

Abstract
Mutations in SPTBN2, the gene encoding β-III spectrin, cause spinocerebellar ataxia type 5 in humans (SCA5), a neurodegenerative disorder resulting in loss of motor coordination. How these mutations give rise to progressive ataxia and what the precise role β-III spectrin plays in normal cerebellar physiology are unknown. We developed a mouse lacking full-length β-III spectrin and found that homozygous mice reproduced features of SCA5 including gait abnormalities, tremor, deteriorating motor coordination, Purkinje cell loss, and cerebellar atrophy (molecular layer thinning). In vivo analysis reveals an age-related reduction in simple spike firing rate in surviving β-III−/− Purkinje cells, whereas in vitro studies show these neurons to have reduced spontaneous firing, smaller sodium currents, and dysregulation of glutamatergic neurotransmission. Our data suggest an early loss of EAAT4- (protein interactor of β-III spectrin) and a subsequent loss of GLAST-mediated uptake may play a role in neuronal pathology. These findings implicate a loss of β-III spectrin function in SCA5 pathogenesis and indicate that there are at least two physiological effects of β-III spectrin loss that underpin a progressive loss of inhibitory cerebellar output, namely an intrinsic Purkinje cell membrane defect due to reduced sodium currents and alterations in glutamate signaling.
Introduction
Dominant spinocerebellar ataxias (SCAs) are a heterogeneous group of inherited neurodegenerative disorders characterized by postural abnormalities, progressive motor incoordination, and cerebellar degeneration (Dueñas et al., 2006; Soong and Paulson, 2007). Recently, using a large kindred descended from the grandparents of President Abraham Lincoln, mutations in SPTBN2, the gene encoding β-III spectrin, were found to cause spinocerebellar ataxia type 5 (Ikeda et al., 2006). However, the mechanism through which the in-frame deletions and missense mutations cause disease is still unknown, with toxic gain-of-function, loss-of-function with a dominant-negative effect on wild-type protein, or haploinsufficiency all being possibilities.
Spectrins, heterotetramers consisting of two α and two β subunits, are important structural components of the plasma membrane skeleton and are thought to play a significant role in restricting and stabilizing membrane-spanning proteins within specific subdomains of the plasma membrane. β-III spectrin is primarily expressed in the nervous system with the highest levels of expression in the cerebellum, where it is found in Purkinje cell soma and dendrites (Sakaguchi et al., 1998; Jackson et al., 2001). However, transcripts are also found in a number of different organs (Stankewich et al., 1998), and low levels of β-III spectrin protein are detected in kidney and liver (our unpublished observation). We have shown that β-III spectrin interacts with EAAT4, the glutamate transporter predominately expressed in Purkinje cells, and stabilizes it at the plasma membrane (Jackson et al., 2001). This implicates β-III spectrin in clearance of glutamate from the synaptic cleft, and consequently both modulation of glutamatergic neurotransmission and prevention of glutamate-mediated neurotoxicity. Other investigators have shown β-III spectrin interacts with ARP1 and is found in a complex with dynactin (Holleran et al., 2001). Since dynactin is the accessory protein that mediates the association of dynein with cytoplasmic vesicles, another suggested function of β-III spectrin is to facilitate protein trafficking by linking the microtubule motor to vesicle-bound cargo. An earlier study also suggested that β-III spectrin associates with Golgi and cytoplasmic vesicles (Stankewich et al., 1998). In the case of Drosophila and Caenorhabditis elegans, which have only one form of β-spectrin, loss of β-spectrin results in destabilization of the neuromuscular junction through loss of synaptic cell-adhesion molecules (Pielage et al., 2005) and axonal breakage (Hammarlund et al., 2007), respectively.
To further investigate the role of β-III spectrin in normal cerebellar development and SCA5 disease pathogenesis, we generated a mouse model in which β-III spectrin expression is disrupted. Here, we show that a functional β-III spectrin knock-out mouse (β-III−/−) develops characteristic features of cerebellar ataxia including progressive motor incoordination, a wider hindlimb gait, tremor, cerebellar atrophy, and Purkinje cell loss, thus resembling clinical cases. Therefore, the β-III−/− mouse is a new model of cerebellar ataxia and, furthermore, our results implicate several physiological defects arising from loss of β-III spectrin in disease pathogenesis.
Materials and Methods
Creation of β-III spectrin-deficient mice.
Mouse β-III spectrin gene was cloned from a 129Sv mouse BAC library (ResGen). To construct the targeting vector, two gene fragments were subcloned into the vector pPNT: a 2.0 kb KpnI fragment containing intron 6 was cloned in between the PGK-neo selection cassette and the PGK-tk cassette to form the 3′ homology region; and a 7.0 kb XhoI-Sse I fragment containing exons 1 and 2 was cloned upstream of the PGK-neo cassette forming the 5′ region of homology. The targeting vector was linearized with NotI and electroporated into 129/Ola embryonic stem (ES) cells (clone E14Tg2a). Stably transfected ES cell clones were isolated after double selection with G418 and ganciclovir, and homologously recombined ES cell clones identified by Southern blotting. Genomic DNA from 700 G418- and ganciclovir-resistant ES clones was digested with SphI and transferred to Hybond-N membrane (GE Healthcare). A 208 bp and a 160 bp intronic fragment, external to vector, were used as 5′ and 3′ probes, respectively. Correctly targeted ES clones were karyotyped and used for blastocyst injections. The resulting chimeric males were bred with C57Bl6J females to identify germ-line transmission by agouti coat color. Heterozygous F1 mice were then backcrossed with C57Bl6J for at least six further generations. All procedures involved in generation and analysis of mutant mice were performed according to the United Kingdom Animals (Scientific Procedures) Act (1986) and other Home Office regulations under specific pathogen-free conditions.
Genotyping.
Animals were genotyped by PCR analysis on genomic DNA extracted from ear notch biopsies using ChargeSwitch gDNA tissue kit (Invitrogen). A common upstream primer (5′-gagcgagaagccgtgcagaag-3′) and primers specific for the wild-type allele (5′-aggatgatggtccacactagcc-3′) and the PGK-neo cassette in the mutant allele (5′-ctaccggtggatgtggaatg-3′) were used for amplification. The 710 bp (from wild-type allele) and 562 bp (from targeted allele) PCR products were resolved by electrophoresis on a 1% agarose gel. β-III spectrin-deficient mice were fully viable and generally born at a ratio consistent with Mendelian inheritance (1:2:1). Litters composed of 41 wild-type (WT), 79 β-III+/−, and 44 β-III−/− animals.
Immunoblotting.
Whole cerebella were homogenized in 400 μl of ice-cold homogenization buffer [20 mm HEPES, pH 7.4, 1 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, and Protease Inhibitor Cocktail Set III (Calbiochem)] with a Teflon-glass homogenizer. Protein concentrations were determined using Coomassie-Plus Reagent and bovine serum albumin as standard (Pierce). Protein samples were resolved by denaturing SDS-PAGE and transferred to nitrocellulose membranes (GE Healthcare, Pharmacia). The membranes were blocked for 1 h at room temperature with 5% w/v nonfat dry milk in Tris-buffered saline/Tween 20 (TBS/T) (20 mm Tris, 17 mm NaCl, pH 7.6, with 0.1% v/v Tween 20). Blots were incubated overnight at 4°C with either rabbit anti-βIII spectrin, -EAAT4, -GLAST (1:200), -GLT1 (1:4000), -GluR1 (1:1000; AbCam), or mouse anti-actin, -calbindin (1:1600; Sigma) in blocking buffer. After washing with TBS/T, the blots were incubated for 1 h at room temperature with either HRP-conjugated donkey anti-rabbit IgG, or HRP-conjugated sheep anti-mouse IgG (1:4000; GE Healthcare, Pharmacia). Immunoreactive proteins were visualized with ECL (Insight Biotechnology).
Immunohistochemistry.
Brains were removed and immersion fixed with 4% paraformaldehyde in 0.1 m sodium phosphate buffer, pH 7.4, overnight at 4°C and cryoprotected by immersion in 0.1 m sodium phosphate buffer, pH 7.4, containing 30% sucrose. Tissue was quick-frozen on dry ice, then 15-μm-thick cerebellar sections were cut and fixed onto microscope slides coated with poly-l-lysine. After air drying for 30 min, sections either were stained for Nissl with cresyl violet (0.25%) or immunostained. All quantification was performed in a blinded manner on Nissl-stained sections by counting the number of Purkinje cells along a 1 mm linear length in folia II, III, IV, VI, and VIII (these folia were found to be the most consistent in shape between animals) and the counts averaged for each animal. The thickness of the molecular layer was measured in each animal at the same three points within each of the five chosen folia and the 15 measurements averaged. For deep cerebellar nuclei (DCN) counts, four areas (150 μm2 each) were measured and the counts averaged for each animal. For immunostaining, sections were incubated for 1 h with blocking solution (5% normal goat serum with 0.4% Triton X-100 in PBS. Rabbit anti-β-III spectrin, anti-EAAT4 or mouse anti-calbindin primary antibody (1:50) (2% normal goat serum/0.1% Triton X-100 in PBS) was applied for 1 h at room temperature. Sections were washed three times in PBS before applying goat anti-rabbit FITC-conjugated secondary antibody (Cappel) or goat anti-mouse Cy-3-conjugated secondary antibody (Jackson ImmunoResearch) for 1 h at room temperature followed by three rinses in PBS and coverslipping with Vectashield (Vector Laboratories). Mouse anti-calbindin primary antibody (1:50) and anti-Neu-N antibody (1:100; Millipore Bioscience Research Reagents) was applied overnight at 4°C and biotinylated universal horse anti-mouse/rabbit IgG, diluted at 1:200 in PBS, was applied for 1 h at room temperature. Detection of biotinylated secondary antibody was performed using ABC method with DAB and H2O2 used as peroxidase substrate (Vector Laboratories). Images were captured with an Olympus IX70 fluorescence microscope using Openlab software (Improvision) or a Zeiss inverted LSM510 confocal laser scanning microscope.
