

Abstract
Structure-based mutational analysis was used to probe the architecture of the glutamine binding pocket in Escherichia coli glutaminyl-tRNA synthetase (GlnRS). Crystallographic studies of several different GlnRS complexes in a lattice that supports catalytic activity have shown that the glutamine amide group makes only ambiguous hydrogen-bonding interactions with a tyrosine hydroxyl and bound water molecule, rather than the highly specific hydrogen-bonding and electrostatic interactions made by the substrate amino acid in all other nonediting tRNA synthetases. Further, the amide oxygen of substrate glutamine accepts a hydrogen bond from the 3′-ribose hydroxyl group of ATP, an unusual distal substrate–substrate interaction also not observed in any other tRNA synthetase complex. Steady-state and pre-steady-state kinetic analysis using a 3′-dATP analogue in place of ATP shows that removal of this distal interaction does not affect Km for the analogue as compared with ATP, yet decreases the efficiency of aminoacylation by 103-fold while significantly elevating Km for glutamine. In other experiments, mutation of eight nearly fully conserved residues in the glutamine binding pocket reveals decreases in kcat/Km ranging from 5- to 400-fold, and in Kd for glutamine of up to at least 60-fold. Amino acid replacements at Tyr211 and Gln255, which participate with substrate glutamine in an antidromic circular arrangement of hydrogen bonds, cause the most severe decreases in catalytic efficiency. This finding suggests that the relative absence of direct hydrogen bonds to glutamine may be in part compensated by additional binding energy derived from the enhanced stability of this circular network. Calculations of electrostatic surface potential in the active site further suggest that a complementary electrostatic environment is also an important determinant of glutamine binding.
Formation of aminoacyl-tRNA depends on the accuracy of aminoacyl-tRNA synthetases (AARS1), which link amino acids with their cognate tRNAs in a two-step reaction that proceeds through an activated aminoacyl adenylate intermediate (1). The 23 known AARS are divided into two classes (class I and class II) featuring topologically distinct active site domains (2), with each enzyme also possessing divergent domains that provide further discrimination against noncognate tRNAs. A remarkable property of all AARS is their ability to selectively match pairs of substrates from structurally distinct subclasses. This, of course, provides the embodiment of the genetic code at the molecular level: the chemical language of nucleic acids is converted into that of proteins on the surfaces of these enzymes. All AARS are selective for one particular amino acid and a subset (usually between one and three in bacteria and archaea) of structurally similar tRNAs possessing matching anticodons.
To meet cellular requirements for high fidelity in protein synthesis, many tRNA synthetases have acquired editing activities by which misactivated amino acids or misacylated aminoacyl-tRNAs are hydrolyzed, sometimes on a structurally distinct domain adjacent to the domain containing the synthetic site (3). In some cases, the need for editing is clear: for example, Pauling predicted over 50 years ago that no enzyme would be able to directly discriminate at a high level between isoleucine and the smaller valine, which differ by a single methyl group (4). Indeed, isoleucyl-tRNA synthetase (IleRS) hydrolyzes both misactivated Val-AMP by a “pre-transfer” editing activity, and misacylated Val-tRNAIle by “post-transfer” editing (5). However, other editing activities have been measured that are less easy to rationalize; for example, PheRS edits tyrosine (6), and AlaRS edits serine (7), despite the expected capacities of their respective synthetic sites to exclude the noncognate substrate on steric grounds.
About half of the AARS are able to distinguish their cognate amino acid substrates without the need for a separate hydrolytic editing mechanism (8). Crystal structures of these enzymes, which are typically specific for charged or polar amino acids, generally reveal binding pockets that provide direct electrostatic, hydrogen-bonding, and steric complementarity with the amino acid that appears sufficient to distinguish all noncognate substrates (9–11). The only known exception is the Escherichia coli glutaminyl-tRNA synthetase (GlnRS), which, while capable of discriminating against the synthesis of Glu-tRNAGln by a factor of 107-fold in kcat/Km (12), appears to make only ambiguous hydrogen-bonding interactions with substrate glutamine (12, 13).
In structures of the GlnRS–tRNAGln complex determined in the presence of either the glutaminyl adenylate analogue 5′-O-[N-(l-glutaminyl)sulfamoyl] adenosine (QSI), or free glutamine and the ATP analogue AMPCPP, the glutamine is oriented such that the side-chain amide −NH2 group donates hydrogen bonds to the hydroxyl moiety of Tyr211 and to a buried water molecule. The amide oxygen is located 3.4 to 4.0 Å from the guanidinium of Arg30, seemingly too far to make a strong hydrogen bond. To rationalize the very high observed specificity against isosteric glutamate, it was proposed that a network of four buried water molecules is specifically oriented by peripheral obligate hydrogen-bond donor and acceptor groups including Asp212, Asp219, and Glu257, such that Tyr211 and one buried water become obligate acceptors from the glutamine −NH2 group (13). This would, presumably, generate an unfavorable electrostatic juxtaposition when the negatively charged glutamate binds. However, the structure of the GlnRS–tRNAGln complex bound to glutamate revealed that one carboxylate oxygen of glutamate occupies the identical position to the −NH2 of glutamine, suggesting that the buried water molecules could easily reorient their hydrogen-bonding functions, and that this network could thus not provide the necessary selectivity. Instead, specificity for glutamine appears to be a more distributed property of the active-site Rossmann fold: extensive mutagenesis of both the primary binding pocket and adjacent surface loops was required to endow GlnRS with 16000-fold enhanced capacity for synthesis of misacylated Glu-tRNAGln (14).
Another aspect of amino acid binding that appears unique to GlnRS is the formation of a hydrogen bond between the amide oxygen of glutamine and the 3′OH ribosyl moiety of ATP (13). No other AARS is known to facilitate such a distal interaction between nonreactive moieties of the ATP and amino acid. This contact is not formed in the cocrystal structure of GlnRS bound to noncognate glutamate, suggesting a role in amino acid selectivity.
GlnRS is also unusual in several other respects. First, it is one of only four tRNA synthetases, including also GluRS, ArgRS, and class I LysRS, that require the presence of tRNA for synthesis of the aminoacyl adenylate (1). The requirement for tRNA to promote active site assembly, which is well-documented by structural analyses (15–17), might conceivably be correlated with the presence of amino acid specificity determinants that are more globally distributed in the enzyme structure. The order of substrate binding to GlnRS in the steady-state is not known, but the absolute requirement for tRNA binding to mediate synthesis of glutaminyl adenylate strongly suggests that tRNA must bind first (18–20). Second, it is known that GlnRS originated late in evolution as a consequence of a duplication in the gene encoding eukaryotic GluRS: the GlnRS enzyme is present now only in eukaryotes and, by horizontal transfer, in some Gram-negative bacteria (21, 22). It is not clear, however, whether such a late differentiation of the glutamine binding pocket within the class I tRNA synthetase Rossmann fold might also be related to the unusual features observed in the crystal structures. Asparaginyl-tRNA synthetase (AsnRS), which is restricted in its phylogenetic distribution similarly to GlnRS, features a much-better differentiated asparagine binding pocket in which the specificity against charged aspartate can clearly be accounted for by direct hydrogen bonding (23, 24). It does appear that water-mediated interactions may be required in AsnRS to distinguish protonated aspartate from asparagine, although such specificity might arise instead from a downward shift in the pKa of aspartate promoted by the electronic environment of the binding pocket.
We sought to examine the unusual network of interactions in the glutamine binding pocket of E. coli GlnRS by systematic mutagenesis coupled to steady-state and transient kinetics of tRNA aminoacylation. We rely here on the recently developed highly sensitive assay in which the 3′-internucleotide linkage of the tRNA is 32P-labeled, and product formation is monitored by thin-layer chromatography (TLC) after digestion of the tRNA with P1 nuclease (25). The use of unlabeled glutamine permits the use of very high concentrations of this amino acid, a necessity for the examination of mutants in which the Km is substantially elevated (14). Using this approach in both steady-state and single-turnover kinetic experiments then permits substantial insight as compared with a steady-state analysis alone. Measurements of Kd (Gln) and kobs for aminoacylation allows us to deduce the importance of specific enzyme–substrate and intramolecular contacts in conferring glutamine binding affinity and proper positioning of the three reactive moieties in the two-step reaction, respectively. Comparison of these mechanistic constants with the steady-state parameters kcat and Km permits further inferences with respect to the rate-limiting step in the reactions of the mutants. Together, these measurements on wild-type GlnRS and a selected set of eight mutant enzymes offer detailed insight into the architectural underpinnings of the genetic code for glutamine.
Experimental Procedures
Mutagenesis
Oligonucleotide primers for mutagenesis were purchased from IDT. The mutations were made using the Quickchange site-directed mutagenesis (Stratagene) protocol with the GlnRS-6His (QRSH) plasmid as template (18), followed by transformation into XL-1 blue supercompetent cells (Stratagene). DNA sequencing of mutant genes was performed by the University of Iowa commercial sequencing facility. After sequence verification, the mutant plasmids were transformed into BL21-DE3 (pLysS) supercompetent cells (Invitrogen) for expression.
