

Abstract
Interest in the design of peptide-based fibrous materials is growing because it opens possibilities to explore fundamental aspects of peptide self-assembly and to exploit the resulting structures—for example, as scaffolds for tissue engineering. Here we investigate the assembly pathway of self-assembling fibers, a rationally designed α-helical coiled-coil system comprising two peptides that assemble on mixing. The dimensions spanned by the peptides and final structures (nanometers to micrometers), and the timescale over which folding and assembly occur (seconds to hours), necessitate a multi-technique approach employing spectroscopy, analytical ultracentrifugation, electron and light microscopy, and protein design to produce a physical model. We show that fibers form via a nucleation and growth mechanism. The two peptides combine rapidly (in less than seconds) to form sticky ended, partly helical heterodimers. A lag phase follows, on the order of tens of minutes, and is concentration-dependent. The critical nucleus comprises six to eight partially folded dimers. Growth is then linear in dimers, and subsequent fiber growth occurs in hours through both elongation and thickening. At later times (several hours), fibers grow predominantly through elongation. This kinetic, biomolecular description of the folding-and-assembly process allows the self-assembling fiber system to be manipulated and controlled, which we demonstrate through seeding experiments to obtain different distributions of fiber lengths. This study and the resulting mechanism we propose provide a potential route to achieving temporal control of functional fibers with future applications in biotechnology and nanoscale science and technology.
Introduction
The rational design of peptide-based fibers is an important and growing area in the fields of protein design, biophysics, and biomaterials. This is because peptide/protein design and engineering provide the acid test for our fundamental understanding of protein folding and assembly (1), and new biomaterials are an underpinning technology for applications such as three-dimensional cell culture and tissue engineering (2). As a consequence, a large number of designed fibrous biomaterials have been obtained, either by engineering of natural systems or by rational peptide and protein design (3–6). Because of the interest in amyloid-like structures and their ability to self-assemble into fibrous and gelling materials, much effort has been directed toward β-structured systems (7–9). However, in recent years, investigators have increasingly been exploring materials based on the α-helix (10,11).
For both α-helical and β-structure materials, the majority of these efforts have focused on how changes to peptide sequence influence the final morphology of the mature assemblies (7,11). Developing an understanding of the relationship between sequence and fiber structure, stability, morphology, and rheology is key, and will drive the rational design of new materials. However, a full understanding of such materials—and, moreover, our ability to control and manipulate them fully—requires an understanding of how and over what timescales they assemble. Although the assembly of natural proteins and synthetic polymers is a mature field (12), and kinetic studies of amyloid formation by natural sequences are becoming established (13,14), few such studies of assembly of de novo designed biomaterials have been reported (15–17).
In a previous work (18), we described the design and experimental characterization of a self-assembling fiber (SAF) system that comprises two complementary, de novo designed, leucine-zipper peptides, each 28 residues long. The leucine zipper is a natural protein-folding domain in which short sequences with a characteristic heptad repeat of polar (P), hydrophobic (H), and leucine (L) residues, HPPLPPP, fold to form amphipathic α-helices; two such helices associate to bury their hydrophobic faces. Unlike natural leucine zippers, where the hydrophobic faces align in-register to yield blunt-ended dimers (19), the SAF peptides were engineered to form a sticky ended heterodimer (18). With the sticky ends designed to interact, the heterodimers are directed to associate and propagate long noncovalently linked α-helical coiled-coil fibrils. Consistent with this, circular dichroism (CD) and linear dichroism (LD) spectroscopy (18,20,21), and x-ray fiber diffraction (XRD) (22) confirm that the SAFs comprise α-helical coiled coils aligned with the long axis of the fibers. Negative-stain transmission electron microscopy (TEM) reveals long (tens of microns), straight fibers that are curiously thicker at ∼50 nm than expected for a single coiled-coil dimer (∼2 nm) (18,22,23). Further, XRD and high-resolution TEM indicate considerable crystalline order in the fibers, with the peptides aligned both longitudinally and laterally, and the coiled coils bundled hexagonally (22). Finally, and consistent with this high level of internal order, fluorescence light microscopy (LM) demonstrates that, at least in its late stages, SAF assembly is polar (24).
Since the development of the original SAF design (18), we have reported redesigns with improved stability (23) and altered morphologies, including thicker (23), thinner (25), kinked, branched, and segmented fibers (26–29). We have also decorated the basic SAF structures (28) and used them as templates for chemical ligation (30) and biomineralization (31). Most recently, the SAF system was altered to produce hydrogelating variants that have been taken forward for use in three-dimensional tissue culture (32).
In all of these cases, we focused on the matured SAF samples, which are prepared by incubating mixtures of SAF peptides, 100 μM in each peptide, at pH 7.4, 20°C for 24 h. Here we present an exploration of the full pathway for assembly of what we term the second-generation SAF design, which comprises peptides SAF-p1 and SAF-p2a (23), under these standard conditions. The time period covered spans tens of seconds to many hours after mixing, leading up to the fully matured fibers. Unfortunately, there are no assays for helical systems comparable to the Congo-Red and Thioflavin-T assays for the assembly and structure of β-sheet-based fibers (33,34). This necessitated a multi-technique approach to probing SAF folding and assembly, and the development of more general tools.
To provide a starting hypothesis to guide our experiments, we discuss the possible phases en route to the assembled, highly ordered α-helical fibers (Fig. 1). The first stage was to probe the initial state of the peptides individually, which we investigated by CD spectroscopy and analytical ultracentrifugation (AUC). The next step was to determine whether assembly proceeded directly from the individual peptides (Fig. 1 B), or whether any spontaneous folding or oligomerization occurred on mixing (Fig. 1 A). This presented some difficulties due to rapid fiber formation when the peptides were mixed, which we overcame with the use of peptide engineering. The third phase covered the early stages of self-assembly, which may be energetically favorable from the start (Fig. 1 C) or may involve an energy barrier and hence require a nucleation process (Fig. 1 D). The final part of the process was the growth phase. Here we investigated the elongation and thickening of the fibers to ascertain whether the latter was due to the association of thin fibers (Fig. 1 E) or the addition of monomers to the surface (Fig. 1 F). To follow the last two stages of assembly, we used a combination of TEM, LM, CD, LD, and NMR spectroscopy to obtain direct kinetic information about fiber formation.