Electron microscopy.
Brains were dissected out and immersion fixed overnight (4°C) with a mixture of 2% paraformaldehyde and 2% glutaraldehyde in 0.1 m sodium cacodylate buffer, pH 7.3. Specimens were postfixed in 1% osmium tetroxide in 0.1 m sodium cacodylate for 45 min, washed three times in 0.1 m sodium cacodylate buffer, and dehydrated in 50%, 70%, 90%, and 100% normal grade acetones for 10 min each, followed by two 10 min changes in analar acetone. Samples were embedded in Araldite resin, and 1-μm-thick sections cut, stained with toluidine blue, and viewed in a light microscope to select suitable areas for investigation. Ultrathin 60-nm-thick sections were cut from selected areas, stained with uranyl acetate and lead citrate, and viewed in a Phillips CM120 transmission electron microscope (FEI). Morphological criteria used were irregular cell body, dark cytoplasm, and shrinkage. Images were taken on a Gatan Orius CCD camera.
Semiquantitative reverse transcriptase-PCR.
Total RNA was extracted from mouse cerebellum using RNeasy Mini kit (Qiagen), and reverse transcriptase (RT)-PCRs were performed using One-Step RT-PCR kit (Qiagen) according to manufacturer's instructions. Primers for RT-PCRs located in exon 1, 7, 34, and 36 of β-III spectrin were: F1 5′-atgagcagcactctgtcacccact-3′; R7 5′-gccaattcttttgccttccacagc-3′; F34 5′-ggcccagggaagtgtggcctt-3′; and R36 5′cttaaagaagctgaatcgtttttctgc-3′. Amplification of the ubiquitously expressed elongation factor α was used to control for RNA levels (Stratagene).
Cell culture and transfection.
β-III spectrin cDNA lacking exons 2–6 was subcloned into the NotI site of pCDNA3.1 (Invitrogen) and a Myc-tagged pRK5 vector. For microscopic observation, HEK293 and Neuro2A cells were plated onto coverslips coated with poly-l-lysine in 35 mm dishes and transfected with 1 μg of each DNA construct using Fugene reagent (Roche) in accordance with the manufacturer's instructions. Anti-c-myc (Ab-1; Calbiochem) and tetramethylrhodamine isothiocyanate-conjugated goat anti-mouse IgG (SouthernBiotech) were used for immunofluorescence. For cell homogenates and biotinylation assays, HEK and HEK-rEAAT4 cells, respectively, were plated onto 35 mm dishes and transfected with 4 μg of each DNA using Lipofectamine 2000 (Invitrogen) according to manufacturer's instructions.
Motor coordination tests.
Footprint patterns were analyzed using a runway (80 cm by 10.5 cm wide) with white paper at the bottom. Hindpaws of animals were dipped in nontoxic, water-soluble black ink (Indian Ink, Winsor & Newton). Three consecutive strides were measured for each animal. Stride length measurements were taken from the base of two consecutive paw prints on the same side, and the base width was measured as the distance between the center of one paw print to the center of the next print on the opposite side. The elevated beam test was performed using a narrow horizontal beam (2 cm wide, 80 cm long, held at a height of 30 cm from the table). The number of hindpaw slips the animal made while traversing the beam were counted. For the hanging wire test, mice were placed on a wire cage lid, which was turned upside down, and the latency to fall measured. A 60 s cutoff time was used. In the rotarod test, the ability of mice to maintain balance on a stationary (maximum time, 60 s) or rotating (3, 5, and 10 rpm) 3-cm-diameter cylinder was assessed (TSE Rotarod) and the time a mouse remained on the accelerating cylinder recorded (maximum, 120 s).
Glutamate uptake assays.
Each cerebellum was homogenized in 800 μl of tissue buffer (5 mm Tris/320 mm sucrose, pH 7.4) with Protease Inhibitor Cocktail Set III (Calbiochem), using Teflon-glass homogenizer. Each sample was split and pellets washed twice in ice-cold Tissue Buffer, before being resuspended in either Na+-containing Krebs buffer (120 mm NaCl, 25 mm NaHCO3, 5 mm KCl, 2 mm CaCl2, 1 mm KH2PO4, 1 mm MgSO4, 10% glucose) or Na+-free Krebs (120 mm choline-Cl and 25 mm Tris-HCl, pH 7.4, substituted for NaCl and NaHCO3, respectively). Samples were then incubated with 5 μm 3H-glutamate for 10 min at 37°C and uptake stopped by placing back on ice. Pellets were washed twice with Wash Buffer (5 mm Tris/160 mm NaCl, pH 7.4) and radioactivity measured using a scintillation counter. Na+-dependent uptake was determined by subtracting Na+-free counts.
In vivo electrophysiology.
Extracellular recordings were obtained from Purkinje cells in the vermis of the posterior lobe (lobules V and VI) in WT and β-III−/− mice anesthetized with intraperitoneal injection of 1.5 g/kg urethane (solution at 12.5%). The head of the animal was immobilized in a stereotaxic frame and the cerebellum exposed by a craniotomy extending from the obex to the lamboidal ridge. The dura was opened along the midline, and the brain covered with a mixture of paraffin oil and vaseline to prevent dessication. Glass microelectrodes filled with 0.9% NaCl were placed ±1 mm on either side of the midline then lowered into the vermis using a hydraulic micromanipulator (Narishige). Recordings of the spontaneous firing activity were made to a depth of 2 mm. Purkinje cells were identified by their characteristic firing of complex spikes followed by a pause in simple spike firing (see Fig. 5). Spike activity was digitized at 10 kHz for a minimum of 3 min for each cell of interest, using Spike2 software (CED). The mean firing rates of complex and simple spikes were quantified using analysis functions in the Spike2 software.
Figure 5.

Reduced in vivo simple spike firing rate in old β-III−/− mice compared with young β-III−/− mice. Top, Representative trace of in vivo Purkinje cell output from 8-month-old WT and β-III−/− animals. Complex spike (dashed box) enlarged on right of trace. Bottom, Firing rates of 12-week-old and 8-month-old β-III−/− mice expressed as percentage of wild-type frequency show reduction in simple but not complex spikes with age. All data are means ± SEM (12 weeks: WT, N = 3, n = 11; β-III−/−, N = 3, n = 12; 8 months: WT, N = 3, n = 8; β-III−/−, N = 3, n = 10; p = 0.02).
Slice electrophysiology.
Cerebella were dissected out into ice-cold modified artificial CSF (ACSF) containing the following (in mm): 60 NaCl, 118 sucrose, 26 NaHCO3, 2.5 KCl, 11 glucose, 1.3 MgCl2, and 1 NaH2PO4 at pH 7.4 when bubbled with 95% O2:5% CO2. The cerebellar vermis was glued to the vibratome cutting platform (Dosaka EM Co) with cyanoacrylate adhesive. Two-hundred-micrometer-thick sagittal slices were cut and incubated for 30 min at 30°C in standard ACSF composed of the following (in mm): 119 NaCl, 2.5 CaCl2, 26 NaHCO3, 2.5 KCl, 11 glucose, 1.3 MgCl2, and 1 NaH2PO4 at pH 7.4 when bubbled with 95% O2:5% CO2. Slices were stored at room temperature until required for recording. Slices were transferred to a submerged recording chamber and superfused with standard ACSF (3–5 ml min−1) at room temperature for voltage-clamp experiments and at 32 ± 2°C for recording spontaneous action potentials. Purkinje cells were visualized with 40× immersion objective and Normarski differential interference contrast optics. Whole-cell recordings were obtained from Purkinje cells using thick-walled borosilicate glass pipettes pulled to 5–8 MΩ. For recording action potentials, the internal solution contained the following (in mm): 125 K-gluconate, 15 KCl, 10 HEPES, 5 EGTA, 2 MgCl2, 0.4 NaGTP, 2 NaATP, and 10 Na-phosphocreatine, adjusted to pH 7.4 with KOH. For parallel fiber (PF)-mediated EPSC (PF-EPSC) measurements, the internal solution contained the following (in mm): 108 Cs-methanesulfonate, 9 NaCl, 9 HEPES, 1.8 EGTA, 1.8 MgCl2, 0.4 NaGTP, 2 MgATP, 63 sucrose, and 5 QX-314, adjusted to pH 7.4 with CsOH. Picrotoxin (50 μm) was added to the ACSF. PF-EPSCs were evoked by placing a patch pipette filled with standard ACSF at the same position in the molecular layer and applying a range of stimuli (1.5–5.0 V, 200 μs duration). Pairs of PF-EPSCs (100 ms apart) were evoked at 0.033 Hz, and a minimum of three PF-EPSCs were averaged under each condition. Series resistances were <15 MΩ and were compensated for by 40–60%. Membrane currents and voltages were filtered at 5 kHz and sampled at 10 and 200 kHz, respectively, for voltage-clamp and current-clamp experiments. Data were acquired using pClamp 9 (Molecular Devices) and analyzed using IGOR Pro (Wavemetrics). The amplitudes and decay time constants of PF-evoked EPSCs were measured using the ChanneLab analysis program (Synaptosoft Inc).
