

Abstract
PARK8/LRRK2 (leucine-rich repeat kinase 2) was recently identified as a causative gene for autosomal dominant Parkinson's disease (PD), with LRRK2 mutation G2019S linked to the most frequent familial form of PD. Emerging in vitro evidence indicates that aberrant enzymatic activity of LRRK2 protein carrying this mutation can cause neurotoxicity. However, the physiological and pathophysiological functions of LRRK2 in vivo remain elusive. Here we characterize two bacterial artificial chromosome (BAC) transgenic mouse strains overexpressing LRRK2 wild-type (Wt) or mutant G2019S. Transgenic LRRK2-Wt mice had elevated striatal dopamine (DA) release with unaltered DA uptake or tissue content. Consistent with this result, LRRK2-Wt mice were hyperactive and showed enhanced performance in motor function tests. These results suggest a role for LRRK2 in striatal DA transmission and the consequent motor function. In contrast, LRRK2-G2019S mice showed an age-dependent decrease in striatal DA content, as well as decreased striatal DA release and uptake. Despite increased brain kinase activity, LRRK2-G2019S overexpression was not associated with loss of DAergic neurons in substantia nigra or degeneration of nigrostriatal terminals at 12 months. Our results thus reveal a pivotal role for LRRK2 in regulating striatal DA transmission and consequent control of motor function. The PD-associated mutation G2019S may exert pathogenic effects by impairing these functions of LRRK2. Our LRRK2 BAC transgenic mice, therefore, could provide a useful model for understanding early PD pathological events.
Introduction
LRRK2 encodes a complex protein (285 kDa) that belongs to the ROCO family defined by the presence of a ROC (Ras/GTPase of complex proteins) domain followed by a COR (C-terminal of ROC) domain of unknown function (Bosgraaf and Van Haastert, 2003; Paisán-Ruíz et al., 2004; Zimprich et al., 2004). The LRRK2 protein contains both kinase and GTPase activities, which are apparently altered by several familial LRRK2 mutations (West et al., 2005; Gloeckner et al., 2006; Smith et al., 2006; Guo et al., 2007; Ito et al., 2007; Li et al., 2007). Despite the controversy over whether all Parkinson's disease (PD)-linked mutations affect the enzymatic activities of LRRK2, the most common familial mutation, G2019S, has been consistently shown to enhance its kinase activity, which has been correlated with neurotoxicity in vitro (Smith et al., 2005, 2006; Greggio et al., 2006; MacLeod et al., 2006; Greggio and Cookson, 2009). Whether this “gain of function” in LRRK2 kinase activity contributes to the pathological process of PD has not been examined in vivo.
The physiological function of LRRK2 is largely unknown. Characterization of a Caenorhabditis elegans mutant with deletion of LRK-1, the paralog of LRRK2, suggests that LRK-1 is not essential for the survival of dopaminergic (DAergic) neurons, but may be involved in axonal sorting of synaptic vesicle proteins (Sakaguchi-Nakashima et al., 2007). In nematode worms, overexpression of either human wild-type (Wt) LRRK2 or LRRK2 G2019S protects against rotenone-mediated toxicity and extends life span (Wolozin et al., 2008). Genetic studies in Drosophila melanogaster show that deletion of dLRRK (LRRK2 paralog in fly) does not affect DAergic neuron viability; however, expression of PD-associated LRRK2 mutants or the corresponding dLRRK mutant causes selective degeneration of DAergic neurons, as well as motor deficits (Lee et al., 2007; Liu et al., 2008; Wang et al., 2008; Ng et al., 2009). Furthermore, overexpression of the PD-pathogenic mutant of dLRRK (but not Wt dLRRK) results in a depletion of brain dopamine (DA), with elevated DA content in flies with dLRRK deletion (Imai et al., 2008). These findings suggest that dLRRK negatively regulates DA homeostasis.
To elucidate LRRK2 function in mammals, as well as dysfunction after LRRK2 PD-associated mutation, we developed bacterial artificial chromosome (BAC) transgenic mice overexpressing either FLAG-tagged murine forms of Wt LRRK2 (LRRK-Wt) or PD-related mutation G2019S in LRRK2 (LRRK2-G2019S). Utilizing BAC mice not only permits transgene expression under the control of an endogenous promoter for cell-type specific with temporal and spatial regulation, but also facilitates the study of gene function in vivo through controlling gene dosage (Heintz, 2001). Here we assessed consequences of overexpression of Wt LRRK2 or the G2019S mutant. Our data show that overexpression of Wt LRRK2 or the G2019S mutant cause distinct effects on striatal DA transmission and consequent motor function. The comparative study of these two transgenic lines sheds light on the physiological function of Wt LRRK2 and provides clues as to how G2019S mutation alters LRRK2 function.
