

Abstract
Overexpression of COX2 appears to be both a marker and an effector of neural damage after a variety of acquired brain injuries, and in natural or pathological aging of the brain. COX2 inhibitors may be neuroprotective in the brain by reducing prostanoid and free radical synthesis, or by directing arachidonic acid down alternate metabolic pathways. The arachidonic acid shunting hypothesis proposes that COX2 inhibitors' neuroprotective effects may be mediated by increased formation of potentially beneficial eicosanoids. Under conditions where COX2 activity is inhibited, arachidonic acid accumulates or is converted to eicosanoids via lipoxygenases and cytochrome P450 (CYP) epoxygenases. Several P450 eicosanoids have been demonstrated to have beneficial effects in the brain and/or periphery. We suspect that arachidonic acid shunting may be as important to functional recovery after brain injuries as altered prostanoid formation per se. Thus, COX2 inhibition and arachidonic acid shunting have therapeutic implications beyond the suppression of prostaglandin synthesis and free radical formation.
Introduction
The role of cyclooxygenase-2 (COX2) and its inhibitors in the brain must be examined in the larger context of its role in arachidonic acid metabolism (Figure 1). Perturbations or insults to the brain activate phospholipases, releasing arachidonic acid from membrane stores (Dumuis et al., 1988; Gardiner et al., 1981). Cyclooxygenase-2 catalyzes the conversion of arachidonic acid and molecular oxygen into vasoactive prostaglandins, producing reactive oxygen free radicals in the process. COX2 is the dominant player in a complex and interlocking metabolic pathway that converts a structural membrane lipid into a plethora of biologically active eicosanoids, many of which have opposing physiological activity. Moreover, there are several other related biomolecules (e.g., docosahexenoic acid and docosanoids, the endocannabinoids anandamide and 2-arachidonoyl glycerol, etc.) that further expand the scope of influence of COX2 in neurophysiological functions.
Figure 1.
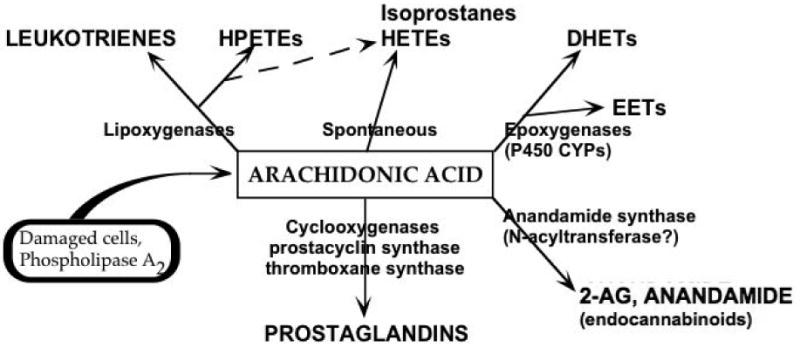
Arachidonic acid metabolism. Cell damage and phospholipase activation release arachidonic acid with subsequent oxidation to a variety of eicosanoids. Arachidonic acid is converted to highly labile prostanoids and leukotrienes by COXs and lipoxygenases, respectively, producing reactive oxygen free radicals in the process. Alternatively, arachidonic acid can be monooxygenated by cytochrome P450 epoxygenases, producing highly labile epoxide regioisomers (5,6-; 8,9-; 11,12-; or 14,15-EET)(Chacos et al., 1982; Oliw et al., 1982). Allylic oxidation is also catalyzed to form HETEs (5-, 8-, 9-, 11-, 12-, 15-, 19-, or 20-HETE)(Capdevila et al., 1982; Oliw et al., 1982). Certain HETEs (e.g., 5-, or 12-HETE) can also be formed via lipoxygenase action from hydroperoxyeicosatetraenoic acid (HPETE) precursors. EETs are metabolized by epoxide hydrolase to the corresponding dihydroxyeicosatrienoic acids (DHETs)(Chacos et al., 1983; Oliw et al., 1982; Yu et al., 2000b; Zeldin et al., 1995). Interestingly, EETs and HETEs are often incorporated in membrane phospholipid, enabling phospholipase-mediated release of these activities (Brezinski and Serhan, 1990; Capdevila et al., 1987; Karara et al., 1991).
Inhibition of COX2 after pathological insult has been shown to benefit recovery in the brain and spinal cord (Nagayama et al., 1999; Resnick et al., 1998). However, the mechanisms of COX2 in neuropathology are not well described. Our working hypothesis is that prolonged COX2 expression in the brain interferes with intrinsic neuroprotective mechanisms, contributing to the establishment of a “vicious cycle” in which cell death, rather than survival pathways dominate; and tissue damage is made worse by propagation of oxidative damage and chemotactic signals. Thus, we propose that COX2 inhibition blocks delayed cell death and neuroinflammation.
That COX2 inhibitors may function in the brain by shunting arachidonic acid down alternate metabolic pathways has been alluded to by Christie et al. (Christie et al., 1999) in a model of opioid-NSAID synergy, who “speculated that blockade of cyclooxygenase and/or 5-lipoxygenase might lead to shunting of arachidonic acid metabolism… [and] enhanced formation of 12-LOX metabolites, thereby enhancing the efficacy of opioids” in the periaqueductal gray. Arachidonic acid can be oxidized to many biologically and chemically active derivatives, the most prevalent being prostaglandins. Thus, under conditions where COX2 activity increases, proportionately more arachidonic acid is converted to prostanoids and less to other metabolites. Conversely, when COX2 activity is inhibited, arachidonic acid, that would otherwise be converted to prostanoids, accumulates or is converted to other eicosanoids (Figure 2, arachidonic acid shunting). Both these conditions are especially germane under conditions where phospholipases are activated, with the resultant increase in free arachidonic acid. The succeeding review examines some observations of the reactions of COX2 to brain injuries, its association with cell death and neuroinflammation, and its response to COX2 inhibitor treatments.
Figure 2.

Arachidonic acid shunting. The action of COX2 inhibitors decreases synthesis of prostanoids and free radicals. However, because it is the dominant metabolic reaction, COX2 inhibition causes arachidonic acid shunting down alternate enzymatic pathways (e.g., cytochrome P450 epoxygenases), resulting in the synthesis of potentially neuroprotective eicosanoids. COX2 and prostanoid levels rise acutely after brain injuries, and remain elevated for days. The extent of COX2 expression may correlate to the severity of the insult. This may be due to a “vicious cycle”, in which secondary injury cascades promulgate COX2 gene expression. Prolonged elevation contributes to inflammation, programmed cell death, free radical-mediated tissue damage, and alterations in cellular metabolism. These, in turn, cause secondary injuries that worsen clinical outcomes. Thus, COX2 inhibitors' beneficial actions are mediated both by reducing prostanoid formation and by arachidonic acid shunting to form antiinflammatory, neuroprotective eicosanoids.
COX2 expression in the brain
Normally, COX2 is rare or absent in most organs of the body. However, significant levels of COX2 mRNA are expressed in mammalian brain (Feng et al., 1993; Seibert et al., 1994). Prostaglandin products of COX2 have also been found in moderate concentrations in postmortem human brain (Ogorochi et al., 1984). Normal brain COX2 protein is found primarily in neuronal cell bodies and dendrites (Breder et al., 1992; Kaufmann et al., 1996; Li et al., 1993b; Strauss et al., 2000a; Tsubokura et al., 1991). Low steady state levels of COX2 are found in neurons of the cerebral cortex, hippocampus and cerebellum, but not in glia or endothelial cells. Localization of gene expression in rat brain by in situ hybridization histochemistry has shown high levels of COX2 mRNA in pyramidal and granule cells of the hippocampus and cerebellar granule cells. Moderate levels are found in the pyramidal cells of piriform cortex, cellular layers of cortex, central n. of amygdala, and several hypothalamic nuclei. Cyclooxygenase-1 is also expressed at low levels in microglia throughout the brain and in subsets of granule neurons (O'Neill and Ford-Hutchinson, 1993; Yermakova et al., 1999), however this isoform is not readily induced by neural perturbations, as is COX2.
Neuronal COX2 is glutamatergically regulated
Steady-state COX2 appears to be regulated by normal glutamatergic synaptic activity in the adult brain (Adams et al., 1996; Chen et al., 1995; Kaufmann et al., 1996; Marcheselli and Bazan, 1996; Yamagata et al., 1993). Several recent in vivo studies have demonstrated specificity of glutamate-mediated COX2 gene regulation in the brain (Candelario-Jalil et al., 2002; Hirst et al., 1999; Koistinaho et al., 1999; Miettinen et al., 1997). Basal COX2 expression in cultured neurons could be suppressed using a variety of glutamatergic receptor antagonists (Strauss and Marini, 2002). Interestingly, only toxic concentrations of glutamate, kainate, and N-methyl-D-aspartate (NMDA) induced overexpression of COX2 mRNA in cultured rat neurons (Hewett et al., 2000; Strauss and Marini, 2002). In contrast, COX2 gene induction in non-neural tissues (endothelial cells, immune cells, fibroblasts, etc.) has been shown to be controlled by glucocorticoids, interleukin-1, or protein kinase C, via several DNA-binding activities (for reviews, see Herschman et al., 1995; Smith and Dewitt, 1996). Immunofluorescence colocalization studies revealed prolonged COX2 expression in neurons and glia of the injured brain (Strauss et al., 2000a), not in vascular endothelial or other lining cells as seen after peripheral inflammatory challenges (Cao et al., 2001; Minghetti, 2004; Parfenova et al., 1997; Sanz et al., 1997; Tzeng et al., 2005). Thus, inflammatory and brain lining cells regulate COX2 via interleukin-1-dependent mechanisms, while neurons and astrocytes do not (Nadjar et al., 2005; Strauss et al., 2000a). COX2 induction in these neural-derived cells appears to be via excitotoxic and oxidative mechanisms.
COX2 overexpression in the brain
Neuronal COX2 overexpression is induced by excitotoxicity in vivo (Adams et al., 1996; Chen et al., 1995; Kaufmann et al., 1996; Koistinaho et al., 1999; Marcheselli and Bazan, 1996; Yamagata et al., 1993) and in vitro (Hewett et al., 2000; Strauss and Marini, 2002; Tocco et al., 1997). In vitro, overactivation of kainate or NMDA receptors results in cell death, killing most cultured neurons (Marini and Paul, 1992; Novelli et al., 1988; Schramm et al., 1990). Kainate strongly induces neuronal COX2 overexpression. The NMDA receptor antagonist, MK-801, completely blocks glutamate and NMDA toxicity. MK-801 also attenuates COX2 mRNA induction in glutamate-treated neural cultures (Hewett et al., 2000; Strauss and Marini, 2002). In contrast, alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) only weakly induces cell death, and metabotropic receptor group 1 agonists are not excitotoxic at all. Neither type of glutamatergic receptor agonist cause increases in COX2 mRNA levels.
Excessive glutamate receptor activation and pathological COX2 induction have been associated with delayed cell death in brain regions specifically associated with functional deficits that occur following brain injuries (Caggiano et al., 1996; Collaco-Moraes et al., 1996; Hewett et al., 2000; Kunz and Oliw, 2001; Strauss et al., 2000a; Strauss and Marini, 2002; Wallace et al., 1998). Molecular findings from our lab indicate that excitotoxic levels of glutamate receptor agonists (kainate ≫ NMDA ≫ AMPA) increase neuronal COX2 expression, implicating COX2 as an effector of neurological deficits. As we shall see, COX2 activity is intimately linked to excitotoxic neuronal death.
Oxidant stress is also an important determinant of COX2 gene expression. The role of oxidative stress in functional outcome after brain injuries has been abundantly demonstrated. Antioxidants such as dimethyl sulfoxide (DMSO), tetramethylthiourea (TMU) (Feng et al., 1995), pyrrolidine dithiocarbamate (PDTC) and rotenone (Tetsuka et al., 1996) block interleukin-1β induction of COX2 protein in vitro. In both studies COX2 protein production was diminished, however, DMSO and TMU inhibited interleukin-1 induction of COX2 mRNA, while PDTC and rotenone did not. This implies a post-transcriptional regulation of COX2 by reactive oxygen species. A byproduct of COX2 activity is reactive oxygen species, adding support to the idea that aberrant COX2 activity results in a vicious cycle of overexpression that contributes to cell death and neuroinflammation.
COX2 and brain injuries
Brain injuries activate multiple cellular pathways; some of these appear to be adaptive, others are pathological, and still others are initially adaptive but persist and become harmful. The response of COX2 to neural injury appears to belong in the latter category. For example, we have found that COX2 levels increase in proximal and distant brain regions following many types of acquired brain injury. Neural COX2 activity and expression are rapidly induced and may remain elevated for many days. Interestingly, COX2 expression is increased for prolonged periods in brain regions specifically associated with functional deficits after neurotrauma (Gopez et al., 2005; Strauss et al., 2000a). COX2 has been associated with worse outcomes after brain injury, as well as early onset dementia (Jantzen et al., 2002; Jellinger, 2004; Oka and Takashima, 1997). Further, COX2 appears at abnormally high levels in the (post mortem) hippocampus of Alzheimer's disease patients (Ho et al., 1999). The discrete localization and distinct regulation of COX2 gene expression in the brain provide a rational basis to suspect that perturbation of COX2 and arachidonic acid metabolism play a significant role in normal and pathological responses.