Dissociated Purkinje cell electrophysiology.
Purkinje cells were isolated from P16–P20 mice using dissociation techniques modified from Raman and Bean (1997). Isolated Purkinje cells were visually identified by their large, pear-shaped soma (due to the stump of the apical dendrite). The control extracellular recording solution contained the following (in mm): 110 TEA-Cl, 25 NaCl, 2 BaCl2, 0.3 CdCl2, 10 HEPES, and 10 glucose, buffered to pH 7.4 with NaOH. Recordings were made at room temperature with borosilicate pipettes (3–5 MΩ) containing the following (in mm): 117 CsCl, 9 EGTA, 9 HEPES, 1.8 MgCl2, 14 Na-phosphocreatine, 4 MgATP, and 0.3 NaGTP, adjusted to pH 7.4 with CsOH. To isolate the TTX-sensitive Na+ current voltage protocols were repeated in the presence of 300 nm TTX and subtracted from the control recordings.
Statistics.
Statistical analysis was performed using Student's t test, two samples assuming unequal variance, apart from densitometry analysis of Western blots where one sample t test was used with a predicted value of 100% for WT and proportion data were arcsine transformed before t test.
Results
Generation of β-III spectrin-deficient mice
We used targeted recombination to knock out expression of β-III spectrin, and our targeting strategy involved replacing exons 3–6 with the neomycin-resistance gene (see Materials and Methods). In the recombinant allele, when exon 2 is spliced onto exon 7 this will disrupt the open reading frame, introducing a premature stop codon at the beginning of exon 7. WT (+/+), heterozygous (β-III+/−;+/−), and homozygous (β-III−/−;−/−) progeny were identified by PCR analysis (see Materials and Methods).
To determine whether we had successfully abolished β-III spectrin expression, we analyzed, using a previously characterized polyclonal antibody raised against a C-terminal epitope of β-III spectrin (Jackson et al., 2001), whole cerebellar homogenates by Western blot analysis. This revealed that full-length β-III spectrin (270 kDa) was absent in β-III−/− mice and its quantity approximately halved (58.4 ± 7.4% of WT) in β-III+/− animals (Fig. 1A). However, a smaller molecular weight protein was detected, at low levels, in β-III−/− mice (18.8 ± 2.2% of full-length β-III spectrin in WT) (Fig. 1A) and β-III+/− mice but not WT animals. Confocal immunofluorescence microscopy revealed that this protein was still located throughout the β-III−/− Purkinje cell dendritic tree but, consistent with results from Western blot analysis, at a substantially reduced level when compared with WT β-III spectrin (Fig. 1B).
Figure 1.

Progressive motor impairment in β-III−/− mice. A, Western blot analysis of whole cerebellar homogenates (10 μg) confirms full-length β-III spectrin (270 kDa) is absent in β-III−/− mice, but a smaller molecular weight protein (∼250 kDa; arrowhead) is expressed at low levels in β-III+/− and β-III−/− animals. Degradation product is identified by an asterisk. Calbindin levels confirm equal protein loading. B, Cerebellar sections, from 3-week-old mice, immunostained with anti-β-III spectrin and anti-calbindin antibody show intense β-III labeling (arrow) of WT (+/+) dendritic tree but faint staining in β-III−/− mice, although still present in proximal and distal dendrites. ML, Molecular layer; PCL, Purkinje cell layer. Scale bar, 50 μm. C, Left, Representative footprints of 18-week-old WT and β-III−/− littermates. Base width shown by double arrow and stride length by solid line. Right, Summary data showing significant increase in β-III−/− base width compared with WT at 18 weeks (p = 0.0078), 6 months (p = 2.82 × 10−6), and 1 year (p = 0.02) of age. D, Time mice remained on stationary rod before falling reveals 6-month-old and 1-year-old β-III−/− mice are impaired (p = 0.004 and 0.0007). Mice were given four consecutive trials, with maximum time of 60 s. E, Latency of 3-week-old animals to fall from rotarod at 3, 5, and 10 rpm. Mice were given four trials per day and allowed a maximum retention time of 120 s per trial. Both genotypes improved performance during consecutive days at 3 and 5 rpm, but difference in latency to fall remained significant in all comparisons, except on day 3 at 3 rpm (p = 0.218). F, 6-month-old β-III−/− mice failed to stay on rotarod at 3 rpm and showed no improvement. G, Number of hindlimb slips β-III−/− mice made when crossing narrow, elevated beam increased from 12 weeks of age (p = 0.001 and 0.0006). All data are means ± SEM (WT, N = 5–12; β-III−/−, N = 4–15). *p < 0.05; **p < 0.01; ***p < 0.001.
We determined that the smaller molecular weight protein arises from exon 1, not exon 2, being spliced onto exon 7 in the targeted but not WT allele (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). This retains the reading frame, and so no premature stop codon is introduced. Therefore, the mutant mice lack full-length β-III spectrin but express, at a low level, a form of β-III spectrin (∼250 kDa) that lacks most of the actin-binding domain encoded by exons 2–6 (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). In vitro studies demonstrate that Δ2-6βIII protein has no obvious gain-of-function or adverse property but, if anything, appears to be less functional than WT (supplemental Fig. 1C–F, available at www.jneurosci.org as supplemental material), providing persuasive evidence that the β-III−/− mouse is a functional knock-out (hypomorph).
β-III spectrin deficiency causes cerebellar ataxia
To test whether β-III−/− mice show signs of ataxia, we first analyzed the footprint patterns of 6-week-old to 1-year-old animals for differences in base width and stride length (Fig. 1C). Hindlimb gait of β-III−/− mice became progressively wider than that of littermate WT mice, but there was no significant alteration in step length at any age. However, variation in walking speed down the runway introduced heterogeneity in stride length that may have masked genotype-dependent differences.
To quantify motor performance and coordination, we used three behavioral tests: stationary rod, rotarod, and elevated beam. We found no significant difference at 3 weeks of age between β-III−/− and WT mice in maintaining balance on a stationary rod, whereas 6-month-old and 1-year-old β-III−/− mice struggled to maintain balance (Fig. 1D). Performance of young β-III−/− mice on a rotating rod (3 rpm) was initially significantly below that of WT animals (Fig. 1E). However, the performance improved on days 3 and 4, demonstrating that β-III−/− mice are able to learn motor tasks and can perform well when the task is relatively easy. When the speed was increased to 5 rpm, again β-III−/− mice fell off more rapidly than WT controls, and at 10 rpm they showed no improvement during consecutive days of testing (Fig. 1E). In contrast, by 6 months of age β-III−/− mice performed much worse and showed no improvement even at 3 rpm (Fig. 1F; see supplemental video, available at www.jneurosci.org as supplemental material). To test whether weakness contributed to poorer rotarod performance, we evaluated muscle strength with a hanging wire test. There was no significant difference between the performance of β-III−/− mice and WT animals at any age, suggesting no muscle weakness (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). Defects in motor performance were also not due to body size variation as there was no significant difference in body weight (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). Finally, an elevated beam test was used to evaluate motor coordination and balance. The beam width was 2 cm, so that impaired coordination and balance would result in hindlimb errors. The β-III−/− mice were generally more hesitant to cross the beam compared with WT animals (see supplemental video, available at www.jneurosci.org as supplemental material) and when turning sometimes fell off the beam, which WT mice never did. Counting the number of slips made by each animal while traversing the beam showed that β-III−/− mice made more, and generally bigger, slips than WT littermates from 12 weeks of age (Fig. 1G). However, β-III−/− mice remain ambulatory and do not fall when walking, resembling a mild form of ataxia that is observed in SCA5 patients, in contrast to SCA1 transgenic mice, which display a more severe form of ataxia (Burright et al., 1995). Finally, a tremor, a characteristic of some forms of cerebellar ataxia, was observed in β-III−/− mice (see supplemental video, available at www.jneurosci.org as supplemental material).
