

Abstract
Secondary injury, the complex cascade of cellular events following spinal cord injury (SCI), is a major source of post-insult neuron death. Experimental work has focused on the details of individual factors or mechanisms that contribute to secondary injury, but little is known about the interactions among factors leading to the overall pathology dynamics that underlie its propagation. Prior hypotheses suggest that the pathology is dominated by interactions, with therapeutic success lying in combinations of neuroprotective treatments. In this study, we provide the first comprehensive, system-level characterization of the entire secondary injury process using a novel relational model methodology that aggregates the findings of ~250 experimental studies. Our quantitative examination of the overall pathology dynamics suggests that, while the pathology is initially dominated by “fire-like,” rate-dependent interactions, it quickly switches to a “flood-like,” accumulation-dependent process with contributing factors being largely independent. Our evaluation of ~20,000 potential single and combinatorial treatments indicates this flood-like pathology results in few highly influential factors at clinically realistic treatment time frames, with multi-factor treatments being merely additive rather than synergistic in reducing neuron death. Our findings give new fundamental insight into the understanding of the secondary injury pathology as a whole, provide direction for alternative therapeutic strategies, and suggest that ultimate success in treating SCI lies in the pursuit of pathology dynamics in addition to individually involved factors.
Key words: combination therapy, neuroprotection, secondary insult, spinal cord injury, therapeutic treatment window, traumatic brain injury
Introduction
Despite their promise, translating in vitro and in vivo experimental spinal cord injury (SCI) treatments into effective and repeatable clinical therapies has been problematic (Hall and Springer, 2004; Blight and Tuszynski, 2006; Faden and Stoica, 2007). It is therefore often concluded that the progression of neuronal death in secondary injury must be dominated by complex interactions, rather than any given single factor, and that the solution must therefore lie in multifaceted treatments aimed at simultaneously targeting several secondary injury factors (Hall and Springer, 2004; Faden and Stoica, 2007). However, the overall dynamics of the processes underlying the pathology remain unknown.
At a conceptual level, the secondary injury process is often thought to behave like a forest fire—that is, a propagating wave of death that results in a slowly expanding lesion, driven by multiple factors, often referred to as the necrotic-apoptotic continuum (PorteraCailliau et al., 1997). Thus, the assumption is that a critical intervention in one or more factors might arrest the propagation, thereby preventing subsequent damage. The most commonly pursued secondary injury factors can be categorized into excitotoxic, energetic, inflammatory, “necro-apoptotic,” and free radical. The excitotoxic factors arise from a cascade originating from the initial mechanical insult, leading to the direct disruption of ion gradients (e.g., sodium, calcium) and the escape of neurotransmitters such as glutamate (Agrawal and Fehlings, 1996; Schwab and Bartholdi, 1996; Park et al., 2004). These effects, in turn, cause activation of metabotropic and ionotropic receptors, further increasing external glutamate and internal calcium concentrations, thereby perpetuating the excitotoxic response (Agrawal and Fehlings, 1997; Park et al., 2004).
Energetic factors arise from the cell's attempt to maintain homeostasis in the face of the above cascade (Ahmed et al., 2002). Thus, cellular respiration falls off as mitochondrial dysfunction occurs (Sullivan et al., 2007) and local ATP concentrations decrease (Anderson et al., 1980), compromising the cell's energy supply (Sullivan et al., 2007) and hampering the ability of ionic pumping mechanisms, such as the Na-K-ATPase transporter (Faden et al., 1987; Li and Stys, 2001), to restore ionic homeostasis. Free radical factors, including nitric oxide (NO) (Merrill et al., 1993; Hamada et al., 1996) and reactive oxygen species (ROS) (Hall and Braughler, 1993), accumulate, damaging DNA. Necro-apoptotic (i.e., necrotic and apoptotic) factors arise from these damaged cells, as well as those with increased membrane permeability (Farkas et al., 2006; Shi and Whitebone, 2006) from membrane damage. Cells that do not die necrotically initiate apoptotic cascades (Crowe et al., 1997; Lu et al., 2000) via caspase and calpain activation (Crowe et al., 1997). Inflammatory factors are activated (Dusart and Schwab, 1994; Beattie, 2004), including microglia (Merrill et al., 1993; Gomes-Leal et al., 2004), macrophages (Giulian and Robertson, 1990), and astrocytes (O'Brien et al., 1994), resulting in the production of proinflammatory cytokines (Bartholdi and Schwab, 1997; Klusman and Schwab, 1997; Pineau and Lacroix, 2007). Other secondary injury factors include demyelination (Totoiu and Keirstead, 2005), oligodendrocyte death (Crowe et al., 1997), and axon damage (Shi and Whitebone, 2006).
Experimental investigation of individual secondary injury factors has resulted in a substantial, yet disparate pool of single factor data, making the interpretation of multi-factor effects difficult. Recently, we have developed a methodology (Mitchell and Lee, 2007) that greatly facilitates pooling disparate data, enabling a novel, comprehensive view into the pathology of secondary injury across time points, preparations, and protocols. We developed a system-wide relational model of secondary injury by aggregating the relevant relationships between factors commonly believed to be involved in the progression of secondary injury from over 250 experimental papers. This relational model represents a comprehensive view of the progression of neuron death following mechanical insult by directly incorporating the literature-derived experimental relationships into a network of time-varying factors. Thus, the dynamics of the entire secondary injury process, including potential treatments, can be quantitatively examined. This systems-based relational modeling approach encompasses and recapitulates experimental data without having to assume the detailed and cumbersome mathematics of numerous unknown mechanisms.
Methods
General strategy
The secondary injury model is characterized as a relational model (Fig. 1a), which uses intrinsic relationships identified in the experimental data to aggregate and recapitulate the findings of hundreds of experimental findings to make predictions regarding pathology dynamics and interactions over time. Based on over 250 research articles, we constructed a 20-output, 26-differential equation, 85-relationship system that transformed the individual experimentally derived relationships into a model that exhibited the known behaviors of secondary injury in the spinal cord.
FIG. 1.

Comprehensive pathology of secondary injury post-spinal cord injury (SCI). (a) The diagram represents the structure of the relational model, which is an embodiment of the published literature. The model permits cross-factor examination of the pathology and treatment responses of the secondary injury process. Boxes represent tracked factors in the model. Categories of factors shown in differing colors represent established theories from the literature regarding secondary injury: necro-apoptosis, energetics, excitotoxicity, free radicals, inflammation, and other. (b) The figure illustrates how the extraction of experimental relationships results in a relational model capable of making clinical and mechanistic predictions. Each arrow in (a) represents an experimentally derived relationship or “gain,” which is extracted from the experimental relationship between two factors. Altogether, these gains are used to form the relational model's differential equations, which transcribe the relationships into a network of time-varying factors.
The general relational modeling strategy utilizes the review-relate-refine technique as summarized in Table 1: review the literature to identify pertinent factors; relate the factors into a map transcribing a system of differential equations; and refine the model to meet validation criteria. Specific methodological and analytical details central to the secondary injury relational model are outlined in the sections of text below. As an aid to the reader, Table 2 summarizes pertinent terminology used to convey dynamical concepts, methodology, illustrations, and treatments, which appear throughout this article.
Table 1.
Relational Modeling Technique: Review-Relate-Refine
| Review |
| 1. Determine criteria for primary literature reference inclusion. |
| 2. Determine a base list of references and system factors for inclusion. |
| 3. Record references, categorized by factor, in an annotated database. |
| 4. Expand the scope of the literature base manual searches. Record new or additional factors. |
| Relate |
| 5. Devise a “map” that illustrates how identified factors are related. Include relevant system output(s). |
| 6. For each two-way relationship, extract a value from the literature that quantitatively describes the relationship (e.g., a gain). |
| 7. Translate the map into a system of equations. |
| Refine |
| 8. Validate using experimental data. |
| 9. Repeat steps 3–7 for areas that need improvement. |
Table 2.
Glossary of Terminology Used to Describe Dynamical Concepts, Methodology, Illustrations, and Treatments
| Dynamical concepts | |
| Fire | Describes rate-dependent dynamics in which a high degree of interaction between factors drives the propagation of the secondary injury process. |
| Flood | Describes accumulation-dependent dynamics in which the accumulation of independent factors drives the propagation of the secondary injury process. |
| Methodology | |
| Relational model | Aggregates multiple two-way experimental relationships at discrete time points to predict the interactions and dynamics of all involved factors over a continuous time frame. |
| Relational analysis | Set of analytical techniques that evaluates and subsequently uses the relationships among parameters, variables, and especially model outputs to hypothesize process dynamics, mechanisms, and/or functions (Mitchell and Lee, 2007). |
| Factor | Quantifiable entity or output that has a measurable impact on the process outcome. |
| Factor category | Set of factors, which have been grouped together as “similar” by scientists in the field, based on their function, mechanism, or impact on process outcome. |
| Factor gain | Value that quantitatively specifies the one-way impact of an inter-related or interacting factor. |
| Factor time constant | Calculated using the factor peak value over its experimentally measured range and used to form the factor's differential equation. |
| Dynamics illustration | |
| Landscape | Matrix of correlations, which quantifies the inter-relatedness of model outputs (or factors) and is representative of their degree of interaction (i.e., a measure of “fire”). |
| Pathology diagram | A map/survey of the overall system operation, including the changes in factor size, impact, and “flow” (i.e., a measure of accumulation). |
| Treatments | |
| Reducing treatment | Targets factor accumulation by reducing the existing factor (e.g., a free radical scavenger actively reduces existing free radicals) |
| Inhibiting treatment | Targets the interactions by inhibiting formation of a factor (e.g., a free radical anti-oxidant inhibits the formation of free radicals). |
| Single [factor] treatment | A single treatment, either reducing or inhibiting, applied independently to target one factor. |
| Combination treatment | An n-number of inhibiting or reducing treatments given in combination targeting n-number different factors. |
Derivation of equations
Differential equations are of the standard Euler form. The derivative for each factor at each time step is calculated using its relationships to the other factors and then integrated numerically. Every arrow pointing to a specific factor in Figure 1 represents a relationship between the two factors (for illustration of extraction method, see Fig. 1b). For example, NMDA activation is mediated by calcium and glutamate. The relationships between factors are taken or measured from the experimental data, and are effectively linear gains denoted by “G” (Table 3). Similarly, time constants for each factor, denoted by τ, are calculated from experimental data and represent the time constants for the acute and subacute secondary injury periods (Table 4). Therefore, for our example with NMDA we have:
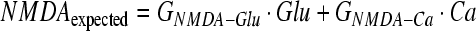 |
(1) |
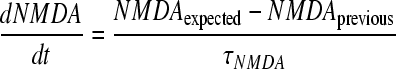 |
(2) |
Table 3.