Materials and Methods
Animals
Mice were housed in the Center for Comparative Medicine at Mount Sinai School of Medicine or the Veterinary Medical Unit at the James J. Peters Veterans Affairs Medical Center. Handling procedures were in accordance with National Institutes of Health guidelines and approved by the Institutional Animal Care and Use Committees of the above institutions and New York University School of Medicine.
Generation of LRRK2-Wt and LRRK2-G2019S BAC transgenic mice
LRRK2-Wt mice were generated using the BAC technique as previously (Li et al., 2007). To generate LRRK2-G2019S mice, the already-modified mouse LRRK2-Wt BAC was used as the template to introduce mutation G2019S. As a result of this sequence change, the MfeI restriction enzyme site was created for the confirmation of mutation in genotyping. The following primers were used for generating homologous arms in BAC modification: primer A forward, 5′-GGGGCGCGCCTGTGTGTACAGGGCTGTGTGC-3′; primer A reverse, 5′-CCTGCAGCAGTACTGTGCAATTGAGTAGTCCGCAATCTTCGC-3′; primer B forward, 5′-GCGAAGATTGCGGACTACTCAATTGCACAGTACTGCTGCAGG-3′; primer B reverse, 5′-GGGTTAATTAA CTATTTCCGTCGTGATCCGGC-3′. The following primers were used to clone the fragments from genomic tail DNA containing the MfeI restriction enzyme site and subsequently digested by MfeI (New England Biolabs): primer C forward, 5′-GATCTTTACGCTTTGAAGCCTG-3′; primer C reverse, 5′-GGGTTAATTAACTATTTCCGTCGTGATCCGGC-3′. The following primers were used to amplify the kinase domain by RT-PCR using mRNA isolated from transgenic and control brains, and the cDNA was purified and digested by MfeI: primer D forward, 5′-AAGATCTGACCAACCAAGGCTCACTATTC-3′; primer D reverse, 5′-CCGCTCGAGTCACTCATAGCTGTATCTTTCCG-3′.
DA and HVA contents
The dorsal striatum from transgenic and littermate control mice (6 months, n = 6–8 per group or 12 months, n = 7 per group) were dissected and quickly frozen on dry ice. Samples were homogenized in 500 μl of HPLC mobile buffer (0.1 m trichloroacetic acid, 0.01 m sodium acetate, 0.1 μm EDTA, and 9% methanol, pH 3.9). Samples were then spun at 13,000 rpm for 20 min, the supernatant was transferred to a new tube, and an aliquot was analyzed by HPLC (Vanderbilt Molecular Neuroscience Center). Protein content from pellets homogenized in 500 μl of 0.1 m HCl was determined by the BCA method (Pierce).
Western blot analysis of brain lysates
Mice were decapitated and dorsal striatum was dissected on ice and quickly immersed in 200 μl of buffer (20 mm HEPES, pH 7.4/1 mm MgCl2/0.25 mm CaCl2/protease inhibitor cocktail/PMSF-pepstatin/phosphatase inhibitor/DNase 80U/ml) and homogenized by Dounce Tissue Homogenizer (Eberbach). Protein was determined by the BCA method (Pierce), and 25 μg of protein per sample was loaded onto gels. Rabbit anti-LRRK2 antibody (1 μg/ml) was produced in our laboratory. Monoclonal anti-tyrosine hydroxylase (TH) (Pel Freeze Biological) was used at 1:500. Rabbit polyclonal anti-vesicular monoamine transporter 2 (VMAT2) (Santa Cruz Biotechnology) was used at 1:200, rat anti-dopamine transporter (DAT) monoclonal antibody (Millipore) was used at 1:2000, mouse anti-D2 DA receptor (D2R) monoclonal antibody (Santa Cruz Biotechnology) was used at 1:200, and rabbit anti-phospho-TH (P-Ser40) (Sigma) and rabbit anti-phospho-TH (PSer31) (Cell Signaling Technology) were used at 1:1000.
TH activity assay
Striatal TH activity was determined from the release of tritium from [3,5-3H]-l-tyrosine [modified from Khan (2004)]. Whole striatum was homogenized in 700 μl of 0.2 m PBS, pH 7.4, containing 0.5% Triton X100. The sample was centrifuged at 20,800 × g for 10 min at 4°C, protein determined by the BCA method, then 200 μg of protein was incubated with 0.8 μCi [3,5-3H]-l-tyrosine (PerkinElmer), 100 U of catalase, 1.0 mm dithiothreitol, and 1.5 mm dl-6-methyl-5,6,7,8-tetrahydropterine (6-MPH4) in a volume of 300 μl for 20 min at 37°C. The reaction was stopped by adding 1 ml of a suspension of 7.5% (w/v) activated charcoal in 0.1 m HCl. The mixture was vortexed and centrifuged at 12,000 × g for 5 min, and 200 μl of supernatant was counted by a standard scintillation method. Reagents were from Sigma.