The severity of neural injury appears to correlate to the degree and duration of COX2 overexpression; mild injuries yield shorter elevations (≤24h) of COX2 mRNA and prostaglandin production, while moderate to severe injuries yield extended elevations (≥3 days) in brain cells. This may be due to a “vicious cycle” (Figure 2), in which secondary injury cascades promulgate COX2 gene expression. In the brain, COX2 appears to propagate the central inflammatory response, in part by modulation of complement component C1qB, which is involved in neurodegenerative processes (Spielman et al., 2002). Increased COX2 expression has been observed with head trauma, cerebral ischemia, spreading depression, and seizures, as well as in several progressive neurodegenerative conditions, e.g., Alzheimer's disease, Parkinson's disease, Huntington's disease, Down's syndrome, etc. (Caggiano et al., 1996; Collaco-Moraes et al., 1996; Dash et al., 2000; Graham et al., 1996; Nakayama et al., 1997; Nogawa et al., 1997; Pasinetti and Aisen, 1998; Sanz et al., 1997; Strauss et al., 2000a; Walton et al., 1997). Overexpression of brain COX2 in these disorders may reflect its role in chronic neuroinflammation and neural cell death.
Early on after moderate brain injuries, neurons show increased COX2 levels that may persist for 1-3 days. At later time points (i.e. ≥3 days), COX2 remains elevated and (at least in rat models of brain injury) appears to be primarily expressed in glial cells proximal to the site of injury. This is in contrast to the neural cell populations that have been found to produce COX2 after a peripherally induced inflammatory challenge (Strauss et al., 2000a).
Interestingly, we have observed a temporal “wave” of COX2 mRNA starting in CA1-2 neurons at 2h to 6h after neurotrauma and appearing in CA3 and dentate gyrus neurons by 6h to 24h postinjury in the rat. In contrast, the induction of COX2 expression following transient global ischemia in the rat appeared to “move” in the opposite direction (dentate gyrus → CA3 → CA2-1) over the same time course (Koistinaho et al., 1999; Nakayama et al., 1998). Whether this reflects on the findings that mainly CA1 neurons are lost to apoptotic cell death after ischemic insult, while mostly CA3 neurons are lost after unilateral traumatic brain injury, remains to be determined.
Eicosanoids and brain injuries
Injuries that result in cell membrane damage activate phospholipases, releasing arachidonic acid from phospholipid membranes (Dumuis et al., 1988; Gardiner et al., 1981). Increases of intracellular free Ca++ also mediate activation of phospholipases that release arachidonic acid from membrane stores (Dhillon et al., 1997; Lewen and Hillered, 1998; Marklund et al., 1997; Prasad et al., 1994; Sevanian and Kim, 1985). Arachidonic acid is converted to eicosanoids producing reactive oxygen species in the process. This can result in lipid peroxidation via formation of highly reactive OONO•-, O2•-, and OH•- free radicals that damage neural membranes (including the blood-brain barrier) and initiate a central inflammatory cascade that includes COX2 upregulation.
Prostaglandins products of COX2 mediate many brain-specific functions, e.g., temperature regulation (Saper and Breder, 1992) and hippocampal long term potentiation (Nishizaki et al., 1999b). Normally, prostaglandin (PG) D2 levels are highest in the brain, however brain injuries induce the production of large amounts of other prostanoids. Prostaglandins, particularly PGE2 levels, rise dramatically after traumatic brain injury (Dewitt et al., 1988; Ellis et al., 1989; Ellis et al., 1981; Shohami et al., 1987). We measured PGE2 changes in the hippocampus and parietal cortex ipsilateral and contralateral to injury. Tissue levels rose by 2h and continued increasing to a maximum by 24h, then returned to baseline over the next several days (Strauss et al. 2000b, 2005). Treatment with the COX2-specific inhibitor DFU (5,5-dimethyl-3(3-fluorophenyl)-4(4-methylsulphonyl) phenyl-2(5H)-furanone, Merck-Frosst, 1 mg/kg, intraperitoneal injections, twice daily for 3 days) attenuated the injury-induced rise in PGE2 at 24 h postinjury by more than 60%.
The induction of COX2 after brain injuries results in increased prostaglandin E2 (PGE2) levels. Surprisingly, the proinflammatory role of prostaglandins in the brain has met with much debate. PGE2 has been reported to suppress proinflammatory tumor necrosis factor (TNF) release, and a few studies support the view that PGE2 facilitates cell survival. However, neural cells are isolated from the periphery by the blood brain barrier, use limited substrates for energy, and have unique anatomical and electrophysiological traits. Consequently, many genes expressed in the central nervous system (CNS) function differently than in the periphery. As we shall see, COX2 is such an effector gene that has come under intense investigation.
The effects of PGE2 on TNF is different in peripheral organs than in the CNS. In many tissues, increased PGE2 levels suppress the intrinsic release of TNF, resulting in immunosuppression (Bertrand et al., 1998; Fennekohl et al., 2002; Flohe et al., 2000; Goncalves de Moraes et al., 1996; Ishaque et al., 2003; Vassiliou et al., 2003). In contrast, PGE2 does not reduce TNF signaling in neural tissues (Quadros et al., 2003; Sacco et al., 1998). Interestingly, unlike primary peritoneal macrophages, cultured RAW cells do not show a decreased TNF response to PGE2 after LPS treatment (Rouzer et al., 2005). The presence of high basal levels of PGD2 (and its receptors) in RAW cells, as in the brain, may help to explain this difference. In fact, this could be the basis for establishment of a vicious cycle of COX2 expression in the brain; the absence of the TNF reduction response to increased PGE2 levels may propagate neural tissue damage.
On the other hand, some of our rodent traumatic brain injury studies indicate acute COX2 activity may be beneficial (Gopez et al., 2005). The immediate production of thromboxane (0.5 to 2h, (Strauss et al., 2000b)), for example, could help in hemostasis and reduce the brain's exposure to toxic blood products. The peak in brain PGE2 levels (12 to 24h) may also be adaptive, perhaps helping to increase blood flow to recovering neural tissue, while minimizing the premature activation of monocytes and infiltration by macrophages (Westcott et al., 1987; Zhang and Rivest, 2001). However, we propose it is prolonged COX2 overexpression and activity that causes harm to the injured brain.
Eicosanoids activate G-protein coupled receptors (Abramovitz et al., 2000), and may act directly as chemokines and neuromodulators. Prostaglandins are vasoactive (constriction, dilatation), alter platelet aggregation, vascular permeability, and act as chemokines (Cao et al., 1995), among numerous other activities (Ellis et al., 1989; Ellis et al., 1979; Kontos et al., 1980a; Kontos et al., 1980b; Wei et al., 1981). Leukotriene products are inflammatory mediators, alter vascular permeability, and also function in the hippocampus and pineal (Manev et al., 2000; Uz and Manev, 1998). HPETEs can alter neuronal potassium currents (Ba-insensitive) (Vaughan et al., 1997). Formation of reactive oxygen species in these reactions and spontaneous oxidation to isoprostanes can promote inflammation in the brain. HETEs and EETs are potently vasoactive and may have proinflammatory properties. However, cytochrome P450 eicosanoids and arachidonic acid derivatives appear to have potentially neuroprotective properties, as well.
Node et al. (Node et al., 1999) demonstrated that certain EETs had unique antiinflammatory properties in a cardiac muscle system. Hampson and Grimaldi (Hampson and Grimaldi, 2002) provided evidence that certain HETEs ameliorate AMPA-type excitotoxic cell death in cultured neurons. And Panikashvili et al. (Panikashvili et al., 2001) clearly demonstrated that 2-arachidonoyl glycerol (2-AG), systemically administered to mice after closed head injury, was neuroprotective and improved functional outcomes. Most recently, Alkayed et al. (Alkayed et al., 2002; Koerner et al., 2005) have presented evidence of cerebral protection mediated by the arachidonic acid epoxygenase metabolites, 11,12- EET and 14,15-EET. They demonstrated that cytochrome P450 (CYP) arachidonate epoxygenase CYP2C11 (Liu and Alkayed, 2005), and its major product 14,15-EET (Koerner et al., unpublished data) mediate neuroprotection in rodent models of stroke and preconditioning. Moreover, this group has furthered our understanding of how neural tissue may utilize short-acting eicosanoids to regulate local blood flow “on demand” (Iliff et al., 2007).
Arachidonic acid shunting
Many studies on the role of COX2 in neuropathology have focused on the specificity of the third generation, highly specific COX2 inhibitors in reducing prostanoid production. A newer hypothesis that we are testing proposes that COX2 inhibitors' beneficial effects may be mediated, at least in part, by the increased formation of potentially neuroprotective and antiinflammatory eicosanoid metabolites, so called “arachidonic acid shunting.” Although the formation of proinflammatory eicosanoids is also a possibility, the net effect of COX2 inhibition after CNS injury has been generally beneficial, or at least not detrimental, to neurological recovery (Dash et al., 2000; Resnick et al., 1998; Shapira et al., 1988; Shohami et al., 1990).
Arachidonic acid shunting may be especially relevant after acute insults that activate phospholipase A2. Massive amounts of arachidonic acid are released from membrane stores (Dumuis et al., 1988; Gardiner et al., 1981). Free arachidonic acid can be converted by cyclooxygenases and lipoxygenases to proinflammatory prostanoids and leukotrienes. When COX2 activity increases after brain injury, proportionately more arachidonic acid is converted to prostanoids and less to other metabolites. Conversely, under conditions of COX2 “deficiency” after brain injuries, arachidonic acid is more effectively metabolized to other eicosanoids.
An alternate pathway for arachidonate metabolism produces epoxyeicosatrienoic acids (EETs), and hydroxyeicosatetraenoic acids (HETEs). These epoxyeicosanoids are synthesized by the cytochrome P450 (CYP) epoxygenases. These mitochondrial enzymes catalyze the monooxygenation of arachidonic acid, producing specific epoxide regioisomers (5,6-EET; 8,9-EET; 11,12-EET; and 14,15-EET). EETs are metabolized by epoxide hydrolase to the corresponding dihydroeicosatrienoic acids (DiHETEs). Allylic oxidation of arachidonic acid is also catalyzed to form HETEs (5-, 8-, 9-, 11-, 12-, 15-, 19-, and 20-HETE). Certain HETEs (e.g., 5-, and 12-HETE) can also be formed via lipoxygenase action from hydroperoxyeicosatetraenoic acid (HPETE) precursors.
The presence of COX2 inhibitors may result in shunting free arachidonic acid down alternate metabolic pathways to potentially neuroprotective eicosanoids. For example, DFU treatments resulted in increased EET and HETE levels in injured rat brain (Gopez et al. 2005, K. Strauss, manuscript in preparation). We began to explore this idea in an effort to reconcile seemingly contradictory results from our laboratory. Administration of either a dehydroepiandrosterone analog (DHEF) or a COX2-specific inhibitor (DFU) significantly improved functional recovery in a rat TBI model. However, DHEF augmented the observed rise in total prostaglandin levels in brain tissue following injury, while DFU potently inhibited this rise. We wondered whether altered arachidonic acid metabolism might be more important to functional recovery than altered prostanoid formation per se.
As described above, several eicosanoids may have beneficial effects in the brain and/or periphery. In fact, arachidonic acid (a small dose, 5 μM) itself reproducibly protected cultured cerebellar neurons against glutamate-mediated excitotoxicity (A. Marini and K. Strauss, unpublished data). Thus, the enzymes necessary to shunt arachidonic acid metabolism toward protective compounds are likely expressed in neurons. Interestingly, NMDA administration induces neuronal arachidonic acid release (Dumuis et al., 1988; Lazarewicz et al., 1990; Lazarewicz et al., 1988; Sanfeliu, 1990); and stimulates prostaglandin D2 production in vivo (Lazarewicz et al., 2000). This link between the glutamate and the arachidonic acid pathways may indicate parallel mechanisms of toxicity and/or neuroprotection (e.g., via the phenomenon of preconditioning).
Induction of the CYP450 epoxygenase pathway has been identified in the normal and pathological brain (Alkayed et al., 1996; Ellis et al., 1991; Harder et al., 1995; Schilter et al., 2000; Schilter and Omiecinski, 1993), as well as in the endovasculature of the heart, kidney (Campbell, 2000; Makita et al., 1994; Node et al., 1999; Yu et al., 2000a), liver and other organs (Capdevila et al., 1988; Capdevila et al., 1982; Ellis et al., 1991; Harder et al., 1995; McGiff and Carroll, 1991). P450 epoxygenase activity has been implicated in the physiology of the febrile response, pancreatic glucagon and insulin release, stimulation of hypothalamic somatostatin release, pituitary vasopressin, oxytocin and luteinizing hormone release, inhibition of arachidonic acid-induced platelet aggregation, inhibition of the activity of Na+K+-ATPase in the nephron and corneal epithelium, regulation of blood pressure, vasodilation of local microcirculation in the kidney, intestine, heart and brain (Fitzpatrick and Murphy, 1988; Kozak et al., 2000b; Makita et al., 1996; McGiff and Carroll, 1991). Many of the CYP enzymes are induced during inflammatory challenge, however recent evidence suggests that the EETs and HETEs may also function in diminishing inflammation (Campbell, 2000; Chen et al., 2001; Kozak et al., 2000b; Node et al., 1999).