Enzyme Purifications
All GlnRS mutants were made as His6 C-terminal-tagged enzymes, and were expressed in BL21-DE3 (pLysS) cells by induction with 1 mM IPTG at OD600 between 0.4 and 0.6. Cells were resuspended in a buffer containing 0.5 M NaCl, 50 mM HEPES (pH 7.2), 10 mM imidazole, and were disrupted by sonication. 100 mM PMSF, 100 mM benzamidine, 50 μL of RQ1 DNase (Promega), and 15 mM MgCl2 were added to the lysate, and the mixture was stirred at room temperature for 20 min. Lysed cells were then centrifuged for 30–45 min at 15,000 rpm, and the pellet was discarded. Enzymes were purified on 5 mL NTA-agarose nickel affinity columns (Amersham Biosciences) equilibrated in 10 mM imidazole, 20 mM HEPES (pH 7.2), 1 mM β-mercaptoethanol, and 0.5 M NaCl; the elution buffer was identical to the equilibration buffer except for the inclusion of 120 mM imidazole. Eluted enzymes were dialyzed for 6–12 h into storage buffer containing 50% glycerol, 20 mM potassium phosphate (pH 7.4), 50 mM KCl, 2 mM DTT. His-tagged GlnRS mutants were recovered at better than 99% purity as judged by SDS-PAGE, and were stored at high concentration at −20 °C, as described for the native enzyme (12). Enzyme concentrations were determined by UV absorbance (ε280 (1 mg/mL) = 1.06). The C-terminal His6 tag is without effect on the catalytic properties of GlnRS (27). Correct folding of the mutants is highly likely based on the high overexpression levels and stabilities in the cellular extract.
tRNA Synthesis and Purification
E. coli G-1 tRNA2Gln (containing a catalytically neutral U1G mutation to promote efficient transcription initiation) was transcribed in high yield from a synthetic DNA template. The template was constructed from complementary synthetic oligonucleotides containing a short central overlapping region, which were extended to form the full-length duplex by treatment with the Klenow fragment of E. coli DNA polymerase I (28). The DNA template incorporated 2′-O methyl sugars at the two 5′ nucleotides in the noncoding DNA strand, which is required in this system to produce a high proportion of enzymatically active tRNA transcripts by T7 RNA polymerase. Transcription of E. coli tRNA2Gln from unmodified DNA templates generates 3′-end heterogeneity, such that the maximum plateau aminoacylation acheivable is 15–20% (28). The transcription reactions were then loaded on a 5 mL DE-52 (Whatman) column, eluted, precipitated with ethanol, dried, and stored at −20 °C. Prior to use the tRNA was resuspended in and dialyzed against highly purified water.
Aminoacylation Assay Using 32P-Labeled tRNA
tRNA was 32P-labeled at the 3′ terminal internucleotide linkage using the exchange reaction of tRNA nucleotidyltransferase, as described (12, 25, 27). To ensure maximal substrate activity, labeled and unlabeled tRNA were refolded by mixing to the appropriate final concentration and heating at 65 °C for 3 min, followed by addition of MgCl2 to 7.5 mM and cooling to ambient temperature. All reactions were quenched in a buffer containing 0.1% SDS and 0.15 M sodium acetate (pH 5.2). P1 nuclease digestions were performed by adding 1–5 μL of the reaction mixture to a microtiter well containing 3–5 μL of 0.1 mg/mL P1 nuclease (Fluka) and 0.15 M sodium acetate (pH 5.2), followed by incubation for 10 min at ambient temperature. Aminoacylated tRNA (as 3′-aminoacylated A76) and the unreacted substrate (as unmodified AMP) were separated by TLC and quantitated by phosphorimaging analysis. Steady-state assays were performed at 10 mM MgCl2, 20 mM Tris (pH 7.4), 5–10 mM ATP, 1 mM DTT and appropriate concentrations of glutamine ranging from 0.025–100 mM. The tRNA concentrations used were 10 μM for C229S, 12.5 μM for mutants Y211F and E257A, 15 μM for mutants R30K, H215N, Q255N, and S227A, and 22.5 μM for D212A. Enzyme concentrations ranged from 3–100 nM depending on the mutant. Steady-state analysis of GlnRS in the presence of 3′-dATP was performed using 50 nM enzyme, 12.5 μM tRNA and 5 mM 3′-dATP. Time courses were adjusted to ensure that maximal product formation remained in the range of 5%–20% of the total acylable tRNA. All steady-state reactions were performed in triplicate at 37 °C. Plateau aminoacylation levels were measured for all preparations of tRNAGln by performing end-point titrations under conditions of molar excess enzyme, and were in the range of 75%–93% for all experiments described herein.
Rapid Chemical Quench Kinetics
All single-turnover measurements to determine kobs were performed using a rapid chemical quench flow apparatus (KinTek RQF-3) at saturating concentrations of all substrates. The temperature was maintained at 37 °C for all experiments. Enzyme and substrates in two 20 μL sample loops were rapidly mixed into a single reaction loop of specified dimensions to control the time of the reaction. The mole ratios of enzyme to tRNAGln were maintained at 3:1 for all experiments. Reactions were quenched by adding 50 μL of a solution containing 400 mM sodium acetate (pH 5.2), 0.1% SDS. Six to eight time points were collected for each kobs determination. The reactions were fit to a first-order exponential equation (Y = A1 e−kobst), where Y represents moles of Gln–tRNAGln product formed, and plotted using Kaleidagraph (Synergy Software). For the 3′-dATP experiments, the analogue (cordycepin) was purchased from Trilink Biotechnologies.
The glutamine concentration dependencies of the single-turnover reactions were determined by titrating the concentration of glutamine while maintaining saturating levels of tRNA as described for the wild-type enzyme (18). To determine the glutamine Kd values, the reactions contained 15 μM enzyme, 5 μM tRNAGln, 7.5 mM ATP, 13.75 μM MgCl2, 50 mM Tris (pH 7.4), 5 mM DTT, and varying glutamine concentrations between 0.1–78 mM, depending on the mutant. For all experiments, the concentration of the 3′-end-labeled tRNAGln was negligible compared to the unlabeled concentration. The kobs values determined at each substrate concentration were fit to a hyperbolic binding equation to derive the Kd value under aminoacylation conditions. All single-turnover data under saturating conditions were acquired in duplicate and plotted using Kaleidagraph.
Electrostatic Surface Potential Calculations
Electrostatic potentials were determined using the Adaptive Poisson–Boltzmann Solver (APBS) run as a plugin for Pymol (29, 30). Due to limitations associated with the program input, for calculations including the tRNA only the active-site proximal six base-pairs of the acceptor stem, and the four bases of the single-stranded 3′-end, were retained. Solvent molecules and other ligands were removed from the structure. The charge of each atom for each pH calculation was determined using the program pdb2pqr (31). The protein dielectric constant was set at 20 and the solvent dielectric was assigned a value of 80. pKa assignments for all ionizable protein groups were determined with the program PROPKA (32). The scale is displayed in units of kbT/e, where kb is Boltzmann's constant, T is temperature, and e is the charge on the electron. The GlnRS structure used for electrostatics calculation corresponds to PDB code 1O0b.
Results
Glutamine–ATP Interaction
We sought to examine the importance of the unusual hydrogen bond formed between the substrate glutamine amide oxygen atom and the 3′-OH moiety of ATP. To accomplish this we made use of the ATP analogue cordycepin, in which the 3′-ribose hydroxyl group is altered to hydrogen, removing the hydrogen-bonding capability. The hydrogen bond with glutamine is formed in both the GlnRS ternary (GlnRS–tRNAGln–QSI) and quaternary (GlnRS–tRNAGln–Gln–AMPCPP) complexes, and appears to be strong based on the observation that the electronegative atoms are 2.5–2.7 Å apart (12, 13) (Figure 1). Replacement of the 3′-OH group with hydrogen also removes an interaction with a bound water molecule that in the QSI complex bridges as well to Ser46, Asn50, and Leu228. In the quaternary complex, the 3′-OH also contacts a water molecule, but the conformation of the ribose and position of this water differ slightly from their positions in the QSI complex, probably because the phosphates of AMPCPP are disordered in that structure (12). All of the ribose contacts made in the QSI structure are identical to those formed in the ternary GlnRS–tRNAGln–ATP complex, except for the missing glutamine (16).
Figure 1.
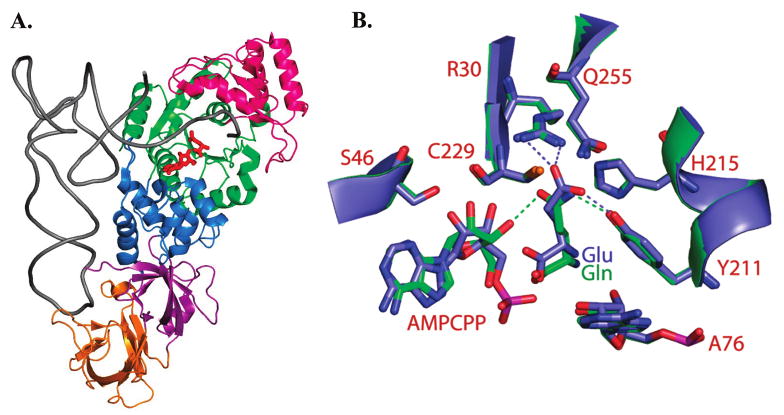
(A) Structure of E. coli GlnRS bound in a quaternary complex with tRNAGln, glutamine, and the ATP analogue AMPCPP (12). The phosphates of AMPCPP are disordered in this structure. The Rossmann fold catalytic domain is colored green, the inserted domain that binds the tRNA acceptor end is bright pink, a helical subdomain following the Rossmann fold in primary sequence is shown in dark blue, and the proximal and distal anticodon binding domains are depicted in magenta and orange, respectively. (B) Superposition of the GlnRS active site in quaternary complexes bound to tRNAGln, AMPCPP and either cognate glutamine (green) or noncognate glutamate (blue). Selected hydrogen bonds are shown as dotted lines. Both bound glutamine and glutamate participate in a hydrogen bond with Tyr211, but only cognate glutamine accepts a hydrogen bond from the 3′-OH group of ATP. Glutamate also makes direct electrostatic hydrogen bonds with Arg30.