Figure 1.
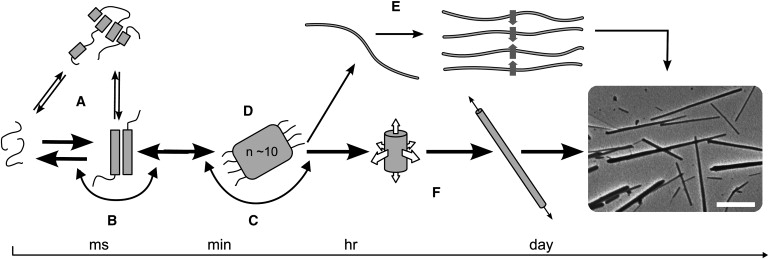
Schematic of possible pathways for fiber formation. (A) The two peptides may interact to form various oligomers that are competent for fibrillogenesis, or (B) the individual peptides may be competent for assembly. (C) Onward assembly may be immediately energetically favorable, or (D) further assembly may only be favorable once a critical nucleus has formed. (E) Fibers may thicken via the bundling of fibrils, or (F) the addition of material in both radial and longitudinal directions yields mature fibers as shown in the electron micrograph (scale bar: 5 μm). The pathway determined from the experiments described herein is shown in bold.
By piecing together information from this range of techniques spanning the various timescales and length scales of the assembly process, we present a kinetic pathway for self-assembly that essentially follows the central mechanism depicted in Fig. 1 (bold).
Materials and Methods
Peptide synthesis and purification
Peptides were synthesized using standard Fmoc-based, solid-phase peptide synthesis and purified by reverse-phase high-performance liquid chromatography as described previously (24).
CD spectroscopy
Measurements were made using a Jasco J815 spectropolarimeter fitted with a Peltier temperature controller. Peptide solutions were prepared in 3-(N-Morpholino)propanesulfonic acid (MOPS) buffer at 20°C, pH adjusted to 7.4 using sodium hydroxide (10 mM MOPS/NaOH, pH 7.4), and measured in a 1 mm pathlength quartz cuvette. Time series of CD spectra were interpreted by fitting to a linear combination of the initial and final spectra (see the Supporting Material).
LD spectroscopy
Couette flow experiments were performed using a Jasco J815 spectropolarimeter (Jasco, Great Dunmow, UK) modified for LD measurements. For these experiments, 60 μL of 100 μM SAF peptides were mixed immediately before measurements were carried out in a microvolume couette cell with a 0.5 mm pathlength at 21°C and a cell rotation speed of 2000 rpm.
TEM
For TEM, 200 μL of 100 μM SAF peptides in 10 mM MOPS buffer, pH 7.4, were allowed to assemble at 20°C. At each time point, 5 μL were applied to carbon-coated copper TEM grids and stained with uranyl acetate. Images were obtained using a JEOL JSM 1200EX TEM with an accelerating voltage of 120 keV, and at magnifications of 60,000× and 120,000×.
NMR spectroscopy
All 1H NMR data were collected on a Varian VNMRS 600 MHz spectrometer equipped with Z-pulsed field gradients and a triple-resonance, salt-tolerant cryoprobe. The assembly of a 650 μL sample of 100 μM SAF peptides in 10 mM MOPS buffer, pH 7.4, in 99.9% D2O at 20°C was observed by means of the 1D1H spectrum over 24 h.
LM
Confocal images were obtained using a Perkin Elmer Ultraview ERS 6FE confocal system (Perkin Elmer, Waltham, MA) attached to a Leica DMI6000 inverted epifluorescence microscope (Leica, Milton Keynes, UK). Samples were initially mixed in Eppendorf tubes and immediately transferred to sealed optical cells. Images were recorded in situ using an excitation wavelength of 488 nm.
AUC
Sedimentation equilibrium experiments were conducted at 20°C in a Beckman-Optima XL-I analytical ultracentrifuge (Beckman Coulter, High Wycombe, UK) fitted with an An-60 Ti rotor.
Results and Discussion
Overview of SAF assembly
We begin with an overview of the behavior of the system with time as probed by three main methods: CD spectroscopy, TEM, and LM. These are developed in the main text and buttressed with additional data from complementary techniques, including LD and NMR spectroscopy, and AUC, as given in the Supporting Material.
To initiate folding and assembly, 200 μM stock solutions of SAF-p1 and SAF-p2a were mixed and incubated at 20°C. The final solution conditions were 10 mM MOPS, pH 7.4, and 100 μM in each peptide. This was done simultaneously for two identical samples in 1 mm quartz cuvettes, which were sampled or measured over 24 h as follows: Samples were taken from one cuvette, applied to TEM grids, and stained with uranyl acetate solution (1% w/v) for visualization. The other cuvette was used directly to measure the CD spectra as a function of time. In parallel experiments with a 1% spike of fluorescein-labeled SAF-p1, but otherwise identical conditions, fibrillogenesis was assessed in situ by confocal fluorescence LM. Data from these experiments recorded at similar time points are presented in Fig. 2.
Figure 2.