COX2 inhibitors and brain injuries
Several COX2-specific inhibitors have been employed to treat brain injuries (e.g., NS398, nimesulide, celecoxib, DFU). Their efficacy, when administered at various doses and times before or after neurological insult has not been entirely consistent, perhaps because of widely different partition coefficients across the blood-brain barrier. However, the overwhelming preponderance of evidence clearly shows that protracted brain COX2 activity mediates a toxic response that worsens functional and neuroanatomical deficits after brain and spinal cord injuries (Candelario-Jalil et al., 2002; Candelario-Jalil et al., 2003; Candelario-Jalil et al., 2005; Dash et al., 2000; Gopez et al., 2005; Resnick et al., 1998; Shapira et al., 1988; Shohami et al., 1990). Further, proof of principle is provided in a study of ischemic stroke using genetically modified COX2 null mutant mice (Iadecola et al., 2001). Compared to wild type littermates, COX2 deficient mice had reduced lesion volumes, attenuation of glutamate neurotoxicity and postischemic inflammation after middle cerebral artery occlusion. Thus, COX2 inhibitors that benefit the injured brain likely produce their effects primarily by reducing COX2 activity rather than by suppressing free radical-mediated brain damage or other non-specific mechanisms.
COX2 inhibitors are potent neuroprotectants in vitro and in vivo. Work from Hewett's group clearly showed that inhibition of COX2 protected neurons in mixed cultures against NMDA excitotoxicity (Hewett et al., 2000). Significantly, COX2-specific inhibition (NS398, 3-30 μM) blocked neuronal cell death, whereas COX1-specific inhibition (valeryl salicylate, 10-100 μM) did not (Hewett et al., 2000). In addition, newly synthesized COX2 appeared to be responsible; irreversible cyclooxygenase inhibition with aspirin pretreatment (100 μM, 1.5h) did not block subsequent excitotoxic cell death. Our results concur with these; in nearly homogeneous cultured cerebellar granule neurons, DFU (1nM to 1μM) protected from glutamate-mediated apoptosis (Strauss and Marini, 2002). In fact, DFU (10nM) protected 100% of vulnerable neurons from glutamate (100μM) mediated excitotoxicity (vital dye staining at 24h). A DNA fragmentation assay yielded identical results; COX2 inhibition attenuated apoptotic cell death in cultured neurons.
In vivo, systemic COX2 inhibitor administration reduces lesion size, neuroinflammation, and cell death in various rat brain injury models. As further proof of principle, in a genetic model, COX2-/- null mutant mice exposed to either ischemia or neurotoxic cerebral NMDA injection showed ∼40% less neural damage 1-4 days after insult compared to wild-type littermates (Iadecola et al., 2001; Sasaki et al., 2003).
COX2 inhibitor treatments reduced apoptosis in vivo as well. The activation of caspase-3 is a good marker of cellular commitment to apoptotic cell death. A biphasic response in the appearance of activated caspase-3 immunoreactive (AC3-ir) cells was observed in injured cortex and hippocampus after lateral cortical impact traumatic brain injury (Gopez et al., 2005). At 6 h postinjury, large numbers of AC3-ir cells were found in the injured cortex and the CA1-2 hippocampal regions, where COX2 levels were also maximal. By 24 h postinjury, AC3-ir cell counts were lower than at 6h or 72h in the injured cortex, ipsilateral CA2 and CA3 hippocampus, and contralateral perirhinal and piriform cortices, but at least double the control numbers. AC3-ir cell counts decreased to sham control levels in all other brain regions. As expected from delayed cell death profiles, of the 3 time points examined, the injured cortex and hippocampal CA3 region exhibited the highest number of AC3-ir cells at 72 h postinjury. DFU treatment attenuated the number of AC3-ir cells at 6 h and 72h postinjury in all cortical regions where an increase was observed. In the hippocampus at 6 h postinjury, DFU did not appreciably reduce AC3-ir cell number in the ipsilateral hippocampus. At 24h and 72h postinjury, when COX2 levels in the most vulnerable hippocampal regions DFU diminished the number of AC3-ir cells in the entire Ammon's horn. The protective effects of DFU in cortex, and particularly in hippocampus, coincided with preestablished profiles of maximal COX2 expression for these areas at the later times. Thus, COX2 “deficiency” is important in acute neurotoxicity and chronic neuroinflammation.
COX2 inhibition improves behavioral recovery
Recently, several groups have demonstrated improved functional recovery with systemic COX2 inhibitor treatments. Nimesulide improved memory (Barnes maze) and motor (rotarod) performance after neurotrauma (Cernak et al., 2001, 2002). Candalario-Jalil and colleagues also observed improved functional recovery (neurological severity score, rotarod) with this COX2 inhibitor in a rat model of cerebral ischemia (Candelario-Jalil et al., 2004; Candelario-Jalil et al., 2005).
DFU treatments effectively improved functional recovery, compared with vehicle-treated injured control rats (Gopez et al., 2005). Neurological reflexes (as measured by observing forelimb flexion, hindlimb extension, and lateral pulsion at 3 days postinjury) improved significantly for both low dose and high dose groups (1 mg/kg, 10 mg/kg) whether the first dose of DFU was administered 30 min, 2 or 6 hours after the injury. Significantly, working spatial memory was also improved after neurotrauma by DFU treatments. Memory performance in the Morris water maze was improved only in the high dose treatment groups and nearly achieved sham levels of performance. In our hands, DFU improved memory and neurological recovery in rats treated up to 6 hours after the injury. No injury-induced changes in swim speed or exploratory behavior were observed between any of the groups.
DFU and nimesulide improved behavioral recovery when administered hours after brain injury (Candelario- Jalil et al., 2002, 2003; Gopez et al., 2005). These findings provide evidence that COX2 inhibitors have an extended window of opportunity to protect vulnerable neurons from secondary damage after neurotrauma. This also correlated with reduced inflammatory proteins and cell death in the cortex and hippocampus. We have set the stage for acute COX2 inhibitor treatment clinical trials in brain injury patients. Notwithstanding their potential adverse cardiovascular side-effects, these findings have compelling implications for the use of COX2 inhibitors in chronic neurodegenerative disorders, as well.
COX2 inhibition attenuates COX2 expression in the injured brain
DFU apparently crossed the blood brain barrier and improved behavioral recovery, so we hypothesized that COX2-specific inhibition might affect COX2 expression to break the proposed “vicious cycle” that results in prolonged upregulation. Immunohistochemical analyses revealed that DFU attenuated both the number and intensity of COX2-immunoreactive (COX2-ir) staining in the cortex and hippocampus after traumatic brain injury, compared to vehicle-treated controls (Gopez et al. 2005). Specifically, there was observably less COX2-ir in the ipsilateral cortex, including cells of the cingulate, injured and adjacent parietal, perirhinal and piriform regions. The COX2-ir decreases observed at 6 h and 24 h persisted through 72 h postinjury, and the decrease in the injured cortex at this time could be demonstrated by immunoblot analysis. COX2 inhibition produced no observable effects on basal levels of COX2 either in vitro or in vivo (Gopez et al., 2005; Strauss and Marini, 2002)
DFU treatment also reduced COX2-ir in the ipsilateral hippocampus at 6 h, 24 h, and 72 h postinjury. At 6 h postinjury, the attenuation occurred primarily in the CA1, CA2 and dentate gyrus regions. In the CA3 region, most vulnerable to delayed cell death, DFU attenuated COX2–ir at 6 h postinjury, and in the dentate gyrus at 24 h postinjury, compared to vehicle-treated controls.
These observations support the “vicious cycle” hypothesis (Figure 2), that brain COX2 activity promotes further COX2 induction and eventual tissue damage. Thus, COX2 inhibitors not only block harmful enzymatic activity, but may also turn down the gain on dysregulated inflammatory gene transcription.
Antiinflammatory and neuroprotective effects of COX2 inhibition
Central inflammation following traumatic brain injuries includes the proliferation of activated astrocytes and microglia proximal to the injury site (Csuka et al., 2000; Morganti-Kossmann et al., 1997) that may exacerbate tissue damage after injury (Yakovlev et al., 1997). Cytokines affect inflammatory cell proliferation and infiltration. Proinflammatory cytokines such as interleukin-1 and interleukin-6 initiate inflammation after TBI, and antiinflammatory interleukin-10 reduces this activity (Knoblach and Faden, 1998; Yakovlev et al., 1997). Interleukin-1 induces COX2 in endothelial, inflammatory, and lining cells of the brain. COX2 may promote inflammatory cell proliferation and infiltration into the CNS after injury (Amruthesh et al., 1993; Bezzi et al., 1998; Blom et al., 1997; Hirst et al., 1999; Scali et al., 2003; Scali et al., 2000), but the role of eicosanoids in this process has not been well described. Thus, prolonged COX2 activity may contribute to brain infiltration by macrophages and leukocytes, implicated in the secondary processes that produce edema (Holmin and Mathiesen, 2000; Mayhan, 2000), cavity and scar formation (Fitch et al., 1999; Fitch and Silver, 1997) at delayed times postinjury (Whalen et al., 1999).
The EP3 receptor is the predominant PGE2 receptor in neurons and is distributed widely in the rat, mouse, and pig brain (Ericsson et al., 1995; Li et al., 1993a; Sugimoto et al., 1994). It has the highest binding affinity to PGE2 among the four EP receptors (Kiriyama et al., 1997). EP3 receptors are strictly confined to IL-1β-responsive neurons in the rat preoptic anterior hypothalamus, suggesting that EP3 receptors may be involved in the cytokine-induced inflammatory response of the CNS (Ericsson et al., 1995).
Our studies showed COX2 inhibition reduced COX2 expression in the cortex and hippocampus 72h after TBI (Gopez et al., 2005). This treatment also reduced interleukin-1β (IL-1β), a proinflammatory cytokine, in the injured brain at the acute 12h time point (S. Lewis and K. Strauss, manuscript in preparation). IL-1β is proteolytically activated by caspase-1 (ICE); thus it is not surprising that its biphasic appearance closely follows that of AC3 (Gopez et al., 2005). Intracerebral microinjections of IL-1β in rats increased inflammatory cells, neuronal death, and vasogenic edema (Holmin and Mathiesen, 2000). Histochemical analyses also indicate a reduction in vascular endothelial adhesion molecule expression (K. Strauss, manuscript in preparation), similar to that described in cardiac vascular endothelial cells (Node et al., 1999).
Cellular adhesion molecules (I-CAM, V-CAM, E-selectin) facilitate the adherence of peripheral inflammatory cells to the cerebrovascular endothelium, the first step in extravasation into the brain. Infiltration and proliferation of peripheral blood cells, e.g., neutrophils and macrophages (Kubo et al., 2000; Weaver et al., 2000) exacerbate brain injury (Bank et al., 1999; Rimpilainen et al., 2000). In a pig model of ischemic brain injury, leukocyte depletion lowered mortality, improved behavioral recovery and neuropathology scores at day 7 postinjury (Rimpilainen et al., 2000). In addition, vasogenic edema is the result of changes in the blood-brain barrier, emanating from the interactions of astrocytic end-feet and the cerebral vascular endothelium. If astrocyte and endothelial cell metabolism is stabilized by P450 eicosanoids, their ability to withstand the injury and retain an intact blood brain barrier may be preserved (Campbell, 2000; Fitch et al., 1999; Medhora and Harder, 1998; Medhora et al., 2001; Zeldin and Liao, 2000).
Activation and/or inhibition of transcription factor NF–κB is a likely candidate mechanism for COX2 inhibitor-mediated neuroprotection. Activated NF-κB increases transcription of COX2 in neural tissue (Kaltschmidt et al., 2002; Lukiw and Bazan, 1998). Its inhibitor, IκB can be inactivated via phosphorylation or by direct oxidation by free radicals (Barr et al., 2007; Heck et al., 1999; Nakai et al., 2000; Post et al., 1998; Vollgraf et al., 1999; Zou and Crews, 2006). By reducing prostaglandin and ROS production, COX2 inhibition may reduce NF-κB activation thus affecting its own transcription, and that of other apoptosis-related genes. In addition, other eicosanoid activities might involve stabilization of NF-κB (Acarin et al., 2001; Acarin, 2002; Hernandez et al., 2001; Lukiw and Bazan, 1998; Nurmi et al., 2004; Yoshida et al., 2003), or its inhibitor IκB (Grilli and Memo, 1999; Node et al., 1999; Richter et al., 2001; Tamatani et al., 2000). Thus, increased brain eicosanoid production and COX2 “deficiency” reduce peripheral inflammatory infiltration (Node et al., 1999), glial proliferation (Stephenson et al., 1996) and scar formation (Fitch et al., 1999; Sasaki et al., 2003).
Some alternative viewpoints
Most studies have found that activation of NF-κB and phosphorylation of its inhibitor IκB, correspond to a proinflammatory stimulus for gene expression. There are contradictory findings in the literature regarding the role of NF-κB in central inflammation. One study, using glutamate excitotoxicity, showed activated NF-κB was associated with cell death, and was reversed by high concentration aspirin (Grilli et al., 1996); while another study showed a rise in activated NF-κB during NMDA-mediated neuroprotection (Lipsky et al., 2001). NMDA-mediated neuroprotection induced the phosphorylation of IκB (Lipsky et al., 2001), whereas peripherally, antiinflammatory agents are known to stabilize IκB (Campbell, 2000). Interestingly, EETs have been shown to block (indirectly) the TNF-α mediated activation of IκB kinase (IKK) in endothelial cells (Campbell, 2000; Node et al., 1999; Stuhlmeier et al., 1997). However, it remains to be determined if EETs exert similar inhibitory effects in astrocytes, microglia, macrophages, and neutrophils in the injured CNS.