3′-dATP functions as a poor mimic of ATP in the glutaminylation reaction. Catalytic efficiency as measured by the steady-state kcat/Km parameter is decreased by over 103-fold as compared with ATP, with the greater part of this effect appearing as a 70-fold reduction in kcat (Table 1). The Km for 3′-dATP is elevated by only 2-fold, suggesting that the hydrogen bonds made by the 3′-OH group are not major determinants of the binding pocket complementarity for ATP. However, the Km for glutamine is elevated by 16-fold in the 3′-dATP reaction, suggesting that the glutamine–ATP hydrogen bond is a significant determinant of the binding pocket complementarity for glutamine. To assess whether the strong kcat effect reflects a decreased rate for the chemical steps of the reaction, we also measured the composite rate constant for the two-step glutaminylation reaction under conditions of enzyme molar excess, and with saturating concentrations of all three substrates. As compared with the native ATP-mediated reaction, glutaminylation mediated by 3′-dATP is decreased in kobs by nearly 50-fold (Table 1), suggesting that the distal hydrogen bond likely also promotes the correct positioning of the reactive groups of the two substrates with each other, and/or with the 2′-OH group of the 3′-A76 tRNA nucleotide. In this single-turnover reaction carried out under conditions in which all three substrates are saturating, kobs represents either kchem, the composite rate of the two-step aminoacylation reaction, or a closely linked first-order physical rearrangement of the complex. Only the overall two-step reaction can be measured, since tRNA is required for synthesis of glutaminyl adenylate.
Table 1.
Kinetic Analysis of Wild-Type and 3′-dATP Reactionsa
| ATP | 3′-dATP | |
|---|---|---|
| kcat (s−1) | 3.2 ± 0.5 | 0.045 ± 0.02 (71)b |
| kobs (s−1) | 28 ± 2 | 0.6 ± 0.08 (47) |
| Km (ATP/3′-dATP) (mM) | 0.2 ± 0.02 | 0.41 ± 0.03 (2) |
| Km (GLN) (mM) | 0.26 ± 0.04 | 4.24 ± 0.01 (16) |
| kcat/Km (GLN) (s−1 M−1) | 1.2 × 104 | 11.3 (1060) |
Values for wild-type GlnRS are taken from ref 26.
Numbers in parentheses designate the fold diminution from wild-type for kcat, kcat/Km, and kobs, and the fold elevation from wild-type for Km.
These data show that a hydrogen-bonding interaction between the distal, nonreactive portions of two bound substrates to GlnRS makes a very significant contribution to the efficiency of aminoacylation. The crystal structure of GlnRS bound to tRNAGln, AMPCPP and glutamate shows that the distance between the ATP 3′-OH and the nearest glutamate carboxylate oxygen atom is increased to 4.6 Å, well outside hydrogen-bonding distance (Figure 1). Although this observation predicts that misacylation of tRNAGln with glutamate might be unaffected by the use of 3′-dATP, we were unable to detect any reaction with the analogue even at very high glutamate concentrations and under single-turnover conditions. Thus, misacylation must be significantly decreased even below the already-low kcat/Km of 9.5 × 10−4 M−1 s−1 characteristic of wild-type GlnRS in the ATP-dependent reaction (12). This suggests that the use of 3′-dATP may perturb the amino acid pocket structure more globally, implying that the 16-fold increase in Km for cognate glutamine might arise in part from a longer-range perturbation of interactions made with Tyr211 or with bound water molecules.
The dependence of GlnRS catalytic efficiency on this distal interaction may be unique among tRNA synthetases. The crystal structure of the closely related GluRS enzyme bound to tRNAGlu and a glutamyl adenylate analogue indicates that the comparable distance between the 3′-OH of ATP and the nearest glutamate carboxylate oxygen atom is 3.6 Å, too far to form an interaction with significant hydrogen bonding character (15). Examination of all other AARS structures in which the adenylate, the free amino acid and ATP, or suitable analogues are bound fails to reveal a comparable distal contact in any active site. It appears then that the required mutual positioning of the reactive moieties of the ATP and amino acid is accomplished solely by interactions with the protein in other AARS, but that the comparable juxtaposition in GlnRS to synthesize glutaminyl adenylate depends as well on the substrate–substrate interaction. This contact may also stabilize the orientation of the mixed anhydride linkage for attack by the 2′-OH of tRNA-A76 in the second step. A further effect on the product release step is possible as well, because, if still present, the additional contact made between the AMP and Gln–tRNAGln would presumably need to be broken during dissociation.
Protein Network Stabilizing Glutamine Binding: Analysis of the Reaction Pathway in Mutant Enzymes
The substrate glutamine is bound in a pocket formed primarily from residues in the second half of the catalytic Rossmann fold domain. Amino acids in the first α-helix and in each of the two parallel β-strands of the α-β-α-β motif contribute to formation of the binding site, while a single residue from the first half of the catalytic domain, Arg30, also participates (Figure 2). The glutamine side chain rests on the imidazole ring of His215, while the lid of the pocket is formed from the phenyl group of Phe233, which, together with Tyr211, in turn stacks directly on the adenine base of the tRNA 3′-A76 nucleotide. At the back of the pocket, the glutamine amide makes direct steric contact with the β-methylene group of Cys229. The amide oxygen accepts no strong direct hydrogen bond from any protein moiety, but lies 3.4 to 4.0 Å from the closest nitrogen of the Arg30 guanidinium group. The amide nitrogen donates hydrogen bonds to the Tyr211 phenol hydroxyl group and to a buried water molecule, completing the primary shell contacts (Figure 3). In general, the absence of an unambiguous hydrogen-bond acceptor (e.g., a carboxylate or main-chain amide oxygen) for the hydrogens located on the glutamine amide nitrogen, and the lack of a strong hydrogen-bond donor to the amide oxygen, creates a pocket with surprisingly meager complementarity for its cognate substrate. However, the reproducibility of these contacts as found in two different liganded complexes, as well as the observation that GlnRS maintains catalytic activity in this crystal lattice (33), suggests that the interactions indeed depict a functional environment capable of very high catalytic efficiency and of 107-fold specificity against misacylation with glutamate (12). Comparison of the glutamine binding site in the GlnRS–tRNAGln–QSI and GlnRS–tRNAGln–Gln–AMPPCP complexes, with the empty site in the GlnRS–tRNAGln–ATP ternary complex, shows that Gln binding in the presence of tRNA causes small, local structural changes, including the rearrangement of water molecules (12, 13, 16).
Figure 2.
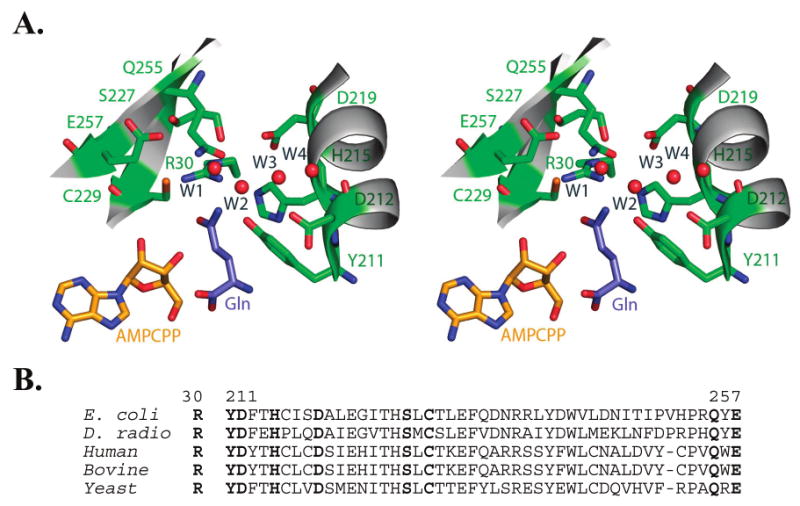
(A) Divergent stereoview of the glutamine binding pocket in E. coli GlnRS. Buried water molecules in the pocket are depicted as red spheres. Nomenclature for the waters follows Bullock et al. (12); see also Figures 3 and 5B herein. The phosphates of AMPCPP are disordered in this quaternary structure bound to substrate glutamine (12). The binding interactions made by the glutamine portion of a glutaminyl adenylate analogue are nearly identical (13). (B) Sequence alignment of selected GlnRS enzymes in the region corresponding to the glutamine binding pocket. Residues 211–260 comprise the second half of the catalytic Rossmann fold. Residues shown in panel A are highlighted in boldface.
