

Abstract
Recent studies have shown that the renal cytochrome P-450 metabolites of arachidonic acid: the vasoconstrictor 20-hydroxyeicosatetraenoic acid (20-HETE), and the vasodilator epoxyeicosatrienoic acids (EETs) play an important role in the pathophysiology of angiotensin II (ANG II)-dependent forms of hypertension and the associated target organ damage. The present studies were performed in Ren-2 renin transgenic rats (TGR) to evaluate the effects of chronic selective inhibition of 20-HETE formation or elevation of the level of EETs, alone or in combination, on the course of hypertension and hypertension-associated end-organ damage. Both young (30 days of age) prehypertensive TGR and adult (190 days of age) TGR with established hypertension were examined. Normotensive Hannover Sprague-Dawley (HanSD) rats served as controls. The rats were treated with N-methylsulfonyl-12,12-dibromododec-11-enamide to inhibit 20-HETE formation and/or with N-cyclohexyl-N-dodecyl urea to inhibit soluble epoxide hydrolase and prevent degradation of EETs. Inhibition in TGR rats of 20-HETE formation combined with enhanced bioavailability of EETs attenuated the development of hypertension, cardiac hypertrophy, proteinuria, glomerular hypertrophy and sclerosis as well as renal tubulointerstitial injury. This was also associated with an attenuation of the responsiveness of the systemic and renal vascular beds to ANG II without modifying their responses to norepinephrine. Our data suggest that altered production and/or action of 20-HETE and EETs plays a permissive role in the development of hypertension and hypertension-associated end-organ damage in this model of ANG II-dependent hypertension. This information provides a basis for a search of new therapeutic approaches to the treatment of hypertension.
Keywords: cytochrome P-450 metabolites, renin-angiotensin system, hypertension, end-organ damage, soluble epoxide hydrolase
Introduction
Chronic kidney disease and end-stage renal disease present severe medical problems; their incidence has been increasing steadily, especially in industrialized countries [1]. For both conditions hypertension remains one of the most important risk factors even though antihypertensive treatment has significantly advanced over the recent decades [2]. It is generally accepted that inappropriate activation of the renin-angiotensin system (RAS) is a major factor in the development of angiotensin II (ANG II)-dependent forms of hypertension [3]. However, the role of the RAS in the pathophysiology of hypertensive target organ damage is still poorly understood.
It is well recognized that cytochrome P-450 (CYP) dependent metabolites of arachidonic acid: 20-hydroxyeicosatetraenoic acid (20-HETE) and the epoxyeicosatrienoic acids (EETs) play an important role in the regulation of renal tubular ion transport and renal and systemic vascular tone [4,5]. Moreover, recent studies strongly suggest that altered production and/or action of CYP dependent metabolites contribute to the development of ANG II-dependent forms of hypertension [6-11].
The hypertensive rat transgenic for the mouse Ren-2 renin gene [TGR; strain name TGR(mRen2)27] represents a unique ANG II-dependent animal model in which the development of hypertension is attributable to a single gene alteration [12]. We have found recently that TGR exhibit increased intrarenal levels of 20-HETE and, simultaneously, an intrarenal deficiency of EETs [13]. 20-HETE is a vasoconstrictor and is commonly regarded as a natriuretic agent, a combination of properties forming the background for both its pro- and antihypertensive potential [4,6,7]. On the other hand, we have recently provided evidence that 20-HETE may induce antinatriuresis in TGR [13]. EETs exhibit anti-hypertensive properties related to their vasodilator and natriuretic potency [4-6].
In the present study we tested the hypothesis that in TGR, a monogenic model of ANG II-dependent hypertension, increased intrarenal 20-HETE combined with intrarenal deficiency of EETs may at least partially account for the enhanced renal and systemic vascular responsiveness to ANG II and thereby contribute to the development and/or maintenance of hypertension. To examine this possibility, we evaluated the possible anti-hypertensive effects of chronic selective inhibition of 20-HETE formation and of an elevation of EET levels in young pre-hypertensive heterozygous TGR rats; the effect of the treatment on the associated hypertension-induced end-organ damage was also examined. In addition, to make the study more relevant to the clinical condition of hypertensive patients, we assessed the effects of chronic selective inhibition of 20-HETE production and of enhanced bioavailability of EETs on blood pressure and target-organs in adult TGR with established hypertension.
Finally, to gain a more detailed insight into the mechanism(s) underlying the potential beneficial (anti-hypertensive) effects of selective pharmacological interventions into the CYP-dependent metabolism of arachidonic acid, experiments were performed in which systemic and renal vascular responses to ANG II and norepinephrine (NE) were evaluated in TGR and HanSD rats. TGR and HanSD rats were either untreated or exposed to chronic reduction of 20-HETE production, to an increase in the level of EETs, or they were subjected to both (combined) maneuvers at the same time.
Materials and Methods
Ethical approval and animals
The studies were performed in accordance with guidelines and practices established by the Animal Care and Use Committee of the Institute for Clinical and Experimental Medicine. All animals used in the present study were bred at the Department of Experimental Medicine of the Institute for Clinical and Experimental Medicine, Prague, from stock animals supplied by the Max Delbruck Center for Molecular Medicine, Berlin, Germany, which is accredited by the Czech Association for Accreditation of Laboratory Animal Care. The animals were kept on a 12-hour/12-hour light/dark cycle. Throughout the experiments rats were fed a normal salt, normal protein diet (0.45 % NaCl, 19-21 % protein) produced by SEMED (Prague, Czech Republic) and had free access to tap water.
Chemicals
N-methylsulfonyl-12,12-dibromododec-11-enamide (DDMS), a selective inhibitor of 20-HETE formation [14], was administered using osmotic minipumps implanted at the dorsal neck; the concentration used allowed the delivery of 2 mg per day. As tested in our preliminary experiments, this dose blocks 20-HETE formation selectively and persistently [13].
N-cyclohexyl-N-dodecyl urea (NCND) selectively inhibits soluble epoxide hydrolase (sEH), the enzyme responsible for the conversion of EETs to dihydroxyeicosatrienoic acids (DHETEs), the biologically inactive metabolites of EETs. NCND was administrated i.p. at a dose of 10 mg. kg-1. day-1, as described previously [15]. It has been demonstrated that this dose selectively blocks sEH activity and increases the bioavailability of EETs [16].
Experimental protocols
Series 1: Inhibition of 20-HETE formation and of sEH activity starting in young pre-hypertensive rats (early treatment protocol)
Male heterozygous TGR rats, aged twenty-eight days, and age-matched male HanSD rats from several litters were randomly assigned to experimental groups, to make sure that the animals from a single litter did not prevail in any of the groups. Beginning from 30 days of age, systolic blood pressure (SBP) was measured every second day in appropriately trained conscious animals by tail-plethysmography, using a tail-cuff apparatus (MC 4000; Hatteras Instruments Co., Cary, NC, USA); in all cases a mean SBP of 4 measurements was taken. This approach is regularly employed in our laboratory and a close correlation between tail-plethysmography and direct blood pressure measurements with an indwelling catheter was established [11,17,18]. At 80 and 140 days of age, the animals were placed in individual metabolic cages and, after appropriate habituation training, their 24-hour urine was collected for sodium and water excretion and protein determination. At the end of the experiment (150 days of age), animals were again placed in metabolic cages, food was withheld to prevent contamination of urine samples, and 12-hour urine samples were collected on dry ice. The samples were stored at -80 °C until assayed. The urinary 20-HETE, EETs and DHETEs concentrations were measured by ELISA using commercially available kits, according to the manufacturer’s instructions (Detroit R&D Inc., Detroit, MI, USA). Thereafter rats were sacrificed by decapitation and ANG II levels in plasma, whole right kidney and left ventricular heart tissue were measured by radioimmunoassay (RIA) as described in detail in our previous studies [19-21]. We used the ratio of left ventricular weight to tibial length (LVW/TL) to evaluate the degree of cardiac hypertrophy, since we demonstrated recently that this is the most suitable index to assess cardiac hypertrophy [21].
To assess the renal glomerular damage, the left kidney was quickly removed, fixed in 4% formaldehyde, dehydrated and embedded in paraffin. The sections stained with hematoxylin – eosin and PAS (periodic acid – Schiff reaction) were examined and evaluated in a blind-test fashion. Fifty glomeruli in each kidney were examined on a semi-quantitative scale as described previously [22]: grade 0, all glomeruli normal; grade 1, sclerotic area up to 25% (minimal sclerosis); grade 2, sclerotic area 25 to 50% (moderate sclerosis); grade 3, sclerotic area 50 to 75% (moderate-to-severe sclerosis); grade 4, sclerotic area 75 to 100% (severe sclerosis). The glomerulosclerosis index (GSI) was calculated using the following formula: GSI = (1 × n1) + (2 × n2) + (3 × n3) + (4 × n4)/n0 + n1 + n2 + n3 + n4, where nx is the number of glomeruli in each grade of glomerulosclerosis.
Cortical tubulointerstitial injury was evaluated as defined by Nakano et al. [23] for inflammatory cell infiltration, tubular dilatation and/or atrophy, or interstitial fibrosis and was graded semi-quantitatively using the following scale: grade 0, no abnormal findings; 1, mild (<25 % of the cortex); 2, moderate (25 – 50 % of the cortex); 3, severe (>50 % of the cortex). Lesions were assessed for at least 30 random and non-overlapping fields in the renal cortex.