DA release and uptake
To decrease experimenter or recording bias, experiments were done blind with a transgenic mouse paired with its respective littermate control examined each experimental day. Coronal striatal slices (350 μm) from male mice (6 or 12 months; 3–4 mice per cohort, 3–4 slices per animal) were cut on a VT1200S vibrating blade microtome (Leica Microsystems) and maintained as previously (Avshalumov et al., 2008). After a 30 min equilibration period at 32°C in a submersion chamber superfused at 1.2 ml/min with bicarbonate-buffered aCSF, FCV with carbon-fiber microelectrodes (Patel et al., 2009) was used to monitor [DA]o evoked by single-pulse stimulation (0.4–0.6 mA amplitude, 0.1 ms duration) at 3–5 sites in dorsolateral striatum per slice. Evoked [DA]o was quantified by postexperimental calibration at 32°C.
To analyze the uptake kinetics of single-pulse evoked [DA]o curves, a segment of the falling phase, beginning 100 ms after the peak, was fitted to the Michaelis–Menten equation to extract Vmax using a fixed Km). The use of single pulses allows release to be quantified in the absence of autoreceptor regulation by endogenous DA (Limberger et al., 1991; Kennedy et al., 1992; Patel et al., 1992; Phillips et al., 2002) and avoided modulation by concurrently released glutamate or GABA (Avshalumov et al., 2003; Chen et al., 2006). As noted previously (e.g., Wu et al., 2001), the falling phase of an evoked DA response is governed by the following:
 |
where Km and Vmax are the Michaelis–Menten uptake constants. This equation assumes that a relatively large volume of tissue surrounding the recording carbon-fiber microelectrode is at the same [DA]o so that diffusion does not contribute to DA clearance [diffusion would necessitate a more complicated analysis (Nicholson, 1995)]. Thus, the peak value of the curve, just before commencement of the falling phase, will have a finite value. The above equation was integrated to yield an implicit equation for [DA]o as a function of time. The equation was solved for a sequence of time points using a standard root-finding algorithm (function “fzero” in MATLAB, MathWorks) and the solution used with a nonlinear curve-fitting simplex algorithm (function ‘fminsearch’ in MATLAB) to determine best values of Vmax (the maximal rate constant for uptake, proportional to the number of functional DATs) and the peak value of [DA]o, for a fixed Km (which is inversely related to the affinity of the DAT for DA). The value of Km was set at 0.9 μm (Schmitz et al., 2001) and assumed not to be altered in the transgenic lines. The goodness of fit was determined from the sum of squares of the differences between measured and predicted values, which in turn provided a coefficient of determination for each fit (r2 value). The above solution had the same shape as the falling phase of the experimental data, i.e., a rapid descent followed by a longer, slower tail; for the chosen Km (0.9 μm) the tail of the computed curve fell more rapidly than the data. Late differences between predicted and actual data suggest that additional factors might play a role in determining the shape of the curve (Schmitz et al., 2001; Schönfuss et al., 2001). Such explanations have not been rigorously explored, however, so were not incorporated into our analysis. For this reason we restricted the curve fitting to the rapid falling phase just after the peak of the curve. It should be noted that although the Vmax values obtained were similar to those previously found for mouse striatal slices (Schmitz et al., 2001; John and Jones, 2007), they should be viewed as a way to compare curves quantitatively rather than definitive Vmax values.
Behavioral testing
Male transgenic mice and their respective littermate controls (6 or 12 months, 15 per group) were tested for general locomotor activity in an open field for 60 min. Motor coordination was assessed on a challenge beam and by gait stride. Because no differences were seen between control groups in any behavioral test, control data were pooled for analysis (n = 30). All testing was performed in a double-blind manner.
Open-field test.
Individual mice were put into the 16 × 16 inch cage of a Versamax monitor system (Accuscan) in a quiet dark room. The mouse horizontal and vertical movement was monitored and recorded for 60 min by a grid of 32 infrared beams at ground level and 16 elevated (3 inch) beams.
Beam test.