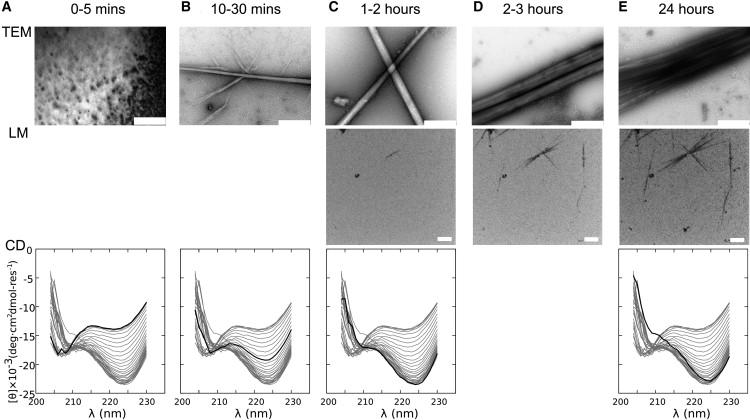
Overview of the SAF folding and assembly probed by TEM, LM, and CD spectroscopy. (A) t = 0–5 min; no change in the CD spectrum, and no fibers by TEM or LM (0% material in fibers). (B) t = 10–30 min; the CD spectra begin to change, and small fibers become visible by TEM but not LM (5–50% material in fibers). (C) t = 1–2 h; the CD spectra continue to change, TEM shows an increase in number and thickness of the fibers, and fibrous material begins to appear by fluorescence microscopy (55–65% material in fibers). (D) t = 2–3 h; fibers are unchanged by TEM (which is insensitive to fiber length), but the LM reports elongation (65–70% material in fibers). (E) t = 24 h; the CD spectrum reaches equilibrium, the TEM shows mature striated fibers, and growth is complete in the LM (70% material in fibers). Scale bar: 500 nm for TEM and 10 μm for LM; heavy lines indicate the CD spectrum for the corresponding time point. The percentages of material in fibers given in brackets were estimated as follows: the final figure of 70% came from estimates of material that remained soluble 1), after matured fibers were pelleted by AUC, and 2), the remaining NMR signal after completion of fibrillogenesis (see the Supporting Material for details regarding both of these experiments). All other figures came from the percentage completion of fibrillogenesis from the time-resolved CD data. The kinetic CD experiments were repeated three times and provided similar qualitative results, indicating a batch-to-batch variation in rates and lag times of ∼10–15%. The LM and TEM experiments were repeated multiple times (more than six) and gave qualitatively similar results.
In the first 5 min after mixing, neither the TEM nor the LM images showed fibers (Fig. 2 A). Nevertheless, from the earliest measurable time point (∼30 s), and for the extent of these first 5 min, the CD spectra showed a constant pronounced double minima at ∼208 and ∼222 nm indicative of significant α-helical content. We estimate this signal to be approximately one-third of that possible for fully folded SAF peptides (35). By TEM, the first fibers were visible at 10–30 min (Fig. 2 B, top). These were thinner than the matured fibers that developed over the next 24 h (Fig. 2, C–E, top), but were otherwise typical of those we have reported elsewhere (18,22,23). By LM, fibers were not clearly visible until 1–2 h, and because of the limits of this technique, we only observed growth along the long fiber axes (Fig. 2, D and E, middle). However, and of importance, this technique afforded direct visualization of fiber growth in solution.
The development of the CD signal over time is noteworthy because it is complex and potentially informative. The spectra obtained immediately after mixing were consistent with a partially folded α-helical system. Within the first ∼5 min, the CD spectra changed very little (Fig. 2 A), indicative of a lag phase. After this, although the spectra became more helical (as judged by increased intensity at 222 nm), they also become distorted from the archetypal α-helical spectrum, with the intensity at 208 nm decreasing and the 222 nm band red shifting (Fig. 2, B–E, bottom). These changes correlate with 1), the appearance of fibers in TEM (Fig. 2, B–E, top); and 2), the formation of elongated helical structures as judged by LD spectroscopy (Fig. S1 B). We previously noted the distorted spectrum for 24-h matured SAFs and attributed it to scattering (36,37). As detailed in Section S2, we explored this phenomenon in more detail and now assign it to chiral scattering from the extended α-helices bundled in the fibers (22). This effect, which is observed for other helical fibers (38,39), gives a wavelength-dependent bias toward positive CD signals. We confirmed this hypothesis by collecting more of the scattered light, which revealed that the fiber spectrum converged to the expected mixed α-helical and random-coil spectrum. The final mature fiber spectrum was a sum of contributions from the remaining soluble unfolded peptides, the α-helical peptide incorporated into fibers, and the chiral scattering from the fibers.
Several conclusions can be drawn from these overview data: 1), there is an initial α-helical folding upon mixing that does not produce fibers visible by TEM or LM; 2), there is a lag time, or a period in which the system does not change spectroscopically (CD or LD) or visibly; 3), there is synchronicity between subsequent changes in the CD spectra and the appearance of fibers in TEM; and 4), once visible fibers appear, they thicken and lengthen over a period of 24 h. The lag phase implies an activation energy barrier for assembly, and that an energetically unfavorable nucleus must form before further association becomes favorable.
Species formed upon initial mixing
CD spectra for 100 μM samples of the individual SAF peptides showed SAF-p1 to be slightly more folded than SAF-p2a (Fig. 3 A); however, the degree of helicity in both cases is marginal and consistent with the essentially unfolded spectra for natural leucine-zipper peptides (40). AUC experiments on these individual peptides indicated that SAF-p1 forms a homodimer at 100 μM, and that SAF-p2a is in a monomer-dimer equilibrium at 100 μM (Fig. S3).
Figure 3.
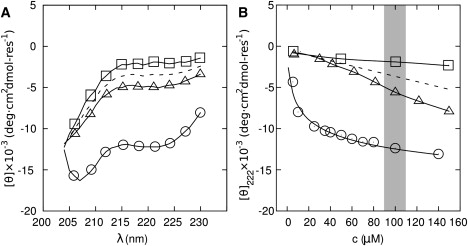
Initial folding of SAF-p1 and SAF-p2a upon mixing. (A) CD spectra of the SAF peptides: 100 μM SAF-p1 (▵), 100 μM SAF-p2a (□), average of these spectra (dashed line); and for the mixture 100 μM in each peptide at t = 0 (○). (B) Concentration dependence of the CD signal at 222 nm for SAF-p1 (▵), SAF-p2a (□), the average of these signals (dashed line), and the initial mixture (○). The curve for the mixture shows a fit to a monomer-dimer equilibrium.