Morphometric evaluation of the glomerular volume was made in the same kidney sections that were examined for morphological changes, using the method developed by Weibel [24] and validated by Lane et al. [25] using Nikon NIS-Elements AR 3.1 morphometric program (Nikon, Tokyo, Japan). Briefly, this method consists of determination of mean glomerular profile area and calculation of mean glomerular volume from the following formula: Glomerular volume = area 1.5 × 1.38/1.01, where 1.38 is β, the shape coefficient for a sphere, and 1.01 is the size distribution coefficient assuming a 10 % coefficient of variation.
Since osmotic minipumps (M 2006, Alzet Co., CA, USA) have an operating time of 42 days, they were replaced by new minipumps containing DDMS on days 68 and 108 of the experiment. Control rats received osmotic minipumps containing saline vehicle and i.p. saline vehicle at the same time as treated rats. The following experimental groups were investigated:
HanSD rats + vehicle (n = 15)
TGR + vehicle (n = 16)
TGR + DDMS (n = 18)
TGR + NCND (n = 16)
TGR + DDMS + NCND (n = 18)
Series 2: Inhibition of 20-HETE formation and of sEH activity starting in adult hypertensive rats (late treatment protocol)
In this series, animals were randomly assigned into experimental groups and remained untreated from weaning until 188 days of age. At the age of 189 days the appropriate treatment(s) was started as described above. At 185, 260 and 300 days of age the animals were placed in individual metabolic cages and their 24-hour urine was collected for protein determination. New minipumps containing DDMS were implanted on days 228 and 268 of the experiment. At the end (310 days of age) the animals were again placed individually in metabolic cages and their 12-hour urine was collected for determination of 20-HETE, EETs and DHETEs as described above. Thereafter the rats were killed for morphological examination and measurements of ANG II levels as described above. The following experimental groups were investigated:
HanSD rats + vehicle (n = 12)
TGR + vehicle (n = 15)
TGR + DDMS (n = 15)
TGR + NCND (n = 15)
TGR + DDMS + NCND (n = 15)
Series 3: Mean arterial pressure (MAP) and renal blood flow (RBF) responses to ANG II and norepinephrine (NE) during inhibition of 20-HETE formation and/or of sEH in the early treatment protocol
In this series TGR and HanSD rats were subjected to the same experimental protocols as described above (series 1). At the end of the experiments (150 days of age), the animals were fasted overnight and anaesthetized with thiopental sodium (50 mg (kg body weight) -1). They were placed on a thermoregulated table to maintain body temperature at 37-37.5°C. A tracheostomy was performed to maintain a patent airway and the exterior end of the tracheal cannula was placed inside a small plastic chamber into which a humidified 95% O2/5% CO2 mixture was continuously passed. The right jugular vein was cannulated with PE-50 tubing for infusion of solutions and of additional anaesthetic as required, and for intravenous drug administration. The right femoral artery was cannulated to allow continuous monitoring of MAP and blood sampling. MAP was monitored with a pressure transducer (model MLT 1050) and recorded on the computer using a computerized data-acquisition system (PowerLab/4SP, AD Instruments, Chalgrove, UK). The left kidney was exposed via a flank incision, isolated from the surrounding tissue, and placed in a lucite cup. A tapered PE-10 catheter was inserted into the left renal artery via the left femoral artery, for selective intrarenal administration. This catheter was kept patent by a continuous infusion of heparinized isotonic saline at a rate of 4 μl min-1 throughout the experiment. A PE-10 catheter was inserted into the left ureter. During surgery, an isotonic saline solution containing bovine serum albumin (6%) was infused at a rate of 20 μl min-1. An ultrasonic transit-time flow probe (1RB)) connected to a Transonic flowmeter (Transonic Systems Inc., Ithaca, NY, USA) was placed around the left renal artery and RBF was recorded using a computerized data-acquisition system. After a 30-minute equilibration period, the experimental protocol was started to assess the responses of MAP and RBF to systemic (intravenous) and intrarenal bolus doses of ANG II or NE. The vasoactive agents were dissolved in 100 μl or 20 μl of isotonic saline for intravenous and intrarenal administration, respectively. After intravenous drug administration, a bolus of 100 μl isotonic saline was injected to ensure a rapid and complete delivery of the agent into the systemic circulation. Just before intrarenal administration of either agent, the intrarenal infusion rate was increased from 4 to 40 μl min-1 to deliver the entire dose rapidly into the kidney; thereafter the basal rate of the intrarenal infusion was restored. We and others have demonstrated previously that a 10-min period between injections is sufficient for MAP and RBF recovery [11,26,27]. MAP responses to intravenous bolus doses of ANG II (50 ng) and thereafter of NE (150 ng) were studied in the following experimental groups in each series:
HanSD rats + vehicle (n = 8)
TGR + vehicle (n = 8)
TGR + DDMS (n = 7)
TGR + NCND (n = 7)
TGR + DDMS + NCND (n = 7)
Thereafter, the RBF responses to intrarenal bolus doses of ANG II (10 ng) and of NE (30 ng) were determined in the same experimental groups.
Series 4: MAP and RBF responses to ANG II and NE during inhibition of 20-HETE formation and/or of sEH activity in the late treatment protocol
In this series TGR and HanSD rats were subjected to the same experimental protocols as described in series 2. At the end of the experiment (301 days of age), the animals were prepared in a manner described above (series 3) and MAP and RBF responses to the same doses of ANG II and NE were determined in the following groups:
HanSD rats + vehicle (n = 6)
TGR + vehicle (n = 7)
TGR + DDMS (n = 8)
TGR + NCND (n = 9)
TGR + DDMS + NCND (n = 7)
Statistical analysis and calculations
Statistical analysis of the data was performed using Graph-Pad Prism software (Graph Pad Software, San Diego, California, USA). All values are expressed as mean ± SEM. Two-way repeated-measures ANOVA was used to detect differences within each experimental group. One-way ANOVA was employed when appropriate. The renal vascular resistance (RVR) was calculated as the MAP/RBF ratio during the 30-minute equilibration period, using a computerized data-acquisition system (PowerLab/4SP, ADInstruments, UK). Statistical significance was defined as p<0.05.
Results
Series 1: Effects of “early” inhibition of 20-HETE formation or/and of sEH activity
As shown in figure 1A, DDMS treatment did not alter the development or the final level of hypertension in TGR whereas NCND did retard its development significantly. The increase in the pressure was less steep in NCND treated rats than in untreated TGR, and at the age of 79 days the SBP levels were 181 ± 4 and 196 ± 4 mmHg, respectively (p<0.05). Thereafter the progress of hypertension was similar and the final SBP did not significantly differ between NCND-treated and untreated TGR (202 ± 6 vs. 199 ± 5 mmHg, respectively). In contrast, the combined treatment with DDMS and NCND both attenuated the progression of hypertension and lowered the final SBP to a level that was significantly different from that in untreated TGR (178 ± 3 vs. 199 ± 5 mmHg, p<0.05). Nevertheless, the level was still distinctly higher than that in HanSD rats which remained normotensive throughout the experiment.
Figure 1.
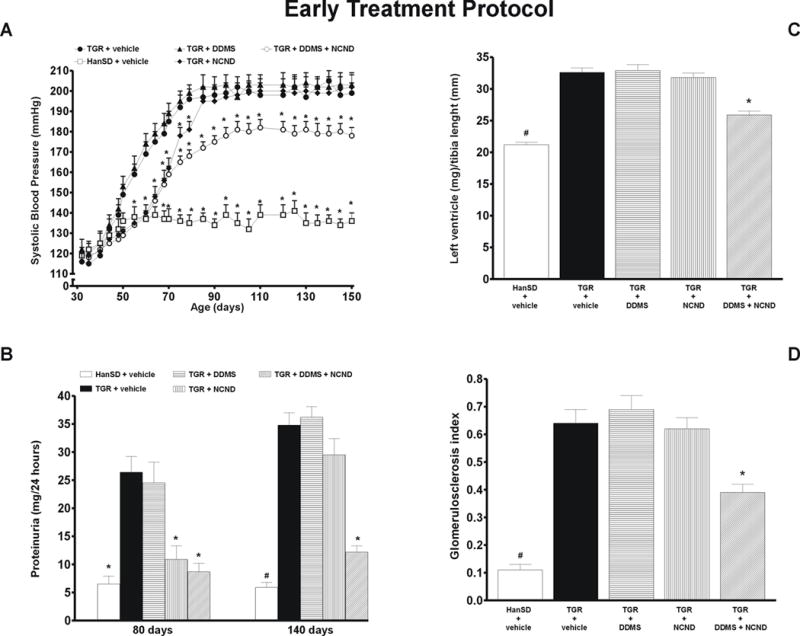
Changes in systolic blood pressure (A), proteinuria (B), heart left ventricle weight to tibia length ratio (C) and glomerulosclerosis index (D) in untreated TGR (heterozygous Ren-2 renin transgenic rats) and HanSD (transgene-negative) rats and in TGR treated either by DDMS or NCND or by their combination. Data are taken from the “early treatment” protocol. * P<0.05 versus untreated TGR. # P<0.05 versus the values in each of TGR groups.
Fig. 1B shows that HanSD rats exhibited only minor proteinuria over the entire duration of the experiment. In TGR a pronounced proteinuria was observed already during the early phase of hypertension, at the age of 80 days (26.4 ± 2.8 mg (24 h)-1, compared with 6.5 ± 1.4 mg (24 h)-1 seen in HanSD rats). The proteinuria further increased to 34.8 ± 2.2 mg (24 h)-1 in the phase of established hypertension (day 140 of age). The treatment with DDMS did not affect the degree of proteinuria in TGR. On the other hand, NCND attenuated proteinuria during the early but not in the established phase of hypertension. In contrast, combined treatment with DDMS and NCND distinctly ameliorated the proteinuria in the early as well as in the established phase of hypertension (8.7 ± 1.5 and 12.2 ± 1.1 mg (24 h)-1, respectively, compared with 26.4 ± 2.8 and 34.8 ± 2.2 mg (24 h)-1 observed in untreated TGR (p<0.05 for both the early and the established phase).
