

Abstract
Biocatalytic reduction of α- or β-alkyl-β-arylnitroalkenes provides a convenient and efficient method to prepare chiral substituted nitroalkanes. Pentaerythritol tetranitrate reductase (PETN reductase) from Enterobacter cloacae st. PB2 catalyses the reduction of nitroolefins such as 1-nitrocyclohexene (1) with steady state and rapid reaction kinetics comparable to other old yellow enzyme homologues. Furthermore, it reduces 2-aryl-1-nitropropenes (4a-d) to their equivalent (S)-nitropropanes 9a-d. The enzyme shows a preference for the (Z)-isomer of substrates 4a-d, providing almost pure enantiomeric products 9a-d (ees up to > 99%) in quantitative yield, whereas the respective (E)-isomers are reduced with lower enantioselectivity (63-89% ee) and lower product yields. 1-Aryl-2-nitropropenes (5a, b) are also reduced efficiently, but the products (R)-10 have lower optical purities. The structure of the enzyme complex with 1-nitrocyclohexene (1) was determined by X-ray crystallography, revealing two substrate-binding modes, with only one compatible with hydride transfer. Models of nitropropenes 4 and 5 in the active site of PETN reductase predicted that the enantioselectivity of the reaction was dependent on the orientation of binding of the (E)- and (Z)-substrates. This work provides a structural basis for understanding the mechanism of asymmetric bioreduction of nitroalkenes by PETN reductase.
Keywords: asymmetric hydrogenation, biocatalysis, bioreduction, nitroalkenes, pentaerythritol tetranitrate reductase, stereocontrol
Introduction
Nitroalkanes are versatile, inexpensive synthons that can be converted readily to the corresponding amines, aldehydes, carboxylic acids, oximes, hydroxylamines, or denitrated compounds.[1] Enantiomerically pure nitroalkanes have been produced by a variety of chemical methods. These include asymmetric conjugate addition to nitroalkenes,[2] asymmetric conjugate addition of nitroalkanes to various α,β-unsaturated bonds (C=C or C=N),[3] asymmetric conjugate reduction of nitroalkenes using chiral transition metal catalysis,[4] or asymmetric transfer hydrogenation of olefins using a Jacobsen-type organocatalyst.[5] Asymmetric reduction is especially attractive as up to two stereogenic centres can be formed. Therefore, the asymmetric reduction of activated C=C bonds by microorganisms or enzymes has recently received a lot of interest since the catalysts are environmentally benign, the reactions can be performed under mild conditions, substrate ranges are broad, and product enantiopurity is generally excellent.[6]
Enzymes from the old yellow enzyme (OYE) family have great potential in industrial biocatalysis as they catalyse a range of synthetically useful reductions.[6-11] These FMN-containing enzymes catalyse the NAD(P)H-dependent reduction of activated α,β-unsaturated alkenes (Scheme 1).[6] Asymmetric bioreduction by OYEs proceeds in a step-wise fashion with a mechanism resembling Michael-type conjugate addition of hydride to activated alkenes[12] with the addition of [H2] proceeding in a trans-fashion,[12] although there is evidence for cis-addition in other related classes of enzymes.[6,13]
Scheme 1.

(a) General mechanism of the biocatalytic reduction of α,β-unsaturated alkenes catalyzed by the OYE family. (b) General mechanism of the biocatalytic reduction of α,β-unsaturated nitroalkenes involving nitronate formation and aci-nitro tautomerisation of the product formed. EWG=electron withdrawing group; *=chiral centre.
Recently, the asymmetric bioreduction of a variety of α,β-unsaturated aldehydes, ketones, maleimides and nitroalkenes was demonstrated with various members of the OYE family.[6-11] Highly enantioselective reduction of β,β-disubstituted nitroalkenes has been shown to occur in organisms such as baker’s yeast,[14] and using OYE homologues from Lycopersicon esculentum (OPR1 and OPR3),[8,10] baker’s yeast Saccharomyces carlsbergensis (OYE1-3),[9] Zymomonas mobilis (NCR),[9] and Bacillus subtilis (YqjM)[8] and a new reductase from Clostridium sporogenes.[15] The enzymes exhibited different substrate selectivities, but, in general, the reactions were catalysed with high enantioselectivity and yield. In contrast, biological reduction of α,β-disubstituted nitroalkenes proceeded with poor enantioselectivity (< 50% ee).[15-17]
Some of the OYE family members are known to catalyse a broad range of chemical reactions. For example, pentaerythritol tetranitrate reductase (PETN reductase) from Enterobacter cloacae st. PB2 not only acts as an ene reductase (e.g., for reduction of 2-cyclohexenone and some steroids),[18] but also catalyses the reduction of explosives, including nitroaromatic compounds (e.g., trinitrotoluene, TNT), and cyclic triazines (e.g., Royal Demolition Explosive).[18,19] The enzyme also cleaves nitrate esters, such as pentaerythritol tetranitrate (PETN) and the vasodilator nitroglycerin.[18-20]
Owing to its broad specificity, we conjectured that PETN reductase might be an excellent candidate for industrial biocatalysis. This enzyme is extremely stable, and can be expressed to high levels in Escherichia coli.[21] It has been characterised extensively at both the mechanistic and structural level with substrates such as 2-cyclohexenone and nitroaromatic explosives.[18] Multiple structures of PETN reductase have been determined to high resolution,[22] both in the presence and absence of bound substrates and inhibitors.[18,22-25] This enabled us to take a structure-driven approach to aid in the prediction of both substrate selectivity and the stereochemical outcome of the reactions. In this paper, we have explored the ability of PETN reductase to catalyse the enantioselective reduction of a variety of both (E)- and (Z)-α/β-alkyl-β-arylnitroalkenes to examine its potential as a generic biocatalyst.
Results and Discussion
Kinetic Analysis of Nitroalkene Reduction
To explore its reactivity with nitroalkenes, PETN reductase was initially tested for reduction of the commercially available substrates 1-nitrocyclohexene (1), (E)-1-nitro-2-phenylethene (2) and (E)-1-nitro-2-(2′-thienyl)-ethene (3) (Scheme 2).
Scheme 2.

Substrates and products of C=C double bond bioreduction catalysed by PETN reductase.
The steady-state kinetic parameters were determined using the stopped-flow method of Gibson et al.[26] monitoring the reduction of the flavin coenzyme directly (~ 450 nm) to avoid the interference from the overlapping spectra of NADPH and the substrate at shorter wavelengths. The catalytic rate for reduction of 1-nitrocyclohexene was twice as fast with PETN reductase as with OYE1,[27] whilst the PETN reductase-catalysed reduction of nitroalkenes (E)-2 and (E)-3 was 4-5 times as rapid, suggesting that PETN reductase would be an excellent candidate for industrial biocatalysis (Table 1a; Supporting Information Figure S1). PETN reductase had 5- to 10-fold higher Km values for the compounds tested compared to OYE1 (Table 1a), making the specificity of the reaction (kcat/Km) between 11- and 45-fold lower for PETN reductase. Nevertheless, the Km values were still < 1 mM, and, therefore, would not place restrictions on industrial exploitation.
Table 1.
(a) Steady state kinetic constants and (b) rate constants for the oxidative half-reaction of PETN reductase with three nitro olefin substrates. In the case of (E)-2-3, kox/Kd was determined from the initial slope of the kobs vs. substrate concentration plot where the substrate concentration is well below Km
| (a) Steady State Compound | ||||||
|---|---|---|---|---|---|---|
| kcat [s-1] | PETN reductase Km [μM] |
kcat/Km [s-1M-1] | kcat [s-1] | OYE1[a] Km [μM] |
kcat/Km [s-1M-1] | |
| 1 | 9.5±1.0 | 971±146 | 9773 | 4.4±0.2 | 10±1 | 440,000 |
| (E)-2 | 20.9±0.8 | 341±42 | 61290 | 5.0±0.1 | 4.2±0.3 | 1,190,476 |
| (E)-3 | 21.4±1.0 | 413±39 | 51815 | 4.3±0.2 | 7.7±0.5 | 558,441 |
| (b) Oxidative Half Reaction Compound | ||||||
|---|---|---|---|---|---|---|
| kox [s-1] | PETN reductase Kd [mM] |
kox/Kd [s-1M-1] | kox [s-1] | OYE1[a] Kd [mM] |
kox/Kd [s-1M-1] | |
| 1 | 41.7±2.1 | 6.4±0.7 | 6502 | - | - | 610,000 |
| (E)-2 | ND | >3 mM | 4250 | 420±20 | 0.15±0.02 | 2,800,000 |
| (E)-3 | ND | >2 mM | 1272 | 170±30 | 0.17±0.03 | 1,000,000 |
Data from[27]
ND=not determined.