Figure 3.
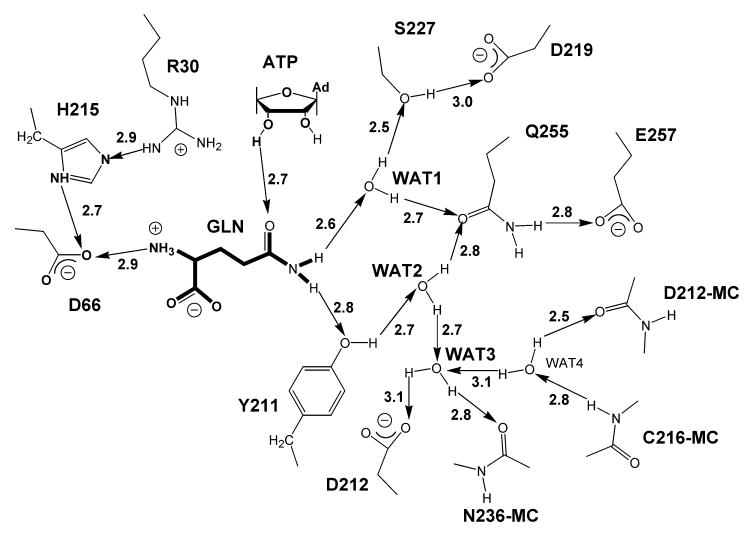
Schematic drawing of a proposed network of hydrogen bonds in the amino acid binding pocket of GlnRS with substrate glutamine bound. Arrowheads point toward the hydrogen bond acceptor of each pair, and numerals indicate the distance in Å between the electronegative atoms of the pair, as estimated from a 2.3 Å crystal structure (12). MC indicates main chain. Nomenclature for the waters follows Bullock et al. (12).
The demonstration that the immediate binding environment is insufficient to confer selectivity against glutamate (12), and the finding that a swap of distal surface loops does confer a capacity for Glu-tRNAGln synthesis (14), focused our attention on the detailed roles of the second and third-shell amino acids in providing cognate Gln recognition as well. In addition to the primary shell, the binding pocket features three other well-ordered water molecules that form hydrogen bonds with each other and with the side chains of Tyr211, Asp212, Gln255, and Ser227, as well as with several main-chain groups (Figure 3). In addition, the side chains of Asp219 and Glu257 provide possibly crucial stabilization of particular rotamers of Ser227 and Gln255, thus making possible an array of proposed hydrogen-bond donor–acceptor pairs ultimately connecting to the bound substrate, for which the orientations of hydrogens and lone electron pairs appear well-defined. Remarkably, all of these amino acids, as well as several others (Arg64, Asp66, Glu222) that make further distal contacts with the above-mentioned residues, are nearly fully conserved among the 71 bacterial and 10 eukaryotic GlnRS enzyme sequences that are available for analysis (the only known exceptions are the substitution of valine at position Gln255 in the enzymes from Dictyostelium discoideum and Lupinus luteus) (Figure 2B). This extraordinary degree of amino acid conservation in regions not contacting substrate, which is present to a much smaller extent among GluRS enzymes, suggests that the GlnRS binding pocket is constructed with an unusual requirement for structural precision, and further motivates interest in exploring the importance of particular interactions in conferring glutamine binding affinity and catalytic efficiency.
To explore the architecture of the glutamine binding pocket in GlnRS, we constructed and expressed a set of eight His-tagged protein mutants (Table 2). The mutant enzymes were separated from endogenous wild-type E. coli GlnRS by affinity chromatography on an immobilized metal ion resin, and were analyzed by a combination of steady-state and single-turnover kinetics to determine the Michaelis parameters, the rate of the combined two-step chemical reaction on the enzyme surface, and the affinity for glutamine (Figure 4). Saturating levels of ATP and tRNAGln were maintained in all experiments. For the single-turnover measurements, mixing controls established that the rate of the chemical steps is independent of how substrates are combined in each syringe of the chemical quench instrument.
Table 2.
Kinetic analysis of wild-type and mutant GlnRS enzymes
| kcat (s−1) | Km (GLN) (mM) | kcat/Km (s−1M−1) | kobs (s−1) | Kd (GLN) (mM) | |
|---|---|---|---|---|---|
| WT GlnRSa | 3.2 ± 0.5 | 0.26 ± 0.04 | 1.2 × 104 | 28 ± 2 | 1.1 ± 0.04 |
| C229S | 0.7 ± 0.2 (4.6)b | 1.2 ± 0.03 (4.6) | 580 (21) | 20.1 ± 5.1 (1.4) | 7.8 ± 2.1 (7.1) |
| H215N | 2.0 ± 0.1 (1.6) | 0.9 ± 0.2 (3.5) | 2220 (5) | 28.6 ± 1.4 (1.0) | 7.9 ± 0.1 7.2 |
| E257A | 2.3 ± 0.1 (1.4) | 2.0 ± 0.1 (7.7) | 1150 (10) | 25.5 ± 7.5 (1.1) | 9.2 ± 2.7 (8.4) |
| S227A | 4.6 ± 0.3 (0.7) | 3.1 ± 0.7 (11.9) | 1480 (8) | 21.0 ± 4.8 (1.3) | 11.2 ± 0.5 (10.2) |
| D212A | 11.6 ± 4.0 (0.3) | 60 ± 4 (230) | 193 (62) | >122c (<0.2) | >70c (>63) |
| Y211F | 1.2 ± 0.1 (2.7) | 35.5 ± 0.5 (140) | 34 (350) | NDd | ND |
| Q255N | 0.5 ± 0.2 (6.4) | 17.1 ± 3.0 (66) | 30 (400) | ND | ND |
| R30K | 2.6 ± 0.3 (1.2) | 6.3 ± 0.7 (24) | 410 (29) | 2.3 ± 0.3 (12.1) | 5.7 ± 3.0 (5.2) |
Values for wild-type GlnRS are taken from ref 26.
Numbers in parentheses designate the fold diminution from wild-type for kcat, kcat/Km, and kobs, and the fold elevation from wild-type for Km and Kd.
No saturation with glutamine could be observed at concentrations up to 70 mM.
Reproducible measurements of kobs and Kd [GLN] could not be made, possibly due to aggregation of the protein.
Figure 4.
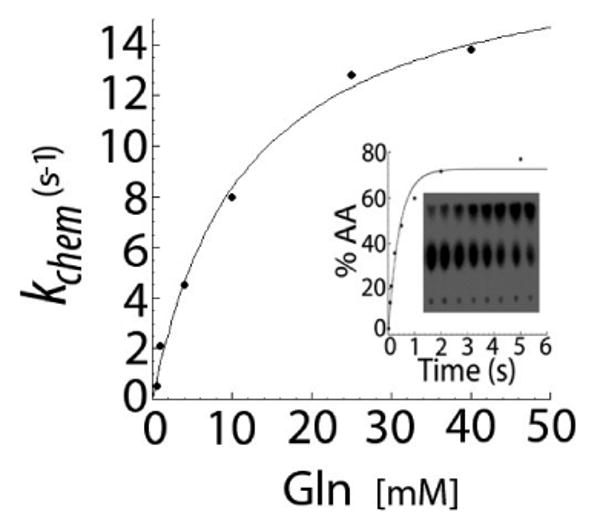
kobs dependence on the glutamine concentration for the E257A GlnRS mutant, fit to a hyperbolic binding equation to determine the kobs(max) and Kd values. The kinetic constant kobs is very close to the wild-type GlnRS value, but the Kd (Gln) is elevated by 8.4-fold (see Table 2). The inset shows a TLC plate demonstrating the separation of substrate (AMP; center of TLC plate) and product (aa-AMP; top of TLC plate). The time points depicted are 0.02, 0.08, 0.12, 0.25, 0.5, 1.0, 2.0, and 5.0 s. The reactions were spotted at the bottom of the plate.
For wild-type GlnRS, we have previously used pre-steady-state kinetics to establish that the rate-limiting step in catalysis corresponds to product dissociation: product formation monitored under conditions of modest substrate excess revealed a rapid burst corresponding to the first turnover on the enzyme (kobs), followed by a slower, linear steady-state rate (kcat) (26). The conclusion that product dissociation is rate-limiting was also reached by comparing the kcat and kobs values measured in separate steady-state and single-turnover experiments (kobs exceeds kcat by 9-fold for wild-type GlnRS; Table 2). It was also shown that Kd for glutamine exceeds Km by 4-fold, consistent with the notion that a step following formation of the ES complex is rate-limiting (35). To examine whether the binding-pocket mutants retain these kinetic characteristics, we compared kcat with kobs, and Km (Gln) with Kd (Gln), for each of the mutants to assess possible alterations in the rate-limiting step. With the exception of R30K, all of the mutants for which single-turnover kinetics could be reliably measured exhibit the property that kobs > kcat, with kobs faster by factors of 3- to 29-fold. Therefore, while the rate of the chemical steps on the enzyme may partially contribute to kcat for S227A (kobs is less than 5-fold greater than kcat), release of aminoacyl-tRNA as the rate-limiting step is generally retained. For R30K, however, kobs and kcat are identical within experimental error, and this is achieved entirely via a 10-fold reduction in kobs. For this mutant the rate-limiting step corresponds to the chemical transformation on the enzyme. With the exception of R30K, all of the mutants also exhibit Kd (Gln) greater than Km (Gln), further confirming that their rate-limiting steps correspond to release of aminoacyl-tRNA (Table 2). For R30K, the rate-limiting chemistry step predicts that Km = Kd, as observed (36). In general, the strength of glutamine binding varies along the reaction coordinate, and the mutants disrupt the interaction at different points. Thus, Arg30 contributes most to the transition state for the enzyme-bound conversion of substrates to products, while the other mutants have greater differential effects on ground-state binding.