As shown in figure 1C, untreated TGR developed a distinct cardiac hypertrophy, expressed as the ratio of LVW/TL, which was not attenuated by DDMS or NCND given alone. In contrast, the combined treatment with DDMS and NCND substantially attenuated the degree of cardiac hypertrophy (25.9 ± 0.6 compared with 32.6 ± 0.7 observed in untreated TGR, p<0.05); however, the levels observed in HanSD rats were not attained.
As shown in figure 1D, untreated TGR exhibited a glomerulosclerosis index that was substantially higher than in HanSD rats (0.64 ± 0.05 vs. 0.11 ± 0.05, p<0.05). The treatment with DDMS or NCND given alone did not prevent the development of glomerulosclerosis. In contrast, the combined treatment with DDMS and NCND significantly reduced the glomerulosclerosis index, even though it did not decrease to the level observed in HanSD rats.
As shown in figure 2A, untreated TGR had markedly higher degree of kidney tubulointerstitial injury, compared with HanSD rats (0.025 ± 0.005 vs. 0.002 ± 0.001, p<0.05). The treatment with DDMS alone did not attenuate the injury in TGR. In contrast, the treatment of TGR with NCND alone markedly inhibited the development of kidney tubulointerstitial injury, compared with untreated TGR, and the combined treatment with DDMS and NCND completely prevented the development of the injury (figure 2A).
Figure 2.

Kidney tubulointerstitial injury score (A) and glomerular volume (B) in untreated TGR (heterozygous Ren-2 renin transgenic rats) and HanSD (transgene-negative) rats and in TGR treated either by DDMS or NCND or by their combination. Data are taken from the “early treatment” protocol. # P<0.05 versus untreated TGR. @ P<0.05 versus the values for TGR + vehicle, TGR + DDMS and TGR + NCND groups.
The glomerular volume in untreated TGR was significantly greater than in HanSD rats (1.43 ± 0.065 vs. 1.08 ± 0.025 106 μm3, p<0.05)(figure 2B). The treatment with DDMS alone did not change the volume in TGR as compared with untreated TGR. In contrast, the treatment of TGR with NCND alone distinctly reduced the volume and the combined treatment with DDMS and NCND normalized it, bringing the values down to the levels observed in HanSD rats.
As shown in figure 3A, the urinary excretion of 20-HETE in untreated TGR was significantly higher than in HanSD rats (1480 ± 165 vs. 460 ± 90 ng (12 h)-1, p<0.05). The treatment with NCND did not alter the urinary excretion of 20-HETE. However, as expected, the treatment with DDMS alone, or the combined treatment with NCND and DDMS significantly reduced 20-HETE excretion down to the levels observed in HanSD rats.
Figure 3.
Urinary excretion of 20-HETE (A), EETs (B), DHETEs (C) and the urinary EETs to DHETEs excretion ratio (D) in untreated TGR (heterozygous Ren-2 renin transgenic rats) and in HanSD (transgene-negative) rats and in TGR treated by DDMS, NCND or by combination of the two agents. Data are taken from the “early treatment” protocol * P<0.05 versus untreated TGR.
In contrast, urinary excretion of EETs was significantly lower in TGR than in HanSD rats (640 ± 120 vs. 1295 ± 110 ng (12 h)-1, p<0.05). Treatment with DDMS did not alter EETs excretion whereas the treatment with NCND, alone or in combination with DDMS, significantly increased it to the levels observed in HanSD rats (figure 3B).
Figure 3C shows that the urinary excretion of DHETEs, the biologically inactive metabolites of EETs, was significantly higher in untreated TGR than in HanSD rats (920 ± 60 vs. 490 ± 60 ng (12 h)-1, p<0.05). The treatment with DDMS did not significantly change DHETEs excretion, but treatment with NCND alone or in combination with DDMS significantly decreased it to the levels observed in HanSD rats.
Figure 3D summarizes the data on the intrarenal availability of biologically active epoxygenase metabolites when expressed as the EETs/DHETEs ratio. This ratio was significantly lower in untreated TGR than in HanSD rats (0.7 ± 0.07 vs. 2.64 ± 0.76 p<0.05). It was not significantly changed by DDMS treatment. However, treatment with NCND, alone or in combination with DDMS, significantly increased this ratio to the levels observed in HanSD rats.
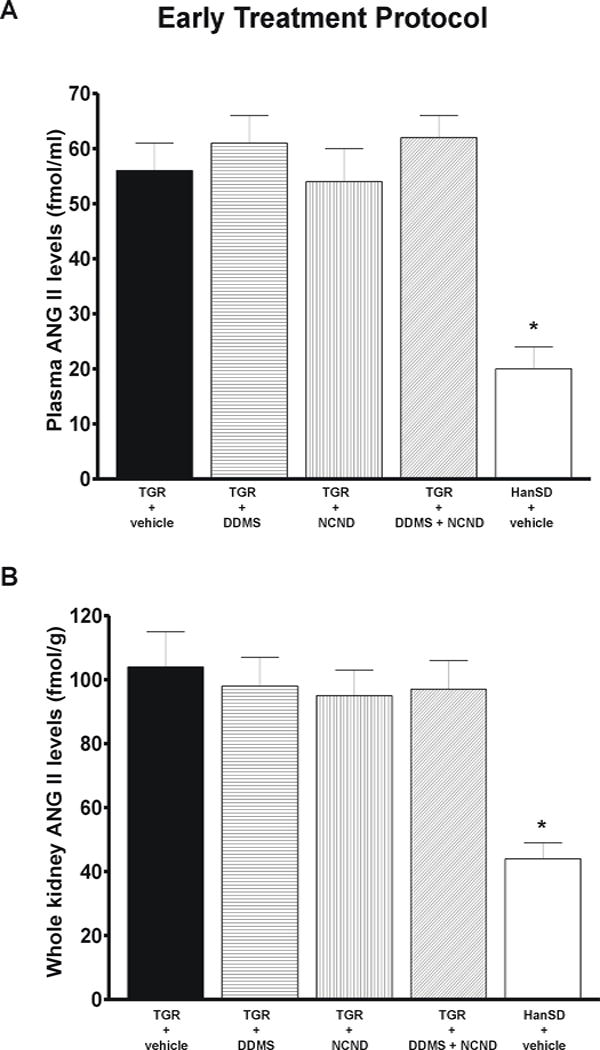
Plasma, kidney and left heart ventricle ANG II levels were significantly higher in untreated TGR than in HanSD rats; in the former the levels were not altered by any of the three treatment modalities. In addition, urinary output and urinary sodium excretion were not significantly different among experimental groups at any time of the experiment (see on-line supplemental table 1).
Series 2: Effects of “late” inhibition of 20-HETE formation and/or sEH activity
As shown in figure 4A, untreated TGR remained distinctly hypertensive throughout the experiment, with the final SBP of 204 ± 5 mmHg, whereas HanSD rats were invariably normotensive, with the final SBP of 138 ± 3 mmHg. In TGR, the treatment with DDMS did not significantly change SBP at any time point. Within the first 8 days of treatment, NCND significantly decreased SBP from 204 ± 6 to 181 ± 3 mmHg (p<0.05), but during the further 10 days it returned to the levels observed in untreated TGR, reaching a final value of 202 ± 7 mmHg. In contrast, the combined treatment with DDMS and NCND resulted in a distinct decrease in SBP from 205 ± 4 to 179 ± 2 mmHg (p<0.05) within 8 days. Thereafter, despite a gradual return of the pressure toward the original hypertensive levels, SBP remained significantly lower than in untreated TGR (191 ± 2 vs. 204 ± 5 mmHg, p<0.05).
Figure 4.
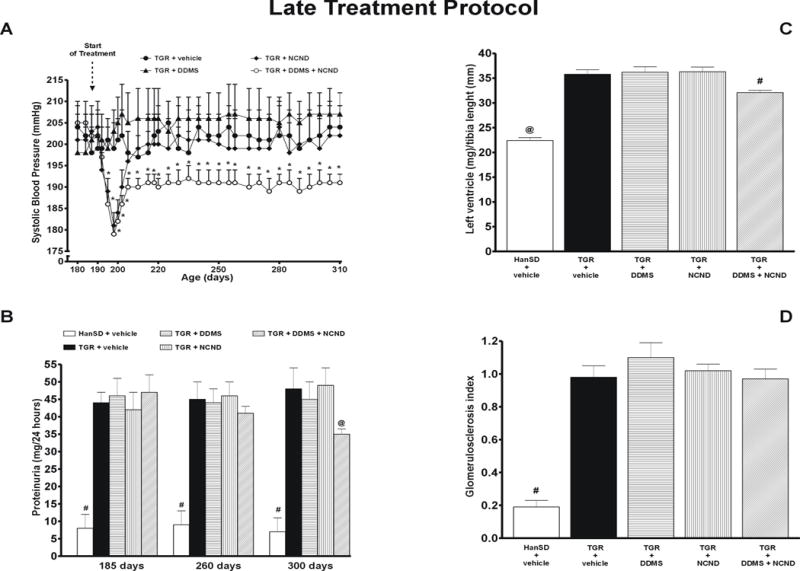
Changes in systolic blood pressure (A), proteinuria (B), heart left ventricle weight to tibia length ratio (C) and glomerulosclerosis index (D) in untreated TGR (heterozygous Ren-2 renin transgenic rats) and HanSD (transgene-negative) rats, and in TGR treated either by DDMS or NCND or by their combination. Data are taken from the “late treatment” protocol. * P<0.05 versus pre-treatment values. # P<0.05 versus untreated TGR. @ P<0.05 versus the values in each of TGR groups.