Mixing the peptides led to a substantial increase in α-helical signal (Fig. 3 A) that was concentration-dependent (Fig. 3 B). These data fitted a monomer-dimer equilibrium with a KD of ∼7 μM. Therefore, in the 100 μM region (which is of interest as our standard fiber-forming condition) the equilibrium is substantially on the folded side and does not change rapidly. When extrapolated to a fully folded spectrum, the shape and size of the CD signal indicate that the dimeric species is ∼45% folded as an α-helix (35).
Equilibrium experiments cannot be performed on SAF mixtures as they form fibers; however, using AUC, we investigated the peptides that remained in solution after fibrillogenesis reached equilibrium. At high speeds, the fibers sedimented completely, leaving soluble material at ∼30 μM in each peptide (Fig. S4 A). We confirmed this proportioning by following the loss of signals by 1H NMR spectra with time (Fig. S5). As the peptides assembled into fibers, their signals broadened, leaving sharp signals for the smaller, soluble peptides. By AUC, these remaining species had a molecular mass of 5230 Da (Fig. S4 B), which compares with predicted masses of 3250 Da for the average monomer, and 6500 Da for the heterodimer. Moreover, these data fitted a monomer-dimer equilibrium of KD 7–10 μM, consistent with the above data.
Together, these data support the conclusion that when SAF peptides are mixed, they initially form a monomer-dimer equilibrium, which tends toward 45% helical. This fits with the original design hypothesis, indicating the formation of a sticky ended heterodimer. To identify which ends of each peptide interact initially, however, we needed a sharper structural probe.
Nature of the initial species
To dissect the SAF-p1/SAF-p2a interaction, we used proline-scanning mutagenesis (41). For each peptide, four single-point mutants were made in which leucine residues of the hydrophobic helix-helix interface were systematically replaced by proline (Fig. 4 A and Fig. S6). Proline disrupts the regular i→i + 4 α-helical hydrogen-bonding network, and therefore destabilizes local helix formation (42). On this basis, we reasoned that 1), proline substitutions in regions involved in initial helix formation would reduce the early time CD signal; 2), prolines located in the disordered regions would not affect the CD signal significantly; and 3), all mutations should disrupt onward fiber assembly. Thus, these mutations should identify the initially helical parts of each peptide, and combinations of them should yield isolable species with which to probe the initial interaction more fully.
Figure 4.
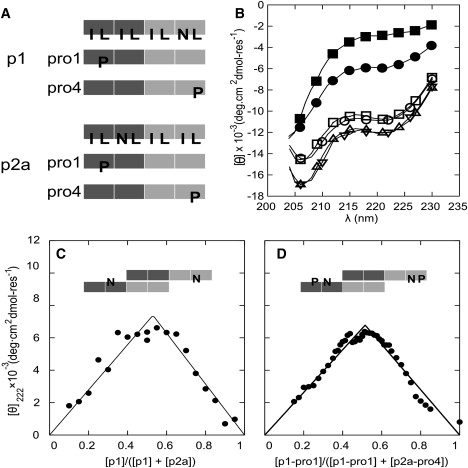
Proline-scanning mutagenesis. (A) Schematics of the proline-mutant peptides. The light and dark blocks represent oppositely charged heptad repeats. In the uppermost schematics, I, L, and N indicate isoleucine, leucine, and asparagine residues at the H-type sites in the HPPHPPP repeats of the parent sequences. In the remaining schematics, P highlights proline residues that have replaced specific L residues, and all of the other H sites remain unchanged. (B) CD spectra from mixtures of proline variants and parent peptides: p1-Pro1:p2a (■), p1:p2a-Pro4 (•), p1:p2a (○), p1-Pro4:p2a (▵), p1:p2a-Pro1, (▿), and p1-Pro4:p2a-Pro1 (□). (C) Job plot for mixtures of SAF-p1 and SAF-p2a, showing a 1:1 binding stoichiometry. (D) Similar Job plot for mixtures of SAF-p1-pro4 and SAF-p2a-pro1, showing an unaltered binding stoichiometry. In both C and D, the total peptide concentration was kept at 200 μM, and lines are fits to the data as described in the Supporting Material.
Each mutant was mixed with the partnering parent peptide (Fig. S7 and Fig. S8). From these eight experiments, two combinations—SAF-p1-Pro4 (proline in the fourth heptad) + SAF-p2a, and SAF-p1 + SAF-p2a-Pro1 (proline in the first heptad)—gave spectra that closely resembled the initial spectrum for the mixture of parent SAF-p1 and SAF-p2a (Fig. 4 B). In marked contrast, all other mixtures showed either no folding or a significantly reduced α-helical structure (Fig. S7 and Fig. S8). Mixing SAF-p1-Pro4 + SAF-p2a-Pro1 also produced the same level of folding as the initial mixture of the parent peptides (Fig. 4 B).
The significance of the competent pairings is that they have the potential to yield sticky ended heterodimers with the largest uninterrupted hydrophobic overlap (Fig. 4, A and D). In the SAF design rationale, the sticky ended heterodimer is specified by interhelical charge-charge and hydrogen-bonded interactions. The latter occur between asparagine residues offset in different halves of each sequence (Fig. 4 A) at what would otherwise be canonical hydrophobic core sites, i.e., the H site of the HPPLPPP sequence repeat. While asparagine residues at the first hydrophobic site of the repeat help to specify parallel dimeric species over alternative oligomer states (43), such inclusions within the hydrophobic core are destabilizing (44). Thus, the halves of the SAF peptides with asparagines are expected to interact less strongly than those without them. This is precisely what the proline scan highlights: proline is more damaging in the non-asparagine-containing halves of the peptides. This confirms the original design hypothesis for the sticky ended dimer, and further ties down the initial interactions made en route to fibers. To probe this in more detail, we conducted additional experiments using the three competently folded, proline-containing pairs described above.