Table of Model Gains for the Calculation of Secondary Injury Factorsa
| Gain | Value | Reference | In vitro | In vivo | TBI | SCI | Other |
|---|---|---|---|---|---|---|---|
| (A) Energetics | |||||||
| ATP-mitochondria | 0.40 | (Nicholls and Budd, 2000) | × | × | |||
| ATP-NaKATPase factor | 1.00 | (Green and Kroemer, 1998) | × | × | |||
| Blood-blood | 0.50 | (Yanase et al., 1995) | × | × | |||
| Glucose-blood | 0.50 | (Anderson et al., 1980) | × | × | |||
| Mitochondria-calcium | 0.10 | (White and Reynolds, 1996) | × | × | |||
| Mitochondria-glutamate | 0.15 | (Ankarcrona et al., 1995) | × | × | |||
| Mitochondria-ROS | 0.05 | (Azbill et al., 1997) | × | × | |||
| Mitochondria-sodium | 0.40 | (Iwai et al., 2002) | × | × | |||
| (B) Excitotoxicity | |||||||
| AMPA-calcium | 0.10 | (Yanase et al., 1995) | × | × | |||
| AMPA-glutamate | 0.55 | (Saftenku, 2005) | × | ||||
| Calcium-AMPA | 0.23 | (Carriedo et al., 1998) | × | × | |||
| Calcium-calcium (uptake) | −3.50 | (Wingrave et al., 2003) | × | × | |||
| Calcium-mitochondria | 1.50 | (Wingrave et al., 2003) | × | × | |||
| Calcium-NMDA | 0.28 | (Carriedo et al., 1998) | × | × | |||
| Calcium-membrane damage | 0.25 | (Yoshioka et al., 1996) | × | × | × | ||
| Glutamate-AMPA | 1.10 | (Saftenku, 2005) | × | ||||
| Glutamate-glutamate (uptake) | −3.50 | (Xu et al., 2004) | × | × | |||
| Glutamate-NMDA | 1.30 | (Mitchell et al., 2007) | × | ||||
| Glutamate-membrane damage | 0.25 | (LaPlaca and Thibault, 1998) | × | × | |||
| Glutamate-ROS | 0.02 | (Volterra et al., 1994) | × | × | |||
| NMDA-calcium | 0.10 | (Zhang et al., 1996) | × | × | |||
| NMDA-glutamate | 0.60 | (Mitchell et al., 2007) | × | ||||
| Sodium-AMPA | 1.10 | (Agrawal and Fehlings, 1996) | × | × | |||
| Sodium-NaKATPase | 1.00 | (Agrawal and Fehlings, 1996) | × | × | |||
| Sodium-NMDA | 1.30 | (Agrawal and Fehlings, 1996) | × | × | |||
| Sodium-membrane damage | 0.10 | (Schwartz and Fehlings, 2001) | × | × | |||
| (C) Inflammation | |||||||
| Astrocyte-calcium | 0.30 | (Schnell et al., 1999) | × | × | × | ||
| Astrocyte-cytokine | 2.00 | (Gomes-Leal et al., 2004) | × | × | |||
| Astrocyte-glutamate | 0.50 | (Schnell et al., 1999) | × | × | × | ||
| Cytokine-astrocyte | 1.20 | (Klusman and Schwab, 1997) | × | × | |||
| Cytokine-microglia | 2.00 | (Klusman and Schwab, 1997) | × | × | |||
| Macrophage-microglia | 4.50 | (Tian et al., 2007) | × | × | |||
| (Popovich et al., 2002) | × | × | × | × | |||
| Microglia-demyelination | 0.33 | (Blight, 1985) | × | × | |||
| Microglia-macrophage | 0.30 | (Dusart and Schwab, 1994) | × | × | |||
| Microglia-NO | 0.20 | (Zhao et al., 2004) | × | × | |||
| Sodium-NaKATPase | 1.00 | (Agrawal and Fehlings, 1996) | × | × | |||
| (D) Free radicals | |||||||
| NO-microglia | 0.27 | (Merrill et al., 1993) | × | × | |||
| NO-oligodendrocyte | 0.57 | (Zhao et al., 2004) | × | × | |||
| ROS-AMPA | 2.50 | (Carriedo et al., 1998) | × | × | |||
| ROS-cytokine | 0.40 | (Hu et al., 1997) | × | × | × | ||
| ROS-mitochondria | 0.20 | (Azbill et al., 1997) | × | × | |||
| ROS-NMDA | 2.00 | (Carriedo et al., 1998) | × | × | |||
| ROS-NO | 0.10 | (Mattiasson, 2004) | × | × | |||
| (E) Necro-apoptosis | |||||||
| Caspase-calcium | 2.00 | (Wingrave et al., 2003) | × | × | |||
| Caspase-glutamate | 0.20 | (Liu et al., 1999) | × | × | |||
| Caspase-mitochondria | 0.20 | (Krajewski et al., 1999) | × | × | × | ||
| Caspase-membrane damage | 1.00 | Kacy Cullen, PhD and Michelle LaPlaca, PhD (unpublished data) | × | × | |||
| Neuron-caspase factor | 2.43 | (Hartmann et al., 2000) | × | × | |||
| Neuron-macrophage | 2.03 | (Tian et al., 2007) | × | × | |||
| Neuron-mitochondria | 0.16 | (Sullivan et al., 2007) | × | × | |||
| Neuron-oligodendrocyte | 0.27 | (Zhao et al., 2004) | × | × | |||
| Neuron-membrane damage | 0.81 | (Cullen and LaPlaca, 2006a) | × | × | |||
| Membrane damage-caspase | 1.00 | (Wingrave et al., 2003) | × | × | |||
| Membrane damage-ROS | 1.00 | (Mattiasson, 2004) | × | ||||
| (F) Other | |||||||
| Axon-microtubule | 2.00 | (Pettus and Povlishock, 1996) | × | × | |||
| Axon-neurofilament | 2.00 | (Pettus and Povlishock, 1996) | × | × | |||
| Axon-membrane damage | 0.20 | (Pettus and Povlishock, 1996) | × | × | |||
| Demyelination-axon | 0.30 | (Lovas et al., 2000) | × | × | |||
| Demyelination-oligodendrocyte | 3.00 | (Kandel et al., 2000) | × | ||||
| Neuron-glutamate | 0.09 | (Ankarcrona et al., 1995) | × | × | |||
| Oligodendrocyte-AMPA | 0.89 | (Yoshioka et al., 1996) | × | × | × | ||
| Oligodendrocyte-cytokine | 0.50 | (Louis et al., 1993) | × | × | |||
| Oliogodendrocyte-demyelination | 0.20 | (Yoshioka et al., 1996) | × | × | × | ||
| Oliogodendrocyte-NO | 0.10 | (Merrill et al., 1993) | × | × | |||
How to read the table: Each gain represents the relationship between two factors as shown in Figure 1. The first part of the hyphenated gain name states the name of the factor being calculated while the second part of the gain name states the influencing factor that the gain relates. See main text (equation 1) for the example with NMDA. TBI, traumatic brain injury; SCI, spinal cord injury; ROS, reactive oxygen species. (A) Energetic gains. (B) Excitotoxicity. (C) Inflammation. (D) Free radicals. (E) Necro-apoptosis. (F) Other.
Table 4.