Figure 4B shows that on day 185 the basal values of proteinuria were markedly higher in TGR than in HanSD rats. This pattern was not reduced by chronic treatment with either DDMS or NCND alone. In contrast, the combined treatment with DDMS and NCND decreased the proteinuria from 47.4 ± 4.8 to 34.9 ± 1.1 mg (24 h)-1 (p<0.05). This was still substantially higher than the small proteinuria observed in HanSD rats throughout the entire experiment.
As shown in figure 4C, untreated TGR had a markedly higher LVW/TL index than observed in HanSD rats (35.8 ± 0.9 vs. 22.4 ± 0.6, p<0.05), which was not altered by DDMS or NCND treatment given alone but was reduced by the combined treatment with DDMS and NCND to 32.1 ± 0.4 (p<0.05). Nevertheless, the LVW/TL index remained significantly higher than in HanSD rats.
As shown in figure 4D, untreated TGR exhibited a significantly higher glomerulosclerosis index than did HanSD rats (0.98 ± 0.07 vs. 0.19 ± 0.04, p<0.05); the index was not reduced by any of the three treatment modalities.
As shown in figure 5A, untreated TGR showed a markedly higher degree of kidney tubulointerstitial injury as compared with HanSD rats (0.19 ± 0.04 vs. 0.02 ± 0.01, p<0.05). The treatment with DDMS or NCND given alone did not reduce the kidney tubulointerstitial injury score in TGR. In contrast, the combined treatment with DDMS and NCND substantially attenuated the injury but did not bring it down to the levels observed in HanSD rats (figure 5A).
Figure 5.
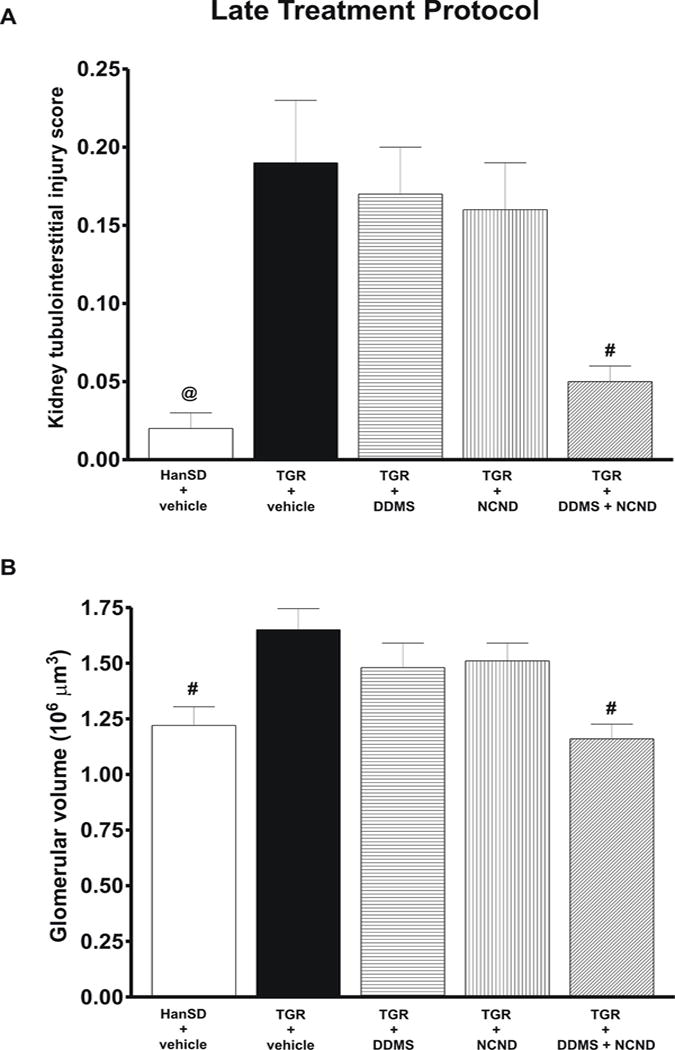
Kidney tubulointerstitial injury score (A) and glomerular volume (B) in untreated TGR (heterozygous Ren-2 renin transgenic rats) and HanSD (transgene-negative) rats and in TGR treated by DDMS or NCND or by their combination. Data are taken from the “late treatment protocol. # P<0.05 versus untreated TGR. @ P<0.05 versus the values for the remaining four groups.
The glomerular volume in untreated TGR was significantly greater in than in HanSD rats (1.65 ± 0.095 vs. 1.22 ± 0.084 106 μm3, p<0.05)(figure 5B). The treatment with DDMS or NCND alone did not change the glomerular volume in TGR as compared with untreated TGR. In contrast, the combined treatment with DDMS and NCND normalized it, bringing the values down to the levels observed in HanSD rats
Figure 6A shows that the urinary excretion of 20-HETE was markedly higher in untreated TGR than in HanSD rats (2142 ± 194 vs. 355 ± 105 ng (12 h)-1, p<0.05). The treatment with DDMS, alone or in combination with NCND, significantly reduced 20-HETE excretion to levels not significantly different from those in HanSD rats. The treatment with NCND alone did not alter 20-HETE excretion.
Figure 6.
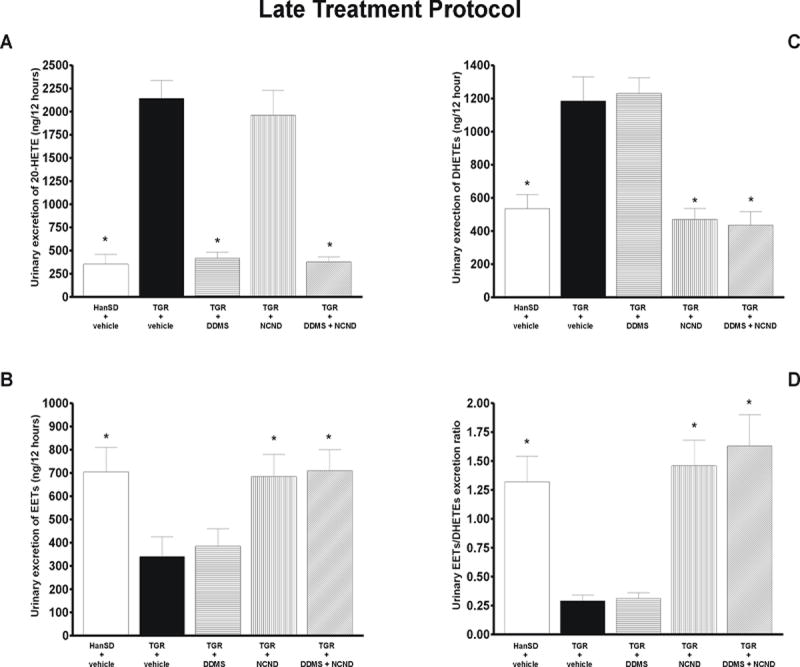
Urinary excretion of 20-HETE (A), EETs (B), DHETEs (C) and the urinary EETs to DHETEs excretion ratio (D) in untreated TGR (heterozygous Ren-2 renin transgenic rats) and HanSD (transgene-negative) rats, and in TGR treated by DDMS or NCND, alone or combined. Data are taken from the “late treatment” protocol. * P<0.05 versus untreated TGR.
As shown in figure 6B, urinary excretion of EETs was significantly lower in TGR than in HanSD rats (340 ± 85 vs. 706 ± 102 ng (12 h)-1, p<0.05) and was not altered by DDMS treatment. In contrast, treatment with NCND, alone or in combination with DDMS, elicited substantial increases of EETs excretion. Urinary excretion of DHETEs was significantly higher in untreated TGR than in HanSD rats (1184 ± 144 vs. 535 ± 85 ng (12 h)-1, p<0.05) and, again, was not altered by DDMS treatment. In contrast, treatment with NCND, alone or in combination with DDMS, significantly reduced DHETEs excretion in TGR to the levels observed in HanSD rats (figure 6C).
Figure 6D shows that the urinary EETs/DHETEs ratio was distinctly lower in untreated TGR than in HanSD rats (0.29 ± 0.05 vs. 1.32 ± 0.22). This was not significantly modified by DDMS treatment, whereas treatment with NCND, alone or in combination with DDMS, significantly increased this ratio to the level observed in HanSD rats.
Plasma, kidney and left heart ventricle ANG II levels were significantly higher in untreated TGR than in HanSD rats and this pattern was not altered by any of the three treatment modalities. Moreover, urinary output and urinary sodium excretion were not significantly different among experimental groups at any time of experiment (see on-line supplemental table 2).
Series 3: MAP and RBF responses to ANG II and NE in the “early treatment” protocol
Basal values for MAP, RBF and RVR (average values from the equilibration period) are summarized in on-line supplemental table 3. As shown in Figure 7A, the responses of MAP to intravenous bolus administration of ANG II, 50 ng, were significantly greater in untreated TGR than in HanSD rats (+32 ± 3 vs. +14 ± 2 mmHg, p<0.05). Treatment with either DDMS or NCND alone did not modify the responses whereas the combined treatment with DDMS and NCND did attenuate them significantly. However, the responses of MAP remained significantly greater than in HanSD rats.
Figure 7.