A study of the reoxidation of PETN reductase by 1-nitrocyclohexene using stopped-flow methods showed a hyperbolic dependence of the rate constant with substrate concentration, as opposed to OYE1 where only a second order rate constant was determined.[27] In contrast, only second order rate constants could be calculated for the oxidative half reaction with nitroalkenes 2-3 due to the low solubility of the substrates and relatively high Kd values compared to OYE1 (Table 1b; Supporting Information Figure S1).
To investigate the biocatalytic potential of PETN reductase we monitored the reduction of a variety of nitroolefins (E)-1-3 and (E)- and (Z)-4-5 to complete substrate depletion under both homogeneous (maximum 5% ethanol in water) and biphasic conditions. Initially the reactions were monitored spectrophotometrically under homogeneous conditions using a glucose 6-phosphate dehydrogenase-NADP+/NADPH cofactor regeneration system to avoid the problem of NADPH spectrum overlap with the substrates.[27] All reactions were performed anaerobically to avoid enzyme inactivation by reactive oxygen species, and to eliminate the competition between the oxidation of the flavin cofactor by the nitroalkene substrates and oxygen.
The spectral changes due to the reduction of (E)- and (Z)-(4′-chlorophenyl)-nitropropenes under homogeneous conditions were monitored continuously until reaction completion (data for substrates 4c and 5b are shown in Figure 1; spectral data for all other compounds are in Supporting information Figure S3). The apparent preference of PETN reductase for substrates under biotransformation conditions (reaction to complete substrate depletion) can be analysed by comparing the 50% depletion time of each substrate (t50; Supporting Information Table S1).
Figure 1.

Spectral changes of nitroalkene reduction by PETN reductase under aqueous conditions. Inset: Time course of absorbance changes upon substrate reduction. The arrows indicate the direction of spectral change. (a) (Z)-2-(4′-chlorophenyl)-1-nitropropene [(Z)-4c]; (b) (E)-2-(4′-chlorophenyl)-1-nitropropene [(E)-4c]; (c) (Z)-1-(4′-chlorophenyl)-2-nitropropene [(Z)-5b]; (d) (E)-1-(4′-chlorophenyl)-2-nitropropene [(E)-5b].
PETN reductase showed a clear preference for the (Z)-β,β-disubstituted nitroalkenes 4a-d compared to their respective (E)-isomers as seen by an approximate 24- to 45-fold increase in the t50values (Supporting Information Table S1). The preference for both (Z)- and (E)-4a, c, d in terms of aromatic ring substituent decreased in the order: Br > Cl > H [~ F for (E)-4b]. This effect was less pronounced with the (E)-substrates, as the apparent rates varied by only 2-fold. When the phenyl ring of the substrate (E)-2 was replaced by thiophene [(E)-3], the turnover rate increased by 5- to 15-fold compared with (E)-2, although the rate with (E)-3 was 3- to 7-fold lower than the (Z)-isomers of substrates 4. This demonstrates that PETNR substrate specificity extends to heteroaromatic derivatives with potential to vary the substituents at Cβ.
Unlike β,β-disubstituted nitroalkenes, the t50 of both (Z)- and (E)-isomers of 1-(4′-chlorophenyl)-2-nitropropene 5b were very similar, with rates comparable to the substrate (Z)-4a. The presence of a methyl group at Cα in substrate (E)-5a resulted in an approximate 75-fold increase in apparent rate compared to the equivalent substrate (E)-4a which has the methyl group at Cβ. The absence of a methyl group at Cβ may have contributed to the increased reaction rate by altering the orientation of substrate binding and/or diminished steric hindrance. Detailed kinetic and structural analyses are required to determine if these trends are due to binding effects and/or the electron-withdrawing effects of the substituted aromatic rings on the Cβ.
Intermediates Formed during Nitroalkene Reduction
The spectral studies performed to determine the rates of reduction of nitroalkenes 1-5 provide some insight into the reaction carried out by PETN reductase. By using a NADPH-regenerating system[27] we were able to follow the spectral changes during substrate reduction in the absence of NADP+/NADPH spectral interference. Previous studies with wild-type/Y196F OYE1[27] and morphinone reductase (MR)[28] showed that hydride transfer and protonation steps during reduction of 1 are decoupled, in contrast to the reaction with 2-cyclohexen-1-one in which these steps are concerted.[29] The decoupling makes it possible to monitor the accumulation of the nitronate intermediate, which results in an increase in absorbance in the region of 238 nm. As expected, nitronate accumulation and slow decay was seen during the reduction of 1-nitrocyclohexene with PETN reductase (Supporting Information Figure S3a) similar to wild-type OYE1.[27] Apparent nitronate accumulation and decay was also seen with substrates (E)-5a and (E)-5b (Supporting Information Figure S3i and Figure 1d, respectively), both of which contain a methyl group at the carbon to be protonated (α-carbon). Furthermore, substrate decay of (E)-5b proceeded at a linear rather than exponential rate. The decay at 238 nm with the other substrates (Supporting Information Figure S3b-h; Figure 1a-c) closely followed the decrease in absorbance due to hydride transfer, suggesting that the hydride transfer and protonation steps may be concerted with these substrates.
Studies with Y186F mutant of OYE1[27] and MR[28] showed either a considerable delay or absence, respectively, of decay of the nitronate intermediate of 1-nitrocyclohexene attributed to protonation by water rather than by the enzyme. Similarly, previous studies with PETN reductase showed that water is the most likely proton donor during the reduction of 2-cyclohexene-1-one.[24] The similarity with the kinetics of nitronate accumulation and decay suggest that 1-nitrocyclohexene (1) and nitroalkenes 5 are reduced via a similar mechanism. However, studies with wild-type OYE1[27] showed that nitronate accumulation with 1-nitrocyclohexene does not rule out the possibility of an enzymatic protonation step. Therefore, comparative studies of reductions of nitroalkenes 1-5 with Y186F PETN reductase mutant[24] are required to determine whether Y186 plays a role in nitronate protonation with these substrates, or if this apparent decoupling of the hydride transfer and protonation steps with (E)-5a and (E)-5b is due to essentially non-catalytic protonation by water.
Stereochemical Outcome of the Reaction
Enoate reductases from the OYE family[6,30] produce opposite enantiomers from (E)- and (Z)-enoates and enals, enabling a substrate-based control of the stereochemical outcome of the reaction. In contrast, the reduction of (E) and (Z)-isomers α- or β-alkyl-β-arylnitroalkenes catalysed by baker’s yeast[14,16] or crude extracts of Clostridium sporogenes,[15] proved to be an enantioconvergent process. There is less information about the stereochemical course of the reduction of (E)- and (Z)-nitroalkenes with purified enzymes of the OYE family, although their enantioselectivity seems to differ widely (Scheme 3). OPR3, YqjM and NCR reduce (E)-4a to the (S)-enantiomer of 9a, although OPR3 and YqjM exhibit rather low enantioselectivities.[8-10] In contrast, OYE1-3 and OPR1 produce the (R)-enantiomer of 9a with 83% ee and 98% ee, respectively.[8,9] Therefore, different enantiomeric products can be obtained by judicious selection of the biocatalyst.
Scheme 3.