Thinning of molecular layer and Purkinje cell loss in β-III−/− mice
We then looked for cerebellar atrophy and Purkinje cell loss, other features of patients with ataxia, in β-III−/− mice. Staining and quantification of cerebellar sections (see Materials and Methods) revealed a thinning of the molecular layer, generally correlated with degeneration of dendrites (He et al., 2006), and a progressive loss of Purkinje cells at 6 months (WT, 50.4 ± 3 cells/mm; β-III−/−, 39 ± 1.9 cells/mm; N = 4 and 3, respectively; p = 0.035; and WT, 156.2 ± 3 μm; β-III−/−, 112.2 ± 3.1 μm; 73 ± 2.5% of WT; p = 1.71 × 10−5) and 1 year of age (WT, 44.7 ± 2.3 cells/mm; β-III−/−, 26.9 ± 1.5 cells/mm; N = 4; p = 0.001; and WT, 150 ± 8.5 μm; β-III−/−, 109.3 ± 4.7 μm; 73 ± 3.1% of WT; p = 0.005). This is not observed in young (3-week-old) β-III−/− animals (WT, 51 ± 3.4 cells/mm; β-III−/−, 50.6 ± 3.8 cells/mm; N = 10 and 9, respectively; p = 0.94; and WT, 157.5 ± 6.4 μm; β-III−/−, 144.1 ± 3.4 μm; p = 0.097) (Fig. 2). There was no obvious variation in the pathology of the lobules examined, with the same degree of neuronal loss and atrophy being detected in all five examined. Immunostaining also highlighted that the remaining Purkinje cell somas in old β-III−/− animals were generally smaller (WT, 14.5 ± 0.13 μm; β-III−/−, 11.6 ± 0.27 μm; N = 3, n = 100/animal; p = 0.0025) and more irregularly shaped compared with WT littermates. It also revealed that in young β-III−/− mice more Purkinje cells were found to lie outside the Purkinje cell layer (WT, 0.28 ± 0.17%; β-III−/− 3 ± 0.34%, N = 3, n = 350–520/animal; p = 0.006) (Fig. 2A), a finding also observed in SCA1 mutant mice (Burright et al., 1995), whereas, we saw no differences in the densities of DCN among genotypes at any age (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Also, no gross morphological change (cell layer thickness) or neuronal loss was observed in hippocampus (CA1 or CA3), cortex, or dentate gyrus (supplemental Table 1, available at www.jneurosci.org as supplemental material).
Figure 2.
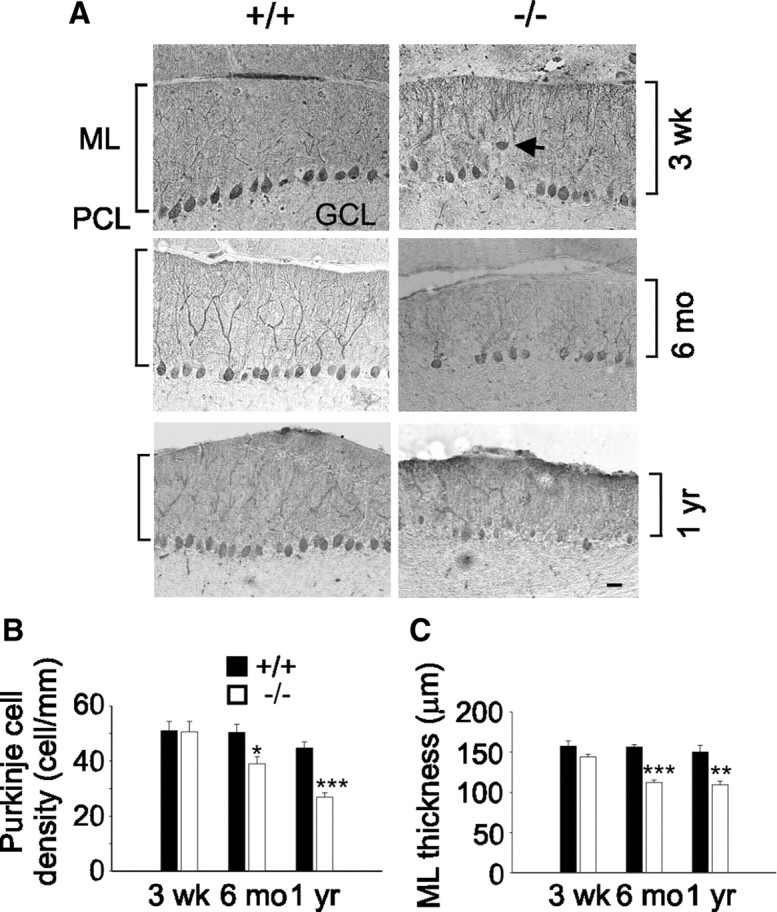
Cerebellar degeneration in old β-III−/− spectrin mice. A, Cerebellar sections immunostained with anti-calbindin D antibody reveal some ectopically expressed Purkinje cells (arrow) in 3-week-old β-III−/− mice and shrunken Purkinje cell soma in 6-month-old and 1-year-old β-III−/− mice. ML, Molecular layer; GCL, granule cell layer; PCL, Purkinje cell layer. Scale bar, 50 μm. B, Mean Purkinje cell density measured from cerebellar folia II-IV, VI, and VIII shows Purkinje cell loss in 6-month-old and 1-year-old but not 3-week-old β-III−/− mice. C, Mean molecular layer thickness shows thinning of molecular layer in old β-III−/− mice. All data are means ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Subsequent ultrastructural analysis using transmission electron microscopy of Purkinje cells confirmed somal shrinkage and revealed an age-dependent accumulation of cells undergoing “dark cell degeneration” (Fig. 3), a process linked to AMPA receptor-elicited delayed excitotoxicity (Garthwaite and Garthwaite, 1991; Turmaine et al., 2000), in β-III−/− mice. In 8-month-old β-III−/− animals, the percentage of Purkinje cells with normal morphology, relative to WT, was 29 ± 2.1% compared with 94.5 ± 1.9% at 8 weeks of age (8 weeks: N = 4, n = 18–35/animal; 8 months: N = 5, n = 7–18/animal; ***p = 7.2 × 10−6). These cells showed irregular somal morphology, increased electron density, dilated cisternae of endoplasmic reticulum denuded of ribosomes, and changes to Golgi apparatus (numerous vesicles, some of which were invaginated, a sign of Golgi fragmentation (Siddhanta et al., 2003)).
Figure 3.
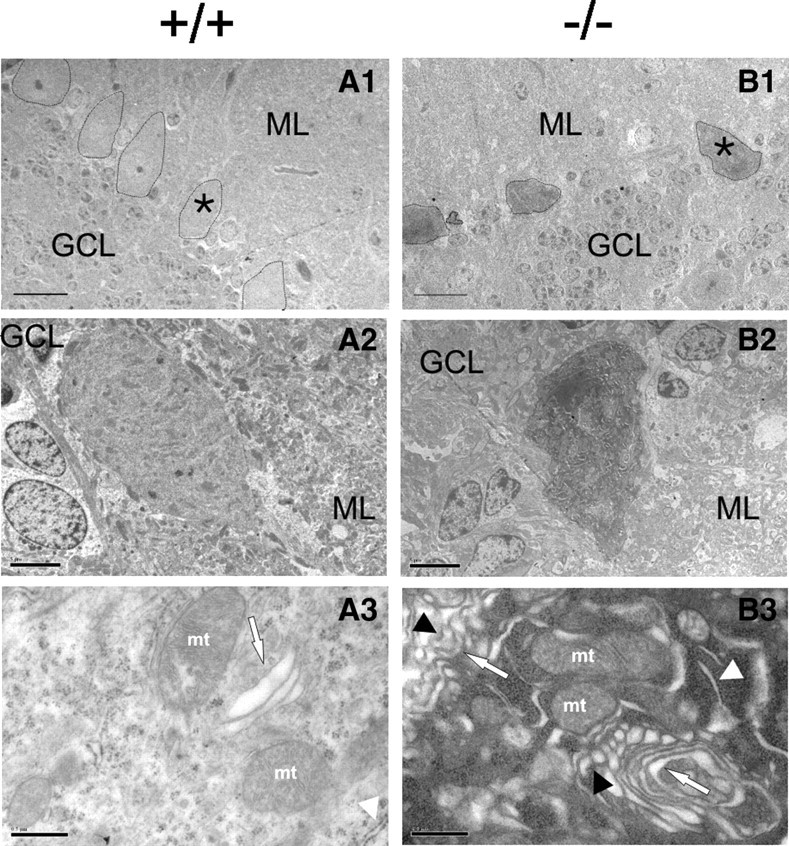
Signs of glutamate-mediated excitotoxicity in β-III−/− Purkinje cells. A1–B2, Ultrastructural analysis of cerebellar sections from 6-month-old mice shows normal cell bodies in WT (A1, A2) but shrunken and electron dense soma in β-III−/− (B1, B2). A1, B1, 170× magnification; scale bar, 20 μm. A2, B2, 750× magnification; scale bar, 5 μm. A3, B3, Electron micrographs (8400× magnification; scale bars, 0.5 μm) show dilated smooth endoplasmic reticulum (ER) (B3, white arrowhead), and fragmented Golgi cisternae (arrow) with increased number of vesicles, some of which are invaginated (black arrowhead) in β-III−/− Purkinje cell somas compared with abundant rough ER (A3, white arrowhead) and normal Golgi apparatus (arrow) in WT. ML, Molecular layer; GCL, granule cell layer; mt, mitochondria; all Purkinje cell bodies outlined. *Denotes from which cell high-magnification images were obtained. Note different cell orientation in A2 and B2 compared with A1 and B1.