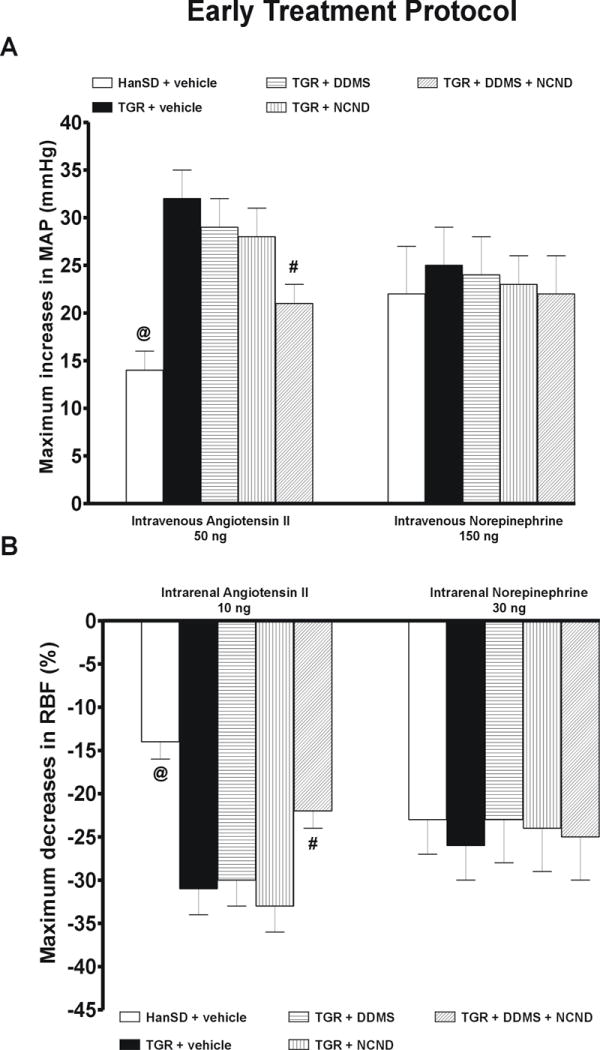
Maximum increases in mean arterial pressure (MAP) in response to intravenous bolus administration of angiotensin II and norepinephrine (A) and maximum decreases in renal blood flow (RBF) in response to intrarenal bolus administration of angiotensin II and norepinephrine (B) in untreated TGR (heterozygous Ren-2 renin transgenic rats) and HanSD (transgene-negative) rats, and in TGR treated by DDMS, NCND, alone or combined. Data are taken from the “early treatment” protocol # P<0.05 versus untreated TGR. @ P<0.05 versus the values in each of TGR groups.
Similarly, as shown in figure 7B, the intrarenal administration of ANG II, 10 ng, decreased RBF significantly more in untreated TGR than in HanSD rats (-31 ± 3 vs. -14 ± 2 %, p<0.05). The treatment with either DDMS or NCND alone did not attenuate the RBF decreases whereas, in contrast, a distinct attenuation was seen with the combined treatment (-22 ± 2 vs. -31 ± 3 % in untreated TGR, p<0.05). As shown in figures 7A and 7B, an intravenous or intrarenal administration of NE elicited similar increases in MAP and decreases in RBF in TGR and HanSD rats; in the former the responses were not altered by any of the three treatment modalities.
Series 4: MAP and RBF responses to ANG II and NE in the “late treatment” protocol
Basal values for MAP, RBF and RVR (average values from the equilibration period) are summarized in on-line supplemental table 3. As shown in figure 8A, an intravenous bolus of 50 ng of ANG II increased MAP significantly more in untreated TGR than in HanSD rats (+35 ± 3 vs. +16 ± 3 mmHg, p<0.05). The treatment with either DDMS or NCND alone did not alter the responses whereas the combined treatment did attenuate them significantly. Similarly, as shown in figure 8B, the intrarenal bolus administration of ANG II, 10 ng, elicited significantly larger decreases in RBF in untreated TGR than in HanSD rats (-35 ± 4 vs. -16 ± 2 %, p<0.05). The responses were not modified by treatment with either DDMS or NCND alone but were significantly reduced by the combined treatment with DDMS and NCND.
Figure 8.

Maximum increases in mean arterial pressure (MAP) in response to intravenous bolus administration of angiotensin II and norepinephrine (A) and maximum decreases in renal blood flow (RBF) in response to intrarenal bolus administration of angiotensin II and norepinephrine (B) in untreated TGR (heterozygous Ren-2 renin transgenic rats) and HanSD (transgene-negative) rats, and in TGR treated by DDMS, NCND, alone or combined. Data are taken from the “late treatment” protocol # P<0.05 versus untreated TGR. @ P<0.05 versus the values in each of TGR groups.
As shown in figures 8A and 8B, an intravenous or intrarenal administration of NE elicited similar increases in MAP and decreases in RBF in TGR and HanSD rats; in the former the responses were not altered by any of the three treatment modalities.
Discussion
After initial controversies regarding the role of the RAS in the pathophysiology of hypertension in TGR [12,28,29], more recent studies have clearly demonstrated that enhanced formation of ANG II leading to elevation of its plasma and kidney tissue levels, and an exaggeration of peripheral and renal vascular responsiveness to ANG II, is the main pathogenetic factors in the development and maintenance of hypertension in TGR [19,20,26,27]. Notwithstanding this conclusion, the important role of CYP-derived eicosanoids in the regulation of renal function and blood pressure as well as in the pathophysiology of hypertension and the associated hypertension-induced end-organ damage has been recently documented for ANG II-dependent forms of hypertension [6-11,16,30-32]. Moreover, disturbed intrarenal production and diminished action of 20-HETE combined with intrarenal deficiency of EETs (a consequence of increased sEH-mediated conversion to biologically inactive DHETEs) have been implicated in the pathophysiology of hypertension in transgenic Ren-2 rats (TGR) [11,13]. Evaluation of the proposed in vivo functional role of 20-HETE and EETs is difficult. One potential approach is to apply pharmacologic interventions aimed at an abolishment or substantial reduction of the agents’ activity. This requires their chronic selective blockade and prolonged follow-up studies of transgenic rats subjected to the treatment and parallel studies of appropriate control animals. The present study evaluates the effect of such blockade on the development of hypertension and of the end-organ damage in TGR.
20-HETE has been shown to promote vasoconstriction via mechanisms involving inhibition of large-conductance, calcium-activated potassium channels [33], activation of calcium channel conductance leading to depolarization of the vascular smooth muscle cell (VSM) [34], and activation of Rho-kinase which causes sensitization of the contractile apparatus to calcium [35]. 20-HETE is also known to augment vascular reactivity to vasoconstrictor agents [36,37] and to play a critical role as a second messenger for ANG II-mediated vasoconstriction [7,8]. Overall, it has been claimed that enhanced formation of 20-HETE in the vasculature is the basis of its prohypertensive action and contributes to the development and maintenance of hypertension in some experimental models [7-9,30,38].
In addition to its action on the vasculature, 20-HETE inhibits sodium reabsorption in the proximal tubule [39] and in the thick ascending limb of the loop of Henle [40]. Accordingly, it has been postulated that increased 20-HETE levels along the renal tubule should promote natriuresis and thereby oppose its constrictor action on the vasculature and the development of hypertension [4,6,30]. The notion that intrarenal 20-HETE exerts also an antihypertensive action is strengthened by the demonstration that chronic blockade of 20-HETE formation promotes the development of salt-sensitive hypertension in otherwise salt-resistant rats [41]. In a clear contrast with the transport inhibitory effect of 20-HETE observed in normotensive rats and in a number of experimental or genetic hypertensive rat models, in a recent study we found that in hypertensive TGR increased intrarenal 20-HETE concentrations cause sodium retention without decreasing renal haemodynamics, which suggests an increase in the tubular sodium transport [13]. Thus, in hypertensive Ren-2 transgenic rats 20-HETE could be pro-hypertensive both owing to its vasoconstrictor and to the sodium retaining activity.
EETs have been consistently shown to cause vasodilatation through stimulation of the large-conductance, calcium-activated potassium channels (KCa) [5]. They have also been identified as an endothelium-derived hyperpolarizing factor (EDHF) that mediates nitric oxide-and prostaglandin-independent vasodilatation, and have been found to oppose the vasoconstrictor actions of ANG II [42-45]. Moreover, EETs inhibit tubular sodium reabsorption in the renal proximal tubule [46] and in the cortical collecting duct [47]. Lastly, an increasing body of evidence suggests that EETs exhibit cardiovascular and renal protective actions [48].
A major finding of the present study is that combined chronic inhibition of 20-HETE synthesis (DDMS) and of sEH activity (NCND), when applied in young pre-hypertensive TGR, significantly attenuated the development of hypertension and had considerable nephroprotective and cardioprotective effects, including a reduction of proteinuria, glomerulosclerosis, kidney tubulointerstitial injury and cardiac hypertrophy. This was associated with a reduction of urinary excretion of 20-HETE and DHETEs, and an elevation of urinary excretion of EETs to the levels observed in normotensive HanSD rats. Of special interest is our finding that nephroprotective effects of the combined treatment with DDMS and NCND were associated with restoration of glomerular volume to values observed in normotensive HanSD rats. Given a strong correlation between the glomerular size and the degree of glomerulosclerosis, these findings are in good agreement with the demonstration that nephroprotective effect of antihypertensive therapy is also associated with the reduction of the size of the glomerulus [49,50]. Remarkably, isolated chronic 20-HETE inhibition did not significantly affect the development of hypertension or of the end-organ damage, even though the urinary excretion of 20-HETE was reduced to levels close to those in the animals treated with the combination of DDMS and NCND. Similarly, chronic isolated inhibition of sEH increased the intrarenal availability of biologically active epoxygenase metabolites 3-fold but elicited only transitory antihypertensive effects and did not alter the extent of the final end-organ damage. Nevertheless, it is important to emphasize that in TGR the treatment with NCND alone exhibited some minor nephroprotective actions, e.g. the degree of tubulointerstial injury was reduced and glomerular hypertrophy was prevented. However, these nephroprotective effects were minimal compared with those of the combined treatment with DDMS and NCND. On the whole, the present findings agree well with those of our previous studies with TGR, which showed that altered production and/or action of CYP-derived metabolites contributes to the development of hypertension and of the associated end-organ damage [11,13]. However, this is the first study clearly showing that only the combination of the blockade of 20-HETE formation and of sEH inhibition exerts long-term antihypertensive and renal and cardiovascular protective actions in the monogenic model of ANG II-dependent hypertension.