It is also of interest to note that for five of the mutants product release remains rate-limiting (kobs > kcat), while the Kd for glutamine is elevated (Table 2). For S227A and D212A, kcat is actually improved as compared with wild-type GlnRS, so that the mutation weakens both glutamine affinity and, by inference, the affinity of Gln–tRNAGln as well (to enable faster product release). For H215N and E257A, this circumstance is only slightly altered: the elevated glutamine Kd values are accompanied by only very small decreases in kcat. In the case of C229S, the 7-fold elevation in glutamine Kd is accompanied by nearly 5-fold decreased kcat, suggesting, perhaps counterintuitively, tighter Gln–tRNAGln binding in this case. These findings may be reconciled by considering that the enzyme-tRNA interactions are likely optimized to balance reasonably tight tRNA binding affinity (Kd (tRNA) = 0.36 μM as deduced from pre-steady-state analysis and global kinetic fitting of the tRNA concentration dependence of product formation during the aminoacylation reaction (18)) with a decreased affinity for the product Gln–tRNAGln, to enable product release at a rate sufficient to sustain in vivo function. In C229S, this balance may be disrupted to generate weaker tRNAGln affinity and tighter Gln–tRNAGln affinity, providing another indication of the cooperative nature of the amino acid and tRNA binding sites in GlnRS (14, 17, 18, 27). A possible rationale for the unusual behavior of C229S could be that the serine hydroxyl group makes additional direct or water-mediated hydrogen bonds with glutamine. In contrast, the more hydrophobic Cys229 thiol is oriented out of the solvated pocket.
Protein Network Stabilizing Glutamine Binding: Structural Determinants of Recognition and Catalysis
In designing mutations to probe the architecture of the binding pocket, we sought to maintain the structural integrity of the enzyme, and particularly of the direct packing contacts to substrate glutamine, to the maximum extent possible. Therefore, all amino acids were altered to isosteric or smaller residues. We describe the results in turn for three subclusters of residues chosen based on physical proximity and mutual interactions: (i) Arg30/His215/Ser227; (ii) Tyr211/Asp212; (iii) Cys229/Gln255/Glu257 (Figure 5). Not all desired mutants could be constructed: we attempted but were unable to successfully express either E257Q or D212N in addition to those studied (Table 2). In the case of E257Q, we found that the mutant enzyme was susceptible to autocatalytic cleavage, as shown by the appearance of two prominent bands after metal ion affinity chromatography. The sizes of the fragments were consistent with a single chain scission at the position of the mutant Gln257 residue. The proposed mechanism is similar to that of asparagine deamidation, and involves attack of the asparagine amide nitrogen on the carbonyl carbon of the adjacent peptide bond to liberate the carboxyl portion of the protein with concomitant formation of a five-membered succinimide ring intermediate (37, 38).
Figure 5.
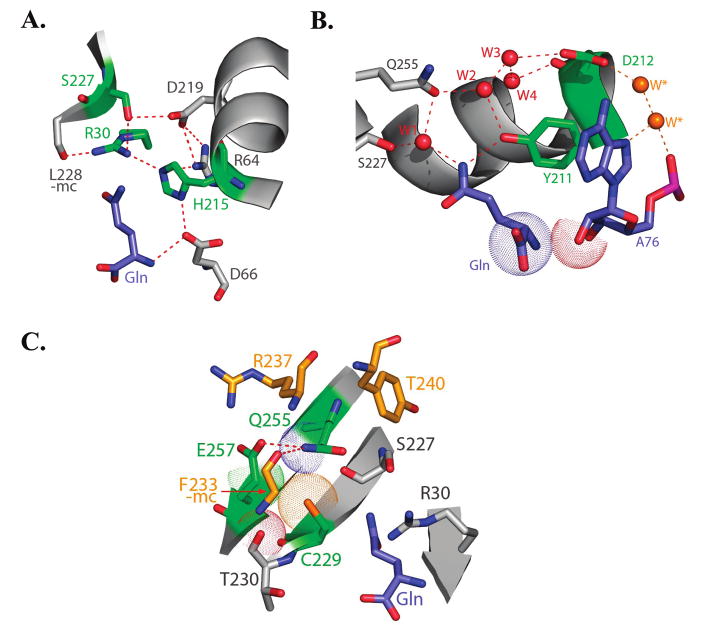
(A) Depiction of a portion of the glutamine binding pocket involving the interactions of Arg30, His215 and Ser227. Substrate glutamine is at the bottom, and hydrogen bonds are depicted in red dashed lines. The tautomer of His215 appears well-determined by interactions with the charged Arg30 and Asp66 groups. (B) Depiction of a portion of the glutamine binding pocket showing interactions of Tyr211 and Asp212. Waters labeled as W*, in yellow, are outside the immediate glutamine binding pocket and bridge Asp212 with the 3′-A76 nucleotide of tRNA. Juxtaposition of the substrate glutamine (bottom) with tRNA-A76 is depicted with van der Waals surfaces (bottom). (C) Depiction of a portion of the glutamine binding pocket showing interactions of Gln255, Cys229 and Glu257. Again, substrate glutamine is at the bottom of the drawing and selected hydrogen bonds are depicted as red dashed lines.
(i) Arg30/His215/Ser227 Cluster
Although substrate glutamine makes at best a weak electrostatic contact with Arg30, the R30K mutation is nonetheless decreased in catalytic efficiency (kcat/Km) by nearly 30-fold. Glutamine binding affinity is weakened by 5-fold, while the rate of chemical transformation on the enzyme is decreased 12-fold (Table 2). Arg30 is stabilized in part through donation of a hydrogen bond to His215 (Figure 5A). The H215N mutant, however, which removes both this interaction and a comparable contact with the Arg64 guanidinium, while retaining the capacity to form an apparently key hydrogen bond with Asp66, retains the wild-type kobs for aminoacylation. Glutamine binding affinity in H215N is decreased 7-fold compared to wild-type GlnRS, and is identical within experimental error to R30K. Therefore, it appears that the Arg30–His215 and Arg64–His215 hydrogen bonds are important mainly in conferring binding affinity. His215 also partially packs against the phenol ring of Tyr211, but the structure suggests that Asn at this position should be able to maintain these packing interactions.
The substantially reduced kobs measured for R30K, then, appears not to arise from disruption of the contact between Arg30 and His215. It may instead be a consequence of breaking other contacts with the backbone carbonyl oxygen group of Leu228, and with the hydroxyl group of Ser227. Ser227 makes a water-mediated contact with the substrate glutamine, implicating this network in proper positioning of the reactive carboxylate group. However, the S227A mutant also exhibits kobs that is only slightly diminished as compared with wild-type GlnRS, suggesting that the Arg30–Ser227 contact, like that between Arg30 and His215, is similarly important in conferring binding affinity (S227A binds Gln 10-fold weaker than wild-type GlnRS; Table 2). Modeling suggests that the terminal NH3+ of Lys30 in the R30K mutant might be capable of interacting with either the Ser227 side-chain hydroxyl or the Leu228 main-chain carbonyl, but likely could not simultaneously donate hydrogen bonds to both, as observed for Arg30. Hence, the Arg30 contact with Leu228 appears as the most likely determinant of the aminoacylation chemistry in this portion of the pocket.
It is also of interest that the R30A mutant is reduced 40-fold further in kcat/Km as compared with R30K (14). Thus, retention of the positive charge at this position, together with the capacity to make a subset of the intramolecular contacts, provides a significant portion of the catalytic benefit conferred by the wild-type residue. Further, the unliganded structure of GlnRS shows that Arg30 is among the amino acids in the pocket (others include the directly interacting Asp66 and Tyr211 side chains; Figure 3) that undergo a local conformational change upon binding of ATP, glutamine, and tRNAGln. In apo GlnRS, Arg30 interacts directly with the carboxylate of Asp219, and this contact is broken and replaced with the hydrogen bond to the Leu228 main-chain carbonyl in the quaternary complex (Figure 5A). Therefore, part of the catalytic deficits in R30K and R30A may arise from impairments in achieving the catalytically productive binding pocket configuration.
(ii) Tyr211/Asp212 Cluster
The only direct contact made by a protein group with substrate glutamine is the hydrogen bond donated by the amide −NH2 group to the phenolic oxygen of Tyr211 (Figure 3). The Tyr211 hydroxyl also participates in the water-mediated hydrogen-bonding network, by serving as a donor to Wat2 (Figure 5B), and the aromatic ring stacks with Phe233, His215, and the 3′-A76 of tRNA. Analysis of the Y211F enzyme, which should preserve the stacking interactions, shows that removal of the capacity for hydrogen bonding at this position results in 350-fold decrease in kcat/Km (Table 2), with most of the effect appearing as a 140-fold elevation in Km. Y211F was previously studied by steady-state kinetics using an assay in which the radiolabel is carried in the amino acid. However, the greatly elevated Km of this mutant renders that analysis unreliable, because radiolabeled glutamine from the commercial source used is available only at relatively low concentrations (39).