One 100 cm beam, consisting of four 25 cm sections of varying width (3.5, 2.5, 1.5 and 0.5 cm), was used (Fleming et al., 2004). To enhance the difficulty of the task, the corresponding width of each section was covered with 1 cm high metal wire mesh (1 cm square). Before the real test, individual mice were trained for 2 d without the wired mesh. On each training day, the mouse was trained two times for home cage assisted running and then five times for unassisted running across the entire length of the beam. On the experimental day, task difficulty was enhanced by placing the mesh on the beam. Animals were then videotaped while traversing the grid-surfaced beam for a total of five trials. The mouse running steps, times, and slips were scored and analyzed.
Gait stride test.
Individual mice were allowed to run the length of a black Plexiglas runway (dimensions 152 cm L × 10 cm W, elevated 13 cm), from an exposed start to the shelter of the home cage. Mice were given three training runs each day for 3 d, assisted by the home cage on the first and unassisted on the next two. On the fourth day (test), the mouse's paws were coated with ink (red ink on the hindpaws, black or blue on the forepaws), and paw prints were recorded on two consecutive runs. The record that displayed the clearest prints and the most consistent gait for five consecutive strides was chosen for analysis. The distance between forepaws and between hindpaws, the left and right stride, the diagonal distance, and fore/hind overlap were recorded.
Kinase assay
The kinase assay was performed using previously described methods (Li et al., 2007).
TH immunohistochemistry and stereology
Transgenic and littermate control mice (n = 6 per group) were anesthetized with ketamine (100 mg/kg) and xylazine (20 mg/ml) before intracardial perfusion with 30 ml of cold PBS and 30 ml of 4% paraformaldehyde (PFA). Brains were postfixed in PFA overnight, then equilibrated in 30% sucrose at 4°C for 48 h. Midbrain sections containing substantia nigra pars compacta (SNc) were sliced on a sliding microtome, collected consecutively, and preserved in an anti-freezing buffer (30% glycerin solution in ethylene glycol) at −20°C. Using the D3V region as a reference, every fourth section was selected, and a total of six sections were stained for TH-DAB. An anti-TH monoclonal antibody (Pel Freeze Biological, 1:500) and VectaStain Elite ABC kit (Vector Laboratories) were used. The substrate was Fast diaminobenzidine tablets (Sigma) (1 tablet into 15 ml of ddH2O). An unbiased stereology protocol (Stereo Investigator 6, MicroBrightField) was used with an Olympus Denmark CAST-Grid system to count TH+ SNc neurons (coefficient of error of p < 0.1).
Quantification and cell counting
DAB staining with anti-PHF-1 antibody (gift from Dr. P. Davis, Albert Einstein College of Medicine, Manhasset, NY) was performed in 30 μm coronal (bregma 0.38 ± 0.12 mm) and sagittal (lateral 1.8 ± 0.24 mm) sections. Cells of interest were quantified in a 0.40 mm2 area of the dorsal–medial striatum. Three sections from each animal (3–4 mice per group) were analyzed using an Olympus BX60 microscope with a 20× lens. Positive cell bodies were quantified statistically.
Statistical analysis
All quantitative data are expressed as means ± SEM, where n is the number of mice or the number of voltammetric DA recording sites. Significance of differences was assessed by Student's t test for comparison of two groups or one-way-ANOVA with Bonferroni's post hoc analysis for comparison of three groups or more. Sustainability of DA release with time was analyzed by two-way-ANOVA with Bonferroni's post hoc analysis.
Results
BAC transgenic mice overexpressing FLAG-tagged LRRK2-Wt or LRRK2-G2019S
We identified a mouse BAC containing the entire genomic sequence of LRRK2. The BAC was genetically engineered by first inserting a FLAG tag after the start codon ATG to induce overexpression of LRRK2-Wt, then introducing a G-to-S mutation at amino acid 2019 to induce G2019S mutant overexpression (LRRK2-G2019S) (Fig. 1A) (supplemental Fig. S1A,B, available at www.jneurosci.org as supplemental material). We performed repeated genetic crosses of these BAC transgenic mice to a nearly isogenic background of C57BL/6J; two mouse lines that expressed similar protein levels for LRRK2-Wt and LRRK-G2019S were selected for further characterization. Immunoblots of total brain lysate with anti-LRRK2 antibody confirmed similar expression levels (approximately sixfold higher than endogenous LRRK2 protein) (Fig. 1B) in these LRRK2-Wt and LRRK2-G2019S lines. The specificity of the anti-LRRK2 antibody used was shown previously (Li et al., 2007) and was further confirmed here using brain tissue from LRRK2 knock-out mice (supplemental Fig. S1C, available at www.jneurosci.org as supplemental material). We also confirmed the presence of the G2019S mutation in LRRK2-G2019S mice by examining messenger RNA (mRNA) using RT-PCR, followed by restriction enzyme digestion (supplemental Fig. S1D, available at www.jneurosci.org as supplemental material). As expected for BAC transgenics, immunohistochemical analysis showed similar brain expression patterns for LRRK2-Wt and LRRK2-G2019S in many areas of brain, such as cerebral cortex, striatum, substantia nigra, internal capsule, and hippocampus, whether stained with anti-LRRK2 (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material) or anti-FLAG antibodies (data not shown). Total transgene expression levels in different regions of brain, analyzed by Western blot analysis, are also comparable between LRRK2-Wt and LRRK2-G2019S mice (supplemental Fig. S2B, available at www.jneurosci.org as supplemental material). Both mouse lines had the usual number of pups and expected gender ratio (data not shown), showing no major impairment in embryonic development. Anatomic evaluation showed no obvious brain abnormalities in either LRRK2-Wt or LRRK2-G2019S mice up to 18 months (supplemental Fig. S2C, available at www.jneurosci.org as supplemental material).