Asymmetric bioreduction of α,β-disubstituted nitroalkenes by OYE enzymes. OYE1-3=old yellow enzymes 1-3; NCR=NCR enoate reductase from Zymomonas mobilis; OPR1 and 3=12-oxophytodienoate reductase isoforms from Lycopersicon esculentum; YqjM=from Bacillus subtilis.[8-11]
In order to investigate the stereochemical outcome and the enantioselectivity of the reaction, the bioreductions were performed in a biphasic reaction system (isooctane/phosphate buffer pH 7.0, 40:60, v/v; Table 2) in the presence of the glucose 6-phosphate dehydrogenase NADP+/NADPH cofactor regeneration system.[15] This allowed us to conduct reactions at higher concentrations of the substrate and enabled facile monitoring of the reaction by chiral HPLC and GC-MS.
Table 2.
Reduction of nitroolefins 1-5 by PETN reductase under biphasic reaction conditions[a]
| Number | Substrate | λmax[b] [nm] | Time[c] [days] | Conversion[d],[e] [%] | Yield[d],[e] [%] | Product[f] | ee[e] [%] |
|---|---|---|---|---|---|---|---|
| 1 |
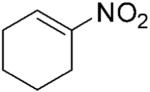
|
264 | 2 | 100 | >99 | 6 | N/A |
| (E)-2 |
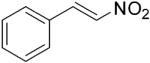
|
319 | 2 | 100[g] | >99[g] | 7 | N/A |
| (E)-3 |
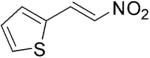
|
357 | 1 | 100 | 82 | 8 | N/A |
| (E)-4a |
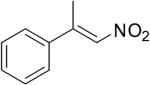
|
306 | 3 | 100[h] | 58[h] | (S)-9a | 89[h] |
| (Z)-4a |
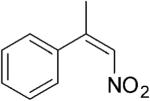
|
208 | 3 | 100[h] | 89[h] | (S)-9a | 96[h] |
| (E)-4b |
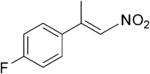
|
312 | 3 | 100 | 86 | (S)-9b | 63 |
| (E)-4c |
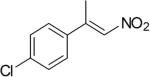
|
310 | 7 | 70 | 18 | (S)-9c | 72 |
| (Z)-4c |
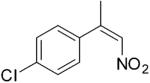
|
221 | 4 | 100 | 91 | (S)-9c | 96 |
| (E)-4d |
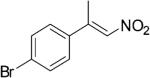
|
314 | 7 | 73 | 35 | (S)-9d | 75 |
| (Z)-4d |
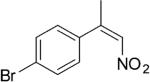
|
224 | 7 | 83 | 76 | (S)-9d | >99 |
| (E)-5a |
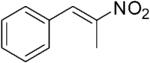
|
316 | 3 | 100[h] | 93[h] | (R)-10a | 14[h] |
| (E)-5b |
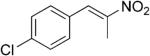
|
321 | 7 | 93 | 84 | (R)-10b | 54 |
| (Z)-5b |
|
230 | 7 | 88 | 80 | (R)-10b | 60 |
Conditions: The reactions contained potassium phosphate buffer (50 mM KH2PO4/K2HPO4 pH 7.0, 9.2 mL) nitroalkene (1.7 mM in 4.8 mL anaerobic isooctane), NADP+ (20 μM), glucose 6-phosphate (14-20 mM), glucose 6-phosphate dehydrogenase (10 units), deoxygenated PETN reductase (2 μM) and sec-butylbenzene (25 μL). Reactions were agitated at 200 rpm for 1-7 days at 30°C in an anaerobic cabinet. N/A=not applicable.
Maximal absorbance wavelength of the substrate used to monitor reduction.
Time of biphasic reaction in days.
Conversions and yields were determined by HPLC from calibration curve of each standard substrate and products and corrected using sec -butylbenzene as internal standard.
Chiralcel OD-H.
Absolute configuration assigned by comparison with literature data.[15]
Phenomenex C-18.
Chiralcel OJ.
Similarly to OPR3, YqjM and NCR, the reduction with (E)-isomers of 4a-d by PETN reductase led to the formation of the products (S)-9a-d (Table 2), although with only moderate and variable enantiopurity (63-89% ee). Furthermore, by-products were formed, which led to decreased product yield (18-58%,Table 2). These products were tentatively assigned by GC-MS to be their equivalent oxime, aldehyde (Nef reaction product) and alcohol. In contrast, the reduction of (Z)-isomers of 4 by PETN reductase gave (S)-enantiomers of the corresponding nitropropanes 9 in almost quantitative yields and with high enantiopurity (≥ 96% ee). Thus, the reduction of (E)- and (Z)-isomers of substrates 4 proceed in a stereoconvergent manner.
Reduction of the α,β-disubstituted nitroalkenes (E)-5a, b (Scheme 2, Table 2), using PETN reductase generated the products (R)-10a and b with a much lower optical purity (14% and 54% ee, respectively), although the reaction rate and yield were generally very good. The reduction of the (Z)-isomer of 5b was stereoconvergent with the reduction of its (E)-counterpart and it led to the formation of the product (R)-10b in a comparable yield, but with a slightly higher optical purity (60% ee). Furthermore, small amounts of by-products were detected during reduction of the compounds 5, which were identified by GC-MS as their respective arylpropanones (Nef reaction products).
The stereoconvergent course of the reduction of both isomers of the substrates 4 and 5 is in striking contrast with the stereodivergence of the reduction of α,β-unsaturated carboxylic acids and carbonyl compounds, which depends on geometry of the substrate (E/Z-isomers).[6,11,31] This unusual behaviour with nitroalkenes could be the result of one or more of the following: (i) the substrate orientation in the active site of an enzyme is not determined by the interaction of the nitro group with H181 and/or H184 as with enoates; (ii) PETN reductase may only react with one isomer of the substrate and non-catalytic E/Z-isomerisation of the substrates ensures the supply of the preferred isomer in the reaction and/or (iii) the reaction proceeds via trans addition[9] of H- and H+ with the α- and β-carbons of both (E)- and (Z)-substrate binding in the same relative orientation (β- and α-carbon above and below FMN N5, respectively).
To investigate this, we followed the time course of reduction of pairs of (E)- and (Z)-isomers of nitroalkenes 4c (Figure 2a and b) and 5b (Figure 2c and d). It should be noted that there was a low rate of nonenzymatic E/Z-isomerisation of each substrate under the reaction conditions with no PETN reductase present (Supporting information Figure S4). Substrate (Z)-4c (Figure 2a) was converted to the product (S)-9c (96% ee) within 4 days. The sample of (Z)-4c contained 7% of the (E)-isomer due to non-catalytic Z- to E-isomerisation prior to the reaction. The concentration of the (E)-isomer decreased, despite the continuous isomerisation of (Z)-4c, indicating that either the (E)-isomer was being consumed or that (E)- to (Z)-isomerisation was occurring. In contrast, while the conversion of (E)-4c initially proceeded quickly, after 5 days the conversion was only about 70%, with only 18% yield of the product (S)-9c (72% ee) (Table 2). In the case of α,β-disubstituted nitroalkenes, reduction of both (Z)- and (E)-isomers of nitroalkenes 5b proceeded with a similar apparent rate, enantioselectivity (54-60% ee) and yield (80-84%). This suggests that the substrates bind in a similar manner, possibly via one or more of the highly conserved H181/H184 residues, as suggested for other OYE enzymes (Scheme 4).[32] The detection of the opposite enantiomeric products in the reduction of (E)-4a-d and (E/Z)-5a-b suggests more than one binding mode of the substrates is possible (Scheme 4).
Figure 2.

Time course of the PETN reductase-catalysed reduction of (Z)- and (E)-isomers of 2-(4′-chlorophenyl)-1-nitropropene 4c (a and b, respectively) under biphasic conditions. Substrates: □=(Z)-4c; ○=(E)-4c; product: ▲=(R)-9c. Time course of the PETNR-catalysed reduction of (Z)- and (E)-isomers of 1-(4′-chlorophenyl)-2-nitropropene 5b (c and d, respectively) under biphasic conditions. Substrates: □=(Z)-5b; ○=(E)-5b; product: ▲=(R)-10b. The % ees throughout the reactions were fairly consistent with that of the final product (% ee was 52-60% and 53-56% for (Z)-5a and (E)-5a, respectively) and dropped by only 1% per day.