First, Job plots derived from CD data for pairings of SAF-p1 and SAF-p2a, and of SAF-p1-pro4 and SAFp2a-pro1 (Fig. 4, C and D) showed a 1:1 stoichiometry of association for both pairings. Second, AUC indicated that though the correctly folded pairs formed small species, they did not associate further into fibers (Fig. S10). The three mixtures showed concentration-dependent averaged molecular masses ranging from 5300 Da at 30 μM to 11,200 Da at 100 μM. Again, at low concentrations the interaction matched that seen by CD spectroscopy, implying helical dimers. The further increases in mass, however, indicate a weaker interaction at higher concentrations. Attempts to separate dimerization and the additional association by adding salt or by changing the pH only resulted in a reduction in overall helicity (Fig. S7 b).
Thus, the initial species formed upon mixing SAF-p1 and SAF-p2a is a helical dimer cemented through the hydrophobic heptads. This associates further, albeit weakly, at higher concentrations. These additional interactions may suggest a back-to-back assembly mechanism of dimers that could potentially influence or even direct fiber elongation and thickening, as described below.
Lag phase
As mentioned above, there is a short period after mixing is completed, and before fibrillogenesis begins, during which the CD signal is unchanged. To illustrate and quantify this, we analyzed the series of CD spectra from Fig. 2, assuming a two-state model (Fig. 5 A and expanded in Fig. S11). Essentially, to estimate the fraction completion of fibrillogenesis at any given time, the relevant spectrum was fitted as a linear combination of the first spectrum and that acquired 24 h after mixing (these being assumed to represent the extremes of the initial and final (equilibrium) spectra, respectively).
Figure 5.
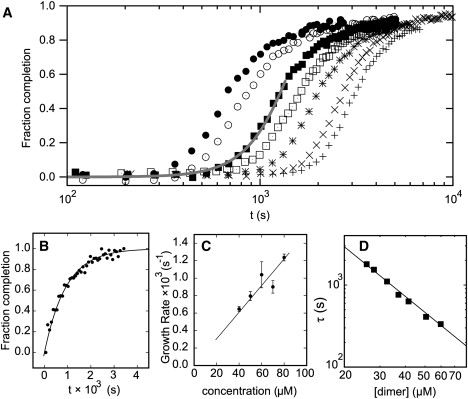
Lag phase, seeded growth, and nature of the nucleus. (A) Dependence of kinetics on initial peptide concentration at 65 μM (+), 70 μM (×), 80 μM (∗), 90 μM (□), 100 μM (■), 120 μM (○), and 140 μM (•) in each peptide. The gray line shows a model fit for the early time points. (B) Fraction completion as a function of time for a seeded sample 80 μM (+); the fit (solid line) is to a single exponential. (C) Fiber growth rate after seeding as a function of initial concentration of peptide. (D) Lag time as a function of the concentration of partially helical dimers (determined from Fig. 3B).
For the standard mixture, 100 μM in each peptide, the plot was sigmoidal with a distinct lag of ∼103 s (∼15 min) before any sharp changes were observed. This is characteristic of nucleation. Because CD spectroscopy reports only on the formation of secondary structure, we confirmed this finding using LD spectroscopy to measure the kinetics of formation of high aspect ratio species (Fig. S1 B), and NMR to measure the rate of loss of small oligomers from solution (Fig. S5 C). Both techniques provided kinetic traces that matched well with those from CD spectroscopy, indicating that the latter is a reliable measure of fiber formation kinetics.
Seeded growth
In a nucleated process, the addition of preformed fibers is expected to act as a seed, removing the lag phase, and producing fastest growth at t = 0. We found this to be the case for the SAF system. Fig. 5 B shows the fraction completion with time for a sample 80 μM in each SAF peptide and with 10% of a vortexed 100 μM matured SAF sample to provide a homogeneous sample of fragmented fibers as seeds. These growth data fit well to single exponentials in time, indicating that seeded growth is predominantly one-dimensional (Section S7). We repeated the seeding experiment at various concentrations of free peptide to explore the nature of the growth process in the absence of nucleation. The growth rate increased linearly with free peptide concentration (Fig. 5 C), confirming that nucleation had been abolished, and the aforementioned initial species—whether dimers or small aggregates thereof—are competent to add onto the preformed fiber directly.
Dimensionality of growth and the size of the nucleus
Analysis of the concentration dependence of the growth curves (Fig. 5 A) can be used to gain further information about the nucleation process, particularly regarding the parameters of dimensionality of growth (d) and the size of the nucleus (n). This section outlines such an analysis, which is described in detail in the Supporting Material.
CD spectra were recorded over time for a range of initial peptide concentrations and analyzed using the two-state method described above to obtain the curves plotted in Fig. 5 A. A general model that might be expected to fit these data can be expressed as:
where α(t) is the normalized progress of fibrillization (α(t) = 1 denotes completion); d reflects the dimensionality or geometry of the growth (1 for rods, 2 for disks, 3 for spheres); and n is the number of components (in our case, fibrillization-competent SAF-p1/SAF-p2 heterodimers) in the nucleus (45) (see the Supporting Material for motivation of equations). A reasonable fit to the early time points of the multiple curves was achieved with 2.5 ≤ d ≤ 3 (Fig. 5 A). This value for the time exponent, and the breakdown of the model for longer time points are plausible in light of the TEM data (Fig. 2). At early times, fibers grew both linearly as rods and by thickening to some extent; hence, the higher value of d. At later times, the fiber width became fixed and growth continued via one-dimensional extension, which would reduce d. This implies that the rod shape of the fibers is not simply due to more-rapid growth longitudinally than radially; rather, the fiber radius reaches a limiting value. The mechanism for this limitation is unclear, though it is a common feature of fiber-forming systems. It is likely to result from twisting or similar distortions of the outer coiled-coil blocks/fibrils, and an associated free-energy penalty that limits growth in this direction (15).