Figure 1.

BAC transgenic LRRK2-Wt and LRRK2–G2019S mouse lines: transgene levels and striatal DA content. A, Schematic showing the original BAC with the mouse LRRK2 gene, modified LRRK2-Wt BAC in which the FLAG tag was inserted in-frame after the ATG translation start codon, and LRRK2–G2019S BAC carrying G-to-S mutation at amino acid 2019. B, Western blot using anti-LRRK2 antibody shows that transgenes LRRK2-Wt and LRRK2–G2019S produce similar protein levels, approximately sixfold higher than endogenous brain LRRK2. C, D, HPLC analysis indicated that striatal DA and HVA contents were ∼25% lower in LRRK2–G2019S than in littermate control mice at 12 months (*p < 0.05 vs control 2, n = 7 mice per group). Striatal DA or HVA in LRRK2-Wt mice did not differ significantly from littermate controls (p > 0.05 vs control 1, n = 7 mice per group). Quantitative data are expressed as mean ± SEM and were analyzed by one-way ANOVA with Bonferroni's post hoc analysis.
Decreased striatal DA content in LRRK2-G2019S mice
Given the key role of striatal DA loss in PD, we first determined tissue striatal DA content in the two BAC transgenic lines. Striatal DA levels, determined by HPLC analysis, were not changed significantly in LRRK2-Wt (224 ± 7 ng/mg protein, n = 6 mice) or LRRK2-G2019S (223 ± 6 ng/mg protein, n = 6 mice) versus control (246 ± 9 ng/mg protein, n = 8 mice) mice at 6 months (p > 0.05). However, at 12 months, LRRK2-G2019S mice had ∼25% lower levels of DA and its metabolite homovanillic acid (HVA) than controls (p < 0.05), whereas levels in LRRK2-Wt mice were again unaltered (Fig. 1C,D). This suggests an age-related decline of striatal DA content in LRRK2-G2019S mice.
To determine whether TH, the biosynthetic enzyme for DA is altered in the transgenic lines, we examined TH protein levels, enzymatic activity, and posttranslational modification of the TH protein associated with its activity. However, we found no differences in striatal TH protein levels, enzymatic activity, or phosphorylation state (P-Ser 31 and P-Ser 40) among LRRK2-Wt, LRRK2-G2019S, and control mice at 10 months (Fig. 2A–C), suggesting that the decrease in striatal DA content is not due to impaired TH activity. Furthermore, we found no changes in the levels of other proteins involved in regulating presynaptic DA levels, including VMAT2, DAT, or D2Rs, by Western blot analysis (Fig. 2A).
Figure 2.

Examination of TH activity and protein levels for VMAT2, DAT, and D2R in LRRK2-Wt and LRRK2–G2019S mice. A, Western blot analysis (left) and quantification (right) of striatal TH, VMAT2, DAT, and D2R protein levels. No significant differences were detected among LRRK2-Wt, LRRK2–G2019S, and littermate control mice (n = 3 mice per group). B, Striatal TH activity, determined by release of tritium from [3,5-3H]-l-tyrosine, was not changed in LRRK2-Wt or LRRK2–G2019S compared to littermate control mice at 9–10 months (n = 3 mice per group). Data are expressed as percentage of control. C, Western blot analysis of striatal tissue from mice at 9–10 months using anti-TH and anti-phospho-TH (P-Ser31 and P-Ser40) antibodies (left), with quantification (right). No significant differences in total TH or phospho-TH (P-Ser31 and P-Ser40) protein levels were detected among control, LRRK2-Wt, and LRRK2–G2019S mice (n = 3 mice per group). Quantitative data are expressed as mean ± SEM and were analyzed by one-way ANOVA with Bonferroni's post hoc analysis.