It is difficult to explain why the beneficial effects were achieved exclusively with the combined DDMS and NCND treatment. Since in most situations 20-HETE has both pro- and antihypertensive properties, one cannot easily predict the consequence of its inhibition on blood pressure [4,6]. However, we have shown earlier that in TGR rats 20-HETE causes sodium retention rather than natriuresis [13] which means that both components, its vascular and tubular actions, should promote the development of hypertension. Therefore we assumed that in this ANG II-dependent model a chronic and selective inhibition of 20-HETE formation should be more effective in attenuating blood pressure elevation and possibly also the associated organ damage than could be expected in other models of hypertension. However, we found that although DDMS treatment lowered the elevated urinary excretion of 20-HETE to the levels observed in normotensive HanSD rats, it did not exhibit any antihypertensive and organ-protective action. Moreover, chronic treatment with DDMS did not alter plasma and kidney ANG II levels and did not attenuate the enhanced systemic and renal vascular responsiveness to ANG II.
Similar to the effects of specific 20-HETE inhibition, in our TGR chronic inhibition of sEH activity alone by NCND did not exhibit long-term antihypertensive and organ-protective actions, even though the availability of biologically active epoxygenase products was substantially increased. Most probably, it is not tissue EETs or tissue 20-HETE but the elevated ANG II concentrations and the augmented vascular responsiveness to ANG II which are the dominant factors in the development of hypertension and in the end-organ damage in this transgenic ANG II-dependent model. This is in contrast to the results obtained in rats rendered hypertensive by chronic infusion of ANG II in whom the endogenous RAS activity is substantially suppressed [3,16]. In these animals isolated pharmacological manipulations of the CYP pathway of arachidonic acid metabolism clearly reduced blood pressure and the end-organ damage [6,32,51]. Overall, the results strengthen our earlier conclusions that the substantial chronic elevation of RAS activity is the crucial step in the pathophysiology of hypertension in the TGR model [19,20,27].
We found that the combination of suppression of 20-HETE production with an elevation of EETs can partially counteract the vasoconstrictor actions of chronically elevated circulating and tissue ANG II levels in TGR and can partly attenuate the development of hypertension and hypertension-induced end-organ damage. The reasons why the combined treatment was generally effective while the isolated treatment, e.g. with NCND, was not, may be complex. It should be kept in mind that in TGR with established hypertension the isolated inhibition of sEH did not lower blood pressure or attenuate proteinuria whereas the combined treatment had appreciable effects. This could be related to the very high basal sEH activity (In TGR the DHETE excretion increased with age) but also to a high rate of 20-HETE synthesis; the urinary excretion of 20-HETE was almost twice that seen in young TGR and 7fold higher than in old HanSD. One might speculate that the extremely high 20-HETE synthesis diminished the amount of the substrate arachidonic acid (AA) available for the synthesis of EETs and decreased their synthesis rate, in accordance with the repeatedly proposed hypothesis assuming “substrate shifts” between the individual pathways of AA metabolism [4,52-55]. At reduced EETs synthesis rate, an inhibition of their degradation with NCND alone would not increase available EETs very effectively. On the other hand, a simultaneous suppression of 20-HETE synthesis (DDMS treatment) would, in addition to the specific direct effect, make the AA substrate available for the epoxygenase subpathway of CYP-dependent AA metabolism and, at high EETs synthesis rate their action, would increase more than with NCND treatment alone; the final result would be an appreciable reduction of blood pressure and proteinuria. It is commonly recognized that antihypertensive therapy is more effective when applied early during the development rather than during established hypertension [56]. Therefore, in a separate series of experiments we evaluated the effects of chronic blockade of 20-HETE formation and sEH inhibition on blood pressure and on proteinuria, an index of kidney damage, in the TGR with sustained elevation of blood pressure i.e. a condition that more closely resembles clinical hypertension. We found that in this setting – similar to the first series of experiments - a reduction of blood pressure, proteinuria and cardiac hypertrophy was seen only with the combined treatment with DMMS and NCND. The improvement was associated with a reduction of the urinary excretion of 20-HETE and DHETEs and an increase of the urinary excretion of EETs to levels observed in normotensive HanSD rats. Thus, the findings strengthen our conclusion based on the data of “early treatment” experiments. It should be noticed, however, that the amelioration of end-organ damage was significantly smaller than that observed when the same treatment was started immediately after weaning.
After the late-onset combined therapy the proteinuria and cardiac hypertrophy were only modestly attenuated whereas the tubulointerstitial injury was markedly attenuated and the glomerular hypertrophy was prevented. There was no difference between untreated TGR and TGR treated with the combination of DDMS and NCND with regard to the index of glomerulosclerosis – this was so despite a similar blood pressure lowering effect observed with the early and late treatment protocol. The failure of the combined treatment to inhibit the progress of glomerulosclerosis could be related to the fact that renal tissue ANG II and 20-HETE progressively increased in TGR and were much higher in the phase of established hypertension than in the young prehypertensive rats. It should be noticed that Ang II, among other actions, activates Ras/mitogen activated protein kinase by increasing the synthesis of CYP4A-derived 20-HETE; this stimulates proliferation of the VSM [57-60]. One might speculate that prolonged exposure of the glomeruli to increasing concentrations of ANG II resulted in proliferation-based changes that could not be reversed by our combined therapy. Interestingly, the cardiac hypertrophy could still be attenuated by the combined treatment, possibly because the plasma concentration, unlike kidney tissue ANG II, did not increase with the age of TGR. Overall, in the TGR with established hypertension, alterations of 20-HETE and EETs activities have only limited effects on end-organ damage. This suggests that the CYP dependent active agents play a “permissive” rather than causal role in the pathophysiology of hypertension and the associated end-organ damage in TGR.
We found that only the combined blockade of 20-HETE formation and of sEH activity significantly attenuated MAP and RBF responses to bolus injections of ANG II; this was similar when examined in young pre-hypertensive TGR and in the rats with established hypertension. The responses to NE were not altered. These data are in accordance with the previous studies showing that TGR display an exaggerated systemic and renal vascular reactivity to ANG II [26,27]. However, our present findings are the first to clearly show that, in contrast to rats rendered hypertensive by chronic subpressor ANG II-infusion [7,8,16,28,32,51], in TGR an augmented vascular responsiveness to ANG II is not attenuated by an isolated blockade of 20-HETE formation or an isolated enhancement of EETs bioavailability. The fact that an alteration of the activities of the two types of CYP metabolites does result in an appreciable attenuation of the vasoconstriction indicates that an augmentation of the vascular responsiveness to ANG II is one of the mechanism(s) underlying the beneficial antihypertensive actions of combined DDMS and NCND treatment in TGR.
In summary, the results show that selective blockade of 20-HETE formation and of sEH activity, when applied chronically and in combination in young pre-hypertensive TGR, suppresses the development of hypertension and provides a substantial protection from the associated cardiac hypertrophy, proteinuria and glomerulosclerosis. Also in adult TGR with established hypertension the combined treatment with DDMS and NCND reduced blood pressure and slightly decreased proteinuria and cardiac hypertrophy. We found also that a reduction of 20-HETE combined with an increase of EETs levels attenuated the enhanced vascular responsiveness to ANG II in TGR. Taken together, the findings strongly suggest that alteration in the production and/or action of 20-HETE and EETs plays a permissive role in the development of hypertension and hypertension-associated end-organ damage in this ANG II-dependent model. This information derived from our present study should be considered in attempts of the development of new therapeutic approaches or tools for the treatment of early stages of human hypertension.
Supplementary Material
Acknowledgments
This study was supported by grant No. 305/08/J006 awarded to L.Č. by Czech Science Foundation (GAČR). A.W. was partly supported from the Center for Cardiovascular Research (1M6798582302) and by the institutional financial support of the Institute for Clinical and Experimental Medicine (MZO 00023001). V.C.Ch. was supported by grant No. NS/10500-3 awarded by the Internal Grant Agency of the Ministry of Health of the Czech Republic. Z.V. is supported by grant No. NS/10499-3 awarded by the Internal Grant Agency of the Ministry of Health of the Czech Republic. L.K. is supported by grant from the Grant Agency of Academy of Science of Czech Republic (GAAV – IAA 511-700-701) and by grant No. NS/9699-4 awarded by the Internal Grant Agency of the Ministry of Health of the Czech Republic. I.V. is supported by grants from GAČR No. 305/07/0167 and No. 305/07/J004. H.J.K. is supported by grants from the German Research Foundation (DFG), Bonn (Kra 436/14-2 and 436 TSE 113/57/0-1). J.D.I. is supported by grants from the National Institutes of Health (NIH) (HL59699 and DK38226) and Advancing a Healthier Wisconsin. J.R.F. is supported by the Robert A. Welch foundation and NIG grant GM31278. B.D.H. is supported by grants NIEHS R01, ES002710 and NIEHS Superfund Program P42 ES004699.