Time Constants Used in the Model
| Factor | Fast (h) | Slow (h) | Reference | In vitro | In vivo | TBI | SCI |
|---|---|---|---|---|---|---|---|
| NMDA activation | 0.33 | 3.35 | (Zhang et al., 1996) | × | × | ||
| AMPA activation | 0.33 | 3.35 | (Goforth et al., 2004) | × | × | ||
| NaKATPase transporter | 0.67 | 24.00 | (Faden et al., 1987) | × | × | ||
| (Anderson et al., 1980) | × | × | |||||
| ROS | 2.5 | 24.0 | (Hall and Braughler, 1993) | × | |||
| (Hamada et al., 1996) | × | × | |||||
| Glutamate concentration | 0.22 | 3.35 | (Xu et al., 2004) | × | × | ||
| (Liu et al., 1999) | × | × | |||||
| Calcium concentration | 0.22 | 3.35 | (LaPlaca and Thibault, 1998) | × | × | ||
| Sodium concentration | 1.00 | 24.00 | (Lemke et al., 1987) | × | × | ||
| (Fehlings and Agrawal, 1995) | × | × | |||||
| Mitochondria dysfunction | 1.00 | 24.00 | (Alano et al., 2002) | × | × | ||
| ATP concentration | 0.67 | 24.00 | (Anderson et al., 1980) | × | × | ||
| Membrane damage | 0.67 | 24.00 | (Shi and Whitebone, 2006) | × | × | ||
| (Cullen and LaPlaca, 2006b) | × | × | |||||
| (Barut et al., 2005) | × | × | |||||
| Microglia activation | — | 4.00 | (Dusart and Schwab, 1994) | × | × | ||
| (Carlson et al., 1998) | × | × | |||||
| (Vela et al., 2002) | × | × | |||||
| Cytokine concentration | — | 12.00 | (Pineau and Lacroix, 2007) | × | × | ||
| (Klusman and Schwab, 1997) | × | × | |||||
| Astrocyte activation | 6.7 | 5.00 | (O'Brien et al., 1994) | × | × | ||
| Macrophage activation | — | 5.00 | (Carlson et al., 1998) | × | × | ||
| (Vela et al., 2002) | × | × | |||||
| (Fleming et al., 2006) | × | × | |||||
| Oligodendrocyte death | — | 12.00 | (Crowe et al., 1997) | × | × | ||
| Demyelination | — | 24.00 | (Totoiu and Keirstead, 2005) | × | × | ||
| Nitric oxide | — | 8.00 | (Xiong et al., 2007) | × | × | ||
| Caspase activation | 5.4 | 24.00 | (Springer et al., 1999) | × | × | ||
| Axonal damage | — | 12.00 | (Pettus and Povlishock, 1996) | × | × | ||
| Neuron death | 1.4 | 12.00 | (Gaviria et al., 2006) | × | × | ||
| 24.00 | (Fujiki et al., 2005) | × | × |
Fast time constants are used for the acute (<1 h post-insult) and slow time constants are used for the sub-acute period (>1 h and <16 h). TBI, traumatic brain injury; SCI, spinal cord injury; ROS, reactive oxygen species.
The one non-linear exception to the form of Equation 1 is for the factor ATP (Equation 3), which reflects the production of ATP by the mitochondria and the consumption of ATP by the Na-K-ATPase pump. However, the Euler differential equation still has the same form as the other factors as shown in Equation 2.
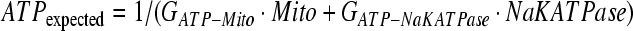 |
(3) |
The model was split into two parts to better mathematically represent the fast or “acute” period (<1 h after injury) and slow or “subacute” (>1 and <16 h after injury). The acute and subacute parts each have their own time constants (Table 4) to represent the changing dynamics seen experimentally between these two time periods. However, depending on a factor's split dynamics, it may only exhibit substantive changes in one period or the other. For a validated comparison between simulated factor values and experimental data, see Table 5. Note that, by splitting the time constants into smaller time frames, the relationship equations, like Equation 1, can be safely approximated as piece-wise linear. However, this linear approximation does not specify that the resulting trajectories of factor values be linear, as shown in the results in Figure 2.
Table 5.
Factor Validation Comparison to Experimental Data
| Factor | Model value | Exp. value | Time point (h) | Reference | In vitro | In vivo | TBI | SCI |
|---|---|---|---|---|---|---|---|---|
| NMDA activation | 2.53 | 2.5 | <1 | (Zhang et al., 1996) | × | × | ||
| AMPA activation | 2.31 | 2.2 | <1 | Goforth et al., 2004 | × | × | ||
| 2.5 | 1 | (Li and Stys, 2001) | × | × | ||||
| NaKATPase transporter | 0.67 | 0.7 | 24 | (Faden et al., 1987) | × | × | ||
| 0.7 | 24 | (Li and Stys, 2001) | × | × | ||||
| ROS | 5.11 | 4–5 | 1 | Hall and Braughler, 1993 | × | |||
| 1.77 | 2.5 | 0.5 | Hamada et al., 1996 | × | × | |||
| Glutamate concentration | 4.1 | 4–7 | 0.75 | (Xu et al., 2004) | × | × | ||
| Calcium concentration | 2.5 | 2.5–3 | 4 | (Wingrave et al., 2003) | × | × | ||
| Sodium concentration | 2.43 | 2 | 1 | Lemke et al., 1987 | × | × | ||
| Mitochondria dysfunction | 1.3 | 1.2 | 6 | (Sullivan et al., 2007) | × | × | ||
| ATP concentration | 0.8 | 0.7 | 1 | Anderson et al., 1980 | × | × | ||
| Membrane damage | 4.5 | 2–7 | <1 | (Choo et al., 2007) | × | × | ||
| Microglia activation | 3.6 | 3 | 6 | (Dusart and Schwab, 1994) | × | × | ||
| Cytokine concentration | 2.6 | 2–5 | 6 | (Bartholdi and Schwab, 1997) | × | × | ||
| Astrocyte activation | 2.1 | 2–4 | 4 | O'Brien et al., 1994 | × | × | ||
| Macrophage activation | 11.1 | 11 | 16 | (Giulian and Robertson, 1990) | × | × | ||
| Oligodendrocyte death | 1.7 | 2 | 6 | Crowe et al., 1997 | × | × | ||
| Demyelination | 2.7 | 2.5 | 12 | Totoiu and Keirstead, 2005 | × | × | ||
| Nitric oxide | 1.5 | 2–5 | 6 | (Merrill et al., 1993) | × | × | ||
| Caspase activation | 2.5 | 3–3.5 | 4 | (Wingrave et al., 2003) | × | × | ||
| 3.3 | 6 | (Springer et al., 1999) | × | × | ||||
| Axonal damage | 2.2 | >2 | 6 | (Pettus and Povlishock, 1996) | × |
The model is “unitless” in that the model generates factor values that are ratios to the baseline values, with all baseline values (values immediately post-insult) starting at one. For example, a factor value of three means that the factor value is three times the baseline value. References listed in bold type indicate primary (i.e., external or independent) validation criteria in which no data from the reference was used to calculate the corresponding validated factor value. References in italic type indicate secondary validation criteria from which only time constant information was extracted. Thus, these secondary references had no impact on their corresponding resulting factor relationships. The remaining references in regular type, with the exception of axonal damage, indicate tertiary validation criteria from which the indicated reference was only one of several references that data was extracted from as part of the calculation of the corresponding factor value. Thus, these tertiary references have only a partial role in determining the impact of their listed factor values and resulting relationships. Limitations imposed by the quantity, applicability and extractability of available data make the prediction and validation of axonal damage more difficult than the other factors. TBI, traumatic brain injury; SCI, spinal cord injury; ROS, reactive oxygen species.
FIG. 2.

Progression of factors over time. For illustration purposes, the trajectories shown are relative to one another in that factors are scaled based on the average of all 19 factors. For comparison of specific factor values to experimental values at specified time points, see Table 5. Each category of factors is highlighted in an individual panel using a monochromatic color scheme that aligns with the category colors used in Fig. 1a. Light gray lines in the background represent the nonhighlighted factors in each respective panel and are shown for the purpose of comparing the different factor and factor category trajectories. (a) Excitotoxic factors. (b) Energetic factors. (c) Inflammatory factors. (d) Necro-apoptotic factors. (e) Free radical factors. (f) Other factors.
Parameter value extraction
When using experimental literature to obtain parameter values (Tables 4 and 5), primary reference selection was based on the quantifiability of the parameter. All parameters were extracted from central nervous system data, and when possible, data measured in the spinal cord and SCI. However, when insufficient quantifiable data was not available from the spinal cord literature, parameters were extracted from the traumatic brain injury literature. This distinction is made in the tables.
Justification for factor inclusion/exclusion
This model is based on what is already known about secondary injury. Therefore, factors included in the model were limited to those known to contribute to secondary injury during the studied time frame and for which there was sufficient experimental data available for obtaining parameter values and validating results. Consequently, mediation factors (factors that mediate cell death), which have been understudied in this early time frame, are difficult to include. Thus, such explicit factors, like remyelination, were excluded from the relational model. Some factors of secondary injury, such as inflammation, are thought to have mediating as well as deleterious effects. In the model presented here, only the deleterious effects are explicitly included, mainly due to the lack of consistent, quantitative information currently available. Thus, it is implicitly assumed that the impact of mediation factors during the time frames examined in this study is negligible. However, we do not discount their potential importance; in the future, as more experimental information becomes available, inclusion may be appropriate.
In general, this study only includes direct factors for which experimental data among various studies is qualitatively consistent. If there was discordant experimental data for a factor, the factor was not directly or explicitly included, but rather implicitly modeled using related indirect factors and/or mechanisms for which experimental data was qualitatively consistent. For example, we recognize the potential importance of cell volume regulation, or edema, in the secondary injury process. However, conflicting experimental results make direct inclusion as an individuated factor very difficult. Several studies have documented a 2–3% change in spinal cord volume post-SCI, and each of these studies states to have reduced this volume change by approximately the same amount using very different methods corresponding to different potential mechanisms—by reducing NMDA receptor activation (Churchwell et al., 1996), macrophage/microglia activation (Tian et al., 2006), ATP depletion (Jurkowitzalexander et al., 1992), and sodium (Ates et al., 2007), to name a few. Thus, in this study, we elect to model the effects of edema indirectly by inclusion of these aforementioned indirect mechanisms. Without a direct connection between edema and neuron death, it is possible that factors likely to contribute to edema, such as sodium, may have their impact on neuron death slightly underestimated, but the actual values of the indirect factors themselves remain in line with experimental data (Table 5).