Asp212 is located on the enzyme surface, where it accepts hydrogen bonds from Asn236 and two water molecules. One of these waters, Wat3, lies within the binding pocket, while the other connects, through a further water-mediated hydrogen bonding linkage, with the N7 ring nitrogen of tRNA A76. Wat3 accepts hydrogen bonds from both Wat2 and Wat4, and further donates a hydrogen bond to the backbone carbonyl oxygen of Asn236, creating a tight network of interactions. Like Y211F, the D212A mutant is severely compromised in the Km for the amino acid, while retaining the ability to effectively catalyze the reaction. The properties of D212A are extreme among the set of eight mutants studied here: both kcat and kobs are actually improved as compared with wild-type GlnRS, while binding affinity is so impaired that no saturation could be detected at 70 mM glutamine (Table 2; only lower bounds can be estimated for kobs and Kd). The high kcat and kobs retained by these mutants suggests that the stacking of the A76 adenine ring is not significantly perturbed, so that the orientation of the catalytic groups is maintained. However, the mutations presumably destabilize or otherwise alter the structure of the binding pocket so as to significantly raise Km and Kd.
It is notable that, with the exception of Glu257, Asp212 is more distant from substrate glutamine than any of the amino acids studied, approaching only to within 8 to 9 Å. Yet the D212A mutant is by far the most perturbed in glutamine binding. This likely reflects an important role for the detailed distribution of charged residues, for the interactions of the water-mediated hydrogen-bonding network in the pocket, or, most likely, for both these features. The E257A mutant is much less perturbed in glutamine binding (Table 2; see below), but the Glu257 negative charge points away from the pocket into solvent on the opposite side of the Rossmann fold. By contrast, the Asp212 carboxylate is connected perhaps more directly with the glutamine substrate, via its contacts with solvent molecules and with Tyr211.
To probe more deeply into the mechanism by which removal of the negatively charged Asp212 carboxylate so severely impacts glutamine binding affinity, we carried out electrostatic surface potential calculations using the APBS module interfaced with PyMol (29, 30). These calculations also returned estimated pKa values for all ionizable protein groups. Among the glutamine binding pocket residues, pKa's were significantly shifted from their solution values for His215 (calculated pKa = 3.3), Arg30 (calculated pKa = 9.1), and Asp212 (calculated pKa = 7.3). The lowered pKa for His215 is consistent with its structural role in accepting a hydrogen bond from Arg30 (Figure 5A). The lowered pKa for Arg30 was unexpected, but remains sufficiently high to maintain the guanidinium group in the positively charged form at physiological pH. The most striking observation is the upward shift for Asp212 by 3.5 pH units. The calculation therefore predicts that the Asp212 carboxylate titrates in the physiological range, a feature not observed for any other binding pocket residue, including Cys229.
We calculated the electrostatic surface potential of GlnRS alone, using coordinates derived from the tRNA cocrystal structure, as well as of the enzyme bound to the six proximal tRNA acceptor stem base-pairs and four single-stranded nucleotides at the 3′-end (tRNA nucleotides 1–6; 67–76). This reveals an acidic protein, consistent with a calculated isoelectric point of 5.73. The tRNA binding orientation is clearly delineated as a relatively positive surface potential across the surface, suggesting that electrostatic guidance may play a significant role in formation of an early enzyme–tRNA encounter complex (Figure 6; panels I and II).
Figure 6.
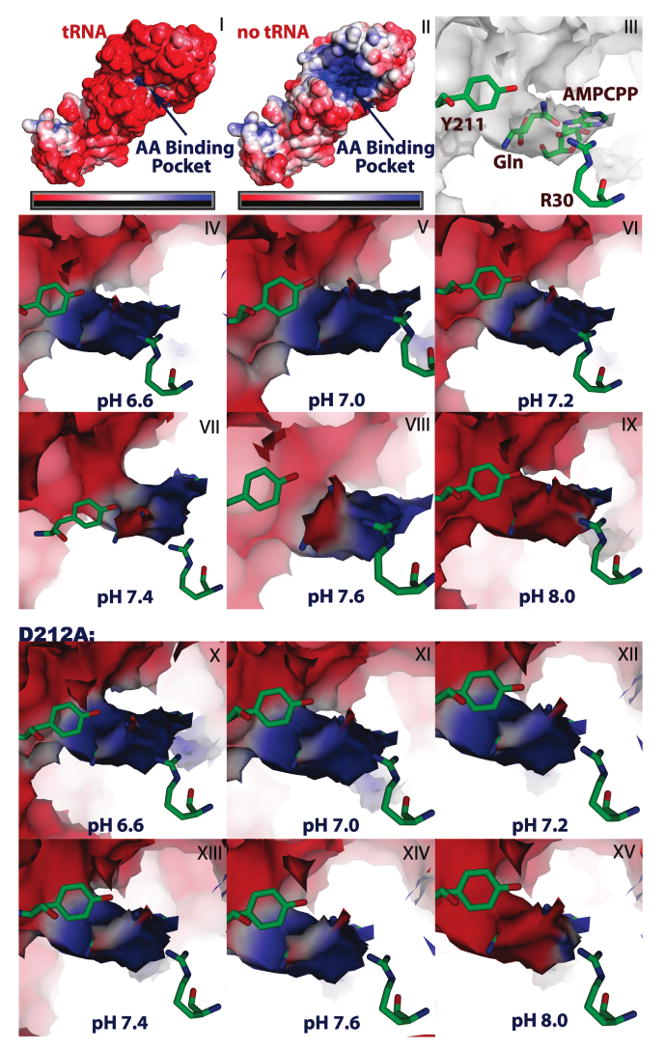
Electrostatic surface potential rendering of the amino acid binding pocket of GlnRS. Panels I and II are a depiction of the potential at the GlnRS surface in the presence and absence of the tRNA acceptor stem. Electronegative surface potential is shown in red and electropositive surface potential is in blue, with the scale ranging from −5kbT/e to +5kbT/e. Panel III is a depiction of the amino acid binding pocket oriented from the inner cavity of the protein looking toward the outer surface. The amino acid and AMPCPP substrates are superimposed on the electrostatic surface potential with residues Y211 and R30 as reference points for visualization. Panels IV through IX demonstrate the change in the electrostatic potential of the amino acid binding cleft as a function of pH for the wild type enzyme, while panels X through XV demonstrate the pH dependency for D212A GlnRS.
The calculations in the presence of tRNA were then systematically performed for pH 6.6, 7.0, 7.2, 7.4, 7.6 and 8.0, for both wild-type GlnRS and the D212A mutant. Coordinates for GlnRS D212A were obtained by truncating the Asp212 side chain to alanine. The data reveal that the potential directly at the glutamine binding surface is very sensitive to pH in this physiologically relevant range, with a shift from strongly positive to strongly negative at pH 6.6 and 8.0, respectively. In particular, the potential at the binding site for the amide −NH2 moiety of substrate glutamine shifts from positive to negative as the pH is raised from 7.2 to 7.4, corresponding to the deprotonation of Asp212 (Figure 6; compare panels VI and VII). At both pH 7.4 and pH 7.6, the surface potential is partitioned between positive and negative contours, in a manner consistent with the polar glutamine amide group: the amide oxygen atom binds in the positively charged region. The corresponding calculation for GlnRS D212A further confirms the prediction, made based on the calculated pKa values, that Asp212 influences the electrostatic potential in the glutamine binding site. At both pH 7.4 and pH 7.6, the electrostatic potential in the cleft is significantly more positive for the mutant, and no longer appears to orient the substrate glutamine amide into a preferred rotamer (Figure 6; panels XIII and XIV). Thus, the sharply weakened glutamine binding affinity in D212A appears likely to have its origin at least partly in an altered electrostatic potential in the binding cleft. This hypothesis may be tested by measuring the detailed pH-dependence of steady-state and pre-steady-state parameters for wild-type GlnRS and D212A. Further, these calculations also suggest the possibility of pH-dependent selectivity against noncognate glutamate, that is also driven by the protonation state of Asp212.
(iii) Cys229/Gln255/Glu257 Cluster
As mentioned above, the Cys229 β-methylene moiety packs directly on the substrate glutamine amide group. The sulfhydryl of Cys229, however, is oriented away from the glutamine substrate, to make packing interactions with the amide of Gln255 and the β-methylene group of Glu257 (Figure 5C). The hydrogen bond between the Gln255 amide and Glu257 carboxylate is one interaction that stabilizes the rotamer of Gln255 for interactions with WATS 1 and 2; a second such interaction is made via a hydrogen bond between the Gln255 amide NH2 group and the main-chain carbonyl oxygen of Phe233 (Figure 5C). Both the C229S and E257A mutants are without effect on kobs and exhibit 7- to 8-fold weaker glutamine binding affinity. These data demonstrate that this network of interactions is of only modest importance in contributing necessary elements of the binding pocket architecture.
Figure 7.