Scheme 4.

Theoretical model for the selectivity of PETN reductase with (E)-4 (a) and (E)-5 (b), adapted from[45]. AA=amino acid residue/s modelled to interact with the substrate.
It has been consistently observed that whereas the reduction of β,β-disubstituted nitroalkenes of structure 4 usually proceeds in a highly enantioselective fashion, the α,β-disubstituted nitroalkenes 5 are reduced with poor enantioselectivity.[6] Low product enantiopurity in the reduction of α,β-disubstituted nitroalkenes 5 was found in the reactions with C. sporogenes[15] and baker’s yeast.[16] Initially, this had been attributed to racemisation of the product under the reaction conditions[6,14] but it is now known that the exchange of the Cα proton is relatively slow.[15,16] Previous studies showed that a 5-day incubation of the product in D2O yielded only about 5% proton exchange, which does not fully account for the observed low enantiopurity of the products 10.[15] Since the enantiopurity of the product of the reduction of the substrates 5 depends on the protonation step of Cα, the low enantioselectivity of PETN reductase could be also due to slow non-enantiospecific interconversion of the aci-form of the corresponding nitronic acid of the bioreduction mechanism (Scheme 1b).[15,16,27] At present the protonation step is not fully understood, however literature data suggest that protonation is the rate-limiting step for bioreductions of nitroalkenes.[27,33] Thus nitronate anions themselves can act as ambident nucleophiles (Scheme 1b), which can be attacked from both soft and hard nucleophiles such as carbon and oxygen, respectively. These can attack soft or hard electrophiles respectively and such behaviour can be seen in the Nef reaction, for example.[34] Therefore, one can also expect the formation of both a nitroalkane (protonation of Cα) and a respective nitronic acid (protonation of O), which will slowly equilibrate to the optically inactive nitroalkane (Scheme 1b). As postulated by Swiderska and Stewart, the pKa values of the nitronate intermediate and the amino acid residue can make proton transfer thermodynamically unfavourable.[35]
In this context, the formation of the Nef reaction by-products during the reduction of nitroalkenes could be a result of an ambident character of nitronate intermediates;[27] however it cannot be concluded whether or not this process involves enzymatic catalysis. It is noteworthy that similar to the OYE family, flavin nitroalkene oxidases[36] catalyse the transformation of nitroalkenes to the respective aldehydes and ketones, in what is an enzymatic equivalent of a Nef reaction.[34] Other enzymes capable of catalysing such reactions are horseradish peroxidase,[37] propionate-3-nitronate oxidase from Penicillium atrovenetum,[38] and 2-nitropropane dioxygenase from Neurospora crassa.[39] These latter reactions also involve the formation of nitronates, which may suggest that Nef by-product formation by PETNR may be a catalytic rather than a chemical decomposition of the intermediate; however, this hypothesis needs to be confirmed by appropriate mechanistic studies. Interestingly, the enzyme CYP83B1 from Arabidopsis thaliana catalyses oxime conversion to thiohydroximic acid via the formation of a nitronate intermediate in the biosynthetic pathway of glucosinolates,[40] which is analogous to the reverse of the reaction catalysed by PETN reductase. While the exact mechanisms of sideproduct formation in PETN reductase are unknown, such comparisons with other known enzymes catalysing similar reactions may give insight into possible mechanisms of action. Additionally, PETNR is known to catalyse additional types of reactions seen by its ability to degrade nitroaromatic explosives such as TNT and picric acids.[18,22] Thus, whilst PETN reductase is a promiscuous biocatalyst, there is a need for (i) detailed evaluation of the mechanism and (ii) caution in inferring mechanistic similarities between various OYE family members and between bioreduction of various α,β-unsaturated compounds and their isomers.
Structure of the PETN Reductase Complex with 1-Nitrocyclohexene 1
We determined the structure of the 1-nitrocyclohexene-bound PETN reductase to 1.34 Å resolution to assist the modelling of aryl-ntiropropenes 4 and 5.We used 1-nitrocyclohexene because the low solubility and apparent high Kd values for the latter substrates prevent structure determination of complexes using X-ray crystallography. The data collection and refinement statistics are found in Supporting Information Table S2. The structure was isomorphous with the previously determined apo-enzyme and substrate-bound complexes with 2-cyclohexenone and steroids,[18,22-24] except for a minor change in the back-bone conformation of a surface loop near the substrate entry point (residues T273 to P280).
The electron density maps are consistent with 1-nitrocyclohexene being bound close to the flavin ring (modelled with a partial occupancy of ~60%) in the enzyme active site (Figure 3a). There was also evidence for partial occupation of a chloride ion and isopropyl alcohol molecule at the position where compound 1-nitrocyclohexene (1) binds, as seen in the PETN reductase Y186F structure (pdb code 2ABB).[24] Inspection of the electron density for the cyclohexene ring revealed that all atoms except for C3 lie approximately in one plane. This suggests that the substrate is bound in two different orientations (in a 1:1 ratio) corresponding to the two different half-chair conformations of the 1-nitrocyclohexene ring. The two binding modes are related by approximately 180° rotation of the cyclohexene ring about the N7-C1 bond with an inversion in the half-chair conformation (Figure 3a). Such multiplicity of binding modes has been shown to occur in PETN reductase with various non-physiological substrates and inhibitors[23] due to the relatively large size of the active site and the presence of few direct interactions between the substrates/inhibitors and the enzyme.
Figure 3.

Active site of PETN reductase containing bound substrate. The figures were generated in Pymol.[56] All residues are shown as atom-coloured sticks with green and yellow carbons for protein and FMN, respectively. Hydrogen bonds are indicated by dotted lines (grey lines indicate the bond is behind the side chain). (a) Stereo view of the 2 |Fo|–|Fc| electron density map contoured at 1 σ and final atomic model of the active site of 1-bound PETN reductase. The catalytic and non-productive positions for 1 are shown as atom-coloured sticks with pink carbons and grey lines, respectively. Dual models of the (b) (E)- and (c) (Z)-isomers of 4c and 5b bound in the active site of PETN reductase. The substrates (E)- and (Z)-4c are shown as atom-coloured sticks with magenta and blue carbons for models 1 and 2, respectively. The substrates (E)- and (Z)-5b are shown as atom-coloured lines with magenta and blue carbons for models 1 and 2, respectively. The hydrogen bonds are colour-coded to indicate the substrate and model, with large and small dots for (E/Z)-4c and (E/Z)-5b substrates, respectively.
1-Nitrocyclohexene (1) binds to the si-face of the flavin and is stabilised by a hydrogen bond to H184, with van der Waals contacts with H181, T26, W102, Y186, and Y351. The electronegative nitro group is stabilised by proximity to positively charged H181 and H184. These residues are known to play a similar role with other substrates such as steroids, 2-cyclohexenone, picric acid and TNT.[18,22-24] In one binding mode, the β-carbon atom C2 (activated for nucleophilic attack through resonance stabilisation with histidine H181 and/or H184)[18] is only 3.7 Å away from the FMN nitrogen atom N5. Superposition of this structure with the PETN reductase-picric acid complex shows that C2 of the cyclohexene moiety is bound in a very similar position to that of the β-carbon of picric acid[18] (results not shown). Furthermore, the angle between atoms N10(FMN)–N5-(FMN)–C2(1-nitrocyclohexene) is 105°, which is close to optimal for hydride transfer.[41] This suggests that the reaction mechanism for the reduction of the C=C double bond of 1-nitrocyclohexene proceeds via a direct nucleophilic attack by hydride from the flavin N5 atom at the Cβ (C2) position of the substrate. In contrast, the second binding mode for 1-nitrocyclohexene (Figure 3a) is thought to be non-productive as the double bond of the cyclohexene ring is not in a suitable orientation and distance from the flavin N5.
Analysis of the productive binding mode of 1-nitrocyclohexene showed that the Cα (C1) of the substrate is only 3.2 Å away from the hydroxy group of Y186 with an angle CZ(Y186)–OH(Y186)–C1(1-nitrocyclohexene) of 125°. This suggests a possible role of Y186 as a potential proton donor to Cα (C1) in the C=C double bond reduction, as seen in OYE,[42] but further kinetic studies are required to determine if water or Y186 is the proton donor.