Other model assumptions and limitations
It is true that there are implicit assumptions with each factor that are inherently associated with the conditions, assumptions and limitations associated with the experiment from which each gain was extracted, including the experimental model type and the time frame of data collection (for a list of such publications, see Table 3). There are also limitations based on the information available for a certain factor. Another implicit assumption lies in the limitation imposed during data extraction from the literature. However, the more general assumptions of the model are: (1) A quantitative experimental correlation specifies an interaction which can be modeled in differential form resulting in both inherent and emergent predictions which reflect the interactive and temporal dynamics of the process. (2) No “events” occur between the discrete time points extracted experimentally, and thus temporal dynamics can be interpolated between discrete points by using an experimentally derived time constant in the translated differential equations.
Sensitivity analysis
A sensitivity analysis was performed by varying each parameter individually by a specified amount (±50% in eight 6.25% increments) to measure its effect on the model output (i.e. factor and neuronal death) values. Sensitivity data was used to obtain the correlations between factors for the landscape. Additionally, the sign of the slope of the linear regression between each gain and neuronal death was used to calculate which direction a gain must be moved to minimize neuron death when specifying inhibiting treatments.
Secondary injury landscapes
The landscapes reveal the inter-relatedness of the factors and thus are illustrative of interaction dynamics. The correlation matrix, which forms the landscape, consists of correlation values obtained by correlating all outputs against one another based on the sensitivity analysis data. Note that landscapes are based on peak or maximal impact of each factor rather than ending impact. “Maximal impact” was defined as the minimum factor value occurrence over 12 h for factors that decrease with neuron death (e.g., the Na-K-ATPase transporter and ATP) and the maximum factor value for the remaining 17 factors, which increase with neuron death. Neuron death values for each sensitivity analysis run were taken at their maximal value (i.e., at 12 h).
Since any given output will correlate perfectly with itself, the correlation matrix contains a diagonal line of identity and is symmetric along the diagonal axis of the square. Correlations range from zero to one, with zero being completely uncorrelated and one completely correlated (for method details see Mitchell and Lee, 2007). The factors were sorted based on their correlation coefficients using hierarchical cluster analysis such that the most correlated factors were located near each other in the landscape. This sorting does not change the correlations in the landscape but rather makes correlations easier to illustrate and readily identifies “groups” of related factors based on correlation.
Pathology diagrams
Pathology diagrams serve not only as a map but also as a survey of the overall system operation, including the changes in factor size, impact, and “flow.” Since the purpose of the diagrams is to provide insight into the operation of the system as a whole, additional scaling was applied to determine, for example, line thicknesses and saturation levels. The overall intent was to scale in a manner that kept all lines and boxes visible and yet still provide meaningful individuation of effect. Qualitatively, greater intensity (darkness/saturation) indicates greater impact on neuron death with the scaling being roughly logarithmic. Thus, each increment in intensity is approximately a 2.5-fold increase in impact on neuron death. In contrast, size (box or line) is an indicator of magnitude. Box size (area) scaling is roughly linear with factor magnitude relative to its peak value. Line thickness scaling is roughly logarithmic as impacts range over approximately five orders of magnitude. Thus, each increment in thickness is worth approximately a 2.7-fold increase in impact.
Inhibiting treatments (treatments that inhibit the growth of a factor) were simulated by co-varying all of the experimentally derived relationships (gains) directly governing a given factor (i.e., all parameters, excluding the time constants, appearing in the mathematical calculation of a factor) by a specified amount or “dose” in a direction that reduces neuron death. For inhibiting treatments, doses were simulated by moving the individual gains, G, that govern a factor or combination of factors between 1% and 95%. The direction that each individual gain must be varied to reduce neuron death was determined from a parameter sensitivity analysis.
Reducing treatments (treatments that directly reduce a factor) were performed by subtracting a factor-dependent “dose,” by multiplying the current factor value at each time step by a “reducing gain” that was roughly based on the sum of all gains (Gtotal, factor) for each factor. However, to facilitate comparison, the exact scaling of reducing gains was set so that reducing and inhibiting resulted in the same effect at very small dosing levels under the premise that reducing and inhibiting should become indistinguishable as dosing approaches zero. The specific scaling point was a 1% inhibiting treatment dose at time zero. Thus, a 1% reducing treatment at time zero was defined as the gain that produced the same change in neuron death as the 1% inhibiting treatment. Reducing gains were then varied between 1% and 1000%. Summarizing by continuing with our example with NMDA, the concentration-dependent dose that would be subtracted from Equation 2 to reduce NMDA is represented by Equation 4, where SNMDA is the applied scaling factor as described above.
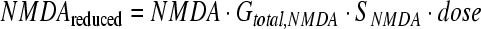 |
(4) |
Combination treatments
A factorial design was used to test single factor and multifactor (simultaneously varying two to five factors) treatment combinations. The maximum of five was determined by a dimensionality analysis of the system (Mitchell and Lee, 2007).
Implementation
The model is implemented in MATLAB R2007a (The Mathworks, Inc., Natick, MA). Secondary injury simulations, sensitivity analyses, cross-correlation analysis for the landscape, and treatments were performed in MATLAB. Hierarchical clustering analysis for the sorting of the factors in the landscape was performed in Systat (Systat Software, Inc., Chicago, IL). Pathology diagrams were created in SmartDraw (www.SmartDraw.com).
Results
Characterization of overall pathology
A key goal of this study was to examine the overall pathology of secondary injury at the system level, including the process dynamics. As a starting point, two generalized mechanisms of secondary injury propagation were examined. The first is a rate-dependent process, similar to a burning forest fire, where damage is driven by interactions between factors. Using this analogy, a fire is critically dependent upon the interactions between, for example, fuel availability, wind speed, and humidity, and even small changes in any one of these can have dramatic effects on the fire's progression and the extent of its damage. The second mechanism is an accumulation-dependent process that is analogous to a rising flood, where damage is driven by the accumulation of factors. Using this analogy, the flood is dependent upon summation over time of, for example, flow rates and geographic contours, and small changes in these factors generally result in only small effects on the overall flood and extent of its damage. The prevailing view of secondary injury would be akin to the fire analogy. Thus, our initial expectation was that the system would be driven by ongoing, rate-dependent interactions of factors.
We began our examination of the secondary injury pathology by investigating the time course of individual factors and neuron death. The primary model output used to signify the propagation of secondary injury is neuron death as a function of time. In actuality, “neuron death” represents the aggregation of all indicators of dead, dying and/or marked for death neurons. As such, it encompasses both the volume affected and the fraction of dying cells within that volume. In the first hour post-insult, the model predicts approximately a threefold increase in neuron death. This first hour shows greatest activity in the excitotoxic factors (Fig. 2) whose relationships were predominately based on in vitro literature. Subsequently, neuron death increases at a slower, but still substantial pace resulting in an additional threefold increase over the next fifteen hours. This subacute period shows substantial activity in the necro-apoptotic and inflammatory factors (Fig. 2) and is based heavily on in vivo literature. The model was validated by comparing its output to experimental data (Table 5), and especially, when possible, to experimental data not used as part of the model construction. Based on comparison to experimental data, the model appears to be valid out to 16–18 h post-insult. However, the dearth of experimental data points between 12 and 24 h make a precise determination difficult. Consequently, we limit our examination to the first 12 h post-insult.
Next, we assessed the simultaneous inter-relationships (i.e., correlations) among the 19 factors and neuron death to obtain “snapshots” of the entire secondary injury process over time (Fig. 3). By looking at how these snapshots change over time, we can visualize the dynamics of propagation. The landscapes (left, Fig. 3) represent both a summary of the two-way, experimentally observed correlations, and the model's predictions regarding broader interactions among the factors. The corresponding pathology diagrams (right, Fig. 3) indicate the relative flow and impact of factors, and thus represent the accumulation of factors over time. Initially, all factors are tightly coupled as denoted by the widespread, intense block of correlations in the landscape. This tight coupling results in a one-dimensional behavior that is indicative of a process that is dominated by interactions (i.e., the fire). However, with time, the system decouples as the effects of interactions diminish. Simultaneously, the effects of factor accumulation rise, eventually dominating the process as indicated in the pathology diagrams, and resulting in a pathology that behaves like multiple independent floods.
FIG. 3.

Analysis of the fire versus flood dynamics of secondary injury pathology. (Left) Landscape of correlations quantifying the strengths of the inter-relationships or interactions among the factors and neuron death. Correlation magnitudes are represented by the grayscale color, and range from zero (white, uncorrelated) to one (black, completely correlated). Colors on the axes represent the category to which the factors belong as denoted in Figure 1. The matrix contains a diagonal line of unity, which has been removed for clarity. (Right) Pathology diagram signifying the “flow” versus accumulation of factors and their impact on neuron death. Arrow line thickness illustrates the effect of one factor on another while line darkness represents the impact of that effect on neuron death. The inner, colored box size illustrates “accumulation” of a factor and is scaled by the factor's maximum, represented by its outline. Box color saturation symbolizes the impact of the factor on neuron death. (a) One-hour snapshot. All factors in the landscape are highly correlated, indicative of the very large interactions associated with a fire, with only minimal factors showing substantial accumulation in the pathology diagram. (b) Two-hour snapshot. The system shows substantial decoupling in the landscape, and an increase in the number of accumulating factors in the diagram, indicating a mixed rate and accumulation-dependent pathology (i.e., fire and flood). (c) Eight-hour snapshot. The system is functionally decoupled, and accumulation clearly dominates, indicative of pathology consisting of several independent floods.