Opposing effects of LRRK2-Wt versus LRRK2-G2019S on striatal DA release and uptake
To examine functional consequence of Wt and mutant LRRK2 overexpression on exocytotic DA release, we used fast-scan cyclic voltammetry (FCV) to monitor evoked DA release in striatal slices from the two BAC transgenic mouse lines at 12 months. Peak extracellular DA concentration ([DA]o) evoked by a single pulse was ∼25% higher in LRRK2-Wt than in paired littermate control mice (p < 0.05 vs control 1) (Fig. 3A). Consistent with the age-dependent decrease of DA content in mice with the G2019S mutation (see above and Fig. 1C), peak evoked [DA]o was ∼35% lower in LRRK2-G2019S mice than in littermate controls (p < 0.001 vs control 2) (Fig. 3A). There was no difference in evoked [DA]o between the two control groups (p > 0.05, control 1 vs 2). Moreover, evoked [DA]o in LRRK2-G2019S mice did not differ from littermate controls at 6 months (1.54 ± 0.08 μm in LRRK2-G2019S vs 1.42 ± 0.11 μm in controls, p > 0.05, n = 32 recording sites from 4 mice per group). This result suggests an age-dependent decrease in evoked DA release caused by LRRK2-G2019S overexpression in mice.
Figure 3.

Opposing regulation of striatal DA release and uptake in LRRK2-Wt versus LRRK2–G2019S mice. A, Comparison of evoked DA release in 12 month transgenic LRRK2 mice (3 mice per group). Left, Averaged single-pulse evoked [DA]o versus time obtained in striatal slices from LRRK2-Wt and littermates (control 1) (n = 43–50 sites) and in LRRK2–G2019S and littermates (control 2) (n = 43–53 sites) (error bars omitted for clarity). Right, Peak [DA]o was ∼25% higher in LRRK2-Wt mice than in control 1 (*p < 0.05), whereas evoked [DA]o was ∼35% lower in LRRK2–G2019S mice than in control 2 (***p < 0.001); control groups did not differ (p > 0.05, control 1 vs 2). B, DA uptake in 12 month transgenic LRRK2 mice. Left, Representative evoked [DA]o from control striatum showing the data points used for curve fitting to extract Vmax. Middle, Fit of representative data points to Michaelis–Menten uptake kinetics with a fixed Km of 0.9 μm gave peak [DA]o = 1.09 μm, Vmax = 4.03 μm/s, and r2 = 0.996. Right, Averaged Vmax values did not differ between LRRK2-Wt and control 1 mice (p > 0.05; n = 41–50 sites), whereas Vmax was lower in LRRK2–G2019S than in control 2 mice (*p < 0.05; n = 45–51 sites), indicating less efficient DA uptake after G2019S mutation. Data are expressed as mean ± SEM and were analyzed by one-way ANOVA with Bonferroni's post hoc analysis.
Evoked [DA]o reflects the net balance between DA release and DA clearance by DAT-mediated uptake. To establish the relative roles of release and uptake in the opposite effects of LRRK2-Wt and LRRK2-G2019S overexpression on evoked [DA]o at 12 months, we fitted the initial falling phase of [DA]o versus time records to the Michaelis–Menten equation to extract the maximum uptake rate, Vmax (Fig. 3B) using a fixed Km of 0.9 μm (Schmitz et al., 2001). The average Vmax pooled from all control mice (control 1 plus control 2) was 3.85 ± 0.11 μm/s (n = 86 sites from 6 mice), which is similar to that reported previously for mouse striatum in vitro (Schmitz et al., 2001; John and Jones, 2007). The Vmax for DA uptake in LRRK2-Wt mice did not differ from that in littermate controls (Fig. 3B). Thus, elevated evoked [DA]o in LRRK2-Wt mice reflects enhanced release, not impaired uptake. In contrast, Vmax was significantly lower in LRRK2-G2019S mice than in littermate controls (p < 0.05 vs control 2) (Fig. 3B). This indicates that decreased evoked [DA]o in LRRK2-G2019S is not from enhanced DA uptake, but implies the converse, that decreased uptake might be a compensatory response to impaired DA release after G2019S mutation.