The concentration dependence of the lag time, τ, was used to estimate n. Polynomial fits to the curves of Fig. 5 A gave τ at a number of total peptide concentrations. Here it is the effective concentration of the partially helical species competent for fiber formation, chelical, that is relevant. Therefore, chelical was calculated from the SAF-p1/SAF-p2 association curve from the initial folding data (Fig. 3 B). The plot of ln(τ) against ln(chelical) has a straight line of gradient 2 (Fig. 5 D). This implies a critical nucleus size of 6–8 (2 × (d + 1)) partially helical dimers, or 12–16 SAF peptides.
It should be noted that, in similarity to amyloid-like fiber formation, the system reproducibly exhibits nucleation and growth kinetics, but the precise values of n and the rate constants are sensitive to subtle changes in the initial conditions, including concentration, temperature, mixing methodology, and peptide purity.
Predictive capability of the model: manipulating fiber-length distributions
The assembly pathway deduced above allows us to make predictions about the response of the system to perturbation. Such control may be of use in nanoscale science and technology to create nano- to micronscale rods with defined length distributions and aspect ratios (46, 47). As shown above, the requirement of SAF nucleation is abolished by adding seeds derived from matured and shattered fibers at the beginning of a series of fibrillogenesis experiments. Therefore, we added different and known amounts of seed to SAF mixtures, with the expectation that more seed would give more and shorter fibers. To maximize the difference in the length distributions, we chose a free-peptide concentration (60 μM) to access the one-dimensional phase of the growth curve (Fig. 5 C), since at this point growth is channeled into elongation as opposed to radial thickening. Reactions were incubated at 20°C for 1 h to allow complete fibrillogenesis (Fig. 5 B). The resulting fibers were imaged in situ by LM to avoid perturbation of the distribution (Fig. 6, A–D). The lengths of the fibers were measured and plotted as histograms and cumulative frequency curves (Fig. 6, D and E). The maximum gradients of the cumulative frequency curves indicate the modal values for the distributions, which increase by a factor of 4 from the 10% to the 1% seed samples. These data indicate that our model is predictive and can be used to influence and control the system.
Figure 6.
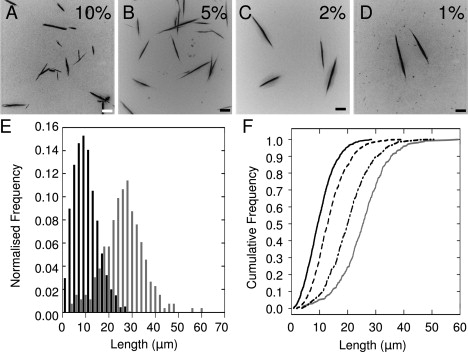
Manipulating SAF length by seeding. Fibers were grown for 1 h from 60 μM samples with (A) 10%, (B) 5%, (C) 2%, and (D) 1% (v/v) of matured fibers from standard 100 μM preparations fragmented after 24 h. Scale bar: 10 μm. (E) Histograms showing the number of fibers as a function of length for the 10% (black) and 1% (gray) seeded samples. (F) Cumulative frequencies for 10% (solid black), 5% (dashed), 2% (dot-dashed), and 1% (gray) seeded samples. Number of observations: 250–1500.
Conclusions
By using a combination of solution-phase biophysical methods, microscopy, and peptide engineering, we were able to determine the pathway for the self-assembly of an α-helical protein fiber. The SAF system comprises two complementary leucine-zipper peptides (SAF-p1 and SAF-p2a) of de novo design. Immediately upon mixing, the folding and assembly processes begin with the establishment of small, mixed oligomers (predominantly with heterodimeric helical interfaces) of SAF-p1 and SAF-p2a with an ∼45% helical conformation (Fig. 1 A). We deduced that the folded, helical region corresponds to a coiled-coil interface comprising unbroken hydrophobic faces spanning ∼2 heptad repeats from each peptide. The next stage of assembly is nucleation, which involves the agglomeration of six or more partially helical dimers that subsequently are more likely to grow than disperse (Fig. 1 D). After nucleation, the initial growth is shown to be self-symmetric and somewhere between 2.5- and 3-dimensional (Fig. 1 F), i.e., initially at least, the fibers grow as thickening cylinders. At later times, the fibers reach an equilibrium width and further growth occurs through elongation only. This model, which is both descriptive (biomolecular) and mathematical, allows the system to be manipulated. We demonstrate this with a series of seeding experiments using fragmented matured fibers, which produce more and shorter fibers with increasing amounts of seed.
By analyzing the kinetics of SAF assembly in detail, we have furthered our understanding of the SAF system and made it one of the best-characterized fiber-forming systems of de novo design. This places us in a strong position to further explore the relationship among the peptide sequence, underlying design features, and final fiber morphology. Furthermore, as we have begun to demonstrate here, the pathway also allows us to intervene with some precision in the assembly process to manipulate the resulting fibrous structures. This should be of interest to those wishing to create soft, self-assembling systems with prescribed nano- to micronscale features. Finally, we present new approaches and techniques for characterizing the kinetics of assembly of α-helix based fibers, which will be of use to others in the field and for the study of peptide fibrillogenesis in general.
Supporting Material
Eleven figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(10)00010-X.
Supporting Material
Acknowledgments
We thank the D. N. Woolfson group for discussions, and professors Enrique De La Cruz and Andrew Miranker (Yale University, New Haven, CT) for sound advice and stimulating discussions at the outset of this project.