Structure-Based Models of Nitroalkene Enzyme-Substrate Complexes
We generated two models each of nitropropenes 4 and 5 bound to the active site of PETN reductase, where we maintained a close to ideal distance and orientation of the β-carbon of the substrate to N5(FMN) (3.71 Å and 105°, respectively), with minimal clashes with surrounding residues. Models 1 and 2 were based on the positions of the 1-nitrocyclohexene and published picric acid-bound structures, respectively, in the productive binding conformation in PETN reductase (Figure 3b and c).[23,24,43] Model 1 of the p-Cl substituted nitroalkene substrates (E)-4c and (E)-5b (Figure 3b) showed the nitro group of the substrate to be in a similar position as the nitro group of the active 1-nitrocyclohexene-PETN reductase complex, where it can potentially form hydrogen bonds with residues H181 and H184.[23] These hydrogen bonds are also seen in the PETN reductase structures containing picric acid and 2,4-dinitrophenol (between histidine and OH groups).[18] In these models, the aromatic rings of the substrates and Y68 are within van der Waals contact, although no π-π stacking is observed. There appears to be a loss of the interaction between the OH groups of Y68 and Y186, with further potential van der Waals contacts with T26, Y186 and S132. The side chain of Y351 was reoriented to accommodate the aromatic ring of the substrate.
In model 2 of substrates (E)-4c and (E)-5b (Figure 3b) a potential loss of interaction between the nitro group O2 and H184 NE2 was seen as well as a putative interaction modelled between the nitro group O1 and T26 OG. Both models for substrate (E)-4c suggest that the methyl group (Cβ) is positioned in close proximity to the side chain of T26, which may hinder productive substrate binding. This may explain why the nitroalkenes (E)-4a-d are poorer substrates compared with (E)-2, since the latter substrate differs by the lack of the methyl group at the Cβ.
Model 1 of the substrates (Z)-4c and (Z)-5b was similar to those for substrates (E)-4c except for minor shifts in position and a reorientation of the aromatic ring (Figure 3c). This enabled a favourable FMN N5-substrate carbon distance and orientation to be maintained, but resulted in the loss of potential interaction/s of the nitro group with H181 and H184 (Figure 3c). However, model 2 of these substrates maintained the nitro group O2 and H184 NE2 interaction, with an additional potential interaction between the Cl atom of the substrate and S132 OG atom (Z)-5b.
The structures of the progesterone-[23,24] and 1-nitrocyclohexene-PETN reductase complexes have shown that the enzyme can bind substrates and inhibitors in non-productive conformations, with potentially a low occupancy of the catalytically active conformations. This is likely to have a significant effect on the turnover rate of the enzyme, and suggests that catalysis (with non-physiological substrates) may require a metastable enzyme-substrate complex rather than preferential binding of the more stable complex/es observed crystallographically.[24] This could explain why modelling the most likely catalytically active conformations based on previously published structures does not necessarily coincide with maximal substrate-enzyme interactions for substrates 4 and 5. Additionally, knowledge is lacking as to the relative contributions of the interactions between the β-carbon and FMN N5 compared to interactions with His181/184, and the role these interactions play in determining the optimal binding mode/s of the substrates for efficient catalysis.
Although we do not have a crystal structure for the complexes of PETN reductase with β,β-or α,β-disubstituted nitroalkenes 4 and 5, the enantioselectivity of the reaction can be predicted from existing knowledge such as (i) the currently accepted OYE flavin-mediated anti-hydrogenation mechanism, (ii) allowances made for water-mediated protonation, (iii) knowledge of the dual binding modes of substrates in OYE1[44] and PETN reductase[23] and (iv) substrate modelling in the active site. A theoretical model for the enantioselectivity of PETN reductase and OYE1 with p-hydroxybenzaldehyde was proposed recently,[45] and has been adapted here for α/β-alkyl-β-arylnitroalkenes (Scheme 4). The mechanism of C=C double bond reduction by OYE1, based on the binding of p-hydroxybenzaldehyde, suggests that α,β-unsaturated substrates stack above the reduced flavin with Cβ aligned optimally with the N5 for hydride transfer from below.[42,46] Tyr196 (Y186 in PETN reductase) lies above the substrate and donates a proton to the Cα in OYE1. Assuming a model where protons are added to the side of the substrate facing away from the flavin, whether from Y186 or water, substrates (E)-4a-d bound in models 1 and 2 (Scheme 4a and Figure 3b) are likely to form the (S)-enantiomeric product (chiral Cβ). Substrates (Z)-4a, b bound in a similar orientation are also likely to produce the same enantiomer (S) of the product (Scheme 4b and Figure 3c). This is in agreement with our data that show that both (E)- and (Z)-isomers of 2-aryl-1-nitropropenes are reduced to form products (S)-9.
Previous docking studies with nitroalkene (E)-4a in OPR1 and OPR3 predicted two substrate binding modes resulting in the (R)- and (S)-enantiomeric products, respectively,[8] with the likelihood of occupancy dependent on the architecture of the active site of each OYE family member. PETN reductase conformations for binding the substrates (E)- and (Z)-4c that would generate the (R)-enantiomeric product are less likely to be populated significantly due to clashes between the aromatic ring of the substrates and the protein (results not shown). Nevertheless, the nitroalkenes (E)-4a-d, unlike their (Z)-counterparts, were reduced with lower enantioselectivity (63-89% ee). This may be accounted for by a small occupancy of the (E)-substrate bound in an alternative binding mode, resulting in a different stereochemical outcome. E- to Z- or Z- to E-isomerisation studies under aqueous conditions (up to 3 h) showed the presence of oxidised PETN reductase (NADPH absent) did not significantly affect the isomerisation rates (results not shown), suggesting the reduced enantioselectivity with substrates (E)-4a-d and (E/Z)-5a-b is unlikely to be due to isomerisation of the enzyme-bound substrate. Therefore, the near 100% ee with (Z)-substrates suggests either a tighter control of the hydride transfer and/or constraints within the active site that restrict the range of catalytic binding modes. The current mechanism of catalysis of OYEs suggests a trans addition of H- and H+,[9] with the protonation step catalysed by Y186.[8] However, if the protonation step is water-mediated for some substrates, it may be possible that reduction of such substrates with PETN reductase could proceed via syn addition, which would generate both enantiomeric products with substrates (E/Z)-5a, b.
Conclusions
We have shown that PETN reductase has a broad substrate specificity for nitroalkene reduction, and exhibits excellent enantioselectivity for reduction of (Z)-isomers of β,β-disubstituted nitroalkenes 4. This makes the enzyme potentially useful in the production of industrially useful enantiopure nitroalkanes. This provides a further example of catalytic promiscuity of the enzyme, which was initially detected by its ability to reduce nitroaromatic explosives via the formation of the Meisenheimer-hydride complex.[19,22] This catalytic flexibility is likely due, at least in part, to the relatively large active site, which allows the enzyme to bind a range of substrates with considerable size variation and to orientate them appropriately for reduction of different functional groups. Most importantly, PETN reductase is a highly expressing, cheap to produce, robust enzyme, making it especially useful as a prospective industrial biocatalyst. Lastly, it crystallises readily leading to atomic resolution structures which can be used to analyse reaction selectivity and mechanism. This information can be used to predict mutations that may enhance catalytic properties, rather than having to rely solely on random mutagenesis to generate useful biocatalysts.
The use of a structure-driven modelling approach in predicting potential substrates for biocatalysis is an invaluable tool as it theoretically reduces the amount of substrate screening required, and instead targets compounds likely to fit in the active site. Modelling techniques based on high-resolution substrate-bound structures in combination with knowledge of the reaction mechanism and idealisation of critical FMN-substrate distances/orientations enables the rational design of substrates by the introduction of new functional groups as well as in some cases predicting the enantioselectivity of the reaction. It could also potentially generate new improved biocatalysts by predicting specific mutations of PETN reductase necessary for more specific biocatalysis of industrially useful synthons. This work emphasises the need for (i) detailed evaluation of the mechanism of catalysis and (ii) caution in inferring mechanistic similarities in structurally related enzymes.