Uptake of released DA via the DAT replenishes vesicles for subsequent DA release (Jones et al., 1998). We then assessed the ability of DAergic axon terminals to sustain release with repetitive stimulation in mice at 12 months. In controls, the amplitude of [DA]o evoked at a given site by single pulses at 2 min intervals decreased with the first few stimulations and then approached a plateau. In either control 1 or control 2 mice peak [DA]o declined by ∼30% by the end of a 20 min monitoring period (p > 0.05, control 1 vs control 2) (Fig. 4A,B, left). This pattern was unaltered in LRRK2-Wt mice (p > 0.05 vs control 1) (Fig. 4A,B, middle). In LRRK2-G2019S mice, however, the decline in evoked [DA]o was amplified (p < 0.001 vs control 2) (Fig. 4A,B, right), indicating less robust release in LRRK2-G2019S mice. To avoid confounding factors that might arise from comparing release sites with differing initial peak evoked [DA]o, we also analyzed a subpopulation of the data that included only release sites with a starting evoked [DA]o of ∼1 μm. We again found no difference in the decline of evoked [DA]o with time in LRRK2-Wt versus control 1 mice (p > 0.05, n = 5 recording sites per group) and confirmed that evoked [DA]o was poorly sustained in LRRK2-G2019S versus control 2 mice p < 0.001, n = 6 sites per group). These results suggest that overexpression of LRRK2-Wt and LRRK2-G2019S cause contrasting effect on DA transmission in mice; the G2019S mutation may impair striatal DA release, resulting in a decrease in extracellular DA levels at striatum.
Figure 4.

Impaired sustainability of striatal DA release in LRRK2–G2019S mice. Sustainability of single-pulse evoked DA release was assessed in 12 month mice (3 mice per group) by comparing the decline in peak [DA]o evoked at 2 min intervals over a 20 min period. A, Representative [DA]o records evoked at a given site in a control 1 and LRRK2-Wt slice (top), as well as in a control 2 and LRRK2–G2019S slice (bottom). The examples shown for each mouse group were selected to have an initial starting peak [DA]o of ∼1 μm to aid visual comparison of the decline in peak [DA]o. B, Average peak [DA]o with time, normalized to peak [DA]o evoked by the first stimulus (P1) of a series at a given recording site taken as 100%. Left, Control 1 (n = 9 sites) and control 2 (n = 11 sites) mice showed a similar decline in peak evoked [DA]o of ∼30% over a 20 min period of stimulation (p > 0.05; control 1 vs control 2). Middle, The average decline in peak [DA]o over a 20 min stimulation period in LRRK2-Wt (n = 10 sites) was similar to control 1 mice (p > 0.05). Right, In contrast, DA release was less well sustained in LRRK2–G2019S (n = 11 sites) versus control 2 mice (***p < 0.001). Quantitative data are expressed as mean ± SEM and were analyzed by two-way ANOVA with Bonferroni's post hoc analysis.
Spontaneous hyperactivity and enhanced motor performance in LRRK2-Wt but not LRRK2-G2019S mice
To investigate how altered striatal DA transmission affects motor function, we performed motor function tests on the two BAC transgenic lines and their respective littermate controls. No differences were seen between the two control groups (p > 0.05 for each test), therefore these data were pooled. In open-field analysis, LRRK2-Wt mice showed an increased number of rearing movements compared to LRRK2-G2019S or control mice (Fig. 5A). Overall motor activity of LRRK2-Wt mice was greater than that of either LRRK2-G2019S or controls, with more movement and longer distances traveled in a given time window (Fig. 5B,C). The difference in motor activity between LRRK2-Wt and LRRK2-G2019S or control mice was more prominent at 12 than at 6 months.
Figure 5.

LRRK2-Wt, but not LRRK2–G2019S, mice show hyperactivity and enhanced motor performance. A–C, In the open-field test, LRRK2-Wt mice displayed increased rearing compared to LRRK2–G2019S or control mice at 6 and 12 months (A), traveled longer distances than LRRK2–G2019S or control mice at 12 months (B), and spent more time moving at 12 months (C). C, All 12 month mice were less active than 6 month mice regardless of genotype. D–F, In the challenge beam test, LRRK2-Wt and LRRK2–G2019S mice used a similar number of steps when crossing a beam covered with metal mesh (D); however, LRRK2-Wt mice had fewer slips (E) and fewer slips per step (F) than LRRK2–G2019S or control mice. Littermate controls for each transgenic line did not differ in any behavioral test; for each age, control data from these animals were therefore pooled (*p < 0.05; **p < 0.01; control, n = 30 mice; LRRK2-Wt, n = 15 mice; LRRK2–G2019S, n = 15 mice). Quantitative data are expressed as mean ± SEM and were analyzed by one-way ANOVA with Bonferroni's post hoc analysis.
Motor coordination was tested using a challenge beam task sensitive to dysfunction of the nigrostriatal pathway (Fleming et al., 2004). While crossing a beam covered with metal mesh, LRRK2-Wt mice showed enhanced ability to traverse the full beam length, indicated by fewer total slips (errors) and slips per step, compared to LRRK2-G2019S or controls. Again, the difference in slips between LRRK2-Wt and LRRK2-G2019S or controls was more pronounced at 12 than 6 months (Fig. 5D–F). In a gait test, the LRRK2-Wt mice showed significantly longer strides, as well as greater diagonal distance between front and hind limbs than LRRK2-G2019S mice (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Thus, enhanced striatal DA transmission may contribute to hyperactivity and better motor performance in LRRK2-Wt mice.