In addition, we would like to point out that V.C.Ch. and A.W. contributed to this study equally and therefore both of them should be considered as first authors.
References
- 1.US Renal Data System: USRDS 2007. Chronic kidney disease. Am J Kidney Dis. 2008;51:S63–S80. [Google Scholar]
- 2.Bakris GL, Ritz E. The message for World Kidney Day 2009: hypertension and kidney disease: a marriage that should be prevented. J Clin Hypertens. 2009;11:144–147. doi: 10.1111/j.1751-7176.2009.00092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007;59:251–287. doi: 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- 4.Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131–185. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- 5.Campbell WB, Falck JR. Arachidonic acid metabolites as endothelium-derived hyperpolarizing factors. Hypertension. 2007;49:590–596. doi: 10.1161/01.HYP.0000255173.50317.fc. [DOI] [PubMed] [Google Scholar]
- 6.Capdevila JH, Falck JR, Imig JD. Role of the cytochrome P450 arachidonic acid monooxygenases in the control of systemic blood pressure and experimental hypertension. Kidney Int. 2007;72:683–689. doi: 10.1038/sj.ki.5002394. [DOI] [PubMed] [Google Scholar]
- 7.Alonso-Galicia M, Maier KG, Greene AS, Cowley AW, Roman RJ. Role of 20-hydroxyeicosatetraenoic acid in the renal and vasoconstrictor actions of angiotensin II. Am J Physiol. 2002;283:R60–R68. doi: 10.1152/ajpregu.00664.2001. [DOI] [PubMed] [Google Scholar]
- 8.Joly E, Seqqat R, Flamion B, Caron N, Michal A, Imig JD, Kramp R. Increased renal vascular reactivity to angiotensin II after unilateral nephrectomy in the rat involves 20-HETE. Am J Physiol. 2006;291:R977–R986. doi: 10.1152/ajpregu.00401.2005. [DOI] [PubMed] [Google Scholar]
- 9.Muthalif MM, Karzoun NA, Gaber L, Khandekar Z, Benter IF, Saeed AF, Parmentier JH, Estes A, Malik KU. Angiotensin II-induced hypertension: contribution of Ras GTPase/mitogen-activated protein kinase and cytochrome P-450 metabolites. Hypertension. 2000;36:604–609. doi: 10.1161/01.hyp.36.4.604. [DOI] [PubMed] [Google Scholar]
- 10.Kaergel E, Muller DN, Honeck H, Theuer J, Shagdarsuren E, Mullally A, Luft FC, Schunck WH. P450-dependent arachidonic acid metabolism and angiotensin II-induced renal damage. Hypertension. 2002;40:273–279. doi: 10.1161/01.hyp.0000029240.44253.5e. [DOI] [PubMed] [Google Scholar]
- 11.Čertíková Chábová V, Kramer HJ, Vaněčková I, Vernerová Z, Eis V, Tesař V, Škaroupková P, Thumová M, Schejbalová S, Husková Z, Vaňourková Z, Kolský A, Imig JD, Červenka L. Effects of chronic cytochrome P-450 inhibition on the course of hypertension and end-organ damage in Ren-2 transgenic rats. Vascular Pharmacol. 2007;47:145–159. doi: 10.1016/j.vph.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 12.Mullins JJ, Peters J, Ganten D. Fulminant hypertension in transgenic rats harboring the mouse Ren-2 gene. Nature. 1990;344:541–544. doi: 10.1038/344541a0. [DOI] [PubMed] [Google Scholar]
- 13.Čertíková Chábová V, Kramer HJ, Vaněčková I, Thumová M, Škaroupková P, Tesař V, Falck JR, Imig JD, Červenka L. The roles of intrarenal 20-hydroxyeicosatetraenoic and epoxyeicosatrienoic acids in the regulation of renal function in hypertensive Ren-2 transgenic rats. Kidney Blood Press Res. 2007;30:335–346. doi: 10.1159/000107710. [DOI] [PubMed] [Google Scholar]
- 14.Wang MH, Brand-Schieber E, Zand BA, Nguyen X, Falck JR, Balu N, Schwartzman ML. Cytochrome P-450-derived arachidonic acid metabolism in the rat kidney: characterization of selective inhibitors. J Pharmacol Exp Ther. 1998;284:966–973. [PubMed] [Google Scholar]
- 15.Morisseau C, Goodrow WH, Dowdy D, Zheng J, Greene JF, Sanborn JR, Hammock BD. Potent urea and carbamate inhibitors of soluble epoxide hydrolase. Proc Nat Acad Sci. 1999;96:8849–8854. doi: 10.1073/pnas.96.16.8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD. Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension. 2002;39:690–694. doi: 10.1161/hy0202.103788. [DOI] [PubMed] [Google Scholar]
- 17.Vaněčková I, Kramer HJ, Bäcker A, Schejbalová S, Vernerová Z, Eis V, Opočenský M, Dvořák P, Červenka L. Early-onset endothelin receptor blockade in hypertensive heterozygous Ren-2 rats. Vascular Pharmacol. 2006;45:163–170. doi: 10.1016/j.vph.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 18.Opočenský M, Kramer HJ, Bäcker A, Vernerová Z, Eis V, Červenka L, Vaněčková I. Late-onset endothelin-A receptor blockade substantially improves survival rate in homozygous hypertensive Ren-2 rats. Hypertension. 2006;48:965–971. doi: 10.1161/01.HYP.0000245117.57524.d6. [DOI] [PubMed] [Google Scholar]
- 19.Husková Z, Kramer HJ, Vaňourková Z, Červenka L. Effects of changes in sodium balance on plasma and kidney angiotensin II levels in anesthetized and conscious Ren-2 transgenic rats. J Hypertens. 2006;24:517–527. doi: 10.1097/01.hjh.0000209988.51606.c7. [DOI] [PubMed] [Google Scholar]
- 20.Husková Z, Kramer HJ, Thumová M, Vaňurková Z, Bürgelová M, Teplan V, Malý J, Červenka L. Effects of anesthesia on plasma and kidney ANG II levels in normotensive and ANG II-dependent hypertensive rats. Kidney Blood Press Res. 2006;29:74–83. doi: 10.1159/000092981. [DOI] [PubMed] [Google Scholar]
- 21.Vaňourková Z, Kramer HJ, Husková Z, Vaněčková I, Opočenský M, Čertíková Chábová V, Tesař V, Škaroupková P, Thumová M, Dohnalová M, Mullins JJ, Červenka L. AT1 receptor blockade is superior to conventional triple therapy in protecting against end-organ damage in Cyp1a1-Ren-2 transgenic rats with inducible hypertension. J Hypertens. 2006;24:2465–2472. doi: 10.1097/01.hjh.0000251909.00923.22. [DOI] [PubMed] [Google Scholar]
- 22.Sandberg MC, Laborde C. Glomerulosclerosis and tubulointerstitial fibrosis are attenuated with 17-beta-estradiol in the aging Dahl salt sensitive rat. J Am Soc Nephrol. 2004;15:1546–1556. doi: 10.1097/01.asn.0000128219.65330.ea. [DOI] [PubMed] [Google Scholar]
- 23.Nakano Y, Hirano T, Uehara K, Nishibayashi S, Hattori K, Aihara M, Yamada Y. New rat model induced by anti-glomerular basement membrane antibody shows sever glomerular adhesion in early stage and quickly progresses to end-stage renal failure. Pathol Int. 2008;58:361–370. doi: 10.1111/j.1440-1827.2008.02237.x. [DOI] [PubMed] [Google Scholar]
- 24.Weibel ER. Practical Methods for Biological Morphometry. London: Academic Press; 1979. Stereological Methods; pp. 44–46. [Google Scholar]
- 25.Lane PH, Steffes MW, Mauer MS. Estimation of glomerular volume: a comparison of four methods. Kidney Int. 1992;41:1085–1089. doi: 10.1038/ki.1992.165. [DOI] [PubMed] [Google Scholar]
- 26.Jacinto SM, Mullins JJ, Mitchell KD. Enhanced renal vascular responsiveness to angiotensin II in hypertensive ren-2 transgenic rats. Am J Physiol. 1999;276:F315–F322. doi: 10.1152/ajprenal.1999.276.2.F315. [DOI] [PubMed] [Google Scholar]
- 27.Kopkan L, Kramer HJ, Husková Z, Vaňourková Z, Škaroupková P, Thumová M, Červenka L. The role of intrarenal angiotensin II in the development of hypertension in Ren-2 transgenic rats. J Hypertens. 2005;23:1531–1539. doi: 10.1097/01.hjh.0000174972.46663.5e. [DOI] [PubMed] [Google Scholar]
- 28.Lee MA, Böhm M, Paul M, Bader M, Ganten U, Ganten D. Physiological characterization of the hypertensive transgenic rat TGR(mRen2)27. Am J Physiol. 1996;270:E919–E929. doi: 10.1152/ajpendo.1996.270.6.E919. [DOI] [PubMed] [Google Scholar]
- 29.Mitchell KD, Jacinto SM, Mullins JJ. Proximal tubular fluid, kidney, and plasma levels of angiotensin II in hypertensive Ren-2 transgenic rats. Am J Physiol. 1997;273:F246–F253. doi: 10.1152/ajprenal.1997.273.2.F246. [DOI] [PubMed] [Google Scholar]
- 30.Sarkis A, Lopez B, Roman RJ. Role of 20-hydroxyeicosatetraenoic acid and epoxyeicosatrienoic acids in hypertension. Curr Opin Nephrol Hypertens. 2004;13:205–214. doi: 10.1097/00041552-200403000-00009. [DOI] [PubMed] [Google Scholar]