A few studies indicate prostaglandins may be protective, but the preponderance of evidence show the opposite. Moreover, the studies in which prostanoids (or their EP2 receptors) are characterized as neuroprotective have never shown improvements in functional outcomes. In our brain injury studies, some data indicate acute COX2 activity may be beneficial (Gopez et al., 2005). However, it is prolonged COX2 overexpression and activity that we propose may cause harm to the injured brain. The “neuroprotective” effects of prostaglandins (PGE2) in neural excitotoxicity models are limited to subacute reductions in propidium iodide staining at 24 h (no protection was seen after 48 h (Liu et al., 2005)). McCullough et al. (2004) show the effects of PGE2 on neuronal viability differ significantly in dispersed neuronal cultures and organotypic cultures, likely because of the preservation of astrocytic/neuronal interactions in organotypic cultures. PGE2, likely via the EP2 receptor, mediates reduced neuroinflammatory responses in cultured neurons, but not in the presence of glia (McCullough et al., 2004). Astrocytic/ neuronal interactions likely account for these differences. For example, high levels of PGE2 stimulate astrocytic glutamate release (Bezzi et al., 1998; Sanzgiri et al., 1999), thus exacerbating excitotoxicity. Interestingly, one recent study has provided evidence that COX2 prostaglandins rather than reactive oxygen species are responsible for COX2-mediated neurotoxicity (Manabe et al., 2004).
COX2 and cognitive function
The contribution of brain COX2 in cognitive function has recently become a topic of intense investigation. Many studies have demonstrated the benefit of COX2-selective inhibitors in improving memory function after brain injuries (see above). Treatments that induce COX2 overexpression in the brain interfere with cognitive function. For example, IL-1β, injected bilaterally into the dorsal hippocampus significantly impaired working memory, and this impairment was attenuated by pretreatment with diclofenac, a partially selective COX2 inhibitor (Matsumoto et al., 2004). Working memory was also impaired in rats administered a bilateral intrahippocampal injection of prostaglandin E2, in a dose-dependent manner. Interestingly, non-selective cyclooxygenase inhibitor treatments resulted in sustained deficits in spatial learning in the Morris watermaze (Shaw et al., 2003). In hippocampal slice preparations, oxygen-glucose deprivation causes a rapid and persistent tissue depolarization within 5 min. These challenges also induce rapid increases in COX2 (Kim et al., 2007). Pretreatment with cyclooxygenase or lipoxygenase inhibitors as well as free radical scavengers improved the latency of postischemic recovery from depolarization, while 14,15-EET or 20-HETE did not (Tanaka et al., 2003). These results suggest that the activation of the arachidonic acid cascade via phospholipase A2 and the free radicals produced by arachidonic acid metabolism contribute to the irreversible depolarization produced by in vitro ischemia. Other findings in hippocampal slices showed selective inhibition of COX-2 (but not COX1) decreased excitatory responses (Slanina and Schweitzer, 2005). Furthermore, the interactions between gene regulation and activation of brain COX2 and phospholipases under basal and pathological conditions is just beginning to be investigated in neural tissues (Sapirstein et al., 2005; Strokin et al., 2004).
Arachidonic acid release via phospholipase A2 has direct effects on brain cholinergic activity (Almeida et al., 1999; Kjome et al., 1998; Nishizaki et al., 1999a), and in the presence of COX2 inhibitors may also affect neurotransmission via catalysis down alternative metabolic pathways. Its eicosanoid metabolites are numerous, possessing a variety of neurophysiological activities. These include energy modulation (5-HPETE (Foley, 1997)), cannabinoid receptor binding (anandamide and 2-arachidonoyl glycerol (Devane et al., 1992; Mechoulam et al., 1995)), lipoxygenase-mediated analgesia (Christie et al., 1999; Mechoulam et al., 1998), circadian modulation (Uz and Manev, 1998) and antiinflammatory effects ([11,12]-EET (Node et al., 1999)), and neuroprotection from glutamate excitotoxicity (12-HETE (Hampson and Grimaldi, 2002)). Shohami's group (Panikashvili et al., 2001) clearly demonstrated that the endocannabinoid 2-arachidonoyl glycerol (2-AG), systemically administered to mice, was neuroprotective, improving functional outcome after closed head injury.
As a final point, COX2 overexpression in the brain may act as an arachidonic acid sink. Increased levels of free arachidonic acid are observed after tissue damage, and COX2 may serve to “sop it up.” While this may be adaptive in the acute time frame (e.g., leading to rapid local thromboxane synthesis and hemostasis), prolonged COX2 activity appears to harm the injured brain, perhaps by diverting arachidonic acid metabolism away from the synthesis of potentially beneficial metabolites and via free radical production. Furthermore, COX2 activity may inactivate neuroprotective eicosanoids, particularly in an environment of increasing COX2 levels, as seen after TBI. Support for this supposition includes work by Kozak and colleagues, and results from our laboratory. For example, COX2 can metabolize arachidonate, docosahexanoate, and other long chain fatty acid derivatives including 2-AG (Kozak et al., 2004; Kozak et al., 2000a). Indeed, neurotrauma reduced brain 2-AG levels in the rat, and the COX2 inhibitor DFU reversed this decrease within 24 hours after injury (Gopez et al. 2005). Moreover, dynamic changes in brain levels of potentially neuroprotective EETs and HETEs after brain injury are enhanced by COX2 inhibition (Gopez et al. 2005, K. Strauss, manuscript in preparation).
Together, these studies implicate a unique role for brain COX2 and its inhibitors in cognition and dementia. Agerelated or pathologically induced COX2 overexpression may play a key role in promoting premature cognitive decline (K. Strauss, manuscript in preparation, Casolini et al., 2002). Thus, COX2 inhibition in the brain has therapeutic utility beyond the suppression of prostaglandin and free radical formation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References Cited
- Abramovitz M, Adam M, Boie Y, Carriere M, Denis D, Godbout C, Lamontagne S, Rochette C, Sawyer N, Tremblay NM, Belley M, Gallant M, Dufresne C, Gareau Y, Ruel R, Juteau H, Labelle M, Ouimet N, Metters KM. The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim Biophys Acta. 2000;1483:285–293. doi: 10.1016/s1388-1981(99)00164-x. [DOI] [PubMed] [Google Scholar]
- Acarin L, Gonzalez B, Castellano B. Triflusal posttreatment inhibits glial nuclear factor-kappaB, downregulates the glial response, and is neuroprotective in an excitotoxic injury model in postnatal brain. Stroke. 2001;32:2394–2402. doi: 10.1161/hs1001.097243. [DOI] [PubMed] [Google Scholar]
- Acarin L, G B, C B. Decrease of proinflammatory molecules correlates with neuroprotective effect of the fluorinated salicylate triflusal after postnatal excitotoxic damage. Stroke. 2002;33:2499–2505. doi: 10.1161/01.str.0000028184.80776.58. [DOI] [PubMed] [Google Scholar]
- Adams J, Collaco-Moraes Y, de Belleroche J. Cyclooxygenase-2 induction in cerebral cortex: an intracellular response to synaptic excitation. Journal of Neurochemistry. 1996;66:6–13. doi: 10.1046/j.1471-4159.1996.66010006.x. [DOI] [PubMed] [Google Scholar]
- Alkayed NJ, Goyagi T, Joh HD, Klaus J, Harder DR, Traystman RJ, Hurn PD. Neuroprotection and P450 2C11 upregulation after experimental transient ischemic attack. Stroke. 2002;33:1677–1684. doi: 10.1161/01.str.0000016332.37292.59. [DOI] [PubMed] [Google Scholar]
- Alkayed NJ, Narayanan J, Gebremedhin D, Medhora M, Roman RJ, Harder DR. Molecular characterization of an arachidonic acid epoxygenase in rat brain astrocytes. Stroke. 1996;27:971–979. doi: 10.1161/01.str.27.5.971. [DOI] [PubMed] [Google Scholar]
- Almeida T, Cunha RA, Ribeiro JA. Facilitation by arachidonic acid of acetylcholine release from the rat hippocampus. Brain Research. 1999;826:104–111. doi: 10.1016/s0006-8993(99)01267-6. [DOI] [PubMed] [Google Scholar]
- Amruthesh SC, Boerschel MF, McKinney JS, Willoughby KA, Ellis EF. Metabolism of arachidonic acid to epoxyeicosatrienoic acids, hydroxyeicosatetraenoic acids, and prostaglandins in cultured rat hippocampal astrocytes. Journal of Neurochemistry. 1993;61:150–159. doi: 10.1111/j.1471-4159.1993.tb03550.x. [DOI] [PubMed] [Google Scholar]
- Bank U, Reinhold D, Schneemilch C, Kunz D, Synowitz HJ, Ansorge S. Selective proteolytic cleavage of IL-2 receptor and IL-6 receptor ligand binding chains by neutrophil-derived serine proteases at foci of inflammation. Journal of Interferon and Cytokine Research. 1999;19:1277–1287. doi: 10.1089/107999099312957. [DOI] [PubMed] [Google Scholar]
- Barr J, Sharma CS, Sarkar S, Wise K, Dong L, Periyakaruppan A, Ramesh GT. Nicotine induces oxidative stress and activates nuclear transcription factor kappa B in rat mesencephalic cells. Mol Cell Biochem. 2007;297:93–99. doi: 10.1007/s11010-006-9333-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand V, Guimbaud R, Tulliez M, Mauprivez C, Sogni P, Couturier D, Giroud JP, Chaussade S, Chauvelot-Moachon L. Increase in tumor necrosis factor-alpha production linked to the toxicity of indomethacin for the rat small intestine. British Journal of Pharmacology. 1998;124:1385–1394. doi: 10.1038/sj.bjp.0701968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, Pozzan T, Volterra A. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature. 1998;391:281–285. doi: 10.1038/34651. [DOI] [PubMed] [Google Scholar]
- Blom MA, van Twillert MG, de Vries SC, Engels F, Finch CE, Veerhuis R, Eikelenboom P. NSAIDS inhibit the IL-1 beta-induced IL-6 release from human post-mortem astrocytes: the involvement of prostaglandin E2. Brain Research. 1997;777:210–218. doi: 10.1016/s0006-8993(97)01204-3. [DOI] [PubMed] [Google Scholar]
- Breder CD, Smith WL, Raz A, Masferrer J, Seibert K, Needleman P, Saper CB. Distribution and characterization of cyclooxygenase immunoreactivity in the ovine brain. J Comp Neurol. 1992;322:409–438. doi: 10.1002/cne.903220309. [DOI] [PubMed] [Google Scholar]
- Brezinski ME, Serhan CN. Selective incorporation of (15S)-hydroxyeicosatetraenoic acid in phosphatidylinositol of human neutrophils: agonist-induced deacylation and transformation of stored hydroxyeicosanoids. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:6248–6252. doi: 10.1073/pnas.87.16.6248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caggiano AO, Breder CD, Kraig RP. Long-term elevation of cyclooxygenase-2, but not lipoxygenase, in regions synaptically distant from spreading depression. Journal of Comparative Neurology. 1996;376:447–462. doi: 10.1002/(SICI)1096-9861(19961216)376:3<447::AID-CNE7>3.0.CO;2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell WB. New role for epoxyeicosatrienoic acids as anti-inflammatory mediators. Trends in Pharmacological Sciences. 2000;21:125–127. doi: 10.1016/s0165-6147(00)01472-3. [DOI] [PubMed] [Google Scholar]
- Candelario-Jalil E, Alvarez D, Castaneda JM, Al-Dalain SM, Martinez-Sanchez G, Merino N, Leon OS. The highly selective cyclooxygenase-2 inhibitor DFU is neuroprotective when given several hours after transient cerebral ischemia in gerbils. Brain Research. 2002;927:212–215. doi: 10.1016/s0006-8993(01)03358-3. [DOI] [PubMed] [Google Scholar]
- Candelario-Jalil E, Alvarez D, Merino N, Leon OS. Delayed treatment with nimesulide reduces measures of oxidative stress following global ischemic brain injury in gerbils. Neuroscience Research. 2003;47:245–253. doi: 10.1016/s0168-0102(03)00184-6. [DOI] [PubMed] [Google Scholar]