Loss of glutamate transporter expression in β-III−/− mice
We therefore examined levels of EAAT4, the Purkinje cell-specific glutamate transporter, in β-III−/− mice, since our previous work showed EAAT4 interacts with and is stabilized by β-III spectrin (Jackson et al., 2001). Moreover, dramatic changes in EAAT4 distribution were seen in SCA5 autopsy tissue (Ikeda et al., 2006). Even at 3 weeks of age, a decrease in EAAT4 protein was obvious in β-III−/− mice compared with WT littermate controls (66.5 ± 3.5% of WT; N = 6 each; p = 0.0002) (Fig. 4A). Values were normalized to calbindin, a Purkinje cell-specific marker. Immunofluorescence microscopy reveals that in β-III−/− mice the remaining EAAT4 is still located throughout the dendritic tree (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). In contrast, no loss of the astroglial glutamate transporters GLAST (103.2 ± 7.4% of WT; p = 0.687) and GLT1 (102.7 ± 5.1% of WT; p = 0.62) was observed at this early stage.
Figure 4.

Loss of neuronal and astroglial glutamate transporters in β-III−/− mice. A, Top, Representative Western blots illustrating loss of EAAT4 protein in β-III−/− mice from 3 weeks of age. Loss of astroglial glutamate transporter GLAST observed in β-III−/− mice from 12 weeks of age. No loss of GLT1 seen at any age. Bottom, Densitometry data quantifying levels. EAAT4 levels normalized with calbindin, a Purkinje cell-specific marker (p < 0.02 for all ages). GLAST and GLT1 normalized with actin. B, Cerebellar sections immunostained with anti-GFAP antibody show no astrogliosis in 1-year-old β-III−/− mice. C, Glutamate uptake assays on whole cerebellar homogenates show reduced uptake in β-III−/− mice from 12 weeks of age. All data are means ± SEM (N = 3–6). *p < 0.05; **p < 0.01.
Western blot analysis and densitometry (Fig. 4A) did reveal a progressive reduction in GLAST protein compared with littermate controls in 12-week-, 6-month-, and 1-year-old animals (81.5 ± 5.5% of WT, p = 0.0206; 64 ± 5% of WT, p = 0.0055; 44.3 ± 13% of WT; p = 0.05; N = 3 each) in addition to the fairly consistent loss of EAAT4 (67.3 ± 6.2% of WT, p = 0.0033; 50.5 ± 5.9% of WT, p = 0.0035; 62 ± 5.5% of WT, p = 0.02). In contrast, no reduction in GLT1 levels was observed at any age (108.3 ± 2.4%; 95.8 ± 2.4%; 111.7 ± 16.9%, respectively). The fact there was no difference in GFAP expression in old β-III−/− animals (Fig. 4B) suggests that the progressive loss of GLAST was not a consequence of global defects in Bergmann glial integrity.
To investigate whether a loss of transporter protein corresponded to a loss in cerebellar glutamate uptake, we measured uptake of radioactively labeled glutamate into crude cerebellar membrane preparations. We found a slight reduction in 12-week-old β-III−/− animals (83.5 ± 6.4% of WT; N = 3 and 4; p = 0.08) when the ataxic phenotype starts to become obvious, and further losses in 6-month-old and 1-year-old animals when the motor deficits are more pronounced (70 ± 7.6% of WT, p = 0.047, and 49.2 ± 9.7% of WT, p = 0.034, respectively; N = 3 each) (Fig. 4C). Together, these results suggest that impaired glutamate uptake, resulting from loss of EAAT4 and GLAST, correlates with neuronal degeneration and plays a role in disease progression.
Reduced in vivo Purkinje cell simple spike firing rate in β-III−/− mice
Purkinje neurons provide the sole output from the cerebellar cortex, in the form of inhibitory inputs to the DCN, and so it is their firing rate that encodes the timing signals essential for motor planning, execution, and coordination. They are one of a few neuronal populations that fire regularly in the absence of synaptic input, but integration of excitatory and inhibitory inputs modulates their output (Häusser and Clark, 1997). We therefore recorded the in vivo firing pattern of Purkinje cells in 12-week-old and 8-month-old β-III−/− and WT mice (Fig. 5). We found that the simple spike firing rate was much less at 8 months than 12 weeks in β-III−/− mice compared with WT animals, indicating an age-related decrease in output from surviving Purkinje cells. There was no difference in the firing rate of complex spikes, but some Purkinje cells from 8-month-old animals appeared to fire only complex spikes, with the number of such cells being greater in β-III−/− mice (WT, 1 of 9 cells; β-III−/−, 5 of 15 cells). These cells were not included in the calculation of the mean simple spike firing rate.
Reduced spontaneous activity in β-III−/− mice
To identify a possible cellular basis for the impaired Purkinje cell output in β-III−/− mice, we first measured spontaneous activity in vitro (Fig. 6A). We found an approximately twofold reduction in cells from 3-week-old β-III−/− animals (18.3 ± 2.6 Hz; N = 2, n = 7) compared with WT cells (43.4 ± 3.5 Hz; N = 3, n = 9; p = 5.22 × 105). The difference in the firing rate was not abolished in the presence of bicuculline, an inhibitor of GABAergic neurotransmission, demonstrating that the observed reduction is not due to increased inhibition. There was also very little effect on firing rate when excitatory inputs were blocked with NBQX, revealing very little tonic excitatory input within the in vitro slice, similar to published data (Häusser and Clark, 1997). Therefore, the observed reduction is due to an intrinsic membrane defect (i.e., altered ion channel activity) in β-III−/− Purkinje cells. There was no further reduction in firing frequency observed in cells from 6-month-old β-III−/− animals (20.8 ± 1.2 Hz; N = 2, n = 16; p = 0.432), suggesting that the intrinsic defect does not underlie the progressive nature of the disease phenotype. No significant difference in cell input resistance, indicating no change in “leak” conductance, or zero current potential was detected between the two genotypes in either 3-week-old (WT, 74 ± 12 MΩ; β-III−/− 78 ± 8 MΩ; and WT, −55 ± 1 mV; β-III−/−, −54 ± 3 mV) or 6-month-old animals (WT, 78 ± 6 MΩ; β-III−/−, 76 ± 6 MΩ; and WT, −56 ± 0.3 mV; β-III−/−, −57 ± 0.8 mV).
Figure 6.

Reduced spontaneous firing due to smaller sodium currents in Purkinje cells from β-III−/− mice. A, Left, Representative traces of Purkinje cell spontaneous firing measured from 3-week-old WT and β-III−/− littermates. Right, Quantification of mean firing frequency shows a reduction in spike frequency in Purkinje cells from 3-week-old β-III−/− mice compared with WT mice. Presence of NBQX or bicuculline does not abolish genotype difference in firing rate. B, Left, Sodium current traces from representative cells evoked with a series of 50 ms depolarizations from a holding potential of −90 mV to potentials ranging from −80 to + 20 mV in 10 mV increments. Right, Current–voltage relationship shows reduced whole-cell current in β-III−/− mice. C, Left, Representative traces of resurgent sodium currents in WT and β-III−/− mice. Currents evoked using a 20 ms step to + 30 mV, followed by repolarizations from –20 to –70 mV. Right, Mean peak resurgent sodium current versus voltage shows reduced resurgent current in β-III−/− mice. All data are means ± SEM. **p < 0.01; ***p < 0.001.
It is known that voltage-gated potassium and sodium channels play important roles in sustaining the high-frequency tonic firing in Purkinje cells (Raman and Bean, 1999; Sacco et al., 2006; Zagha et al., 2008). There was no difference in action potential properties from WT and β-III−/− Purkinje cells (half-width: WT, 0.24 ± 0.05 ms; β-III−/−, 0.23 ± 0.009 ms, p = 0.287; peak: WT, 10.9 ± 1.6 mV; β-III−/−, 11.9 ± 2.1 mV, p = 0.483; hyperpolarization peak: WT, −60.6 ± 1.9 mV; β-III−/−, −60.3 ± 0.9 mV, p = 0.337; n = 7–10 for all analyses), suggesting that the intrinsic firing defect was not due to a potassium channel; whereas, when we recorded whole-cell sodium currents from acutely dissociated P16-20 Purkinje neurons, they were found to be significantly smaller in β-III−/− Purkinje cells compared with WT cells (Fig. 6B). The current–voltage relationships were similar for both genotypes, indicating no difference in voltage dependence of channel activation, and there was no difference in cell capacitance (WT, 10.1 ± 0.5 pF; β-III−/−, 10.0 ± 0.4 pF; p = 0.92), suggesting that the growth and morphology of mutant Purkinje cells was the same as wild-type Purkinje cells. In addition, the resurgent sodium current amplitude, which is an important aspect of the fast repetitive Purkinje cell firing, accelerating depolarization between action potentials, was also found to be smaller in β-III−/− cells (Fig. 6C). These results suggest that it is a sodium channel defect that underlies the changes in the intrinsic membrane properties of β-III−/− Purkinje cells and that β-III spectrin may play a role in maintaining a high density of sodium channels within the soma and dendrites, similar to β-IV spectrin in axons (Komada and Soriano, 2002).