Experimental Section
General Remarks
NMR spectra were recorded on 300 MHz or 400 MHz spectrometers and referenced to the solvent, unless stated otherwise. The chemical shifts are reported in ppm and the coupling constants (J) are given in hertz (Hz). GC-MS analysis was performed using a DBWAX column (30 m, 0.25 mm, 0.25 μm thick, JW Scientific) and an MS unit equipped with an ion trap. IR spectra were recorded neat on NaCl plates. HPLC analysis was performed using an instrument equipped with a UV detector. Yield and conversion, as well as ee of the products 6 and 7, were determined by HPLC using Chiralcel OD-H or Chiralcel OJ columns (ø 4.6 mm × 250 mm) and for the compound (E)-2 using Phenomenex Gemini C-18 (5 μm, ø 4.6 mm × 250 mm, MeOH:H2O, 85:15, v/v, 1mLmin -1). Absolute configuration of the nitroalkanes was determined by comparison of chiral chromatography data with the literature.[15] Preparative HPLC, used for the separation of (E) and (Z) isomers, was carried out on an HPLC system equipped with a UV detector, using a Rainin Dynamax 60 A silica 41.4 mm × 25 cm column coupled with a 41.4 mm × 5 cm guard column. UV-visible data were recorded with either a spectrophotometer situated in a glove box operating at less than 1 ppm oxygen or under anaerobic conditions with a diode array spectrophotometer. For isomerisation of nitroalkenes (E)-4 and (E)-5, a medium-pressure photochemical ultra-violet lamp was used. The sonication reactions were carried out in an ultrasound bath at highfield frequency 35 kHz. All reactions were monitored by GC-MS and HPLC. Substrates 1-3, product 6 and other reagents were obtained from commercial sources, and the solvents were of analytical grade.
Synthesis of Substrates and Reference Materials
Compounds (E)-4a-d, (Z)-4c,(E)-5a and b and model racemic nitroalkanes 7-10 were synthesised according to procedures previously described.[15] (Z)-Isomers of nitroalkenes 4a, 4d and 5b were synthesised by isomerisation of the respective (E) isomers by UV light, as previously described.[15]
(Z)-1-Nitro-2-phenylpropene [(Z)-4a]
Yellow oil; purified by chromatography (hexane/Et2O, 95:5); 1H NMR (CDCl3, 300 MHz): δ=2.20 (d, J=1.5 Hz, 3 H), 7.08 (q, J=1.5 Hz, 1 H), 7.19-7.23 (m, 2 H), 7.40-7.42 (m, 3 H); 13C NMR (CDCl3, 75 MHz): δ=24.1, 126.3, 128.5, 128.8, 134.4, 137.2, 148.4; MS (EI): m/z=163 (1%, M+), 115 (100%); HR-MS (CI): m/z=197.0243, calcd for [M]+ C9H8NO2Cl: 197.0238; IR (neat): ν=1340, 1519, 1636 cm -1.
(Z)-2-(4′-Bromophenyl)-1-nitropropene [(Z)-4d]
Yellow oil; purified by chromatography (hexane/Et2O, 95:5); 1H NMR (CDCl3, 300 MHz): δ=2.18 (d, J=1.4 Hz, 3 H), 7.07 (s, 1 H), 7.08 (d, J=8.2 Hz, 2 H), 7.53 (d, J=8.2 Hz, 2H); 13C NMR (CDCl3, 75 MHz): δ=23.9, 123.1, 128.0, 131.8, 134.8, 136.0, 147.1; MS (EI): m/z=243 (5%, M+), 241 (5%, M+), 115 (100%); HR-MS [ESI(−)]: m/z=239.9671, calcd for [M−H]+ C9H7NO2Br: 239.9666; IR (neat): ν=826, 1010, 1338, 1521, 1637 cm -1.
(Z)-1-(4′-Chloro-phenyl)-2-nitro-propene [(Z)-5b]
Pale yellow crystals: 1H NMR (CDCl3, 300 MHz): δ=2.36 (s, 3 H), 6.44 (s, 1 H), 7.17–7.20 (m, 2 H), 7.31–7.34 (m, 2 H); 13C NMR (CDCl3, 75 MHz): δ=23.9, 123.1, 128.0, 131.8, 134.8, 136.0, 147.1; HR-MS (EI): m/z=197.0241, calcd for [M]+ C9H7NO2Cl: 197.0238; anal. calcd for C9H8ClNO2:C 54.70, H 4.08, Cl 17.94, N 7.09; found: C 54.58, H 4.05, N 7.07; IR (neat): ν=867, 1356, 1490, 1525 cm -1.
Steady State Kinetics
Purified PETN reductase was prepared as described previously.[23] All kinetics measurements were performed within an anaerobic glove box (Belle Technology Ltd) under a nitrogen atmosphere (<5 ppm oxygen). The following extinction coefficients were used to calculate the concentration of substrates and enzyme: NADPH (ε340=6,220 M-1 cm-1); 1-nitrocyclohexene (1; ε270=6,100 M-1 cm-1); (E)-2-nitro-1-phenylethene [(E)-2; ε320=15,500 M-1 cm-1]; (E)-2-thienyl-1-nitroethene [(E)-3; ε364=17,900 M-1 cm-1]; and PETN reductase (ε464=11.3 × 103 M-1 cm-1). PETN reductase is stable in air, but was deoxygenated by passage through a BioRad 10DG column equilibrated in anaerobic reaction buffer to avoid unproductive flavin reoxidation during nitroalkene reduction.
Steady state measurements were performed using the enzyme monitored turnover method[26] which was originally developed for the reactions catalysed by glucose oxidase. Data were collected in an Applied Photophysics SX.18MV stopped-flow spectrophotometer. Reactions were performed in potassium phosphate buffer (50 mM KH2PO4/K2HPO4 pH 7.0) containing NADPH (200 μM) and substrate (0-500 μM). Enzyme reduction (10 μM) was monitored continuously at 464 nm until completely reoxidised at multiple substrate concentrations. The experiments were analysed using kinetic constants calculated as previously described.[26]
Rapid Reaction Kinetics of the Oxidative Half Reaction
Oxidative half reaction kinetic experiments were performed anaerobically using an Applied Photophysics SX.18MV stopped-flow spectrophotometer. The transients were analysed using non-linear least squares regression analysis on an Acorn Risc PC microcomputer using Spectra kinetics software (Applied Photophysics). PETN reductase was titrated with sodium dithionite to the two-electron level then mixed with substrate in potassium phosphate buffer (50 mM KH2PO4/K2HPO4 pH 7.0). Absorption change of the dithionite-reduced enzyme (15-20 μM) was monitored continuously at the flavin absorption maxima (464 nm) with a range of substrate concentrations at 25°C. The concentrations of substrates were always at least 10-fold greater than the enzyme concentration, ensuring pseudo-first order conditions. Each data point is the average of at least 5 transients. The transients were fitted to a double exponential plot (single exponential plot for 4) from which observed rates were determined using the rapid equilibrium formalism of Strickland et al[47] [Eq. (1)] for the following kinetic scheme [Eq. (2)].
| (1) |
| (2) |
Spectrophotometric Studies on Bioreduction of Nitroalkenes 1-5
Biotransformations in homogeneous, aqueous reaction mixtures were performed using a Jasco UV/VIS spectrophotometer. The reductions were carried out according to a modification of the protocol of Meah and Massey,[27] under anaerobic conditions in potassium phosphate buffer (50 mM KH2PO4/K2HPO4 pH 7.0) containing NADP+ (20 μM), glucose 6-phosphate (20 mM), glucose 6-phosphate dehydrogenase (10 units), substrate (50-100 μM) and PETN reductase (50-200 nM) at 25°C. The presence of the enzyme glucose 6-phosphate dehydrogenase was necessary to recycle NADP+ to the electron donor NADPH required for PETN reductase flavin reduction. The reaction was followed by periodically scanning the reaction mixture between 200 and 700 nm.