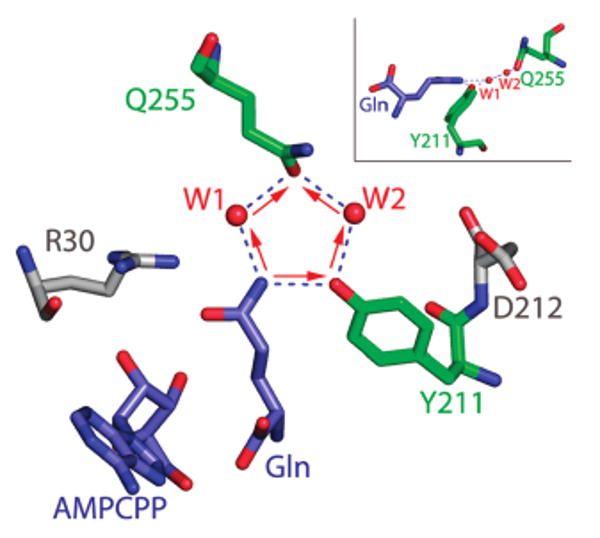
Circular hydrogen-bonding array involving the side-chain −NH2 group of substrate glutamine, Tyr211, Gln255, and buried waters WAT1 and WAT2. Hydrogen bonds are depicted as dashed blue lines. The polarity of the hydrogen bonds is indicated by the red arrows, in the direction from donor to acceptor. The antidromic nature of the circular hydrogen bonding network can be seen in the opposing directions of the individual arrows, which converge on the Gln255 oxygen acceptor. The pentagon of hydrogen bonds is also shown edgewise at upper right. This shows that the −NH2 moiety of substrate glutamine is slightly out of the plane formed by the other four groups.
Gln255 occupies a central position in the binding pocket, and has been shown to be a key determinant conferring selectivity against glutamate (14). In addition to contacts with Cys229, Glu257, the Phe233 backbone carbonyl group, and WATS 1 and 2, portions of the Gln255 side chain also pack against Arg237 and Tyr240 (Figure 5C). The mutation of Gln255 to asparagine was chosen for its ability to disrupt the hydrogen-bonding network, while retaining the capacity to make most or all of the packing interactions. Modeling of Asn255 at this position suggests that shifting the branch-point position closer to the backbone should not disrupt the protein structure, because there are no nearby main-chain atoms, and because the adjacent Glu257 side-chain and water molecules appear to be easily reoriented. We were not able to measure reliable single-turnover kinetic parameters for Q255N, possibly because of enzyme aggregation at the high concentrations required. However, reproducible steady-state parameters could be obtained. Q255N is reduced in kcat/Km by 400-fold as compared with wild-type GlnRS, a catalytic deficit greater than for all other mutants with the exception of Y211F. Most of the decrease in kcat/Km appears as a 70-fold increase in Km for glutamine (Table 2). Because replacement of Glu257 with alanine does not diminish kobs, it appears likely that disruption of the Gln255 hydrogen bond with the Phe233 main-chain carbonyl group, and with WATS 1 and 2, account for the strongly decreased activity of Q255N.
Discussion
Circular Hydrogen Bonds in the Glutamine Binding Pocket
The Y211F and Q255N mutants are each severely attenuated in activity by 350-fold and 400-fold in kcat/Km, respectively, with the majority of the deficits appearing as elevations in Km. The decrease in Y211F activity might most simply be interpreted as arising from the removal of the hydrogen bond with substrate. However, inspection of the structure suggests that disruption of a circular network of hydrogen bonds involving substrate glutamine, Tyr211, Gln255, and WATS 1 and 2 may provide a more comprehensive explanation for why Q255N and Y211F are attenuated in a severe and quantitatively similar manner. The Tyr211 hydroxyl group, WATS 1 and 2, the carbonyl oxygen of the Gln255 side-chain amide, and the −NH2 moiety of substrate glutamine form a five-membered circular hydrogen bonding array in which the polarity of the hydrogen bonds consists of two counter-running chains, an arrangement designated as antidromic by Saenger and colleagues (40, 41) (Figure 7). Hydrogen bond lengths in this circular array range from 2.6 Å to 2.8 Å, and bond angles are in the range 102° to 109°, very close to the ideal 108° adopted in a symmetric pentagon. Quantum mechanical studies suggested that circular hydrogen bonding arrangements are energetically favored above individual hydrogen bonds due to a cooperative effect that provides greater hydrogen bonding activity of an −OH group if it is already forming or donating a hydrogen bond (41). Although described by Saenger in crystals of cyclodextrins in which all of the hydrogen bonds contain oxygen, the increased stability of the circular arrangement may be preserved in mixed antidromic arrangements containing both oxygen and nitrogen. If this is so, the participation of the glutamine substrate −NH2 in this network might then explain why Km is elevated similarly for both the Q255N and Y211F mutants, since the circularity of the array would be destroyed in each case. The strongly destabilized binding of glutamine to D212A may also have its origins partially in the destabilization of Wat2 in the circular array, since Asp212 makes a water-mediated interaction with Wat2 through the intermediary of Wat3. The influence of the Asp212 negative charge on the binding pocket potential, however, suggests that this residue may exert its influence through multiple mechanisms (Figure 6).
Interestingly, the observed binding of one noncognate glutamate carboxylate oxygen in the position of the glutamine −NH2 would convert the antidromic circular hydrogen-bonding array to a heterodromic circular array with randomly oriented hydrogen bond polarities (12) (Figure 7). Saenger suggested that pentagonal circular arrays in general lower the total energy of the five included hydrogen bonds by 2–4 kcal as compared with the corresponding individual interactions (41). Although possible differences in stabilization energies among antidromic hydrogen-bonding circles, heterodromic circles, and homodromic circles (all hydrogen bonds having the same polarity) have not been examined, such variations might in principle provide part of the explanation for how GlnRS excludes glutamate from the binding pocket. The binding affinity of GlnRS for glutamine is at least 700-fold tighter than for glutamate, yet the crystal structures show that the binding pocket is actually more complementary for the noncognate substrate: only Glu makes a direct interaction with Arg30 (12, 18). If noncognate glutamate were to bind in the protonated state, a heterodromic circular network could still form, and the need for discrimination would presumably be greater since there would no longer be a possibility for selectivity based on electrostatic charge. Computational studies based on the coordinates of the cognate and noncognate GlnRS complexes may provide insight into this question, and into the stabilities of circular hydrogen bonds generally.
Interdependent Binding of Glutamine and tRNAGln
The interdependence of glutamine and tRNAGln binding can be probed by comparing the glutamine binding affinity of a particular mutant, with the effect of the mutation at the catalytic step as measured by the kobs parameter. Retention of a fast kobs that approaches the wild-type GlnRS value of 28 s−1 suggests that the glutamine binding pocket mutation has not perturbed the necessary precise juxtaposition of the reactive moieties of ATP, glutamine and tRNAGln in the active site—and thus does not implicate a pathway of communication through which binding of glutamine propagates changes at the tRNA binding interface. The existence of such a pathway, but not the molecular details, is demonstrated by the finding that binding of noncognate glutamate to GlnRS weakens tRNAGln binding affinity by 35-fold (18). Here, all mutants for which Kd [Gln] could be reliably determined do show significant losses of affinity (from 5-fold to over 60-fold; Table 2), but with the exception of R30K, no decreases in kobs were observed. In previous work, we found that mutations at both Glu34 and Glu73 at the opposite periphery of the active site significantly decreased the affinity for glutamine, but only the Glu73 mutants also affected kobs for aminoacylation (42). When we probed interactions in the anticodon region, we found that contacts at position U35, but not G36, significantly perturbed both Kd (Gln) and kobs (27). In this case, the enzyme contacts at U35 were clearly propagated through the enzyme–tRNA complex structure by some 40 Å to reach the active site (34). Clearly, further series of experiments are required next in order to examine how tRNAGln affinities may be altered in these mutants, so that it becomes possible to determine how tRNA affinity and tRNA 3′-end positioning are correlated with changes in the glutamine binding pocket.
Footnotes
Supported by NIH Grant GM63713 (to J.J.P.).
Abbreviations: AARS, aminoacyl-tRNA synthetases; GlnRS, glutaminyl-tRNA synthetase; GluRS, glutamyl-tRNA synthetase; IPTG, iso-propyl-thio galactoside; TLC, thin-layer chromatography; PMSF, phenyl methyl sulfonyl fluoride.