Altered glutamatergic transmission in β-III−/− mice
Next, since the tonic simple spike firing rate is increased by PF inputs, we examined, in cerebellar slices, PF-EPSCs. We found that PF-EPSC amplitudes, at various stimuli, were considerably larger in 6-week-old β-III−/− mice compared with WT (Fig. 7A,B). However, no enhancement in PF-EPSC peak amplitude was seen in cells from 6-month-old β-III−/− animals, and recordings from 1-year-old β-III−/− animals reveal a decrease compared with WT cells (Fig. 7A,B). This indicates Purkinje cell excitation is initially enhanced, which would partly offset some of the intrinsic Purkinje cell defect, but then progressively declines in β-III−/− mice, compounding such deficits.
Figure 7.

Altered PF-EPSCs in β-III−/− mice. A, Left, Representative EPSC waveforms (4 V stimulus) from 6-week-old, 6-month-old, and 1-year-old WT and β-III−/− littermates. Right, Mean PF-EPSC amplitudes versus stimulus intensity shows consistent differences between WT and β-III−/− cells at different stimuli. B, Mean peak amplitude of EPSCs at 4 V stimulus shows changes in PF-evoked currents with age in β-III−/− cells (6 weeks: N = 3, n = 10, p = 0.001; 6 months: WT, N = 2, n = 10; β-III−/−, N = 2, n = 9, p = 0.44; 1 year: WT, N = 1, n = 6; β-III−/−, N = 2, n = 11, p = 0.05). C, Western blot analysis shows loss of GluR1 in 1-year-old but no change in 6-week-old β-III−/− mice.
Western blot analysis of cerebellar tissue from these mice shows no difference between genotypes in the level of GluR1, an AMPA receptor subunit, to account for the enhancement (97.7 ± 1.8% of littermate controls; N = 3 and 4; p = 0.3) (Fig. 7C). There is also no difference in paired-pulse facilitation between young β-III−/− and WT mice (1.25 ± 0.14 and 1.25 ± 0.02, respectively; p = 0.92) at 100 ms intervals, suggesting that a difference in release probability does not underlie the larger EPSCs. Instead, we found, similar to other published data (Takayasu et al., 2004), that application of TBOA, a global glutamate transporter antagonist, enhanced EPSC amplitude in young WT cells such that there was no longer a significant difference in amplitude between genotypes (p = 0.162). In contrast, a loss of AMPA receptors (64.5 ± 10% of WT; p = 0.038) (Fig. 7C) and a slight difference in paired-pulse facilitation (WT 1.4 ± 0.03; β-III−/− 1.29 ± 0.03; p = 0.02) is observed in 1-year-old β-III−/− animals.
Together, the results indicate that the decreased spontaneous firing rate arising from loss of β-III spectrin is, in young animals, partly offset by greater excitation. However, this declines in older animals, likely due to dendritic degeneration, and compounds the intrinsic defect. Ultimately, the combination of Purkinje cell death and reduced output from the surviving neurons leads to less inhibition onto the DCN and a progressive ataxic phenotype.
Discussion
Loss of function in disease pathogenesis
In this study, we show that loss of β-III spectrin produces a phenotype that resembles that of SCA5 patients with gait abnormalities, progressive motor coordination deficits, thinning of the molecular layer, and Purkinje cell loss (Stevanin et al., 1999; Bürk et al., 2004; Ikeda et al., 2006). Therefore, these results suggest that loss of β-III spectrin function underlies SCA5 pathogenesis, providing mechanistic insights into this autosomal-dominant disease. Although autosomal-dominant diseases were originally thought to result from toxic gain-of-function properties, recently several studies have implicated loss-of-function in the pathogenesis of other dominant neurodegenerative diseases (Van Raamsdonk et al., 2005; Thomas et al., 2006; Lim et al., 2008).
To date, heterozygous animals have displayed no obvious ataxic phenotype (our unpublished observation), arguing against haploinsufficiency as a disease mechanism and pointing toward the human mutations possessing dominant-negative effects on wild-type β-III spectrin function. Additional models in which β-III spectrin with a known SCA5 mutation is expressed should address this issue.
Sodium channel dysfunction may play a role in SCA5
Results from our in vitro current-clamp experiments revealed a substantial reduction in spontaneous Purkinje neuron activity in β-III−/− mice. It is known that several ion channels are essential for the sustained high-frequency tonic firing of Purkinje cells (Raman and Bean, 1999; Sacco et al., 2006; Zagha et al., 2008). Moreover, their loss in mice has been shown to cause ataxia (Sausbier et al., 2004; Akemann and Knopfel, 2006; Levin et al., 2006; Walter et al., 2006), and, more importantly, loss of ion channel function has been identified as a genetic defect in several SCA subtypes (Browne et al., 1994; Ophoff et al., 1996; Zhuchenko et al., 1997; Waters et al., 2006).
Here, we report that there are reduced sodium currents in acutely dissociated Purkinje neurons from β-III−/− mice. Of note, a mutation in the gene that encodes Nav1.6 has been found in a patient with cerebellar atrophy and ataxia (Trudeau et al., 2006), and βIV-spectrin has been shown to be required for correct localization and stabilization of voltage-gated sodium and potassium channels within axons and its loss results in progressive ataxia (Parkinson et al., 2001; Komada and Soriano, 2002). Together, these findings suggest that sodium channel dysfunction is a factor in the pathogenesis of some forms of SCA.
Spectrin is known to be a key factor in forming specialized membrane domains by linking transmembrane proteins such as ion channels and cell-adhesion molecules to membrane phospholipids and the actin cytoskeleton (Baines, 2009). Future analysis of β-III−/− mice will delineate the role β-III spectrin plays in assembling such domains as in its absence, or that of associated proteins (Jenkins and Bennett, 2001), sodium channels may no longer be maintained within specific subdomains, potentially contributing to Purkinje cell dysfunction. Alternatively, given β-III spectrin's putative role in facilitating protein transport (Holleran et al., 2001) sodium channels may be incorrectly trafficked.
Role for GLAST dysfunction in Purkinje cell degeneration
We found that in older β-III−/− mice, when motor deficits are more pronounced, there is, in addition to the loss of EAAT4 seen in young β-III−/− mice, a loss of the astroglial transporter GLAST and corresponding reductions in glutamate uptake. These results and the evidence of glutamate-mediated excitotoxicity in Purkinje cells of β-III−/− mice from 6 months onward suggest that it may be the delayed and progressive loss of GLAST activity that leads to Purkinje cell degeneration and the worsening ataxic phenotype. Therefore, a non-cell-autonomous mechanism involving GLAST dysfunction might be a pathogenic mechanism common to several SCAs. A complete loss-of-function mutation in EAAT1 (GLAST), with a dominant-negative effect on wild-type protein, was recently identified in a child with episodic and progressive ataxia (Jen et al., 2005), and this is in accordance with the finding that a GLAST knock-out mouse possesses motor deficits (Watase et al., 1998). Furthermore, Purkinje cell degeneration is also seen when expanded ataxin-7 is expressed solely in Bergmann glia, resulting in impaired glutamate uptake and loss of GLAST (Custer et al., 2006). Therefore, glutamate-mediated excitotoxicity, involving GLAST dysfunction, appears to play a critical role in Purkinje cell degeneration. Exactly how loss of β-III spectrin gives rise to this is not yet determined as β-III spectrin does not appear to be expressed in Bergmann glia (our unpublished observation). One possibility is that β-III spectrin, by orchestrating specialized microdomains in Purkinje cells, assembles a multiprotein transmembrane complex that maintains cell-to-cell contact with Bergmann glia and retains GLAST at the glial membrane. Alternatively, given the role of spectrin in vesicular transport, it may be that β-III spectrin is involved in the trafficking and secretion of trophic factors from Purkinje cells that modulate GLAST protein levels.
Cumulative effects of different insults important for disease progression
It is known that enhanced DCN hyperexcitability is sufficient to induce ataxia (Shakkottai et al., 2004), and so any reduction in GABAergic Purkinje cell output will contribute to motor deficits by reducing inhibition onto the DCN. However, although we identified a reduction in Purkinje cell intrinsic spontaneous firing rate in β-III−/− mice, this loss in output could not account for the increasing disease severity as the same degree of reduction was seen in 3-week-old and 6-month-old animals, which show mild and more severe motor deficits, respectively.
Therefore, it is apparent that additional factors are involved in modifying Purkinje cell output and affecting disease severity. Furthermore, these findings highlight the cellular complexity of cerebellar degeneration and ataxia. From the present study one contributing factor would appear to be altered excitatory inputs. Although previous work suggests EAAT4 to affect current decay (Takayasu et al., 2005), our results show that in young β-III−/− mice, loss of EAAT4, and not GLAST (Fig. 4A) corresponds with increased EPSCs (Fig. 7A,B). One possibility for the discrepancy is that there is less compensation from the other transporter subtypes in β-III−/− mice compared with EAAT4 knock-out mice. It may also be that GLAST is not in close opposition with dendritic spines, reducing rapid removal of glutamate. In contrast, prolonged EPSCs in 1-year-old β-III−/− animals correlate with a loss of GLAST protein, similar to published data (Marcaggi et al., 2003; Stoffel et al., 2004; Takayasu et al., 2005; Takatsuru et al., 2006). These recordings also confirm that the removal of synaptically released glutamate is retarded in old β-III−/− mice and raises the possibility that accumulation of glutamate results in AMPA receptor desensitization. Unexpectedly, given the loss of EAAT4, PF-EPSCs appear to decay faster in cells from young β-III−/− mice. Although both astroglial transporters are slightly elevated in young β-III−/− mice this is unlikely to be the sole reason for the faster decay, due to the small extent of protein increase and lack of increased glutamate uptake. Previous work has shown the sodium channel inhibitor QX-314 may not block all channel subtypes equally (Yeh, 1978). We saw no difference in decay constants between young WT and β-III−/− mice when recordings were performed in the absence of sodium channel blockers. It is possible, therefore, that in β-III−/− mice, due to the loss of sodium currents (Fig. 6B,C) there may be less of a contaminating current, precluding detection of slowing of the EPSC decay. A similar finding of briefer PF-EPSC decay times was observed in Nav1.6 knock-out animals (Levin et al., 2006).