Bioreduction of Nitroalkenes 1-5 at Analytical Scale in Biphasic Systems
Reactions were carried out anaerobically in 30-mL screwtop vials sealed with PTFE-silicon septa. Each reaction mixture (12 mL) contained potassium phosphate buffer (50 mM KH2PO4/K2HPO4 pH 7.0) containing nitroalkene (1.7 mM) dissolved in 4.8 mL anaerobic isooctane, PETN reductase (~3 μM), NADP (20 μM) glucose 6-phosphate (14-20 mM), glucose 6-phosphate dehydrogenase (10 units) and sec-butylbenzene (25 μL), the latter as the internal standard for chromatography. Reactions were agitated at 200 rpm for 1-7 days at 30°C in an anaerobic cabinet. The organic layer was analysed by HPLC, and also by GC-MS to verify the identity of the product(s). Enantiomeric excess, yields and conversions were determined by HPLC using Chiralcel OD-H or Chiralcel OJ columns (ø 4.6 mm × 250 mm, hexane:i-PrOH, 90:10, v/v, 1 mL min-1). Products formed from compound (E)-2 were analysed using a Phenomenex Gemini C-18 column (5 μm, ø 4.6 mm × 250 mm, MeOH:H2O, 85:15, v/v, 1 mL min-1).
Crystallogenesis and Data Collection
Crystals of oxidised PETN reductase were grown using the sitting-drop method as described previously.[23] The crystal complex of oxidised PETN reductase with substrate 1 was produced by soaking these crystals in mother liquor containing 30 mM of 1-nitrocyclohexene 1. The soaked crystals were flash-frozen in liquid nitrogen in the absence of additional cryoprotectant, and a full 1.3 Å X-ray diffraction data set was collected from a single crystal at the European Synchrotron Radiation Facility (Grenoble, France) on Station ID 14.1 (wavelength 0.93 Å; 100 K) using an ADSC CCD detector.
Structure Determination and Refinement
Data were processed and scaled using the programs MOSFLM[48] and Scala.[49] The structure was solved via molecular replacement using the coordinates for the PETN reductase/picric acid complex[18] (PDB entry 1VYR) with picric acid and water molecules removed. Positional and anisotropic B-factor refinement was performed using REFMAC5,[50] with alternate rounds of manual rebuilding of the model in TURBO-FRODO.[51] The positions of water molecules were found automatically using REFMAC combined with ARP/wARP.[52] The final model was refined to 1.34 Å resolution giving a final Rfactor of 0.114 and Rfree of 0.145. The atomic coordinates and structure factors (pdb code 3F03) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
Molecular Modelling
The substrates 4 and 5 were modelled into the active site of PETN reductase using the method of Sketcher[49] and Coot,[53] based on the position of the substrate in the structure containing bound 1-nitrocyclohexene 1. The protein was subject to 1000 steps of energy minimisation using the steepest descent method within AMBER9 software.[54] Prior to this, hydrogen atoms were appended to the crystallographic coordinates according to standard geometries and each protein was explicitly solvated in a box of pre-equilibrated TIP3P water molecules. Each system was neutralised by addition of the appropriate number of counter ions. As flavin is a non-standard residue, the oxidised cofactor was assigned general AMBER force field or GAFF[55] atom types. Cofactor parameters not accounted for in the canonical GAFF parameter set were empirically calculated using the ANTECHAMBER tool distributed with AMBER9 software. All other atoms (protein, water and counter-ions) were modelled with the AMBER ff03 force field. Partial charges of the isoalloxazine atoms were derived from population analysis of lumiflavin. Charge and multiplicity values were specified 0 and 1, respectively. Lumiflavin coordinates were first optimised in the gas phase at the B3LYP/6-31G(d) level of theory using Gaussian. Local minima were confirmed with vibrational frequency calculations, consistent with the parameterisation of both the GAFF and the AMBER ff03 force fields. Charges were derived with the Merz-Sing-Kollmann (MK) scheme using the keyword/value pair ‘pop=mk’, this comprises fitting of atomic charges to reproduce the molecular electrostatic potential (ESP) around the van der Waals envelope. As the latter methods do not recognise C-halide bonds, the minimised structures were further refined using the structure idealisation function of REFMAC5[50] to normalise the bond angles and distances of the substrates and to remove minor clashes.
Supplementary Material
Acknowledgements
We thank Ben Sattelle for his help in preparing the initial energy minimisation models and Chandrani Mukherjee for assistance in some of the analytical and synthesis work. This work was funded by the UK Biotechnology and Biological Sciences Research Council via grants BBE0107171 and BBD0028261. AR is a Wellcome Trust Career Development Fellow, DL is a Royal Society University Research Fellow and NSS is a BBSRC Professorial Research Fellow.
References
- [1].Ono N. The nitro group in organic synthesis. Wiley-VCH; New York, Chichester: 2001. [Google Scholar]
- [2].Mampreian DM, Hoveyda AH. Org. Lett. 2004;6:2829–2832. doi: 10.1021/ol0488338. [DOI] [PubMed] [Google Scholar]; Wu J, Mampreian DM, Hoveyda AH. J. Am. Chem. Soc. 2005;127:4584–4585. doi: 10.1021/ja050800f. [DOI] [PubMed] [Google Scholar]
- [3].Ballini R, Bosica G, Fiorini D, Palmieri A, Petrini M. Chem. Rev. 2005;105:933–972. doi: 10.1021/cr040602r. [DOI] [PubMed] [Google Scholar]
- [4].Czekelius C, Carreira EM. Angew. Chem. Int. Ed. 2003;42:4793–4795. doi: 10.1002/anie.200352175. [DOI] [PubMed] [Google Scholar]; Czekelius C, Carreira EM. Org. Lett. 2005;6:4575–4577. doi: 10.1021/ol048035h. [DOI] [PubMed] [Google Scholar]
- [5].Martin NJA, Ozores L, List B. J. Am. Chem. Soc. 2007;129:8976–8977. doi: 10.1021/ja074045c. [DOI] [PubMed] [Google Scholar]
- [6].Stuermer R, Hauer B, Hall M, Faber K. Curr. Opin. Chem. Biol. 2007;11:203–213. doi: 10.1016/j.cbpa.2007.02.025. [DOI] [PubMed] [Google Scholar]
- [7].Chaparro-Riggers JF, Rogers TA, Vazquez-Figueroa E, Polizzi KM, Bommarius AS. Adv. Synth. Catal. 2007;349:1521–1531. [Google Scholar]; Hall M, Hauer B, Stuermer R, Kroutil W, Faber K. Tetrahedron: Asymmetry. 2006;17:3058–3062. [Google Scholar]
- [8].Hall M, Stueckler C, Ehammer H, Pointner E, Oberdorfer G, Gruber K, Hauer B, Stuermer R, Kroutil W, Macheroux P, Faber K. Adv. Synth. Catal. 2008;350:411–418. [Google Scholar]
- [9].Hall M, Stueckler C, Hauer B, Stuermer R, Friedrich T, Breuer M, Kroutil W, Faber K. Eur. J. Org. Chem. 2008:1511–1516. [Google Scholar]
- [10].Hall M, Stueckler C, Kroutil W, Macheroux P, Faber K. Angew. Chem. Int. Ed. 2007;46:3934–3937. doi: 10.1002/anie.200605168. [DOI] [PubMed] [Google Scholar]