- Candelario-Jalil E, González-Falcón A, García-Cabrera M, León OS, Fiebich BL. Wide therapeutic time window for nimesulide neuroprotection in a model of transient focal cerebral ischemia in the rat. Brain Res. 2004;1007:98–108. doi: 10.1016/j.brainres.2004.01.078. [DOI] [PubMed] [Google Scholar]
- Candelario-Jalil E, Mhadu NH, Gonzalez-Falcon A, Garcia-Cabrera M, Munoz E, Leon OS, Fiebich BL. Effects of the cyclooxygenase-2 inhibitor nimesulide on cerebral infarction and neurological deficits induced by permanent middle cerebral artery occlusion in the rat. J Neuroinflammation. 2005;2:3. doi: 10.1186/1742-2094-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C, Matsumura K, Shirakawa N, Maeda M, Jikihara I, Kobayashi S, Watanabe Y. Pyrogenic cytokines injected into the rat cerebral ventricle induce cyclooxygenase-2 in brain endothelial cells and also upregulate their receptors. European Journal of Neuroscience. 2001;13:1781–1790. doi: 10.1046/j.0953-816x.2001.01551.x. [DOI] [PubMed] [Google Scholar]
- Cao C, Matsumura K, Yamagata K, Watanabe Y. Induction by lipopolysaccharide of cyclooxygenase-2 mRNA in rat brain; its possible role in the febrile response. Brain Research. 1995;697:187–196. doi: 10.1016/0006-8993(95)00839-i. [DOI] [PubMed] [Google Scholar]
- Capdevila J, Gil L, Orellana M, Marnett LJ, Mason JI, Yadagiri P, Falck JR. Inhibitors of cytochrome P-450-dependent arachidonic acid metabolism. Archives of Biochemistry & Biophysics. 1988;261:257–263. doi: 10.1016/0003-9861(88)90340-2. [DOI] [PubMed] [Google Scholar]
- Capdevila J, Marnett LJ, Chacos N, Prough RA, Estabrook RW. Cytochrome P-450-dependent oxygenation of arachidonic acid to hydroxyicosatetraenoic acids. Proceedings of the National Academy of Sciences of the United States of America. 1982;79:767–770. doi: 10.1073/pnas.79.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capdevila JH, Kishore V, Dishman E, Blair IA, Falck JR. A novel pool of rat liver inositol and ethanolamine phospholipids contains epoxyeicosatrienoic acids (EETs) Biochemical & Biophysical Research Communications. 1987;146:638–644. doi: 10.1016/0006-291x(87)90576-6. [DOI] [PubMed] [Google Scholar]
- Casolini P, Catalani A, Zuena AR, Angelucci L. Inhibition of COX-2 reduces the age-dependent increase of hippocampal inflammatory markers, corticosterone secretion, and behavioral impairments in the rat. Journal of Neuroscience Research. 2002;68:337–343. doi: 10.1002/jnr.10192. [DOI] [PubMed] [Google Scholar]
- Cernak I, O'Connor C, Vink R. Activation of cyclo-oxygenase-2 contributes to motor and cognitive dysfunction following diffuse traumatic brain injury in rats. Clinical & Experimental Pharmacology & Physiology. 2001;28:922–925. doi: 10.1046/j.1440-1681.2001.03549.x. [DOI] [PubMed] [Google Scholar]
- Cernak I, O'Connor C, Vink R. Inhibition of cyclooxygenase 2 by nimesulide improves cognitive outcome more than motor outcome following diffuse traumatic brain injury in rats. Experimental Brain Research. 2002;147:193–199. doi: 10.1007/s00221-002-1245-z. [DOI] [PubMed] [Google Scholar]
- Chacos N, Capdevila J, Falck JR, Manna S, Martin-Wixtrom C, Gill SS, Hammock BD, Estabrook RW. The reaction of arachidonic acid epoxides (epoxyeicosatrienoic acids) with a cytosolic epoxide hydrolase. Arch Biochem Biophys. 1983;223:639–648. doi: 10.1016/0003-9861(83)90628-8. [DOI] [PubMed] [Google Scholar]
- Chacos N, Falck JR, Wixtrom C, Capdevila J. Novel epoxides formed during the liver cytochrome P-450 oxidation of arachidonic acid. Biochemical & Biophysical Research Communications. 1982;104:916–922. doi: 10.1016/0006-291x(82)91336-5. [DOI] [PubMed] [Google Scholar]
- Chen J, Marsh T, Zhang JS, Graham SH. Expression of cyclo-oxygenase 2 in rat brain following kainate treatment. Neuroreport. 1995;6:245–248. [PubMed] [Google Scholar]
- Chen JK, Capdevila J, Harris RC. Cytochrome p450 epoxygenase metabolism of arachidonic acid inhibits apoptosis. Molecular & Cellular Biology. 2001;21:6322–6331. doi: 10.1128/MCB.21.18.6322-6331.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie MJ, Vaughan CW, Ingram SL. Opioids, NSAIDs and 5-lipoxygenase inhibitors act synergistically in brain via arachidonic acid metabolism. Inflammation Research. 1999;48:1–4. doi: 10.1007/s000110050367. [DOI] [PubMed] [Google Scholar]
- Collaco-Moraes Y, Aspey B, Harrison M, de Belleroche J. Cyclo-oxygenase-2 messenger RNA induction in focal cerebral ischemia. Journal of Cerebral Blood Flow & Metabolism. 1996;16:1366–1372. doi: 10.1097/00004647-199611000-00035. [DOI] [PubMed] [Google Scholar]
- Csuka E, Hans VH, Ammann E, Trentz O, Kossmann T, Morganti-Kossmann MC. Cell activation and inflammatory response following traumatic axonal injury in the rat. Neuroreport. 2000;11:2587–2590. doi: 10.1097/00001756-200008030-00047. [DOI] [PubMed] [Google Scholar]
- Dash P, Mach S, Moore A. Regional expression and role of cyclooxygenase-2 following experimental traumatic brain injury. J Neurotrauma. 2000;17:69–81. doi: 10.1089/neu.2000.17.69. [DOI] [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [comment] [DOI] [PubMed] [Google Scholar]
- Dewitt D, Kong D, Lyeth B, Jenkins L, Hayes R, Wooten E, Prough D. Experimental traumatic brain injury elevates brain prostaglandin E2 and thromboxane B2 levels in rats. J Neurotrauma. 1988;5:303–313. doi: 10.1089/neu.1988.5.303. [DOI] [PubMed] [Google Scholar]
- Dhillon HS, Dose JM, Scheff SW, Prasad MR. Time course of changes in lactate and free fatty acids after experimental brain injury and relationship to morphologic damage. Experimental Neurology. 1997;146:240–249. doi: 10.1006/exnr.1997.6524. [DOI] [PubMed] [Google Scholar]
- Dumuis A, Sebben M, Haynes L, Pin J, Bockaert J. NMDA receptors activate the arachidonic acid cascade system in striatal neurons. Nature. 1988;336:68–70. doi: 10.1038/336068a0. [DOI] [PubMed] [Google Scholar]
- Ellis EF, Amruthesh SC, Police RJ, Yancey LM. Brain synthesis and cerebrovascular action of cytochrome P-450/monooxygenase metabolites of arachidonic acid. Advances in Prostaglandin, Thromboxane, & Leukotriene Research. 1991;21A:201–204. [PubMed] [Google Scholar]
- Ellis EF, Police RJ, Rice LY, Grabeel M, Holt S. Increased plasma PGE2, 6-keto-PGF1 alpha, and 12-HETE levels following experimental concussive brain injury. Journal of Neurotrauma. 1989;6:31–37. doi: 10.1089/neu.1989.6.31. [DOI] [PubMed] [Google Scholar]
- Ellis EF, Wei EP, Kontos HA. Vasodilation of cat cerebral arterioles by prostaglandins D2, E2, G2, and I2. American Journal of Physiology. 1979;237:H381–385. doi: 10.1152/ajpheart.1979.237.3.H381. [DOI] [PubMed] [Google Scholar]
- Ellis EF, Wright KF, Wei EP, Kontos HA. Cyclooxygenase products of arachidonic acid metabolism in cat cerebral cortex after experimental concussive brain injury. Journal of Neurochemistry. 1981;37:892–896. doi: 10.1111/j.1471-4159.1981.tb04476.x. [DOI] [PubMed] [Google Scholar]
- Ericsson A, Ek M, Lindefors N. Distribution of prostaglandin E2 receptor (EP3 subtype) mRNA containing cells in the rat central nervous system. Soc Neurosci Abs. 1995;21:98. [Google Scholar]
- Feng L, Sun W, Xia Y, Tang WW, Chanmugam P, Soyoola E, Wilson CB, Hwang D. Cloning two isoforms of rat cyclooxygenase: differential regulation of their expression. Archives of Biochemistry & Biophysics. 1993;307:361–368. doi: 10.1006/abbi.1993.1601. [DOI] [PubMed] [Google Scholar]
- Feng L, Xia Y, Garcia G, Hwang D, Wilson C. Involvement of reactive oxygen intermediates in cyclooxygenase-2 expression induced by interleukin-1, tumor necrosis factor-alpha, and lipopolysaccharide. J Clin Invest. 1995;95:1669–1675. doi: 10.1172/JCI117842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fennekohl A, Sugimoto Y, Segi E, Maruyama T, Ichikawa A, Puschel GP. Contribution of the two Gs-coupled PGE2-receptors EP2-receptor and EP4-receptor to the inhibition by PGE2 of the LPS-induced TNFalpha-formation in Kupffer cells from EP2-or EP4-receptor-deficient mice. Pivotal role for the EP4-receptor in wild type Kupffer cells. Journal of Hepatology. 2002;36:328–334. doi: 10.1016/s0168-8278(01)00277-x. [DOI] [PubMed] [Google Scholar]
- Fitch MT, Doller C, Combs CK, Landreth GE, Silver J. Cellular and molecular mechanisms of glial scarring and progressive cavitation: in vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. Journal of Neuroscience. 1999;19:8182–8198. doi: 10.1523/JNEUROSCI.19-19-08182.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch MT, Silver J. Activated macrophages and the blood-brain barrier: inflammation after CNS injury leads to increases in putative inhibitory molecules. Experimental Neurology. 1997;148:587–603. doi: 10.1006/exnr.1997.6701. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick FA, Murphy RC. Cytochrome P-450 metabolism of arachidonic acid: formation and biological actions of “epoxygenase”-derived eicosanoids. Pharmacological Reviews. 1988;40:229–241. [PubMed] [Google Scholar]
- Flohe S, Ackermann M, Reuter M, Nast-Kolb D, Schade FU. Sublethal hemorrhagic shock reduces tumor necrosis factor-alpha-producing capacity in different cell compartments. European Cytokine Network. 2000;11:420–426. [PubMed] [Google Scholar]
- Foley TD. 5-HPETE is a potent inhibitor of neuronal Na+, K(+)-ATPase activity. Biochemical & Biophysical Research Communications. 1997;235:374–376. doi: 10.1006/bbrc.1997.6790. [DOI] [PubMed] [Google Scholar]
- Gardiner M, Nilsson B, Rehncrona S, Siesjo BK. Free fatty acids in the rat brain in moderate and severe hypoxia. Journal of Neurochemistry. 1981;36:1500–1505. doi: 10.1111/j.1471-4159.1981.tb00592.x. [DOI] [PubMed] [Google Scholar]
- Goncalves de Moraes VL, Boris Vargaftig B, Lefort J, Meager A, Chignard M. Effect of cyclooxygenase inhibitors and modulators of cyclic AMP formation on lipopolysaccharide-induced neutrophil infiltration in mouse lung. British Journal of Pharmacology. 1996;117:1792–1796. doi: 10.1111/j.1476-5381.1996.tb15356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopez JJ, Yue H, Vasudevan R, Malik AS, Fogelsanger LN, Lewis S, Panikashvili D, Shohami E, Jansen SA, Narayan RK, Strauss KI. Cyclooxygenase-2-specific inhibitor improves functional outcomes, provides neuroprotection, and reduces inflammation in a rat model of traumatic brain injury. Neurosurgery. 2005;56:590–604. doi: 10.1227/01.NEU.0000154060.14900.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham S, Nakayama M, Zhu R, Chen J. Cyclooxygenase 2 and the pathogenesis of delayed neuronal death after global ischemia. Neurology. 1996;46:406. [Google Scholar]
- Grilli M, Memo M. Possible role of NF-kappaB and p53 in the glutamate-induced pro-apoptotic neuronal pathway. Cell Death & Differentiation. 1999;6:22–27. doi: 10.1038/sj.cdd.4400463. [DOI] [PubMed] [Google Scholar]
- Grilli M, Pizzi M, Memo M, Spano P. Neuroprotection by aspirin and sodium salicylate through blockade of NF-kappaB activation. Science. 1996;274:1383–1385. doi: 10.1126/science.274.5291.1383. [DOI] [PubMed] [Google Scholar]
- Hampson AJ, Grimaldi M. 12-hydroxyeicosatetrenoate (12-HETE) attenuates AMPA receptor-mediated neurotoxicity: evidence for a G-protein-coupled HETE receptor. Journal of Neuroscience. 2002;22:257–264. doi: 10.1523/JNEUROSCI.22-01-00257.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder DR, Campbell WB, Roman RJ. Role of cytochrome P-450 enzymes and metabolites of arachidonic acid in the control of vascular tone. Journal of Vascular Research. 1995;32:79–92. doi: 10.1159/000159080. [DOI] [PubMed] [Google Scholar]
- Heck S, Lezoualc'h F, Engert S, Behl C. Insulin-like growth factor-1-mediated neuroprotection against oxidative stress is associated with activation of nuclear factor kappaB. J Biol Chem. 1999;274:9828–9835. doi: 10.1074/jbc.274.14.9828. [DOI] [PubMed] [Google Scholar]