Therefore, loss of EAAT4 may coincidentally have a biphasic effect on Purkinje cell physiology: initially, the loss is compensatory, maintaining Purkinje cell output and downstream DCN inhibition, resulting in only a mild ataxic phenotype. However, with time it elicits AMPA receptor-elicited excitotoxicity, resulting in dendritic degeneration and eventual cell death, both leading to progressively less Purkinje cell output and hyperexcitability of DCN. Although there are no reports that EAAT4 knock-out mice develop ataxia, a downregulation of EAAT4 is seen before any behavioral phenotype in two other models of ataxia, SCA1 transgenic mouse (Lin et al., 2000; Serra et al., 2004) and staggerer (Gold et al., 2003). Together, the results from these three different mouse ataxia models suggest that EAAT4 may indeed be a common factor in early disease pathogenesis. It appears that for normal glutamatergic neurotransmission a loss of EAAT4 is not deleterious, as GLAST is able to fully compensate, but when GLAST is lost (as here), the cumulative effect becomes pathogenic. A similar concerted role of EAAT4 and GLAST is apparent in brain ischemia where Purkinje cells with low EAAT4 levels are selectively lost in GLAST knock-out mice (Yamashita et al., 2006).
In conclusion, the β-III−/− mouse described here represents a new model of cerebellar ataxia, showing not only progressive behavioral abnormalities but also Purkinje cell degeneration. This is similar to clinical cases, therefore making it a valuable disease model for future study. The data also reveal that at least two separate disease pathways are involved in the loss of inhibitory cerebellar output underlying the ataxic phenotype. The first is a decrease in the tonic firing rate of Purkinje cells due to altered intrinsic membrane properties arising from reduced sodium currents. The second is a reduction in excitatory inputs and Purkinje cell death, possibly a consequence of glutamate-mediated excitotoxicity, arising from loss of EAAT4- and GLAST-mediated glutamate uptake. Therefore, β-III spectrin appears to play an important part in the prevention of glutamate-mediated neurotoxicity and to have a critical role in maintaining Purkinje cell tonic firing. Future analysis of this model will provide a greater understanding of what may be convergent disease mechanisms for various SCAs (i.e., ion channel dysfunction, glutamate-mediated excitotoxicity, and protein-trafficking defects). Insights into the human disease will be obtained by addressing whether known SCA5 mutations have similar detrimental effects to the present mouse model on either or both of the sodium channels or the astroglial glutamate transporter GLAST.
Footnotes
This work was supported by grants from The Wellcome Trust (077946), the National Institutes of Health (NS056158), and Research Councils UK (T27983). We thank Vladimir Buchman and Natalia Ninkina for the gift of ES cells and advice on their culturing, Peter Brophy and Diane Sherman for advice and assistance with blastocyst injections, Seth Grant for the mouse BAC library, Stephen Mitchell for assistance with electron microscopy, Trudi Gillespie from the IMPACT Imaging facility at the University of Edinburgh for technical assistance with microscopy, and Paul Skehel, Matt Nolan, and Laura Ranum for help and useful discussions.
References
- Akemann W, Knöpfel T. Interaction of Kv3 potassium channels and resurgent sodium current influences the rate of spontaneous firing of Purkinje neurons. J Neurosci. 2006;26:4602–4612. doi: 10.1523/JNEUROSCI.5204-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines AJ. Evolution of spectrin function in cytoskeletal and membrane networks. Biochem Soc Trans. 2009;37:796–803. doi: 10.1042/BST0370796. [DOI] [PubMed] [Google Scholar]
- Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA, Kramer P, Litt M. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet. 1994;8:136–140. doi: 10.1038/ng1094-136. [DOI] [PubMed] [Google Scholar]
- Bürk K, Zühlke C, König IR, Ziegler A, Schwinger E, Globas C, Dichgans J, Hellenbroich Y. Spinocerebellar ataxia type 5: clinical and molecular genetic features of a German kindred. Neurology. 2004;62:327–329. doi: 10.1212/01.wnl.0000103293.63340.c1. [DOI] [PubMed] [Google Scholar]
- Burright EN, Clark HB, Servadio A, Matilla T, Feddersen RM, Yunis WS, Duvick LA, Zoghbi HY, Orr HT. SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell. 1995;82:937–948. doi: 10.1016/0092-8674(95)90273-2. [DOI] [PubMed] [Google Scholar]
- Custer SK, Garden GA, Gill N, Rueb U, Libby RT, Schultz C, Guyenet SJ, Deller T, Westrum LE, Sopher BL, La Spada AR. Bergmann glia expression of polyglutamine-expanded ataxin-7 produces neurodegeneration by impairing glutamate transport. Nat Neurosci. 2006;9:1302–1311. doi: 10.1038/nn1750. [DOI] [PubMed] [Google Scholar]
- Dueñas AM, Goold R, Giunti P. Molecular pathogenesis of spinocerebellar ataxias. Brain. 2006;129:1357–1370. doi: 10.1093/brain/awl081. [DOI] [PubMed] [Google Scholar]
- Garthwaite G, Garthwaite J. Mechanisms of AMPA neurotoxicity in cerebellar and hippocampal slices. Eur J Neurosci. 1991;3:729–736. doi: 10.1111/j.1460-9568.1991.tb01669.x. [DOI] [PubMed] [Google Scholar]
- Gold DA, Baek SH, Schork NJ, Rose DW, Larsen DD, Sachs BD, Rosenfeld MG, Hamilton BA. RORalpha coordinates reciprocal signaling in cerebellar development through sonic hedgehog and calcium-dependent pathways. Neuron. 2003;40:1119–1131. doi: 10.1016/s0896-6273(03)00769-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarlund M, Jorgensen EM, Bastiani MJ. Axons break in animals lacking beta-spectrin. J Cell Biol. 2007;176:269–275. doi: 10.1083/jcb.200611117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häusser M, Clark BA. Tonic synaptic inhibition modulates neuronal output pattern and spatiotemporal synaptic integration. Neuron. 1997;19:665–678. doi: 10.1016/s0896-6273(00)80379-7. [DOI] [PubMed] [Google Scholar]
- He Y, Zu T, Benzow KA, Orr HT, Clark HB, Koob MD. Targeted deletion of a single Sca8 ataxia locus allele in mice causes abnormal gait, progressive loss of motor coordination, and Purkinje cell dendritic deficits. J Neurosci. 2006;26:9975–9982. doi: 10.1523/JNEUROSCI.2595-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holleran EA, Ligon LA, Tokito M, Stankewich MC, Morrow JS, Holzbaur EL. βIII spectrin binds to the Arp1 subunit of dynactin. J Biol Chem. 2001;276:36598–36605. doi: 10.1074/jbc.M104838200. [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Dick KA, Weatherspoon MR, Gincel D, Armbrust KR, Dalton JC, Stevanin G, Dürr A, Zühlke C, Bürk K, Clark HB, Brice A, Rothstein JD, Schut LJ, Day JW, Ranum LP. Spectrin mutations cause spinocerebellar ataxia type 5. Nat Genet. 2006;38:184–190. doi: 10.1038/ng1728. [DOI] [PubMed] [Google Scholar]
- Jackson M, Song W, Liu MY, Jin L, Dykes-Hoberg M, Lin CI, Bowers WJ, Federoff HJ, Sternweis PC, Rothstein JD. Modulation of the neuronal glutamate transporter EAAT4 by two interacting proteins. Nature. 2001;410:89–93. doi: 10.1038/35065091. [DOI] [PubMed] [Google Scholar]
- Jen JC, Wan J, Palos TP, Howard BD, Baloh RW. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia and seizures. Neurology. 2005;65:529–534. doi: 10.1212/01.wnl.0000172638.58172.5a. [DOI] [PubMed] [Google Scholar]
- Jenkins SM, Bennett V. Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J Cell Biol. 2001;155:739–746. doi: 10.1083/jcb.200109026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komada M, Soriano P. βIV-spectrin regulates sodium channel clustering through ankyrin-G at axon initial segments and nodes of Ranvier. J Cell Biol. 2002;156:337–348. doi: 10.1083/jcb.200110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin SI, Khaliq ZM, Aman TK, Grieco TM, Kearney JA, Raman IM, Meisler MH. Impaired motor function in mice with cell-specific knockout of sodium channel Scn8a (Nav1.6) in cerebellar Purkinje neurons and granule cells. J Neurophysiol. 2006;96:785–793. doi: 10.1152/jn.01193.2005. [DOI] [PubMed] [Google Scholar]