- [11].Stueckler C, Hall M, Ehammer H, Pointner E, Kroutil W, Macheroux P, Faber K. Org. Lett. 2007;9:5409–5411. doi: 10.1021/ol7019185. [DOI] [PubMed] [Google Scholar]
- [12].Williams RE, Bruce NC. Microbiology. 2002;148:1607–1614. doi: 10.1099/00221287-148-6-1607. [DOI] [PubMed] [Google Scholar]
- [13].Bougioukou DJ, Stewart JD. J. Am. Chem. Soc. 2008;130:7655–7658. doi: 10.1021/ja800200r. [DOI] [PubMed] [Google Scholar]
- [14].Ohta H, Kobayashi N, Ozaki K. J. Org. Chem. 1989;54:1802–1804. [Google Scholar]
- [15].Fryszkowska A, Fisher K, Gardiner JM, Stephens GM. J. Org. Chem. 2008;73:4295–4298. doi: 10.1021/jo800124v. [DOI] [PubMed] [Google Scholar]
- [16].Kawai Y, Inaba Y, Tokitoh N. Tetrahedron: Asymmetry. 2001;12:309–318. [Google Scholar]
- [17].Sakai K, Nakazawa A, Kondo K, Ohta H. Agric. Biol. Chem. 1985;49:2331–2335. [Google Scholar]
- [18].Khan H, Harris RJ, Barna T, Craig DH, Bruce NC, Munro AW, Moody PC, Scrutton NS. J. Biol. Chem. 2002;277:21906–21912. doi: 10.1074/jbc.M200637200. [DOI] [PubMed] [Google Scholar]
- [19].Williams RE, Rathbone DA, Scrutton NS, Bruce NC. Appl. Environ. Microbiol. 2004;70:3566–3574. doi: 10.1128/AEM.70.6.3566-3574.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Vanek T, Gerth A, Vakrikova Z, Podlipna R, Soudek P. In: Advanced science and technology for biological decontamination of sites affected by chemical and radiological nuclear agents. Marmiroli N, editor. Springer; Berlin, Heidelberg: 2007. pp. 209–225. [Google Scholar]
- [21].French CE, Nicklin S, Bruce NC. J. Bact. 1996;178:6623–6627. doi: 10.1128/jb.178.22.6623-6627.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Khan H, Barna T, Harris RJ, Bruce NC, Barsukov I, Munro AW, Moody PC, Scrutton NS. J. Biol. Chem. 2004;279:30563–30572. doi: 10.1074/jbc.M403541200. [DOI] [PubMed] [Google Scholar]
- [23].Barna TM, Khan H, Bruce NC, Barsukov I, Scrutton NS, Moody PC. J. Mol. Biol. 2001;310:433–447. doi: 10.1006/jmbi.2001.4779. [DOI] [PubMed] [Google Scholar]
- [24].Khan H, Barna T, Bruce NC, Munro AW, Leys D, Scrutton NS. FEBS J. 2005;272:4660–4671. doi: 10.1111/j.1742-4658.2005.04875.x. [DOI] [PubMed] [Google Scholar]
- [25].Moody PC, Shikotra N, French CE, Bruce NC, Scrutton NS. Acta Cryst. D. 1998;54:675–677. doi: 10.1107/s0907444997017836. [DOI] [PubMed] [Google Scholar]
- [26].Gibson QH, Swoboda BE, Massey V. J. Biol. Chem. 1964;239:3927–3934. [PubMed] [Google Scholar]
- [27].Meah Y, Massey V. Proc. Natl. Acad. Sci. USA. 2000;97:10733–10738. doi: 10.1073/pnas.190345597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Messiha HL, Munro AW, Bruce NC, Barsukov I, Scrutton NS. J. Biol. Chem. 2005;280:10695–10709. doi: 10.1074/jbc.M410595200. [DOI] [PubMed] [Google Scholar]
- [29].Basran J, Harris RJ, Sutcliffe MJ, Scrutton NS. J. Biol. Chem. 2003;278:43973–43982. doi: 10.1074/jbc.M305983200. [DOI] [PubMed] [Google Scholar]
- [30].Rohdich F, Wiese A, Feicht R, Simon H, Bacher A. J. Biol. Chem. 2001;276:5779–5787. doi: 10.1074/jbc.M008656200. [DOI] [PubMed] [Google Scholar]; Steinbacher S, Stumpf M, Weinkauf S, Rohdich F, Bacher A, Simon H. In: Flavins and Flavoproteins. Chapman S, Percham R, Scrutton NS, editors. 2002. pp. 941–949. [Google Scholar]
- [31].Utaka M, Konishi S, Mizuoka A, Ohkubo T, Sakai T, Tsuboi S, Takeda A. J. Org. Chem. 1989;54:4989–4992. [Google Scholar]
- [32].Muller A, Hauer B, Rosche B. Biotechnol. Bioeng. 2007;98:22–29. doi: 10.1002/bit.21415. [DOI] [PubMed] [Google Scholar]
- [33].Messiha HL, Bruce NC, Sattelle BM, Sutcliffe MJ, Munro AW, Scrutton NS. J. Biol. Chem. 2005;280:27103–27110. doi: 10.1074/jbc.M502293200. [DOI] [PubMed] [Google Scholar]
- [34].Ballini R, Petrini M. Tetrahedron. 2004;60:1017–1047. [Google Scholar]
- [35].Swiderska MA, Stewart JD. Org. Lett. 2006;8:6131–6133. doi: 10.1021/ol062612f. [DOI] [PubMed] [Google Scholar]
- [36].Kurtz KA, Fitzpatrick PF. J. Am. Chem. Soc. 1997;119:1155–1156. [Google Scholar]; Daubner SC, Gadda G, Valley MP, Fitzpatrick PF. Proc. Natl. Acad. Sci. USA. 2002;99:2702–2707. doi: 10.1073/pnas.052527799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Porter DJ, Bright HJ. J. Biol. Chem. 1983;258:9913–9924. [PubMed] [Google Scholar]
- [38].Porter DJ, Bright HJ. J. Biol. Chem. 1987;262:14428–14434. [PubMed] [Google Scholar]
- [39].Francis K, Russell B, Gadda G. J. Biol. Chem. 2005;280:5195–5204. doi: 10.1074/jbc.M411249200. [DOI] [PubMed] [Google Scholar]
- [40].Hansen CH, Du L, Naur P, Olsen CE, Axelsen KB, Hick AJ, Pickett JA, Halkier BA. J. Biol. Chem. 2001;276:24790–24796. doi: 10.1074/jbc.M102637200. [DOI] [PubMed] [Google Scholar]
- [41].Fraaije MW, Mattevi A. Trends Biochem. Sci. 2000;25:126–132. doi: 10.1016/s0968-0004(99)01533-9. [DOI] [PubMed] [Google Scholar]
- [42].Kohli RM, Massey V. J. Biol. Chem. 1998;273:32763–32770. doi: 10.1074/jbc.273.49.32763. [DOI] [PubMed] [Google Scholar]
- [43].Muller A, Sturmer R, Hauer B, Rosche B. Angew. Chem. Int. Ed. 2007;46:3316–3318. doi: 10.1002/anie.200605179. [DOI] [PubMed] [Google Scholar]
- [44].Vaz AD, Chakraborty S, Massey V. Biochemistry. 1995;34:4246–4256. doi: 10.1021/bi00013a014. [DOI] [PubMed] [Google Scholar]
- [45].Fuganti C, Chiringhell D, Grasselli Piero. J. Chem. Soc. Chem. Commun. 1975:846–847. [Google Scholar]
- [46].Fox KM, Karplus PA. Structure. 1994;2:1089–1105. [PubMed] [Google Scholar]
- [47].Strickland S, Palmer G, Massey V. J. Biol. Chem. 1975;250:4048–4052. [PubMed] [Google Scholar]
- [48].Leslie AGW. Joint CCP4 & ESRF-EACBM Newsletter on Protein Crystallography. Vol. 26. Serc Laboratory; Daresbury, Warrington, U.K.: 1992. [Google Scholar]
- [49].Collaborative Computational Project number 4 Acta. Cryst. D. 1994;50:760–763. [Google Scholar]
- [50].Murshudov GN, Vagin AA, Dodson EJ. Acta. Cryst. D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- [51].Roussel A, Cambillau C. Silicon Graphics Geometry Partners Directory. Vol. 86. Silicon Graphics; Mountain View, CA, USA: 1991. [Google Scholar]
- [52].Perrakis A, Morris R, Lamzin VS. Nat. Struct. Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- [53].Emsley P, Cowtan K. Acta. Cryst. D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- [54].Case DA, Cheatham I, Darden TE, Gohlke H, Luo R, Merz J, Onufriev KMA, Simmerling C, Wang B, Woods R. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wang RM, Wolf JW, Caldwell PA, Case DA. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- [56].DeLano WL. The PyMOL User’s Manual. DeLano Scientific; Palo Alto, CA, USA: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.