- Hernandez M, de Arriba AF, Merlos M, Fuentes L, Crespo MS, Nieto ML. Effect of 4-trifluoromethyl derivatives of salicylate on nuclear factor kappaB-dependent transcription in human astrocytoma cells. British Journal of Pharmacology. 2001;132:547–555. doi: 10.1038/sj.bjp.0703820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschman HR, Xie W, Reddy S. Inflammation, reproduction, cancer and all that…. The regulation and role of the inducible prostaglandin synthase. Bioessays. 1995;17:1031–1037. doi: 10.1002/bies.950171207. [DOI] [PubMed] [Google Scholar]
- Hewett S, Uliasz T, Vidwans A, Hewett J. Cyclooxygenase-2 contributes to N-methyl-D-aspartate-mediated neuronal cell death in primary cortical cell culture. J Pharm Exp Ther. 2000;293(2):417–425. [PubMed] [Google Scholar]
- Hirst WD, Young KA, Newton R, Allport VC, Marriott DR, Wilkin GP. Expression of COX-2 by normal and reactive astrocytes in the adult rat central nervous system. Molecular & Cellular Neurosciences. 1999;13:57–68. doi: 10.1006/mcne.1998.0731. [DOI] [PubMed] [Google Scholar]
- Ho L, Pieroni C, Winger D, Purohit DP, Aisen PS, Pasinetti GM. Regional distribution of cyclooxygenase-2 in the hippocampal formation in Alzheimer's disease. Journal of Neuroscience Research. 1999;57:295–303. doi: 10.1002/(SICI)1097-4547(19990801)57:3<295::AID-JNR1>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Holmin S, Mathiesen T. Intracerebral administration of interleukin-1beta and induction of inflammation, apoptosis, and vasogenic edema. J Neurosurgery. 2000;92:108–120. doi: 10.3171/jns.2000.92.1.0108. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Niwa K, Nogawa S, Zhao X, Nagayama M, Araki E, Morham S, Ross ME. Reduced susceptibility to ischemic brain injury and N-methyl-D-aspartate-mediated neurotoxicity in cyclooxygenase-2-deficient mice. Proc Nat Acad Sci USA. 2001;98:1294–1299. doi: 10.1073/pnas.98.3.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Close LN, Selden NR, Alkayed NJ. A novel role for P450 eicosanoids in the neurogenic control of cerebral blood flow in the rat. Exp Physiol. 2007;92:653–658. doi: 10.1113/expphysiol.2006/036889. [DOI] [PubMed] [Google Scholar]
- Ishaque A, Dunn MJ, Sorokin A. Cyclooxygenase-2 inhibits tumor necrosis factor alpha-mediated apoptosis in renal glomerular mesangial cells. Journal of Biological Chemistry. 2003;278:10629–10640. doi: 10.1074/jbc.M210559200. [DOI] [PubMed] [Google Scholar]
- Jantzen PT, Connor KE, DiCarlo G, Wenk GL, Wallace JL, Rojiani AM, Coppola D, Morgan D, Gordon MN. Microglial activation and beta -amyloid deposit reduction caused by a nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. Journal of Neuroscience. 2002;22:2246–2254. doi: 10.1523/JNEUROSCI.22-06-02246.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA. Head injury and dementia. Curr Opin Neurol. 2004;17:719–723. doi: 10.1097/00019052-200412000-00012. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt B, Linker RA, Deng J, Kaltschmidt C. Cyclooxygenase-2 is a neuronal target gene of NF-kappaB. BMC Mol Biol. 2002;3:16. doi: 10.1186/1471-2199-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karara A, Dishman E, Falck JR, Capdevila JH. Endogenous epoxyeicosatrienoyl-phospholipids A novel class of cellular glycerolipids containing epoxidized arachidonate moieties. Journal of Biological Chemistry. 1991;266:7561–7569. [PubMed] [Google Scholar]
- Kaufmann W, Worley P, Pegg J, Bremer M, Isakson P. COX-2, a synaptically induced enzyme, is expressed by excitatory neurons at postsynaptic sites in rat cerebral cortex. Proc Nat Acad Sci USA. 1996;93:2317–2321. doi: 10.1073/pnas.93.6.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Raval AP, Defazio RA, Perez-Pinzon MA. Ischemic preconditioning via epsilon protein kinase C activation requires cyclooxygenase-2 activation in vitro. Neuroscience. 2007;145:931–941. doi: 10.1016/j.neuroscience.2006.12.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiriyama M, Ushikubi F, Kobayashi T, Hirata M, Sugimoto Y, Narumiya S. Ligand binding specificities of the eight types and subtypes of the mouse prostanoid receptors expressed in Chinese hamster ovary cells. Br J Pharmacol. 1997;122:217–224. doi: 10.1038/sj.bjp.0701367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjome JR, Swenson KA, Johnson MN, Bordayo EZ, Anderson LE, Klevan LC, Fraticelli AI, Aldrich SL, Fawcett JR, Venters HD, Jr, Ala TA, Frey WH., 2nd Inhibition of antagonist and agonist binding to the human brain muscarinic receptor by arachidonic acid. Journal of Molecular Neuroscience. 1998;10:209–217. doi: 10.1007/BF02761775. [DOI] [PubMed] [Google Scholar]
- Knoblach SM, Faden AI. Interleukin-10 improves outcome and alters proinflammatory cytokine expression after experimental traumatic brain injury. Experimental Neurology. 1998;153:143–151. doi: 10.1006/exnr.1998.6877. [DOI] [PubMed] [Google Scholar]
- Koerner IP, Zhang W, Hurn PD, Alkayed NJ. Society for Neuroscience Meeting Abstracts. Abstract Viewer/Itinerary Planner. Washington, D.C.: 2005. Neuroprotective effect of 14,15-epoxyeicosatrienoic acid. [Google Scholar]
- Koistinaho J, Koponen S, Chan PH. Expression of cyclooxygenase-2 mRNA after global ischemia is regulated by AMPA receptors and glucocorticoids. Stroke. 1999;30:1900–1905. doi: 10.1161/01.str.30.9.1900. [DOI] [PubMed] [Google Scholar]
- Kontos HA, Dietrich WD, Wei EP, Ellis EF, Povlishock JT. Abnormalities of the cerebral microcirculation after traumatic injury: the relationship of hypertension and prostaglandins. Advances in Experimental Medicine & Biology. 1980a;131:243–256. doi: 10.1007/978-1-4684-3752-2_19. [DOI] [PubMed] [Google Scholar]
- Kontos HA, Wei EP, Povlishock JT, Dietrich WD, Magiera CJ, Ellis EF. Cerebral arteriolar damage by arachidonic acid and prostaglandin G2. Science. 1980b;209:1242–1245. doi: 10.1126/science.7403881. [DOI] [PubMed] [Google Scholar]
- Kozak KR, Prusakiewicz JJ, Marnett LJ. Oxidative metabolism of endocannabinoids by COX-2. Curr Pharm Des. 2004;10:659–667. doi: 10.2174/1381612043453081. [DOI] [PubMed] [Google Scholar]
- Kozak KR, Rowlinson SW, Marnett LJ. Oxygenation of the endocannabinoid, 2-arachidonylglycerol, to glyceryl prostaglandins by cyclooxygenase-2. Journal of Biological Chemistry. 2000a;275:33744–33749. doi: 10.1074/jbc.M007088200. [DOI] [PubMed] [Google Scholar]
- Kozak W, Kluger MJ, Kozak A, Wachulec M, Dokladny K. Role of cytochrome P-450 in endogenous antipyresis. American Journal of Physiology - Regulatory Integrative & Comparative Physiology. 2000b;279:R455–460. doi: 10.1152/ajpregu.2000.279.2.R455. [DOI] [PubMed] [Google Scholar]
- Kubo Y, Suzuki M, Kudo A, Yoshida K, Suzuki T, Ogasawara K, Ogawa A, Kurose A, Sawai T. Thrombin inhibitor ameliorates secondary damage in rat brain injury: suppression of inflammatory cells and vimentin-positive astrocytes. J Neurotrauma. 2000;17:163–172. doi: 10.1089/neu.2000.17.163. [DOI] [PubMed] [Google Scholar]
- Kunz T, Oliw E. The selective cyclooxygenase-2 inhibitor rofecoxib reduces kainate-induced cell death in the rat hippocampus. Eur J Neurosci. 2001;13:569–575. doi: 10.1046/j.1460-9568.2001.01420.x. [DOI] [PubMed] [Google Scholar]
- Lazarewicz J, Salinska E, Stafiej A, Ziembowicz A, Zieminska E. NMDA receptors and nitric oxide regulate prostaglandin D2 synthesis in the rabbit hippocampus in vivo. Acta Neurobiologiae Experimentalis. 2000;60:427–435. doi: 10.55782/ane-2000-1362. [DOI] [PubMed] [Google Scholar]
- Lazarewicz J, Wroblewski J, Costa E. N-Methyl-D-aspartate-sensitive glutamate receptors induce calcium-mediated arachidonic acid release in primary cultures of cerebellar granule cells. J Neurochem. 1990;55:1875–1881. doi: 10.1111/j.1471-4159.1990.tb05771.x. [DOI] [PubMed] [Google Scholar]
- Lazarewicz JW, Wroblewski JT, Palmer ME, Costa E. Activation of N-methyl-D-aspartate-sensitive glutamate receptors stimulates arachidonic acid release in primary cultures of cerebellar granule cells. Neuropharmacology. 1988;27:765–770. doi: 10.1016/0028-3908(88)90088-3. [DOI] [PubMed] [Google Scholar]
- Lewen A, Hillered L. Involvement of reactive oxygen species in membrane phospholipid breakdown and energy perturbation after traumatic brain injury in the rat. J Neurotrauma. 1998;15:521–530. doi: 10.1089/neu.1998.15.521. [DOI] [PubMed] [Google Scholar]
- Li DY, Varma DR, Chatterjee TK, Fernandez H, Abran D, Chemtob S. Fewer PGE2 and PGF2 alpha receptors in brain synaptosomes of newborn than of adult pigs. J Pharmacol Exp Ther. 1993a;267:1292–1297. [PubMed] [Google Scholar]
- Li SR, Wu KK, Anggard E, Ferns G. Localization of prostaglandin G/H synthase gene expression in rat brain by in situ hybridization. Biological Signals. 1993b;2:77–83. doi: 10.1159/000109479. [DOI] [PubMed] [Google Scholar]
- Lipsky RH, Xu K, Zhu D, Kelly C, Terhakopian A, Novelli A, Marini AM. Nuclear factor kappaB is a critical determinant in N-methyl-D-aspartate receptor-mediated neuroprotection. J Neurochem. 2001;78:254–264. doi: 10.1046/j.1471-4159.2001.00386.x. [DOI] [PubMed] [Google Scholar]
- Liu D, Wu L, Breyer R, Mattson MP, Andreasson K. Neuroprotection by the PGE2 EP2 receptor in permanent focal cerebral ischemia. Ann Neurol. 2005;57:758–761. doi: 10.1002/ana.20461. [DOI] [PubMed] [Google Scholar]
- Liu M, Alkayed NJ. Hypoxic preconditioning and tolerance via hypoxia inducible factor (HIF) 1alpha-linked induction of P450 2C11 epoxygenase in astrocytes. J Cereb Blood Flow Metab. 2005;25:939–948. doi: 10.1038/sj.jcbfm.9600085. [DOI] [PubMed] [Google Scholar]
- Lukiw WJ, Bazan NG. Strong nuclear factor-kappaB-DNA binding parallels cyclooxygenase-2 gene transcription in aging and in sporadic Alzheimer's disease superior temporal lobe neocortex. Journal of Neuroscience Research. 1998;53:583–592. doi: 10.1002/(SICI)1097-4547(19980901)53:5<583::AID-JNR8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Makita K, Falck JR, Capdevila JH. Cytochrome P450, the arachidonic acid cascade, and hypertension: new vistas for an old enzyme system. FASEB Journal. 1996;10:1456–1463. doi: 10.1096/fasebj.10.13.8940291. [DOI] [PubMed] [Google Scholar]
- Makita K, Takahashi K, Karara A, Jacobson HR, Falck JR, Capdevila JH. Experimental and/or genetically controlled alterations of the renal microsomal cytochrome P450 epoxygenase induce hypertension in rats fed a high salt diet. Journal of Clinical Investigation. 1994;94:2414–2420. doi: 10.1172/JCI117608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manabe Y, Anrather J, Kawano T, Niwa K, Zhou P, Ross ME, Iadecola C. Prostanoids, not reactive oxygen species, mediate COX-2-dependent neurotoxicity. Ann Neurol. 2004;55:668–675. doi: 10.1002/ana.20078. [DOI] [PubMed] [Google Scholar]
- Manev H, Uz T, Qu T. 5-Lipoxygenase and cyclooxygenase mRNA expression in rat hippocampus:early response to glutamate receptor activation by kainate. Exp Gerontol. 2000;35:1201–1209. doi: 10.1016/s0531-5565(00)00152-2. [DOI] [PubMed] [Google Scholar]
- Marcheselli VL, Bazan NG. Sustained induction of prostaglandin endoperoxide synthase-2 by seizures in hippocampus Inhibition by a platelet-activating factor antagonist. Journal of Biological Chemistry. 1996;271:24794–24799. doi: 10.1074/jbc.271.40.24794. [DOI] [PubMed] [Google Scholar]
- Marini A, Paul S. N-Methyl-D-aspartate receptor-mediated neuroprotection in cerebellar granule cells requires new RNA and protein synthesis. Proc Natl Acad Sci USA. 1992;89:6555–6559. doi: 10.1073/pnas.89.14.6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marklund N, Salci K, Lewen A, Hillered L. Glycerol as a marker for post-traumatic membrane phospholipid degradation in rat brain. Neuroreport. 1997;8:1457–1461. doi: 10.1097/00001756-199704140-00026. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Yamaguchi T, Watanabe S, Yamamoto T. Involvement of arachidonic acid cascade in working memory impairment induced by interleukin-1 beta. Neuropharmacology. 2004;46:1195–1200. doi: 10.1016/j.neuropharm.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Mayhan WG. Leukocyte adherence contributes to disruption of the blood-brain barrier during activation of mast cells. Brain Research. 2000;869:112–120. doi: 10.1016/s0006-8993(00)02376-3. [DOI] [PubMed] [Google Scholar]