- Lim J, Crespo-Barreto J, Jafar-Nejad P, Bowman AB, Richman R, Hill DE, Orr HT, Zoghbi HY. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature. 2008;452:713–718. doi: 10.1038/nature06731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Antalffy B, Kang D, Orr HT, Zoghbi HY. Polyglutamine expansion downregulates specific neuronal genes before pathologic changes in SCA1. Nat Neurosci. 2000;3:157–163. doi: 10.1038/72101. [DOI] [PubMed] [Google Scholar]
- Marcaggi P, Billups D, Attwell D. The role of glial glutamate transporters in maintaining the independent operation of juvenile mouse cerebellar parallel fibre synapses. J Physiol. 2003;552:89–107. doi: 10.1113/jphysiol.2003.044263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Ommen GJ, Hofker MH, Ferrari MD, Frants RR. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543–552. doi: 10.1016/s0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- Parkinson NJ, Olsson CL, Hallows JL, McKee-Johnson J, Keogh BP, Noben-Trauth K, Kujawa SG, Tempel BL. Mutant β-spectrin 4 causes auditory and motor neuropathies in quivering mice. Nat Genet. 2001;29:61–65. doi: 10.1038/ng710. [DOI] [PubMed] [Google Scholar]
- Pielage J, Fetter RD, Davis GW. Presynaptic spectrin is essential for synapse stabilization. Curr Biol. 2005;15:918–928. doi: 10.1016/j.cub.2005.04.030. [DOI] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci. 1997;17:4517–4526. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J Neurosci. 1999;19:1663–1674. doi: 10.1523/JNEUROSCI.19-05-01663.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacco T, De Luca A, Tempia F. Properties and expression of Kv3 channels in cerebellar Purkinje cells. Mol Cell Neurosci. 2006;33:170–179. doi: 10.1016/j.mcn.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Sakaguchi G, Orita S, Naito A, Maeda M, Igarashi H, Sasaki T, Takai Y. A novel brain-specific isoform of β spectrin: isolation and its interaction with Munc13. Biochem Biophys Res Commun. 1998;248:846–851. doi: 10.1006/bbrc.1998.9067. [DOI] [PubMed] [Google Scholar]
- Sausbier M, Hu H, Arntz C, Feil S, Kamm S, Adelsberger H, Sausbier U, Sailer CA, Feil R, Hofmann F, Korth M, Shipston MJ, Knaus HG, Wolfer DP, Pedroarena CM, Storm JF, Ruth P. Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc Natl Acad Sci U S A. 2004;101:9474–9478. doi: 10.1073/pnas.0401702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra HG, Byam CE, Lande JD, Tousey SK, Zoghbi HY, Orr HT. Gene profiling links SCA1 pathophysiology to glutamate signaling in Purkinje cells of transgenic mice. Hum Mol Genet. 2004;13:2535–2543. doi: 10.1093/hmg/ddh268. [DOI] [PubMed] [Google Scholar]
- Shakkottai VG, Chou CH, Oddo S, Sailer CA, Knaus HG, Gutman GA, Barish ME, LaFerla FM, Chandy KG. Enhanced neuronal excitability in the absence of neurodegeneration induces cerebellar ataxia. J Clin Invest. 2004;113:582–590. doi: 10.1172/JCI20216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddhanta A, Radulescu A, Stankewich MC, Morrow JS, Shields D. Fragmentation of the golgi apparatus: a role for βIII spectrin and synthesis of phosphatidylinositol 4,5-bisphosphate. J Biol Chem. 2003;278:1957–1965. doi: 10.1074/jbc.M209137200. [DOI] [PubMed] [Google Scholar]
- Soong BW, Paulson HL. Spinocerebellar ataxias: an update. Curr Opin Neurol. 2007;20:438–446. doi: 10.1097/WCO.0b013e3281fbd3dd. [DOI] [PubMed] [Google Scholar]
- Stankewich MC, Tse WT, Peters LL, Ch'ng Y, John KM, Stabach PR, Devarajan P, Morrow JS, Lux SE. A widely expressed βIII spectrin associated with Golgi and cytoplasmic vesicles. Proc Natl Acad Sci U S A. 1998;95:14158–14163. doi: 10.1073/pnas.95.24.14158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevanin G, Herman A, Brice A, Dürr A. Clinical and MRI findings in spinocerebellar ataxia type 5. Neurology. 1999;53:1355–1357. doi: 10.1212/wnl.53.6.1355. [DOI] [PubMed] [Google Scholar]
- Stoffel W, Körner R, Wachtmann D, Keller BU. Functional analysis of glutamate transporters in excitatory synaptic transmission of GLAST1 and GLAST1/EAAC1 deficient mice. Mol Brain Res. 2004;128:170–181. doi: 10.1016/j.molbrainres.2004.06.026. [DOI] [PubMed] [Google Scholar]
- Takatsuru Y, Takayasu Y, Iino M, Nikkuni O, Ueda Y, Tanaka K, Ozawa S. Roles of glial glutamate transporters in shaping EPSCs at the climbing fiber-Purkinje cell synapses. Neurosci Res. 2006;54:140–148. doi: 10.1016/j.neures.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Takayasu Y, Iino M, Ozawa S. Roles of glutamate transporters in shaping excitatory synaptic currents in cerebellar Purkinje cells. Eur J Neurosci. 2004;19:1285–1295. doi: 10.1111/j.1460-9568.2004.03224.x. [DOI] [PubMed] [Google Scholar]
- Takayasu Y, Iino M, Kakegawa W, Maeno H, Watase K, Wada K, Yanagihara D, Miyazaki T, Komine O, Watanabe M, Tanaka K, Ozawa S. Differential roles of glial and neuronal glutamate transporters in Purkinje cell synapses. J Neurosci. 2005;25:8788–8793. doi: 10.1523/JNEUROSCI.1020-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PS, Jr, Fraley GS, Damian V, Woodke LB, Zapata F, Sopher BL, Plymate SR, La Spada AR. Loss of endogenous androgen receptor protein accelerates motor neuron degeneration and accentuates androgen insensitivity in a mouse model of X-linked spinal and bulbar muscular atrophy. Hum Mol Genet. 2006;15:2225–2238. doi: 10.1093/hmg/ddl148. [DOI] [PubMed] [Google Scholar]
- Trudeau MM, Dalton JC, Day JW, Ranum LP, Meisler MH. Heterozygosity for a protein truncation mutation of sodium channel SCN8A in a patient with cerebellar atrophy, ataxia and mental retardation. J Med Genet. 2006;43:527–530. doi: 10.1136/jmg.2005.035667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turmaine M, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW. Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington's disease (2000) Proc Natl Acad Sci U S A. 2000;97:8093–8097. doi: 10.1073/pnas.110078997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Pearson J, Rogers DA, Bissada N, Vogl AW, Hayden MR, Leavitt BR. Loss of wild-type huntingtin influences motor dysfunction and survival in the YAC128 mouse model of Huntington disease. Hum Mol Genet. 2005;14:1379–1392. doi: 10.1093/hmg/ddi147. [DOI] [PubMed] [Google Scholar]
- Walter JT, Alviña K, Womack MD, Chevez C, Khodakhah K. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci. 2006;9:389–397. doi: 10.1038/nn1648. [DOI] [PubMed] [Google Scholar]
- Watase K, Hashimoto K, Kano M, Yamada K, Watanabe M, Inoue Y, Okuyama S, Sakagawa T, Ogawa S, Kawashima N, Hori S, Takimoto M, Wada K, Tanaka K. Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur J Neurosci. 1998;10:976–988. doi: 10.1046/j.1460-9568.1998.00108.x. [DOI] [PubMed] [Google Scholar]
- Waters MF, Minassian NA, Stevanin G, Figueroa KP, Bannister JP, Nolte D, Mock AF, Evidente VG, Fee DB, Müller U, Dürr A, Brice A, Papazian DM, Pulst SM. Mutations in voltage-gated potassium channel KCNC3 cause degenerative and development central nervous system phenotypes. Nat Genet. 2006;38:447–451. doi: 10.1038/ng1758. [DOI] [PubMed] [Google Scholar]
- Yamashita A, Makita K, Kuroiwa T, Tanaka K. Glutamate transporters GLAST and EAAT4 regulate postischemic Purkinje cell death: an in vivo study using a cardiac arrest model in mice lacking GLAST or EAAT4. Neurosci Res. 2006;55:264–270. doi: 10.1016/j.neures.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Yeh JZ. Sodium inactivation mechanism modulates QX-314 block of sodium channels in squid axons. Biophys J. 1978;24:569–574. doi: 10.1016/S0006-3495(78)85403-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagha E, Lang EJ, Rudy B. Kv3.3 channels at the Purkinje cell soma are necessary for generation of the classical complex spike waveform. J Neurosci. 2008;28:1291–1300. doi: 10.1523/JNEUROSCI.4358-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, Dobyns WB, Subramony SH, Zoghbi HY, Lee CC. Autosomal-dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet. 1997;15:62–69. doi: 10.1038/ng0197-62. [DOI] [PubMed] [Google Scholar]