Our examination of secondary injury during the first 12 h post-insult has revealed that, while the commonly held view of the pathology as a propagating fire is consistent with the system behavior initially, it quickly transitions into “flood” dynamics where the accumulation of factors over time dominates neuron death (Fig. 4). Notably, this transition occurs relatively early, as a substantial majority of neuron death occurs during the flood phase.
FIG. 4.

Summary of secondary injury pathology dynamics: acute fire versus the subacute flood. The pathology dynamics consist of an early, acute fire of interactions chiefly dominated by excitotoxic factors followed by a larger, subacute flood of accumulating factors chiefly dominated by necro-apoptotic and inflammatory factors. The relative size of the arrows indicates the relative impact of the corresponding factor category on neuron death. Excitotoxicity, necro-apoptosis, and inflammation all have a substantial impact on neuron death, while energetics, free radicals, and other factors have a still significant but smaller impact on neuron death. Time of impact is indicated in parentheses.
Single factor treatments
With the above view of the overall pathology in mind, we began our examination of hypothetical treatments by inhibiting the growth of single factors by doses ranging from 10% to 95% inhibition (e.g., a 50% dose would be expected to reduce the growth of the factor by 50% if all else remained the same). To determine the maximum possible impact of each factor on neuron death, calculated single factor treatments were initiated at time zero (i.e., simultaneous with the insult; Fig. 5A). Based on a 50% inhibiting dose, the impact of these single factor treatments ranges from negligible to nearly a 20% decrease in neuron death. However, treatment efficacy drops rapidly with time post-insult (Fig. 5B). Notably, while the impact of all factors decreased substantially with treatment time, several factors that had been highly effective when treatment was initiated at time zero drop precipitously, making them low prospects as the basis for clinical treatment. The net result is that the top five single factor treatments in the 2–8-h treatment initiation window are phagocytes (e.g., macrophages, neutrophils), immune activation (e.g., microglia), apoptotic mediators (e.g., caspase, calpain), membrane damage, and cytosolic calcium.
FIG. 5.

Evaluation and ranking of various hypothetical single, reducing, inhibiting, and combination treatments. (a) Impact ranking of individual secondary injury factors on neuron death as determined by immediate post-insult treatment initiation (zero hours) that inhibits single factors by 50%. (b) Impact of time of treatment initiation on neuron death over clinically relevant time frames. The factors relevant at later, clinically relevant time points contrast from those shown at time zero. The top five single factors are shown in their respective category color (from Fig. 1): phagocytes, immune activation, apoptotic cascades, membrane damage, and calcium. The remaining factors are shown in black. (c) The effect of reducing and inhibiting single and combination treatments as a function of aggressiveness of treatment. The impact of treatment, especially combination treatments, is greatly increased by reducing paradigms (solid lines) which allow for much more aggressive treatment than inhibiting treatments (dashed lines). Aggressiveness of treatment measured relative to maximum (i.e., 100%) inhibiting treatment. Thus, inhibiting treatments max out at 1.0 while reducing treatments can be much more aggressive. (d) Quantified advantage of reducing versus inhibiting combination treatment paradigms. The order of factors for each n-factor combination shown in (c) and (d) are as shown: phagocytes, immune activation, necro-apoptosis, membrane damage, and calcium. As shown in both (c) and (d), the majority of effective impact of combination treatments is contained within three factors.
Combination treatments
To examine the supposition that multi-factor treatments would be more effective in treating secondary injury, we tested combinations of treating two to five factors. The maximum of five was based on a statistical analysis of significance based on the system dimensionality (for dimensionality assessment method, see Mitchell and Lee, 2007). The results indicate the effects of combination therapy during the first 12 h post-insult are substantially sublinear, rather than synergistic. That is, of the approximately 20,000 possible combinations, none performed better and most performed worse than what would be expected by adding the dose-proportional effects of single treatments together (Fig. 5C). Although more effect is gained with each additional factor treated, the majority of the impact resides in treating three factors.
Inhibiting versus reducing treatments
Based on our analysis of the overall system behavior, we explored alternative treatments beyond those that simply inhibit factor growth. Most clinical therapies are aimed at decreasing neuron death by inhibiting the growth of a factor by acting via a specific mechanistic pathway. For example, a common experimental treatment is to inhibit NMDA activation using a receptor antagonist. These inhibiting treatments target interactions by preventing the rate-dependent growth of a factor and its subsequent interaction with other factors. In contrast to this inhibiting treatment paradigm, we also examined reducing treatment paradigms to decrease neuron death (Fig. 5C, D). This paradigm targets the accumulation-dependent nature of the system by directly reducing a factor in a manner similar to adding a “drain” to the flood analogy. An example of a reducing treatment is a free radical scavenger, which actively seeks to “mop up” free radicals, rather than prevent their formation.
The switch from a rate-dependent propagation of secondary injury (i.e., the fire) to an accumulation-dependent process (i.e., the flood) is evident in the performance of the respective inhibiting and reducing treatment paradigms. During the acute period, inhibiting treatments outperform their reducing counterparts, particularly at less aggressive treatment doses. However, this difference becomes negligible by the second hour for three-factor combination treatments and by the fourth hour for two-factor combinations (Fig. 5D). By the eighth hour, reducing treatments outperform inhibiting treatments in all treatment scenarios.
Pathology-driven therapeutic strategies
Based on our results, the switch from the acute, highly interactive, rate-dependent pathology to a subacute lower interaction, accumulation-dependent pathology determines two critical aspects of secondary injury: (1) the strength of relationships governing a factor's impact on neuron death (Fig. 6) and (2) the time frame over which factors are relevant, referred to as the factor “treatment window” (Fig. 4). Since a truly effective treatment must take into account both of these components, ultimately only a few factors are highly influential at clinically relevant time frames. This would appear to explain the disconnect between promising experimental studies, which pre-treat or treat acute factors within minutes of insult (Tolias and Bullock, 2004; Blight and Tuszynski, 2006; Faden and Stoica, 2007), and clinical studies where treatment time frames are typically 4–8 h (Tator and Fehlings, 1999) as the effective treatment window for acute factors has been surpassed. Furthermore, the lack of synergism predicted by the model for combination treatments is also a direct result of the early switch to a diverging, flood-like pathology since synergism is derived from the sustained presence of strong interactions.
FIG. 6.

Summary of the secondary injury pathology dynamics and the top model-predicted therapeutic strategies at clinically relevant time frames. The diagram summarizes the flows and effects of factors on neuron death and the corresponding higher-impact inhibiting and reducing treatments over the 2–8-h time frame. Circular arrows represent the best targets for reducing treatments: membrane damage, apoptotic cascades, and phagocytes. Inhibiting treatments are indicated by an “x” through the targeted interaction: interaction between NO and immune activation, non-neuronal death and immune activation, immune activation and phagocytes, calcium and apoptotic cascades, and membrane damage and apoptotic cascades. Arrow line thickness to and from factors represents relative “flow” and is indicative of the relative rates coming into and out of factor, while line darkness represents the impact of “flow” on neuron death. Box color saturation symbolizes the impact of a factor on neuron death.
Summary of predictions
As shown, the presented secondary injury relational model is able to transcribe literature-extracted relationships into a network of time-varying factors that reproduce a number of experimental results. However, an important aspect of any model is the ability to characterize previously unknown dynamics, mechanisms, or functions. In this work, we have made several predictions regarding the previously uncharacterized dynamics of secondary injury and its response to numerous hypothetical single and multi-factor combination treatment types. A summary of the model's testable predictions is given in Table 6.
Table 6.
Summary of Secondary Injury Dynamics and Therapeutic Predictions Over the 0–12-h Simulated Time Period
| Dynamical Predictions |
| • Dynamical time course: Hours 1–2 are dominated by an acute “fire” of rate-dependent interactions. Hours 2–6 exhibit a mixture of fire-like interactions and the flood-like accumulation of independent factors. Hours 6–12 reveal a nearly decoupled system analogous to a flood. |
| • Factor category time course: Excitotoxicity and energetics peak in hours 1–2. Free radical and necro-apoptosis peak during hours 2–6. Inflammation and “other” peak in the last hours of the sub-acute period (>6 h). |
| • Factor impact on neuron death: Excitotoxicity and energetics impact neuron death in the acute periods. Impact of imflammation is in the sub-acute periods. Impacts of necro-apoptosis, free radicals, and “other” can be seen throughout the entire time course. |
| Therapeutic Predictions |
| • Single factor treatments: Best treatments during clinically relevant time frames (hours 2–8) are phagocytes, immune activation, apoptotic cascades, membrane damage, and calcium. |
| • Combination treatments: Are additive rather than synergistic. Majority of treatment impact obtained with three factors. |
| • Inhibiting treatments: Target interactions are most effective at 0–4 h post-insult. |
| • Reducing treatments: Target accumulation of factors are most effective at >4 h post-insult. Can have much higher doses compared to inhibiting treatments, making them the superior general clinical strategy. |
Discussion
At first, it may seem that this characterization of the secondary injury pathology simply adds to the already disheartening picture painted by a host of failed clinical trials. However, our results may indicate quite the opposite. The pathology characterization presented here identifies positive current and future directions to pursue based on fundamental pathology dynamics. While multi-factor treatment combinations do not provide the much hoped-for synergistic effects, our results do suggest that some combinations would be functionally additive, namely factors with longer treatments windows, such as necro-apoptotic and inflammatory factors. Furthermore, the effects of combination treatments can be amplified with very aggressive reducing treatment paradigms. Such paradigms may be possible with careful selection of existing pharmaceuticals. Thus, multifactor treatments may still play a role in treating SCI, but expectations regarding their effectiveness should remain realistic with continued exploration being pragmatic.