- McCullough L, Wu L, Haughey N, Liang X, Hand T, Wang Q, Breyer RM, Andreasson K. Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia. J Neurosci. 2004;24:257–268. doi: 10.1523/JNEUROSCI.4485-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGiff JC, Carroll MA. Cytochrome P450-dependent arachidonate metabolites, renal function and blood pressure regulation. Adv Prostaglandin Thromboxane Leukot Res. 1991;21B:675–682. [PubMed] [Google Scholar]
- Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochemical Pharmacology. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Fride E, Di Marzo V. Endocannabinoids. European Journal of Pharmacology. 1998;359:1–18. doi: 10.1016/s0014-2999(98)00649-9. [DOI] [PubMed] [Google Scholar]
- Medhora M, Harder D. Functional role of epoxyeicosatrienoic acids and their production in astrocytes: approaches for gene transfer and therapy (review) International Journal of Molecular Medicine. 1998;2:661–669. doi: 10.3892/ijmm.2.6.661. [DOI] [PubMed] [Google Scholar]
- Medhora M, Narayanan J, Harder D. Dual regulation of the cerebral microvasculature by epoxyeicosatrienoic acids. Trends in Cardiovascular Medicine. 2001;11:38–42. doi: 10.1016/s1050-1738(01)00082-2. [DOI] [PubMed] [Google Scholar]
- Miettinen S, Fusco FR, Yrjanheikki J, Keinanen R, Hirovonen T, Roivainen R, Narhi M, Hokfelt T, Koistinaho J. Spreading depression and focal brain ischemia induce cyclooxygenase-2 in cortical neurons through N-methyl-D-aspartic acid-receptors and phospholipase A(2) Proc Natl Acad Sci U S A. 1997;94:6500–6505. doi: 10.1073/pnas.94.12.6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minghetti L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J Neuropathol Exp Neurol. 2004;63:901–910. doi: 10.1093/jnen/63.9.901. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Lenzlinger PM, Hans V, Stahel P, Csuka E, Ammann E, Stocker R, Trentz O, Kossmann T. Production of cytokines following brain injury: beneficial and deleterious for the damaged tissue. Molecular Psychiatry. 1997;2:133–136. doi: 10.1038/sj.mp.4000227. [DOI] [PubMed] [Google Scholar]
- Nadjar A, Tridon V, May MJ, Ghosh S, Dantzer R, Amedee T, Parnet P. NFkappaB activates in vivo the synthesis of inducible Cox-2 in the brain. J Cereb Blood Flow Metab. 2005;25:1047–1059. doi: 10.1038/sj.jcbfm.9600106. [DOI] [PubMed] [Google Scholar]
- Nagayama M, Niwa K, Nagayama T, Ross ME, Iadecola C. The cyclooxygenase-2 inhibitor NS-398 ameliorates ischemic brain injury in wild-type mice but not in mice with deletion of the inducible nitric oxide synthase gene. Journal of Cerebral Blood Flow & Metabolism. 1999;19:1213–1219. doi: 10.1097/00004647-199911000-00005. [DOI] [PubMed] [Google Scholar]
- Nakai M, Qin Z, Wang Y, Chase TN. NMDA and non-NMDA receptor-stimulated IkappaB-alpha degradation: differential effects of the caspase-3 inhibitor DEVD.CHO, ethanol and free radical scavenger OPC-14117. Brain Res. 2000;859:207–216. doi: 10.1016/s0006-8993(00)01959-4. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Imamura S, Miyanohara O, Kawaguchi K, Chen J, Graham S. Cyclooxygenase-2 inhibition decreases infarction after temporary focal ischemia in rats. Soc Neurosci Abs. 1997;23:849. [Google Scholar]
- Nakayama M, Uchimura K, Zhu RL, Nagayama T, Rose ME, Stetler RA, Isakson PC, Chen J, Graham SH. Cyclooxygenase-2 inhibition prevents delayed death of CA1 hippocampal neurons following global ischemia. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:10954–10959. doi: 10.1073/pnas.95.18.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizaki T, Nomura T, Matsuoka T, Enikolopov G, Sumikawa K. Arachidonic acid induces a long-lasting facilitation of hippocampal synaptic transmission by modulating PKC activity and nicotinic ACh receptors. Molecular Brain Research. 1999a;69:263–272. doi: 10.1016/s0169-328x(99)00117-5. [DOI] [PubMed] [Google Scholar]
- Nishizaki T, Nomura T, Matsuoka T, Tsujishita Y. Arachidonic acid as a messenger for the expression of long-term potentiation. Biochem Biophys Res Comm. 1999b;254:446–449. doi: 10.1006/bbrc.1998.9961. [DOI] [PubMed] [Google Scholar]
- Node K, Huo YQ, Ruan XL, Yang BC, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–1279. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogawa S, Fangyi Z, Ross ME, Iadecola C. Cyclo-oxygenase-2 gene expression in neurons contributes to ischemic brain damage. J Neuroscience Res. 1997;17:2746–2755. doi: 10.1523/JNEUROSCI.17-08-02746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novelli A, Reilly J, Lysko P, Henneberry R. Glutamate becomes neurotoxic via the N-methyl-D-aspartate receptor when intracellular energy levels are reduced. Brain Res. 1988;451:205–212. doi: 10.1016/0006-8993(88)90765-2. [DOI] [PubMed] [Google Scholar]
- Nurmi A, Lindsberg PJ, Koistinaho M, Zhang W, Juettler E, Karjalainen-Lindsberg ML, Weih F, Frank N, Schwaninger M, Koistinaho J. Nuclear factor-kappaB contributes to infarction after permanent focal ischemia. Stroke. 2004;35:987–991. doi: 10.1161/01.STR.0000120732.45951.26. [DOI] [PubMed] [Google Scholar]
- O'Neill GP, Ford-Hutchinson AW. Expression of mRNA for cyclooxygenase-1 and cyclooxygenase-2 in human tissues. FEBS Letters. 1993;330:156–160. doi: 10.1016/0014-5793(93)80263-t. [DOI] [PubMed] [Google Scholar]
- Ogorochi T, Narumiya S, Mizuno N, Yamashita K, Miyazaki H, Hayaishi O. Regional distribution of prostaglandins D2, E2, and F2 alpha and related enzymes in postmortem human brain. J Neurochem. 1984;43:71–82. doi: 10.1111/j.1471-4159.1984.tb06680.x. [DOI] [PubMed] [Google Scholar]
- Oka A, Takashima S. Induction of cyclo-oxygenase 2 in brains of patients with Down's syndrome and dementia of Alzheimer type: specific localization in affected neurones and axons. Neuroreport. 1997;8:1161–1164. doi: 10.1097/00001756-199703240-00020. [DOI] [PubMed] [Google Scholar]
- Oliw EH, Guengerich FP, Oates JA. Oxygenation of arachidonic acid by hepatic monooxygenases Isolation and metabolism of four epoxide intermediates. Journal of Biological Chemistry. 1982;257:3771–3781. [PubMed] [Google Scholar]
- Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R, Shohami E. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature. 2001;413:527–531. doi: 10.1038/35097089. [DOI] [PubMed] [Google Scholar]
- Parfenova H, Eidson TH, Leffler CW. Upregulation of COX-2 in cerebral microvascular endothelial cells by smooth muscle cell signals. Am J Physiol. 1997;273:C277–288. doi: 10.1152/ajpcell.1997.273.1.C277. [DOI] [PubMed] [Google Scholar]
- Pasinetti G, Aisen P. Cyclooxygenase-2 expression is increased in frontal cortex of Alzheimer's disease brain. J Neuroscience. 1998;87:319–324. doi: 10.1016/s0306-4522(98)00218-8. [DOI] [PubMed] [Google Scholar]
- Post A, Holsboer F, Behl C. Induction of NF-kappaB activity during haloperidol-induced oxidative toxicity in clonal hippocampal cells: suppression of NF-kappaB and neuroprotection by antioxidants. J Neurosci. 1998;18:8236–8246. doi: 10.1523/JNEUROSCI.18-20-08236.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad M, Dhillon H, Carbary T, Dempsey R, Scheff S. Enhanced phosphodiestric breakdown of phophotidylinositol bisphosphate after experimental brain injury. J Neurochem. 1994;63:773–776. doi: 10.1046/j.1471-4159.1994.63020773.x. [DOI] [PubMed] [Google Scholar]
- Quadros A, Patel N, Crescentini R, Crawford F, Paris D, Mullan M. Increased TNFalpha production and Cox-2 activity in organotypic brain slice cultures from APPsw transgenic mice. Neuroscience Letters. 2003;353:66–68. doi: 10.1016/j.neulet.2003.08.076. [DOI] [PubMed] [Google Scholar]
- Resnick D, Graham S, Dixon C, Marion D. Role of cyclooxygenase 2 in acute spinal cord injury. J Neurotrauma. 1998;15:1005–1013. doi: 10.1089/neu.1998.15.1005. [DOI] [PubMed] [Google Scholar]
- Richter M, Weiss M, Weinberger I, Furstenberger G, Marian B. Growth inhibition and induction of apoptosis in colorectal tumor cells by cyclooxygenase inhibitors. Carcinogenesis. 2001;22:17–25. doi: 10.1093/carcin/22.1.17. [DOI] [PubMed] [Google Scholar]
- Rimpilainen J, Pokela M, Kiviluoma K, Anttila V, Vainionpaa V, Hirvonen J, Ohtonen P, Mennander A, Remes E, Juvonen T. Leukocyte filtration improves brain protection after a prolonged period of hypothermic circulatory arrest: A study in a chronic porcine model. Journal of Thoracic and Cardiovascular Surgery. 2000;120:1131–1141. doi: 10.1067/mtc.2000.111050. [DOI] [PubMed] [Google Scholar]
- Rouzer CA, Jacobs AT, Nirodi CS, Kingsley PJ, Morrow JD, Marnett LJ. RAW264.7 cells lack prostaglandin-dependent autoregulation of tumor necrosis factor-alpha secretion. Journal of Lipid Research. 2005;46:1027–1037. doi: 10.1194/jlr.M500006-JLR200. [DOI] [PubMed] [Google Scholar]
- Sacco S, Agnello D, Sottocorno M, Lozza G, Monopoli A, Villa P, Ghezzi P. Nonsteroidal anti-inflammatory drugs increase tumor necrosis factor production in the periphery but not in the central nervous system in mice and rats. Journal of Neurochemistry. 1998;71:2063–2070. doi: 10.1046/j.1471-4159.1998.71052063.x. [DOI] [PubMed] [Google Scholar]
- Sanfeliu C, H A, P AJ. Exposure to N-methyl-D-aspartate increases release of arachidonic acid in primary cultures of rat hippocampal neurons and not in astrocytes. Brain Research. 1990;526:241–248. doi: 10.1016/0006-8993(90)91228-9. [DOI] [PubMed] [Google Scholar]
- Sanz O, Estrada A, Ferrer I, Planas AM. Differential cellular distribution and dynamics of HSP70, cyclooxygenase-2, and c-Fos in the rat brain after transient focal ischemia or kainic acid. Neuroscience. 1997;80:221–232. doi: 10.1016/s0306-4522(97)00089-4. [DOI] [PubMed] [Google Scholar]
- Sanzgiri RP, Araque A, Haydon PG. Prostaglandin E(2) stimulates glutamate receptor-dependent astrocyte neuromodulation in cultured hippocampal cells. Journal of Neurobiology. 1999;41:221–229. [PubMed] [Google Scholar]
- Saper CB, Breder CD. Endogenous pyrogens in the CNS: role in the febrile response. Prog Brain Res. 1992;93:419–428. 428–419. doi: 10.1016/s0079-6123(08)64587-2. discussion. [DOI] [PubMed] [Google Scholar]
- Sapirstein A, Saito H, Texel SJ, Samad TA, O'Leary E, Bonventre JV. Cytosolic phospholipase A2alpha regulates induction of brain cyclooxygenase-2 in a mouse model of inflammation. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1774–1782. doi: 10.1152/ajpregu.00815.2004. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Kitagawa K, Sugiura S, Omura-Matsuoka E, Tanaka S, Yagita Y, Okano H, Matsumoto M, Hori M. Implication of cyclooxygenase-2 on enhanced proliferation of neural progenitor cells in the adult mouse hippocampus after ischemia. Journal of Neuroscience Research. 2003;72:461–471. doi: 10.1002/jnr.10595. [DOI] [PubMed] [Google Scholar]
- Scali C, Giovannini MG, Prosperi C, Bellucci A, Pepeu G, Casamenti F. The selective cyclooxygenase-2 inhibitor rofecoxib suppresses brain inflammation and protects cholinergic neurons from excitotoxic degeneration in vivo. Neuroscience. 2003;117:909–919. doi: 10.1016/s0306-4522(02)00839-4. [DOI] [PubMed] [Google Scholar]
- Scali C, Prosperi C, Vannucchi MG, Pepeu G, Casamenti F. Brain inflammatory reaction in an animal model of neuronal degeneration and its modulation by an anti-inflammatory drug: implication in Alzheimer's disease. European Journal of Neuroscience. 2000;12:1900–1912. doi: 10.1046/j.1460-9568.2000.00075.x. [DOI] [PubMed] [Google Scholar]