References
- 1.Ibba M, Söll D. Aminoacyl-tRNA synthesis. Annu Rev Biochem. 2000;69:617–650. doi: 10.1146/annurev.biochem.69.1.617. [DOI] [PubMed] [Google Scholar]
- 2.Eriani G, Delarue M, Poch O, Gangloff J, Moras D. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature. 1990;347:203–206. doi: 10.1038/347203a0. [DOI] [PubMed] [Google Scholar]
- 3.Jakubowski H. Accuracy of aminoacyl-tRNA synthetases: proofreading of amino acids. In: Ibba M, Francklyn C, Cusack S, editors. The Aminoacyl-tRNA Synthetases. Landes-Biosciences/Eurekah.com; 2004. pp. 384–396. [Google Scholar]
- 4.Pauling L. Festschrift für Arthur Stoll Siebzigsten Geburtstag. 1958:597–602. [Google Scholar]
- 5.Fersht AR. Editing mechanisms in protein synthesis. Rejection of valine by the isoleucyl-tRNA synthetase. Biochemistry. 1977;16:1025–1030. doi: 10.1021/bi00624a034. [DOI] [PubMed] [Google Scholar]
- 6.Roy H, Ling J, Irnov M, Ibba M. Post-transfer editing in vitro and in vivo by the beta subunit of phenylalanyl-tRNA synthetase. EMBO J. 2004;23:4639–4648. doi: 10.1038/sj.emboj.7600474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beebe K, Ribas De Pouplana L, Schimmel P. Elucidation of tRNA-dependent editing by a class II tRNA synthetase and significance for cell viability. EMBO J. 2003;22:668–675. doi: 10.1093/emboj/cdg065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Splan KE, Musier-Forsyth K, Boniecki M, Martinis SA. In vitro assays for the determination of aminoacyl-tRNA synthetase editing activity. Methods. 2008;44:119–128. doi: 10.1016/j.ymeth.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cavarelli J, Delagouette B, Eriani G, Gangloff J, Moras D. L-arginine recognition by yeast arginyl-tRNA synthetase. EMBO J. 1998;17:5438–5448. doi: 10.1093/emboj/17.18.5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brick P, Bhat TN, Blow D. Structure of tyrosyl-tRNA synthetase refined at 2.3 Å resolution. Interaction of the enzyme with the tyrosyl adenylate intermediate. J Mol Biol. 1989;208:83–98. doi: 10.1016/0022-2836(89)90090-9. [DOI] [PubMed] [Google Scholar]
- 11.Newberry KJ, Hou YM, Perona JJ. Structural origins of amino acid selection without editing by cysteinyl-tRNA synthetase. EMBO J. 2002;21:2778–2787. doi: 10.1093/emboj/21.11.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bullock TL, Uter NT, Nissan TA, Perona JJ. Amino acid discrimination by a class I aminoacyl-tRNA synthetase specified by negative determinants. J Mol Biol. 2003;328:395–408. doi: 10.1016/s0022-2836(03)00305-x. [DOI] [PubMed] [Google Scholar]
- 13.Rath VL, Silvian LF, Beijer B, Sproat BS, Steitz TA. How glutaminyl-tRNA synthetase selects glutamine. Structure. 1998;6:439–449. doi: 10.1016/s0969-2126(98)00046-x. [DOI] [PubMed] [Google Scholar]
- 14.Bullock TL, Rodriguez-Hernandez A, Corigliano EM, Perona JJ. A rationally engineered misacylating aminoacyl-tRNA synthetase. Proc Natl Acad Sci USA. 2008;105:7428–7433. doi: 10.1073/pnas.0711812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sekine S, Shichiri M, Bernier S, Chenevert R, LaPointe J, Yokoyama S. Structural bases of transfer RNA-dependent amino acid recognition and activation by glutamyl-tRNA synthetase. Structure. 2006;14:1791–1799. doi: 10.1016/j.str.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 16.Rould MA, Perona JJ, Söll D, Steitz TA. Structure of E. coli glutaminyl-tRNA synthetase complexed with tRNA(Gln) and ATP at 2.8 Å resolution. Science. 1989;246:1135–1142. doi: 10.1126/science.2479982. [DOI] [PubMed] [Google Scholar]
- 17.Sherlin LD, Perona JJ. tRNA-dependent active site assembly in a class I aminoacyl-tRNA synthetase. Structure. 2003;11:591–603. doi: 10.1016/s0969-2126(03)00074-1. [DOI] [PubMed] [Google Scholar]
- 18.Uter NT, Gruic-Sovulj I, Perona JJ. Amino acid-dependent transfer RNA affinity in a class I aminoacyl-tRNA synthetase. J Biol Chem. 2005;280:23966–23977. doi: 10.1074/jbc.M414259200. [DOI] [PubMed] [Google Scholar]
- 19.Gruic-Sovulj I, Uter NT, Bullock TL, Perona JJ. tRNA-dependent aminoacyl-adenylate hydrolysis by a nonediting class I aminoacyl-tRNA synthetase. J Biol Chem. 2005;280:23978–23986. doi: 10.1074/jbc.M414260200. [DOI] [PubMed] [Google Scholar]
- 20.Ravel JM, Wang SF, Heinemeyer C, Shive W. Glutamyl and glutaminyl ribonucleic acid synthetases of Escherichia coli W. J Biol Chem. 1965;240:432–438. [PubMed] [Google Scholar]
- 21.Woese CR, Olsen GJ, Ibba M, Söll D. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol Mol Biol Rev. 2000;64:202–236. doi: 10.1128/mmbr.64.1.202-236.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siatecka M, Rozek M, Barciszewslo J, Mirande M. Modular evolution of the Glx-tRNA synthetase family-rooting of the evolutionary tree between the bacteria and archaea/eukarya branches. Eur J Biochem. 1998;256:80–87. doi: 10.1046/j.1432-1327.1998.2560080.x. [DOI] [PubMed] [Google Scholar]
- 23.Berthet-Colominas C, Seignovert L, Hartlein M, Grotli M, Cusack S, Leberman R. The crystal structure of asparaginyl-tRNA synthetase from Thermus thermophilus and its complexes with ATP and asparaginyl-adenylate: the mechanism of discrimination between asparagine and aspartic acid. EMBO J. 1998;17:2947–2960. doi: 10.1093/emboj/17.10.2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iwasaki W, Sekine S, Kuroishi C, Kuramitsu S, Shirouzu M, Yokoyama S. Structural basis of the water-assisted asparagine recognition by asparaginyl-tRNA synthetase. J Mol Biol. 2006;360:329–342. doi: 10.1016/j.jmb.2006.04.068. [DOI] [PubMed] [Google Scholar]
- 25.Wolfson AD, Uhlenbeck OC. Modulation of tRNAAla identity by inorganic pyrophosphatase. Proc Natl Acad Sci USA. 2002;99:5965–5970. doi: 10.1073/pnas.092152799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fersht AR. Dissection of the structure and activity of the tyrosyl-tRNA synthetase by site-directed mutagenesis. Biochemistry. 1987;26:8031–8037. doi: 10.1021/bi00399a001. [DOI] [PubMed] [Google Scholar]
- 27.Uter NT, Perona JJ. Long-range intramolecular signaling in a tRNA synthetase complex revealed by pre-steady state kinetics. Proc Natl Acad Sci USA. 2004;101:14396–14401. doi: 10.1073/pnas.0404017101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sherlin LD, Bullock TL, Nissan TA, Perona JJ, Lariviere F, Uhlenbeck OC, Scaringe S. Chemical and enzymatic synthesis of tRNAs for high-throughput crystallization. RNA. 2001;7:1671–1678. [PMC free article] [PubMed] [Google Scholar]
- 29.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–1004. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA: 2002. [Google Scholar]
- 31.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H, Robertson AD, Jensen JH. Very fast empirical prediction and rationalization of protein pKa values. Proteins: Struct, Funct, Bioinf. 2005;61:704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 33.Perona JJ, Rould MA, Steitz TA. Structural basis for transfer RNA aminoacylation by E. coli glutaminyl-tRNA synthetase. Biochemistry. 1993;32:8758–8771. doi: 10.1021/bi00085a006. [DOI] [PubMed] [Google Scholar]
- 34.Hong KW, Ibba M, Weygand-Durasevic I, Rogers MJ, Thomann HU, Söll D. Transfer RNA-dependent cognate amino acid recognition by an aminoacyl-tRNA synthetase. EMBO J. 1996;15:1983–1991. [PMC free article] [PubMed] [Google Scholar]
- 35.Fersht AR. Structure and Mechanism in Protein Science. W. H. Freeman; New York: 1999. pp. 106–111. [Google Scholar]
- 36.Johnson KA. Transient-state kinetic analysis of enzyme reaction pathways. Enzymes. 1991;20:1–61. [Google Scholar]
- 37.Voorter CEM, de Haard-Hoekman WA, van den Oetelaar PJM, Bloemendal H, deJong WW. Spontaneous peptide bond cleavage in aging α-crystallin through a succinimide intermediate. J Biol Chem. 1988;263:19020–19025. [PubMed] [Google Scholar]
- 38.Wright HT. Nonenzymatic deamidation of asparaginyl and glutaminyl residues in proteins. Crit Rev Biochem Mol Biol. 1991;26:1–52. doi: 10.3109/10409239109081719. [DOI] [PubMed] [Google Scholar]
- 39.Liu J, Ibba M, Hong KW, Söll D. The terminal adenosine of tRNAGln mediates tRNA-dependent amino acid recognition by glutaminyl-tRNA synthetase. Biochemistry. 1998;37:9836–9842. doi: 10.1021/bi980704+. [DOI] [PubMed] [Google Scholar]
- 40.Saenger W, Lindner K. OH clusters with homodromic circular arrangement of hydrogen bonds. Angew Chem, Int Ed Engl. 1980;19:398–399. [Google Scholar]
- 41.Saenger W. Circular hydrogen bonds. Nature. 1979;279:343–344. [Google Scholar]
- 42.Uter NT, Perona JJ. Active-site assembly in glutaminyl-tRNA synthetase by tRNA-mediated induced fit. Biochemistry. 2006;45:6858–6865. doi: 10.1021/bi052606b. [DOI] [PMC free article] [PubMed] [Google Scholar]