More importantly, our results suggest that the way forward may lie in pursuing the detailed dynamics of how the secondary injury process propagates rather than just the factors involved in that propagation. For example, treatment of secondary injury based on a flood paradigm opens up therapeutic avenues not currently explored. There are three possible ways to “treat” a flood: (1) “wall off” the flood by building a containment dam; (2) repair the source; and (3) distribute the flooding over a larger area/volume thereby minimizing its impact. In the case of secondary injury, each has its pros and cons. The physiological mechanism seems to be to wall off the area via inflammation and glial scarring (Fawcett and Asher, 1999). However, this approach sacrifices any surviving cells remaining within the walled area. Repairing the source, which could involve repairing the damaged cells, possibly through membrane re-sealing (Liu-Snyder et al., 2007), may have a limited feasible treatment window but may still result in long-term success. More radical would be attempting to distribute the flood in a regulated manner, possibly through controlled activation and inactivation of inflammatory factors over time to minimize overall damage. This last approach could potentially leverage the positive, mediating aspects of inflammation while minimizing the negative, sacrificial effects. A key challenge for many of these alternative approaches lies in the ability to experimentally characterize and analyze the changing spatial and temporal dynamics of the pathology, such as the ability to differentiate early rate-dependent damage from later, accumulation-dependent damage.
While this model does provide the first, preliminary systems-level view of the secondary injury process and possible hypothetical treatments as a whole based on the current state of the field, it is merely scratching the surface. Thus, admittedly, there are multiple factors, details, and mechanisms that will likely need to be added or modified in the future as new experimental findings allow us to hone in closer to the roots, inner-workings, and related systems, which specify the underlying pathology and ultimately the efficacy of very specific treatments. Specific examples of possible refinements include the addition of the mediating effects of inflammation and membrane re-sealing and a more detailed examination of underspecified factors, such as axonal damage, where useable, available data is scarce. Finally, in addition to the excluded mediating and discordant direct factors stated in the justification for factor inclusion/exclusion section, this model does not account for secondary injury occurring at the level of the organism (hypoxia and hypotension) resulting from dysfunction of other organ systems. In the future, such aforementioned refinements will provide further confidence in our ability to predict clinical outcomes.
We foresee this and similar forms of modeling and analysis, perhaps better classified as “theoretical physiology,” to be an invaluable complementary tool to the details and mechanisms identified and validated by SCI experimental and clinical studies by allowing a comprehensive, holistic view into the pathology dynamics and interactions. Ultimately, with continued refinement, modeling may provide a high-throughput screening process from which potential experiments, treatments, and detailed protocols can be tested for feasibility and prioritization, thus speeding the time between therapeutic discovery and clinical success.
Acknowledgments
This work is supported by the National Science Foundation (NSF) via a Graduate Research Fellowship and an Integrative Graduate Education and Research Traineeship Fellowship (DGE-0333411) to C.S.M. Additional support was provided by the Human Brain Project (NINDS, NIMH, NIBIB NS046851) to R.H.L.
Author Disclosure Statement
No competing financial interests exist.
References
- Agrawal S.K. Fehlings M.G. Mechanisms of secondary injury to spinal cord axons in vitro: role of Na+, Na+-K+-ATPase, the N+-H+ exchanger, and the Na+-Ca2+ exchanger. J. Neurosci. 1996;16:545–552. doi: 10.1523/JNEUROSCI.16-02-00545.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal S.K. Fehlings M.G. Role of NMDA and non-NMDA ionotropic glutamate receptors in traumatic spinal cord axonal injury. J. Neurosci. 1997;17:1055–1063. doi: 10.1523/JNEUROSCI.17-03-01055.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed S.M. Weber J.T. Liang S. Willoughby K.A. Sitterding H.A. Rzigalinski B.A. Ellis E.F. NMDA receptor activation contributes to a portion of the decreased mitochondrial membrane potential and elevated intracellular free calcium in strain-injured neurons. J. Neurotrauma. 2002;19:1619–1629. doi: 10.1089/089771502762300274. [DOI] [PubMed] [Google Scholar]
- Alano C.C. Beutner G. Dirksen R.T. Gross R.A. Sheu S.S. Mitochondrial permeability transition and calcium dynamics in striatal neurons upon intense NMDA receptor activation. J. Neurochem. 2002;80:531–538. doi: 10.1046/j.0022-3042.2001.00738.x. [DOI] [PubMed] [Google Scholar]
- Anderson D.K. Means E.D. Waters T.R. Spears C.J. Spinal cord energy metabolism following compression trauma to the feline spinal cord. J. Neurosurg. 1980;53:375–380. doi: 10.3171/jns.1980.53.3.0375. [DOI] [PubMed] [Google Scholar]
- Ankarcrona M. Dypbukt J.M. Bonfoco E. Zhivotovsky B. Orrenius S. Lipton S.A. Nicotera P. Glutamate-induced neuronal death—a succession of necrosis or apoptosis depending on mitochondrial-function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- Ates O. Cayli S.R. Gurses I. Turkoz Y. Tarim O. Cakir C.O. Kocak A. Comparative neuroprotective effect of sodium channel blockers after experimental spinal cord injury. J. Clin. Neurosci. 2007;14:658–665. doi: 10.1016/j.jocn.2006.03.023. [DOI] [PubMed] [Google Scholar]
- Azbill R.D. Mu X. Bruce-Keller A.J. Mattson M.P. Springer J.E. Impaired mitochondrial function, oxidative stress and altered antioxidant enzyme activities following traumatic spinal cord injury. Brain Res. 1997;765:283–290. doi: 10.1016/s0006-8993(97)00573-8. [DOI] [PubMed] [Google Scholar]
- Bartholdi D. Schwab M.E. Expression of pro-inflammatory cytokine and chemokine mRNA upon experimental spinal cord injury in mouse: an in situ hybridization study. Eur. J. Neurosci. 1997;9:1422–1438. doi: 10.1111/j.1460-9568.1997.tb01497.x. [DOI] [PubMed] [Google Scholar]
- Barut S. Unlu Y.A. Karaoglan A. Tuncdemir M. Dagistanli F.K. Ozturk M. Colak A. The neuroprotective effects of z-DEVD.fmk, a caspase-3 inhibitor, on traumatic spinal cord injury in rats. Surg. Neurol. 2005;64:213–220. doi: 10.1016/j.surneu.2005.03.042. [DOI] [PubMed] [Google Scholar]
- Beattie M.S. Inflammation and apoptosis: linked therapeutic targets in spinal cord injury. Trends Mol. Med. 2004;10:580–583. doi: 10.1016/j.molmed.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Blight A.R. Delayed demyelination and macrophage invasion: a candidate for secondary cell damage in spinal cord injury. Cent. Nerv. Syst. Trauma. 1985;2:299–315. doi: 10.1089/cns.1985.2.299. [DOI] [PubMed] [Google Scholar]
- Blight A.R. Tuszynski M.H. Clinical trials in spinal cord injury. J. Neurotrauma. 2006;23:586–593. doi: 10.1089/neu.2006.23.586. [DOI] [PubMed] [Google Scholar]
- Carlson S.L. Parrish M.E. Springer J.E. Doty K. Dossett L. Acute inflammatory response in spinal cord following impact injury. Exp. Neurol. 1998;151:77–88. doi: 10.1006/exnr.1998.6785. [DOI] [PubMed] [Google Scholar]
- Carriedo S.G. Yin H.Z. Sensi S.L. Weiss J.H. Rapid Ca2+ entry through Ca2+-permeable AMPA/kainate channels triggers marked intracellular Ca2+ rises and consequent oxygen radical production. J. Neurosci. 1998;18:7727–7738. doi: 10.1523/JNEUROSCI.18-19-07727.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo A.M. Liu J. Lam C.K. Dvorak M. Tetzlaff W. Oxland T.R. Contusion, dislocation, and distraction: primary hemorrhage and membrane permeability in distinct mechanisms of spinal cord injury. J. Neurosurg. Spine. 2007;6:255–266. doi: 10.3171/spi.2007.6.3.255. [DOI] [PubMed] [Google Scholar]