- 31.Zhao X, Pollock DM, Zeldin DC, Imig JD. Salt-sensitive hypertension after exposure to angiotensin is associated with inability to upregulate renal epoxygenases. Hypertension. 2003;42:775–780. doi: 10.1161/01.HYP.0000085649.28268.DF. [DOI] [PubMed] [Google Scholar]
- 32.Jung O, Brandes RP, Kim I-H, Schweda F, Schmidt R, Hammock BD, Busse R, Fleming I. Soluble epoxide hypdrolase is a main effector of angiotensin II-induced hypertension. Hypertension. 2005;45:759–765. doi: 10.1161/01.HYP.0000153792.29478.1d. [DOI] [PubMed] [Google Scholar]
- 33.Zou AP, Fleming JT, Falck JR, Jacobs ER, Gebremedhin D, Harder DR, Roman RJ. 20-HETE is an endogenous inhibitor of the large-conductance Ca2+-activated K+ channel in renal arterioles. Am J Physiol. 1996;270:R228–R237. doi: 10.1152/ajpregu.1996.270.1.R228. [DOI] [PubMed] [Google Scholar]
- 34.Gebremedhin D, Lange AR, Narayanan J, Aebly MR, Jacobs ER, Harder DR. Cat cerebral arterial smooth muscle cells express cytochrome P450 4A2 enzyme and produce the vasoconstrictor 20-HETE which enhances L-type Ca2+ current. J Physiol. 1998;507:771–781. doi: 10.1111/j.1469-7793.1998.771bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Randriamboavonjy V, Busse R, Fleming I. 20-HETE-induced contraction of small coronary arteries depends on the activation of Rho-Kinase. Hypertension. 2003;41:801–806. doi: 10.1161/01.HYP.0000047240.33861.6B. [DOI] [PubMed] [Google Scholar]
- 36.Imig JD, Pham BT, LeBlanc EA, Reddy KM, Falck JR, Inscho EW. Cytochrome P450 and cyclooxygenase metabolites contribute to the endothelin-1 afferent arteriolar vasoconstrictor and calcium responses. Hypertension. 2000;35:307–312. doi: 10.1161/01.hyp.35.1.307. [DOI] [PubMed] [Google Scholar]
- 37.Oyekan AO, McGiff JC. Cytochrome P-450-derived eicosanoids participates in the renal functional effects of ET-1 in the anaesthetized rat. Am J Physiol. 1998;274:R52–R61. doi: 10.1152/ajpregu.1998.274.1.R52. [DOI] [PubMed] [Google Scholar]
- 38.Singh H, Schwartzman ML. Renal vascular cytochrome P450-derived eicosanoids in androgen-induced hypertension. Pharmacol Rep. 2008;60:29–37. [PubMed] [Google Scholar]
- 39.Quigley R, Baum M, Reddy KM, Griener JC, Falck JR. Effects of 20-HETE and 19(S)-HETE on rabbit proximal straight tubule volume transport. Am J Physiol. 2000;278:F949–F953. doi: 10.1152/ajprenal.2000.278.6.F949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu M, Lopez B, Dos Santos EA, Falck JR, Roman RJ. Effects of 20-HETE on Na+-K+-ATPase activity in the thick ascending loop of Henle. Am J Physiol. 2007;292:R2400–R2405. doi: 10.1152/ajpregu.00791.2006. [DOI] [PubMed] [Google Scholar]
- 41.Hoagland KM, Flasch AK, Roman RJ. Inhibitors of 20-HETE formation promote salt-sensitive hypertension in rats. Hypertension. 2003;42:669–673. doi: 10.1161/01.HYP.0000084634.97353.1A. [DOI] [PubMed] [Google Scholar]
- 42.Imig JD, Zhao X, Falck JR, Wei S, Capdevila JH. Enhanced renal mircovascular reactivity to angiotensin II in hypertension is ameliorated by the sulfonimide analog of 11,12-epoxyeicosatrienoic acid. J Hypertens. 2001;19:983–992. doi: 10.1097/00004872-200105000-00020. [DOI] [PubMed] [Google Scholar]
- 43.Imig JD, Falck JR, Wei S, Capdevila JH. Epoxygenase metabolites contribute to nitric oxide-independent afferent arteriolar vasodilatation in response to bradykinin. J Vasc Res. 2001;38:247–255. doi: 10.1159/000051053. [DOI] [PubMed] [Google Scholar]
- 44.Wang D, Borrego-Conde LJ, Falck JR, Sharma KK, Wilcox CS, Umans JG. Contribution of nitric oxide, EDHF, and EETs to endothelium-dependent relaxation in renal afferent arterioles. Kidney Int. 2003;63:2187–2193. doi: 10.1046/j.1523-1755.2003.00036.x. [DOI] [PubMed] [Google Scholar]
- 45.Kohagure K, Endo Y, Ito O, Arima S, Omata K, Ito S. Endogenous nitric oxide and epoxyeicosatrienoic acids modulate angiotensin II-induced constriction in the rabbit afferent arteriole. Acta Physiol Scand. 2000;168:107–112. doi: 10.1046/j.1365-201X.2000.00638.x. [DOI] [PubMed] [Google Scholar]
- 46.Madhun ZT, Goldthwait DA, McKay D, Hopfer U, Douglas JG. An epoxygenase metabolite of arachidonic acid mediates angiotensin II-induced rises in cytosolic calcium in rabbit proximal tubule epithelial cells. J Clin Invest. 1991;88:456–461. doi: 10.1172/JCI115325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sakairi Y, Jacobson HR, Noland DT, Capdevila JH, Falck JR, Breyer MD. 5,6-EET inhibits ion transport in collecting duct by stimulating endogenous prostaglandin synthesis. Am J Physiol. 1995;268:F931–F939. doi: 10.1152/ajprenal.1995.268.5.F931. [DOI] [PubMed] [Google Scholar]
- 48.Imig JD. Epoxide hydrolase and epoxygenase metabolites as therapeutic targets for renal diseases. Am J Physiol. 2005;289:F496–F503. doi: 10.1152/ajprenal.00350.2004. [DOI] [PubMed] [Google Scholar]
- 49.Yoshida Y, Kawamura T, Ikoma M, Fogo A, Ichikawa I. Effects of antihypertensive drugs on glomerular morphology. Kidney Int. 1989;36:626–635. doi: 10.1038/ki.1989.239. [DOI] [PubMed] [Google Scholar]
- 50.Dworkin LD, Feiner HD, Parker M, Tolbert E. Effects of nifedipine and enalapril on glomerular structure and function in uninephrectomized SHR. Kidney Int. 1991;39:1112–1117. doi: 10.1038/ki.1991.141. [DOI] [PubMed] [Google Scholar]
- 51.Imig JD, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov. 2009;8:794–805. doi: 10.1038/nrd2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McGiff JC. Cytochrome P-450 metabolism of arachidonic acid. Ann Rev Pharmacol Toxicol. 1991;31:339–169. doi: 10.1146/annurev.pa.31.040191.002011. [DOI] [PubMed] [Google Scholar]
- 53.Makita K, Falck JR, Capdevila JH. Cytochrome P450, the arachidonic acid cascade, and hypertension: new vistas for old enzyme system. FASEB. 1996;10:1456–1463. doi: 10.1096/fasebj.10.13.8940291. [DOI] [PubMed] [Google Scholar]
- 54.Sakuma S, Usa K, Fujimoto Y. 15-hydroperoxyeicosapentaenoic acid, but not eicosapentaenoic acid, shifts arachidonic acid away from cyclooxygenase pathway into acyl-CoA synthetase pathway in rabbit kidney medulla microsomes. ProstaglandinsLeukot Essent Fatty Acids. 2006;75:69–74. doi: 10.1016/j.plefa.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 55.Spector AA. Arachidonic acid cytochrome P450 epoxygenase pathway. J Lipid Res. 2009;50:S52–S60. doi: 10.1194/jlr.R800038-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zicha J, Kuneš J. Ontogenetic aspects of hypertension development: analysis in the rat. Physiol Rev. 1999;79:1227–1282. doi: 10.1152/physrev.1999.79.4.1227. [DOI] [PubMed] [Google Scholar]
- 57.Muthalif MM, Benter LF, Karzoun N, Fatima S, Harper J, Uddin MR, Malik KU. 20-Hydroxyeicosatetraenoic acid mediates calcium/calmodulin dependent protein kinase II-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. Proc Natl Acad Sci USA. 1998;95:12701–12706. doi: 10.1073/pnas.95.21.12701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parmentier J-H, Muthalif MM, Nishimoto AT, Malik KU. 20-Hydroxyeicosatetraenoic acid mediates angiotensin II-induced phospholipase D activation in vascular smooth muscle cells. Hypertension. 2001;37:623–629. doi: 10.1161/01.hyp.37.2.623. [DOI] [PubMed] [Google Scholar]
- 59.Yaghini FA, Zhang C, Parmentier JH, Estes AM, Jafari N, Schaefer SA, Malik KU. Contribution of arachidonic acid metabolites derived via cytochrome P4504A to angiotensin II-induced neointimal growth. Hypertension. 2005;45:1182–1187. doi: 10.1161/01.HYP.0000168051.04275.ea. [DOI] [PubMed] [Google Scholar]
- 60.Camara NO, Martins JO, Landgraf RG, Jancar S. Emerging roles for eicosanoids in renal diseases. Curr Opin Nephrol Hypertens. 2009;18:21–27. doi: 10.1097/MNH.0b013e32831a9df7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.