;61:2677–2686. doi: 10.1007/s00018-004-4239-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soubrier F, Alhenc-Gelas F, Hubert C, Allegrini J, John M, Tregear G, Corvol P. Two putative active centers in human angiotensin I-converting enzyme revealed by molecular cloning. Proc Natl Acad Sci U S A. 1988;85:9386–9390. doi: 10.1073/pnas.85.24.9386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamamoto D, Takai S, Miyazaki M. Prediction of interaction mode between a typical ACE inhibitor and MMP-9 active site. Biochem Biophys Res Commun. 2007;354:981–984. doi: 10.1016/j.bbrc.2007.01.088. [DOI] [PubMed] [Google Scholar]
- 43.Volpert OV, Ward WF, Lingen MW, Chesler L, Solt DB, Johnson MD, Molteni A, Polverini PJ, Bouck NP. Captopril inhibits angiogenesis and slows the growth of experimental tumors in rats. J Clin Invest. 1996;98:671–679. doi: 10.1172/JCI118838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reinhardt D, Sigusch HH, Hensse J, Tyagi SC, Korfer R, Figulla HR. Cardiac remodelling in end stage heart failure: upregulation of matrix metalloproteinase (MMP) irrespective of the underlying disease, and evidence for a direct inhibitory effect of ACE inhibitors on MMP. Heart. 2002;88:525–530. doi: 10.1136/heart.88.5.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sakata Y, Yamamoto K, Mano T, Nishikawa N, Yoshida J, Hori M, Miwa T, Masuyama T. Activation of matrix metalloproteinases precedes left ventricular remodeling in hypertensive heart failure rats: its inhibition as a primary effect of Angiotensin-converting enzyme inhibitor. Circulation. 2004;109:2143–2149. doi: 10.1161/01.CIR.0000125741.88712.77. [DOI] [PubMed] [Google Scholar]
- 46.Brower GL, Levick SP, Janicki JS. Inhibition of matrix metalloproteinase activity by ACE inhibitors prevents left ventricular remodeling in a rat model of heart failure. Am J Physiol Heart Circ Physiol. 2007;292:H3057–H3064. doi: 10.1152/ajpheart.00447.2006. [DOI] [PubMed] [Google Scholar]
- 47.Yamamoto D, Takai S, Jin D, Inagaki S, Tanaka K, Miyazaki M. Molecular mechanism of imidapril for cardiovascular protection via inhibition of MMP-9. J Mol Cell Cardiol. 2007;43:670–676. doi: 10.1016/j.yjmcc.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 48.Newby AC, Johnson JL. Genetic strategies to elucidate the roles of matrix metalloproteinases in atherosclerotic plaque growth and stability. Circ Res. 2005;97:958–960. doi: 10.1161/01.RES.0000193565.23357.c0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.