- Churchwell K.B. Wright S.H. Emma F. Rosenberg P.A. Strange K. NMDA receptor activation inhibits neuronal volume regulation after swelling induced by veratridine-stimulated Na+ influx in rat cortical cultures. J. Neurosci. 1996;16:7447–7457. doi: 10.1523/JNEUROSCI.16-23-07447.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe M.J. Bresnahan J.C. Shuman S.L. Masters J.N. Beattie M.S. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat. Med. 1997;3:73–76. doi: 10.1038/nm0197-73. [DOI] [PubMed] [Google Scholar]
- Cullen D.K. Vernekar V. LaPlaca M.C. Abstracts: The effects of shear versus compressive loading in 3-D neuronal-astrocytic co-cultures. J Neurotrauma. 2006a;23:986. [Google Scholar]
- Cullen D.K. LaPlaca M.C. Neuronal response to high rate shear deformation depends on heterogeneity of the local strain field. J. Neurotrauma. 2006b;23:1304–1319. doi: 10.1089/neu.2006.23.1304. [DOI] [PubMed] [Google Scholar]
- Dusart I. Schwab M.E. Secondary cell-death and the inflammatory reaction after dorsal hemisection of the rat spinal-cord. Eur. J. Neurosci. 1994;6:712–724. doi: 10.1111/j.1460-9568.1994.tb00983.x. [DOI] [PubMed] [Google Scholar]
- Faden A.I. Chan P.H. Longar S. Alterations in lipid metabolism, Na+,K+-ATPase activity, and tissue water content of spinal cord following experimental traumatic injury. J. Neurochem. 1987;48:1809–1816. doi: 10.1111/j.1471-4159.1987.tb05740.x. [DOI] [PubMed] [Google Scholar]
- Faden A.I. Stoica B. Neuroprotection: challenges and opportunities. Arch. Neurol. 2007;64:794–800. doi: 10.1001/archneur.64.6.794. [DOI] [PubMed] [Google Scholar]
- Farkas O. Lifshitz J. Povlishock J.T. Mechanoporation induced by diffuse traumatic brain injury: An irreversible or reversible response to injury? J. Neurosci. 2006;26:3130–3140. doi: 10.1523/JNEUROSCI.5119-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett J.W. Asher R.A. The glial scar and central nervous system repair. Brain Res. Bull. 1999;49:377–391. doi: 10.1016/s0361-9230(99)00072-6. [DOI] [PubMed] [Google Scholar]
- Fehlings M.G. Agrawal S. Role of sodium in the pathophysiology of secondary spinal cord injury. Spine. 1995;20:2187–2191. doi: 10.1097/00007632-199510001-00002. [DOI] [PubMed] [Google Scholar]
- Fleming J.C. Norenberg M.D. Ramsay D.A. Dekaban G.A. Marcillo A.E. Saenz A.D. Pasquale-Styles M. Dietrich W.D. Weaver L.C. The cellular inflammatory response in human spinal cords after injury. Brain. 2006;129:3249–3269. doi: 10.1093/brain/awl296. [DOI] [PubMed] [Google Scholar]
- Fujiki M. Furukawa Y. Kobayashi H. Abe T. Ishii K. Uchida S. Kamida T. Geranylgeranylacetone limits secondary injury, neuronal death, and progressive necrosis and cavitation after spinal cord injury. Brain Res. 2005;1053:175–184. doi: 10.1016/j.brainres.2005.06.055. [DOI] [PubMed] [Google Scholar]
- Gaviria M. Bonny J.M. Haton H. Jean B. Teigell M. Renou J.P. Privat A. Time course of acute phase in mouse spinal cord injury monitored by ex vivo quantitative MRI. Neurobiol. Dis. 2006;22:694–701. doi: 10.1016/j.nbd.2006.01.011. [DOI] [PubMed] [Google Scholar]
- Giulian D. Robertson C. Inhibition of mononuclear phagocytes reduces ischemic injury in the spinal cord. Ann. Neurol. 1990;27:33–42. doi: 10.1002/ana.410270107. [DOI] [PubMed] [Google Scholar]
- Goforth P.B. Ellis E.F. Satin L.S. Mechanical injury modulates AMPA receptor kinetics via an NMDA receptor-dependent pathway. J. Neurotrauma. 2004;21:719–732. doi: 10.1089/0897715041269704. [DOI] [PubMed] [Google Scholar]
- Gomes-Leal W. Corkill D.J. Freire M.A. Picanco-Diniz C.W. Perry V.H. Astrocytosis, microglia activation, oligodendrocyte degeneration, and pyknosis following acute spinal cord injury. Exp. Neurol. 2004;190:456–467. doi: 10.1016/j.expneurol.2004.06.028. [DOI] [PubMed] [Google Scholar]
- Green D. Kroemer G. The central executioners of apoptosis: caspases or mitochondria? Trends Cell Biol. 1998;8:267–271. doi: 10.1016/s0962-8924(98)01273-2. [DOI] [PubMed] [Google Scholar]
- Hall E.D. Braughler J.M. Free radicals in CNS injury. Res. Publ. Assoc. Res. Nerv. Ment. Dis. 1993;71:81–105. [PubMed] [Google Scholar]
- Hall E.D. Springer J.E. Neuroprotection and acute spinal cord injury: a reappraisal. NeuroRx. 2004;1:80–100. doi: 10.1602/neurorx.1.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada Y. Ikata T. Katoh S. Tsuchiya K. Niwa M. Tsutsumishita Y. Fukuzawa K. Roles of nitric oxide in compression injury of rat spinal cord. Free Radic. Biol. Med. 1996;20:1–9. doi: 10.1016/0891-5849(95)02017-9. [DOI] [PubMed] [Google Scholar]
- Hartmann A. Hunot S. Michel P.P. Muriel M.P. Vyas S. Faucheux B.A. Mouatt-Prigent A. Turmel H. Srinivasan A. Ruberg M. Evan G.I. Agid Y. Hirsch E.C. Caspase-3: a vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson's disease. Proc. Natl. Acad. Sci. USA. 2000;97:2875–2880. doi: 10.1073/pnas.040556597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S. Peterson P.K. Chao C.C. Cytokine-mediated neuronal apoptosis. Neurochem. Int. 1997;30:427–431. doi: 10.1016/s0197-0186(96)00078-2. [DOI] [PubMed] [Google Scholar]
- Iwai T. Tanonaka K. Inoue R. Kasahara S. Motegi K. Nagaya S. Takeo S. Sodium accumulation during ischemia induces mitochondrial damage in perfused rat hearts. Cardiovasc. Res. 2002;55:141–149. doi: 10.1016/s0008-6363(02)00282-1. [DOI] [PubMed] [Google Scholar]
- Jurkowitzalexander M.S. Altschuld R.A. Hohl C.M. Johnson J.D. Mcdonald J.S. Simmons T.D. Horrocks L.A. Cell swelling, blebbing, and death are dependent on atp depletion and independent of calcium during chemical hypoxia in a glial-cell line (Roc-1) J. Neurochem. 1992;59:344–352. doi: 10.1111/j.1471-4159.1992.tb08910.x. [DOI] [PubMed] [Google Scholar]
- Kandel E.R. Schwartz J.H. Jessell T.M. Principles of Neural Science. 4th. McGraw-Hill; New York: 2000. [Google Scholar]
- Klusman I. Schwab M.E. Effects of pro-inflammatory cytokines in experimental spinal cord injury. Brain Res. 1997;762:173–184. doi: 10.1016/s0006-8993(97)00381-8. [DOI] [PubMed] [Google Scholar]
- Krajewski S. Krajewska M. Ellerby L.M. Welsh K. Xie Z. Deveraux Q.L. Salvesen G.S. Bredesen D.E. Rosenthal R.E. Fiskum G. Reed J.C. Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc. Natl. Acad. Sci. USA. 1999;96:5752–5757. doi: 10.1073/pnas.96.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPlaca M.C. Thibault L.E. Dynamic mechanical deformation of neurons triggers an acute calcium response and cell injury involving the N-methyl-D-aspartate glutamate receptor. J. Neurosci. Res. 1998;52:220–229. doi: 10.1002/(SICI)1097-4547(19980415)52:2<220::AID-JNR10>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Lemke M. Demediuk P. McIntosh T.K. Vink R. Faden A.I. Alterations in tissue Mg2+, Na+ and spinal cord edema following impact trauma in rats. Biochem. Biophys. Res. Commun. 1987;147:1170–1175. doi: 10.1016/s0006-291x(87)80192-4. [DOI] [PubMed] [Google Scholar]
- Li S. Stys P.K. Na+-K+-ATPase inhibition and depolarization induce glutamate release via reverse Na+-dependent transport in spinal cord white matter. Neuroscience. 2001;107:675–683. doi: 10.1016/s0306-4522(01)00385-2. [DOI] [PubMed] [Google Scholar]
- Liu D. Xu G.Y. Pan E. McAdoo D.J. Neurotoxicity of glutamate at the concentration released upon spinal cord injury. Neuroscience. 1999;93:1383–1389. doi: 10.1016/s0306-4522(99)00278-x. [DOI] [PubMed] [Google Scholar]
- Liu-Snyder P. Logan M.P. Shi R. Smith D.T. Borgens R.B. Neuroprotection from secondary injury by polyethylene glycol requires its internalization. J. Exp. Biol. 2007;210:1455–1462. doi: 10.1242/jeb.02756. [DOI] [PubMed] [Google Scholar]
- Louis J.C. Magal E. Takayama S. Varon S. CNTF protection of oligodendrocytes against natural and tumor necrosis factor-induced death. Science. 1993;259:689–692. doi: 10.1126/science.8430320. [DOI] [PubMed] [Google Scholar]
- Lovas G. Szilagyi N. Majtenyi K. Palkovits M. Komoly S. Axonal changes in chronic demyelinated cervical spinal cord plaques. Brain. 2000;123:308–317. doi: 10.1093/brain/123.2.308. [DOI] [PubMed] [Google Scholar]