This work was supported by the UK Engineering and Physical Sciences Research Council (GR/T09224), the UK Biotechnology and Biological Sciences Research Council (E022359), Unilever Ltd., and the University of Bristol.
References
- 1.Woolfson D.N. The design of coiled-coil structures and assemblies. Adv. Protein Chem. 2005;70:79–112. doi: 10.1016/S0065-3233(05)70004-8. [DOI] [PubMed] [Google Scholar]
- 2.Branco M.C., Schneider J.P. Self-assembling materials for therapeutic delivery. Acta Biomater. 2009;5:817–831. doi: 10.1016/j.actbio.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yeates T.O., Padilla J.E. Designing supramolecular protein assemblies. Curr. Opin. Struct. Biol. 2002;12:464–470. doi: 10.1016/s0959-440x(02)00350-0. [DOI] [PubMed] [Google Scholar]
- 4.MacPhee C.E., Woolfson D.N. Engineered and designed peptide-based fibrous biomaterials. Curr. Opin. Solid State Mat. Sci. 2004;8:141–149. [Google Scholar]
- 5.Fairman R., Akerfeldt K.S. Peptides as novel smart materials. Curr. Opin. Struct. Biol. 2005;15:453–463. doi: 10.1016/j.sbi.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 6.Scheibel T. Protein fibers as performance proteins: new technologies and applications. Curr. Opin. Biotechnol. 2005;16:427–433. doi: 10.1016/j.copbio.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 7.Ulijn R.V., Smith A.M. Designing peptide based nanomaterials. Chem. Soc. Rev. 2008;37:664–675. doi: 10.1039/b609047h. [DOI] [PubMed] [Google Scholar]
- 8.Gazit E. Self-assembled peptide nanostructures: the design of molecular building blocks and their technological utilization. Chem. Soc. Rev. 2007;36:1263–1269. doi: 10.1039/b605536m. [DOI] [PubMed] [Google Scholar]
- 9.Rajagopal K., Schneider J.P. Self-assembling peptides and proteins for nanotechnological applications. Curr. Opin. Struct. Biol. 2004;14:480–486. doi: 10.1016/j.sbi.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 10.Woolfson D.N., Ryadnov M.G. Peptide-based fibrous biomaterials: some things old, new and borrowed. Curr. Opin. Chem. Biol. 2006;10:559–567. doi: 10.1016/j.cbpa.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 11.Gunasekar S.K., Haghpanah J.S., Montclare J.K. Assembly of bioinspired helical protein fibers. Polym. Adv. Technol. 2008;19:454–468. [Google Scholar]
- 12.Morris A.M., Watzky M.A., Finke R.G. Protein aggregation kinetics, mechanism, and curve-fitting: A review of the literature. Biochim. Biophys. Acta. 2009;1794:375–397. doi: 10.1016/j.bbapap.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 13.Xue W.F., Homans S.W., Radford S.E. Systematic analysis of nucleation-dependent polymerization reveals new insights into the mechanism of amyloid self-assembly. Proc. Natl. Acad. Sci. USA. 2008;105:8926–8931. doi: 10.1073/pnas.0711664105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruschak A.M., Miranker A.D. Fiber-dependent amyloid formation as catalysis of an existing reaction pathway. Proc. Natl. Acad. Sci. USA. 2007;104:12341–12346. doi: 10.1073/pnas.0703306104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aggeli A., Nyrkova I.A., Boden N. Hierarchical self-assembly of chiral rod-like molecules as a model for peptide β-sheet tapes, ribbons, fibrils, and fibers. Proc. Natl. Acad. Sci. USA. 2001;98:11857–11862. doi: 10.1073/pnas.191250198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams R.J., Smith A.M., Ulijn R.V. Enzyme-assisted self-assembly under thermodynamic control. Nat. Nanotechnol. 2009;4:19–24. doi: 10.1038/nnano.2008.378. [DOI] [PubMed] [Google Scholar]
- 17.Yucel T., Micklitsch C.M., Pochan D.J. Direct observation of early-time hydrogelation in β-hairpin peptide self-assembly. Macromolecules. 2008;41:5763–5772. doi: 10.1021/ma702840q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pandya M.J., Spooner G.M., Woolfson D.N. Sticky-end assembly of a designed peptide fiber provides insight into protein fibrillogenesis. Biochemistry. 2000;39:8728–8734. doi: 10.1021/bi000246g. [DOI] [PubMed] [Google Scholar]
- 19.O'Shea E.K., Klemm J.D., Alber T. X-ray structure of the GCN4 leucine zipper, a two-stranded, parallel coiled coil. Science. 1991;254:539–544. doi: 10.1126/science.1948029. [DOI] [PubMed] [Google Scholar]
- 20.Bulheller B.M., Rodger A., Hirst J.D. Flow linear dichroism of some prototypical proteins. J. Am. Chem. Soc. 2009;131:13305–13314. doi: 10.1021/ja902662e. [DOI] [PubMed] [Google Scholar]
- 21.Dafforn T.R., Rajendra J., Rodger A. Protein fiber linear dichroism for structure determination and kinetics in a low-volume, low-wavelength couette flow cell. Biophys. J. 2004;86:404–410. doi: 10.1016/S0006-3495(04)74116-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papapostolou D., Smith A.M., Woolfson D.N. Engineering nanoscale order into a designed protein fiber. Proc. Natl. Acad. Sci. USA. 2007;104:10853–10858. doi: 10.1073/pnas.0700801104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith A.M., Banwell E.F., Woolfson D.N. Engineering increased stability into self-assembled protein fibers. Adv. Funct. Mater. 2006;16:1022–1030. [Google Scholar]