- Schilter B, Andersen MR, Acharya C, Omiecinski CJ. Activation of cytochrome P450 gene expression in the rat brain by phenobarbital-like inducers. Journal of Pharmacology & Experimental Therapeutics. 2000;294:916–922. [PubMed] [Google Scholar]
- Schilter B, Omiecinski CJ. Regional distribution and expression modulation of cytochrome P-450 and epoxide hydrolase mRNAs in the rat brain. Molecular Pharmacology. 1993;44:990–996. [PubMed] [Google Scholar]
- Schramm M, Eimerl S, C E. Serum and depolarizing agents cause acute neurotoxicity in cultured cerebellar granule cells: Role of the glutamate receptor responsive to N-methyl-D-aspartate. Proc Natl Acad Sci USA. 1990;87:1193–1197. doi: 10.1073/pnas.87.3.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibert K, Zhang Y, Leahy K, Hauser S, Masferrer J, Perkins W, Lee L, Isakson P. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc Natl Acad Sci USA. 1994;91:12013–12017. doi: 10.1073/pnas.91.25.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevanian A, Kim E. Phospholipase A2 dependent release of fatty acids from peroxidized membranes. J Free Radical Biol Med. 1985;1:263–271. doi: 10.1016/0748-5514(85)90130-8. [DOI] [PubMed] [Google Scholar]
- Shapira Y, Shohami E, Sidi A, Soffer D, Freeman S, Cotev S. Experimental closed head injury in rats: mechanical, pathophysiologic, and neurologic properties. Crit Care Med. 1988;16:258–265. doi: 10.1097/00003246-198803000-00010. [DOI] [PubMed] [Google Scholar]
- Shaw KN, Commins S, O'Mara SM. Deficits in spatial learning and synaptic plasticity induced by the rapid and competitive broad-spectrum cyclooxygenase inhibitor ibuprofen are reversed by increasing endogenous brain-derived neurotrophic factor. Eur J Neurosci. 2003;17:2438–2446. doi: 10.1046/j.1460-9568.2003.02643.x. [DOI] [PubMed] [Google Scholar]
- Shohami E, Cotev S, Shapira Y. The role of eicosanoids in the development of delayed brain damage. Journal of Basic & Clinical Physiology & Pharmacology. 1990;1:203–206. doi: 10.1515/jbcpp.1990.1.1-4.203. [DOI] [PubMed] [Google Scholar]
- Shohami E, Shapira Y, Sidi A, Cotev S. Head injury induces increased prostaglandin synthesis in rat brain. Journal of Cerebral Blood Flow & Metabolism. 1987;7:58–63. doi: 10.1038/jcbfm.1987.8. [DOI] [PubMed] [Google Scholar]
- Slanina KA, Schweitzer P. Inhibition of cyclooxygenase-2 elicits a CB1-mediated decrease of excitatory transmission in rat CA1 hippocampus. Neuropharmacology. 2005;49:653–659. doi: 10.1016/j.neuropharm.2005.04.019. [DOI] [PubMed] [Google Scholar]
- Smith W, Dewitt D. Prostaglandin endoperoxide H synthases-1 and -2. Adv Immunology. 1996;62:167–215. doi: 10.1016/s0065-2776(08)60430-7. [DOI] [PubMed] [Google Scholar]
- Spielman L, Winger D, Ho L, Aisen PS, Shohami E, Pasinetti GM. Induction of the complement component C1qB in brain of transgenic mice with neuronal overexpression of human cyclooxygenase-2. Acta Neuropathologica. 2002;103:157–162. doi: 10.1007/s004010100447. [DOI] [PubMed] [Google Scholar]
- Stephenson DT, Lemere CA, Selkoe DJ, Clemens JA. Cytosolic phospholipase A2 (cPLA2) immunoreactivity is elevated in Alzheimer's disease brain. Neurobiology of Disease. 1996;3:51–63. doi: 10.1006/nbdi.1996.0005. [DOI] [PubMed] [Google Scholar]
- Strauss KI, Barbe MF, Marshall RM, Raghupathi R, Mehta S, Narayan RK. Prolonged cyclooxygenase-2 induction in neurons and glia following traumatic brain injury in the rat. J Neurotrauma. 2000a;17:695–711. doi: 10.1089/089771500415436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss KI, Marini AM. Cyclooxygenase-2 inhibition protects cultured cerebellar granule neurons from glutamate-mediated excitotoxic cell death. J Neurotrauma. 2002;19:627–638. doi: 10.1089/089771502753754091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss KI, Meagher RJ, Narayan RK. Hippocampal prostaglandin changes following TBI-treatment with COX2 inhibitors. Restor Neurol Neurosci. 2000b;16:263, P298. [Google Scholar]
- Strokin M, Sergeeva M, Reiser G. Role of Ca2+-independent phospholipase A2 and n-3 polyunsaturated fatty acid docosahexaenoic acid in prostanoid production in brain: perspectives for protection in neuroinflammation. Int J Dev Neurosci. 2004;22:551–557. doi: 10.1016/j.ijdevneu.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Stuhlmeier KM, Kao JJ, Bach FH. Arachidonic acid influences proinflammatory gene induction by stabilizing the inhibitor-kappaBalpha/nuclear factor-kappaB (NF-kappaB) complex, thus suppressing the nuclear translocation of NF-kappaB. Journal of Biological Chemistry. 1997;272:24679–24683. doi: 10.1074/jbc.272.39.24679. [DOI] [PubMed] [Google Scholar]
- Sugimoto Y, Shigemoto R, Namba T, Negishi M, Mizuno N, Narumiya S, Ichikawa A. Distribution of the messenger RNA for the prostaglandin E receptor subtype EP3 in the mouse nervous system. Neuroscience. 1994;62:919–928. doi: 10.1016/0306-4522(94)90483-9. [DOI] [PubMed] [Google Scholar]
- Tamatani M, Mitsuda N, Matsuzaki H, Okado H, Miyake S, Vitek MP, Yamaguchi A, Tohyama M, Grilli M, Memo M. A pathway of neuronal apoptosis induced by hypoxia/reoxygenation: roles of nuclear factor-kappaB and Bcl-2. Journal of Neurochemistry. 2000;75:683–693. doi: 10.1046/j.1471-4159.2000.0750683.x. [DOI] [PubMed] [Google Scholar]
- Tanaka E, Niiyama S, Sato S, Yamada A, Higashi H. Arachidonic acid metabolites contribute to the irreversible depolarization induced by in vitro ischemia. J Neurophysiol. 2003;90:3213–3223. doi: 10.1152/jn.00542.2003. [DOI] [PubMed] [Google Scholar]
- Tetsuka T, Baier LD, Morrison AR. Antioxidants inhibit interleukin-1-induced cyclooxygenase and nitric-oxide synthase expression in rat mesangial cells Evidence for post-transcriptional regulation. J Biol Chem. 1996;271:11689–11693. doi: 10.1074/jbc.271.20.11689. [DOI] [PubMed] [Google Scholar]
- Tocco G, Freire-Moar J, Schreiber SS, Sakhi SH, Aisen PS, Pasinetti GM. Maturational regulation and regional induction of cyclooxygenase-2 in rat brain: implications for Alzheimer's disease. Experimental Neurology. 1997;144:339–349. doi: 10.1006/exnr.1997.6429. [DOI] [PubMed] [Google Scholar]
- Tsubokura S, Watanabe Y, Ehara H, Imamura K, Sugimoto O, Kagamiyama H, Yamamoto S, Hayaishi O. Localization of prostaglandin endoperoxide synthase in neurons and glia in monkey brain. Brain Res. 1991;543:15–24. doi: 10.1016/0006-8993(91)91043-z. [DOI] [PubMed] [Google Scholar]
- Tzeng SF, Hsiao HY, Mak OT. Prostaglandins and cyclooxygenases in glial cells during brain inflammation. Curr Drug Targets Inflamm Allergy. 2005;4:335–340. doi: 10.2174/1568010054022051. [DOI] [PubMed] [Google Scholar]
- Uz T, Manev H. Circadian expression of pineal 5-lipoxygenase mRNA. Neuroreport. 1998;9:783–786. doi: 10.1097/00001756-199803300-00003. [DOI] [PubMed] [Google Scholar]
- Vassiliou E, Jing H, Ganea D. Prostaglandin E2 inhibits TNF production in murine bone marrow-derived dendritic cells. Cellular Immunology. 2003;223:120–132. doi: 10.1016/s0008-8749(03)00158-8. [DOI] [PubMed] [Google Scholar]
- Vaughan CW, Ingram SL, Connor MA, Christie MJ. How opioids inhibit GABA-mediated neurotransmission. Nature. 1997;390:611–614. doi: 10.1038/37610. [see comments] [DOI] [PubMed] [Google Scholar]
- Vollgraf U, Wegner M, Richter-Landsberg C. Activation of AP-1 and nuclear factor-kappaB transcription factors is involved in hydrogen peroxide-induced apoptotic cell death of oligodendrocytes. J Neurochem. 1999;73:2501–2509. doi: 10.1046/j.1471-4159.1999.0732501.x. [DOI] [PubMed] [Google Scholar]
- Wallace C, Lyford G, Worley P, Steward O. Differential intracellular sorting of immediate early gene mRNAs depends on signals in the mRNA sequence. J Neuroscience Res. 1998;18:26–35. doi: 10.1523/JNEUROSCI.18-01-00026.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton M, Sirimanne E, Williams C, Gluckman PD, Keelan J, Mitchell MD, Dragunow M. Prostaglandin H synthase-2 and cytosolic phospholipase A2 in the hypoxic-ischemic brain: role in neuronal death or survival? Molecular Brain Research. 1997;50:165–170. doi: 10.1016/s0169-328x(97)00181-2. [DOI] [PubMed] [Google Scholar]
- Weaver KD, Branch CA, Hernandez L, Miller CH, Quattrocchi KB. Effect of leukocyte-endothelial adhesion antagonism on neutrophil migration and neurologic outcome after cortical trauma. Journal of Trauma. 2000;48:1081–1090. doi: 10.1097/00005373-200006000-00014. [DOI] [PubMed] [Google Scholar]
- Wei EP, Kontos HA, Dietrich WD, Povlishock JT, Ellis EF. Inhibition by free radical scavengers and by cyclooxygenase inhibitors of pial arteriolar abnormalities from concussive brain injury in cats. Circulation Research. 1981;48:95–103. doi: 10.1161/01.res.48.1.95. [DOI] [PubMed] [Google Scholar]
- Westcott JY, Murphy RC, Stenmark K. Eicosanoids in human ventricular cerebrospinal fluid following severe brain injury. Prostaglandins. 1987;34:877–887. doi: 10.1016/0090-6980(87)90068-2. [DOI] [PubMed] [Google Scholar]
- Whalen MJ, Carlos TM, Kochanek PM, Clark RS, Heineman S, Schiding JK, Franicola D, Memarzadeh F, Lo W, Marion DW, Dekosky ST. Neutrophils do not mediate blood-brain barrier permeability early after controlled cortical impact in rats. Journal of Neurotrauma. 1999;16:583–594. doi: 10.1089/neu.1999.16.583. [DOI] [PubMed] [Google Scholar]
- Yakovlev AG, Knoblach SM, Fan L, Fox GB, Goodnight R, Faden AI. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. Journal of Neuroscience. 1997;17:7415–7424. doi: 10.1523/JNEUROSCI.17-19-07415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF. Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron. 1993;11:371–386. doi: 10.1016/0896-6273(93)90192-t. [DOI] [PubMed] [Google Scholar]
- Yermakova AV, Rollins J, Callahan LM, Rogers J, O'Banion MK. Cyclooxygenase-1 in human Alzheimer and control brain: quantitative analysis of expression by microglia and CA3 hippocampal neurons. Journal of Neuropathology & Experimental Neurology. 1999;58:1135–1146. doi: 10.1097/00005072-199911000-00003. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Kubota T, Krueger JM. A cyclooxygenase-2 inhibitor attenuates spontaneous and TNF-alpha-induced non-rapid eye movement sleep in rabbits. American Journal of Physiology - Regulatory Integrative & Comparative Physiology. 2003;285:R99–109. doi: 10.1152/ajpregu.00609.2002. [DOI] [PubMed] [Google Scholar]
- Yu Z, Huse LM, Adler P, Graham L, Ma J, Zeldin DC, Kroetz DL. Increased CYP2J expression and epoxyeicosatrienoic acid formation in spontaneously hypertensive rat kidney. Molecular Pharmacology. 2000a;57:1011–1020. [PubMed] [Google Scholar]
- Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circulation Research. 2000b;87:992–998. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- Zeldin DC, Liao JK. Reply: New role for epoxyeicosatrienoic acids as anti-inflammatory mediators. Trends in Pharmacological Sciences. 2000;21:127–128. doi: 10.1016/s0165-6147(00)01472-3. [DOI] [PubMed] [Google Scholar]
- Zeldin DC, Wei S, Falck JR, Hammock BD, Snapper JR, Capdevila JH. Metabolism of epoxyeicosatrienoic acids by cytosolic epoxide hydrolase: substrate structural determinants of asymmetric catalysis. Archives of Biochemistry & Biophysics. 1995;316:443–451. doi: 10.1006/abbi.1995.1059. [DOI] [PubMed] [Google Scholar]
- Zhang J, Rivest S. Anti-inflammatory effects of prostaglandin E2 in the central nervous system in response to brain injury and circulating lipopolysaccharide. Journal of Neurochemistry. 2001;76:855–864. doi: 10.1046/j.1471-4159.2001.00080.x. [DOI] [PubMed] [Google Scholar]
- Zou J, Crews F. CREB and NF-kappaB transcription factors regulate sensitivity to excitotoxic and oxidative stress induced neuronal cell death. Cell Mol Neurobiol. 2006;26:385–405. doi: 10.1007/s10571-006-9045-9. [DOI] [PMC free article] [PubMed] [Google Scholar]