- Lu J. Ashwell K.W. Waite P. Advances in secondary spinal cord injury: role of apoptosis. Spine. 2000;25:1859–1866. doi: 10.1097/00007632-200007150-00022. [DOI] [PubMed] [Google Scholar]
- Mattiasson G. Analysis of mitochondrial generation and release of reactive oxygen species. Cytometry A. 2004;62A:89–96. doi: 10.1002/cyto.a.20089. [DOI] [PubMed] [Google Scholar]
- Merrill J.E. Ignarro L.J. Sherman M.P. Melinek J. Lane T.E. Microglial cell cytotoxicity of oligodendrocytes is mediated through nitric oxide. J. Immunol. 1993;151:2132–2141. [PubMed] [Google Scholar]
- Mitchell C.S. Feng S.S. Lee R.H. An analysis of glutamate spillover on the N-methyl-D-aspartate receptors at the cerebellar glomerulus. J. Neural Eng. 2007;4:276–282. doi: 10.1088/1741-2560/4/3/013. [DOI] [PubMed] [Google Scholar]
- Mitchell C.S. Lee R.H. Output-based comparison of alternative kinetic schemes for the NMDA receptor within a glutamate spillover model. J. Neural Eng. 2007;4:380–389. doi: 10.1088/1741-2560/4/4/004. [DOI] [PubMed] [Google Scholar]
- Nicholls D.G. Budd S.L. Mitochondria and neuronal survival. Physiol. Rev. 2000;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- O'Brien M.F. Lenke L.G. Lou J. Bridwell K.H. Joyce M.E. Astrocyte response and transforming growth factor-beta localization in acute spinal cord injury. Spine. 1994;19:2321–2329. doi: 10.1097/00007632-199410150-00012. [DOI] [PubMed] [Google Scholar]
- Park E. Velumian A.A. Fehlings M.G. The role of excitotoxicity in secondary mechanisms of spinal cord injury: a review with an emphasis on the implications for white matter degeneration. J. Neurotrauma. 2004;21:754–774. doi: 10.1089/0897715041269641. [DOI] [PubMed] [Google Scholar]
- Pettus E.H. Povlishock J.T. Characterization of a distinct set of intra-axonal ultrastructural changes associated with traumatically induced alteration in axolemmal permeability. Brain Res. 1996;722:1–11. doi: 10.1016/0006-8993(96)00113-8. [DOI] [PubMed] [Google Scholar]
- Pineau I. Lacroix S. Proinflammatory cytokine synthesis in the injured mouse spinal cord: multiphasic expression pattern and identification of the cell types involved. J. Comp. Neurol. 2007;500:267–285. doi: 10.1002/cne.21149. [DOI] [PubMed] [Google Scholar]
- Popovich P.G. Guan Z. McGaughy V. Fisher L. Hickey W.F. Basso D.M. The neuropathological and behavioral consequences of intraspinal microglial/macrophage activation. J Neuropathol Exp Neurol. 2002;61:623–633. doi: 10.1093/jnen/61.7.623. [DOI] [PubMed] [Google Scholar]
- PorteraCailliau C. Price D.L. Martin L.J. Non-NMDA and NMDA receptor-mediated excitotoxic neuronal deaths in adult brain are morphologically distinct: further evidence for an apoptosis-necrosis continuum. J. Comp. Neurol. 1997;378:88–104. [PubMed] [Google Scholar]
- Saftenku E.E. Modeling of slow glutamate diffusion and AMPA receptor activation in the cerebellar glomerulus. J. Theoret. Biol. 2005;234:363–382. doi: 10.1016/j.jtbi.2004.11.036. [DOI] [PubMed] [Google Scholar]
- Schnell L. Fearn S. Klassen H. Schwab M.E. Perry V.H. Acute inflammatory responses to mechanical lesions in the CNS: differences between brain and spinal cord. Eur. J. Neurosci. 1999;11:3648–3658. doi: 10.1046/j.1460-9568.1999.00792.x. [DOI] [PubMed] [Google Scholar]
- Schwab M.E. Bartholdi D. Degeneration and regeneration of axons in the lesioned spinal cord. Physiol. Rev. 1996;76:319–370. doi: 10.1152/physrev.1996.76.2.319. [DOI] [PubMed] [Google Scholar]
- Schwartz G. Fehlings M.G. Evaluation of the neuroprotective effects of sodium channel blockers after spinal cord injury: improved behavioral and neuroanatomical recovery with riluzole. J. Neurosurg. 2001;94:245–256. doi: 10.3171/spi.2001.94.2.0245. [DOI] [PubMed] [Google Scholar]
- Shi R. Whitebone J. Conduction deficits and membrane disruption of spinal cord axons as a function of magnitude and rate of strain. J. Neurophysiol. 2006;95:3384–3390. doi: 10.1152/jn.00350.2005. [DOI] [PubMed] [Google Scholar]
- Springer J.E. Azbill R.D. Knapp P.E. Activation of the caspase-3 apoptotic cascade in traumatic spinal cord injury. Nat. Med. 1999;5:943–946. doi: 10.1038/11387. [DOI] [PubMed] [Google Scholar]
- Sullivan P.G. Krishnamurthy S. Patel S.P. Pandya J.D. Rabchevsky A.G. Temporal characterization of mitochondrial bioenergetics after spinal cord injury. J. Neurotrauma. 2007;24:991–999. doi: 10.1089/neu.2006.0242. [DOI] [PubMed] [Google Scholar]
- Tator C.H. Fehlings M.G. Review of clinical trials of neuroprotection in acute spinal cord injury. Neurosurg. Focus. 1999;6:e8. [PubMed] [Google Scholar]
- Tian D.S. Yu Z.Y. Xie M.J. Bu B.T. Witte O.W. Wang W. Suppression of astroglial scar formation and enhanced axonal regeneration associated with functional recovery in a spinal cord injury rat model by the cell cycle inhibitor olomoucine. J. Neurosci. Res. 2006;84:1053–1063. doi: 10.1002/jnr.20999. [DOI] [PubMed] [Google Scholar]
- Tian D.S. Xie M.J. Yu Z.Y. Zhang Q. Wang Y.H. Chen B. Chen C. Wang W. Cell cycle inhibition attenuates microglia induced inflammatory response and alleviates neuronal cell death after spinal cord injury in rats. Brain Res. 2007;1135:177–185. doi: 10.1016/j.brainres.2006.11.085. [DOI] [PubMed] [Google Scholar]
- Tolias C.M. Bullock M.R. Critical appraisal of neuroprotection trials in head injury: what have we learned? NeuroRx. 2004;1:71–79. doi: 10.1602/neurorx.1.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totoiu M.O. Keirstead H.S. Spinal cord injury is accompanied by chronic progressive demyelination. J. Comp. Neurol. 2005;486:373–383. doi: 10.1002/cne.20517. [DOI] [PubMed] [Google Scholar]
- Vela J.M. Yanez A. Gonzalez B. Castellano B. Time course of proliferation and, elimination of microglia/macrophages in different neurodegenerative conditions. J. Neurotrauma. 2002;19:1503–1520. doi: 10.1089/089771502320914723. [DOI] [PubMed] [Google Scholar]
- Volterra A. Trotti D. Tromba C. Floridi S. Racagni G. Glutamate uptake inhibition by oxygen free radicals in rat cortical astrocytes. J. Neurosci. 1994;14:2924–2932. doi: 10.1523/JNEUROSCI.14-05-02924.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R.J. Reynolds I.J. Mitochondrial depolarization in glutamate-stimulated neurons: an early signal specific to excitotoxin exposure. J. Neurosci. 1996;16:5688–5697. doi: 10.1523/JNEUROSCI.16-18-05688.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingrave J.M. Schaecher K.E. Sribnick E.A. Wilford G.G. Ray S.K. Hazen-Martin D.J. Hogan E.L. Banik N.L. Early induction of secondary injury factors causing activation of calpain and mitochondria-mediated neuronal apoptosis following spinal cord injury in rats. J. Neurosci. Res. 2003;73:95–104. doi: 10.1002/jnr.10607. [DOI] [PubMed] [Google Scholar]
- Xiong Y. Rabchevsky A.G. Hall E.D. Role of peroxynitrite in secondary oxidative damage after spinal cord injury. J. Neurochem. 2007;100:639–649. doi: 10.1111/j.1471-4159.2006.04312.x. [DOI] [PubMed] [Google Scholar]
- Xu G.Y. Hughes M.G. Ye Z. Hulsebosch C.E. McAdoo D.J. Concentrations of glutamate released following spinal cord injury kill oligodendrocytes in the spinal cord. Exp. Neurol. 2004;187:329–336. doi: 10.1016/j.expneurol.2004.01.029. [DOI] [PubMed] [Google Scholar]
- Yanase M. Sakou T. Fukuda T. Role of N-methyl-D-aspartate receptor in acute spinal-cord injury. J. Neurosurg. 1995;83:884–888. doi: 10.3171/jns.1995.83.5.0884. [DOI] [PubMed] [Google Scholar]
- Yoshioka A. Bacskai B. Pleasure D. Pathophysiology of oligodendroglial excitotoxicity. J. Neurosci. Res. 1996;46:427–437. doi: 10.1002/(SICI)1097-4547(19961115)46:4<427::AID-JNR4>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Zhang L. Rzigalinski B.A. Ellis E.F. Satin L.S. Reduction of voltage-dependent Mg2+ blockade of NMDA current in mechanically injured neurons. Science. 1996;274:1921–1923. doi: 10.1126/science.274.5294.1921. [DOI] [PubMed] [Google Scholar]
- Zhao W. Xie W. Le W. Beers D.R. He Y. Henkel J.S. Simpson E.P. Yen A.A. Xiao Q. Appel S.H. Activated microglia initiate motor neuron injury by a nitric oxide and glutamate-mediated mechanism. J. Neuropathol. Exp. Neurol. 2004;63:964–977. doi: 10.1093/jnen/63.9.964. [DOI] [PubMed] [Google Scholar]