- 24.Smith A.M., Acquah S.F.A., Woolfson D.N. Polar assembly in a designed protein fiber. Angew. Chem. Int. Ed. 2005;44:325–328. doi: 10.1002/anie.200461599. [DOI] [PubMed] [Google Scholar]
- 25.Papapostolou D., Bromley E.H.C., Woolfson D.N. Electrostatic control of thickness and stiffness in a designed protein fiber. J. Am. Chem. Soc. 2008;130:5124–5130. doi: 10.1021/ja0778444. [DOI] [PubMed] [Google Scholar]
- 26.Ryadnov M.G., Woolfson D.N. Engineering the morphology of a self-assembling protein fibre. Nat. Mater. 2003;2:329–332. doi: 10.1038/nmat885. [DOI] [PubMed] [Google Scholar]
- 27.Ryadnov M.G., Woolfson D.N. Introducing branches into a self-assembling peptide fiber. Angew. Chem. Int. Ed. 2003;42:3021–3023. doi: 10.1002/anie.200351418. [DOI] [PubMed] [Google Scholar]
- 28.Ryadnov M.G., Woolfson D.N. Fiber recruiting peptides: noncovalent decoration of an engineered protein scaffold. J. Am. Chem. Soc. 2004;126:7454–7455. doi: 10.1021/ja048144r. [DOI] [PubMed] [Google Scholar]
- 29.Ryadnov M.G., Woolfson D.N. MaP peptides: programming the self-assembly of peptide-based mesoscopic matrices. J. Am. Chem. Soc. 2005;127:12407–12415. doi: 10.1021/ja052972i. [DOI] [PubMed] [Google Scholar]
- 30.Ryadnov M.G., Woolfson D.N. Self-assembled templates for polypeptide synthesis. J. Am. Chem. Soc. 2007;129:14074–14081. doi: 10.1021/ja072960s. [DOI] [PubMed] [Google Scholar]
- 31.Holmström S.C., King P.J.S., Woolfson D.N. Templating silica nanostructures on rationally designed self-assembled peptide fibers. Langmuir. 2008;24:11778–11783. doi: 10.1021/la802009t. [DOI] [PubMed] [Google Scholar]
- 32.Banwell E.F., Abelardo E.S., Woolfson D.N. Rational design and application of responsive α-helical peptide hydrogels. Nat. Mater. 2009;8:596–600. doi: 10.1038/nmat2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klunk W.E., Jacob R.F., Mason R.P. Quantifying amyloid by congo red spectral shift assay. Methods Enzymol. 1999;309:285–305. doi: 10.1016/s0076-6879(99)09021-7. [DOI] [PubMed] [Google Scholar]
- 34.LeVine H. Thioflavine T interaction with synthetic Alzheimer's disease β-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 1993;2:404–410. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Myers J.K., Pace C.N., Scholtz J.M. A direct comparison of helix propensity in proteins and peptides. Proc. Natl. Acad. Sci. USA. 1997;94:2833–2837. doi: 10.1073/pnas.94.7.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bustamante C., Tinoco I., Maestre M.F. Circular differential scattering can be an important part of the circular dichroism of macromolecules. Proc. Natl. Acad. Sci. USA. 1983;80:3568–3572. doi: 10.1073/pnas.80.12.3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patterson C.W., Singham S.B., Bustamante C. Circular intensity differential scattering of light by hierarchical molecular structure. J. Chem. Phys. 1985;84:1916–1921. [Google Scholar]
- 38.Frost D.W.H., Yip C.M., Chakrabartty A. Reversible assembly of helical filaments by de novo designed minimalist peptides. Biopolymers. 2005;80:26–33. doi: 10.1002/bip.20188. [DOI] [PubMed] [Google Scholar]
- 39.Potekhin S.A., Melnik T.N., Kajava A.V. De novo design of fibrils made of short α-helical coiled coil peptides. Chem. Biol. 2001;8:1025–1032. doi: 10.1016/s1074-5521(01)00073-4. [DOI] [PubMed] [Google Scholar]
- 40.O'Shea E.K., Rutkowski R., Kim P.S. Evidence that the leucine zipper is a coiled coil. Science. 1989;243:538–542. doi: 10.1126/science.2911757. [DOI] [PubMed] [Google Scholar]
- 41.Wood S.J., Wetzel R., Hurle M.R. Prolines and amyloidogenicity in fragments of the Alzheimer's peptide β/A4. Biochemistry. 1995;34:724–730. doi: 10.1021/bi00003a003. [DOI] [PubMed] [Google Scholar]
- 42.Woolfson D.N., Williams D.H. The influence of proline residues on α-helical structure. FEBS Lett. 1990;277:185–188. doi: 10.1016/0014-5793(90)80839-b. [DOI] [PubMed] [Google Scholar]
- 43.Gonzalez L., Woolfson D.N., Alber T. Buried polar residues and structural specificity in the GCN4 leucine zipper. Nat. Struct. Biol. 1996;3:1011–1018. doi: 10.1038/nsb1296-1011. [DOI] [PubMed] [Google Scholar]
- 44.Acharya A., Rishi V., Vinson C. Stability of 100 homo and heterotypic coiled-coil a-a′ pairs for ten amino acids (A, L, I, V, N, K, S, T, E, and R) Biochemistry. 2006;45:11324–11332. doi: 10.1021/bi060822u. [DOI] [PubMed] [Google Scholar]
- 45.Avrami M. Kinetics of phase change. II. Transformation-time relations for random distribution of nuclei. J. Chem. Phys. 1940;8:212–224. [Google Scholar]
- 46.Wang X.S., Guerin G., Winnik M.A. Cylindrical block copolymer micelles and co-micelles of controlled length and architecture. Science. 2007;317:644–647. doi: 10.1126/science.1141382. [DOI] [PubMed] [Google Scholar]
- 47.Cui H.G., Chen Z.Y., Pochan D.J. Block copolymer assembly via kinetic control. Science. 2007;317:647–650. doi: 10.1126/science.1141768. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.