In contrast, LRRK2-G2019S overexpression did not alter motor performance, despite similar brain levels and distribution to that seen in LRRK2-Wt mice (Fig. 5). This indicates that 25% lower striatal DA content and release levels (Figs. 1, 3) do not lead to motor deficits in LRRK2-G2019S mice up to 12 months, consistent with the usual finding that a decrease in striatal DA content of >50% is typically required before motor deficits appear in human PD or in parkinsonian animal models (Zigmond et al., 1990).
Lack of PD-associated pathophysiology with LRRK2-G2019S overexpression, despite elevated kinase activity
Previous studies indicate that LRRK2 protein purified from brain has higher kinase activity than that from other tissues or cultured cells (Li et al., 2007). To examine whether LRRK2-G2019S mutation also stimulates kinase activity over LRRK2-Wt, as reported for human LRRK2 in vitro (West et al., 2005; Gloeckner et al., 2006; Smith et al., 2006; Jaleel et al., 2007), we assayed kinase activities of LRRK2-Wt and LRRK2-G2019S proteins purified from transgenic brains by the FLAG affinity method. Brain kinase activity in LRRK2-G2019S was significantly higher than in LRRK2-Wt, assessed using either phosphorylation of myelin basic protein (MBP) (2.4-fold increase) or autophosphorylation (2.7-fold increase) (Fig. 6A). These results suggest a highly conserved mechanism whereby G2019S mutation alters the biochemical (and likely structural) properties of LRRK2, regardless of the origin of mammalian LRRK2 (e.g., species or tissue).
Figure 6.
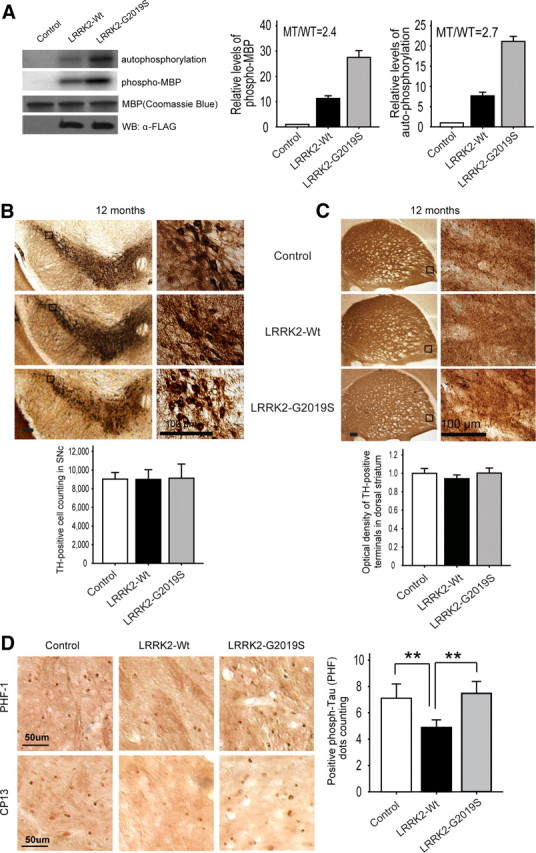
Enhanced brain kinase activity in LRRK2–G2019S does not cause loss of DAergic neurons, but correlates with increased of striatal phospho-tau staining versus LRRK2-Wt. A, Higher kinase activity of transgenic brain LRRK2–G2019S protein versus LRRK2-Wt. Left, Kinase activity was assayed using MBP phosphorylation (2.4-fold increase) and autophosphorylation (2.7-fold increase) with equal protein levels. Right, Quantification of MBP phosphorylation and LRRK2 autophosphorylation from three independent experiments. B, No loss of SNc TH+ neurons with stereological cell counting or change in cell morphology was seen even at 60× magnification in either mouse line at 12 months (n = 6 per group). Scale bar, 100 μm. C, TH staining in the striatum of LRRK2-Wt and LRRK2–G2019S mice at 12 months. Optical density analysis indicates no difference in TH staining among the different genotypes (n = 6 per group). Scale bar, 100 μm. D, Immunohistochemical staining with anti-phospho-tau antibodies (PHF-1 and CP13) in dorsal striatum. LRRK2-Wt mice had fewer PHF-1- and CP13-positive cells than control or LRRK2–G2019S mice. Scale bar, 50 μm. Quantitative data show attenuation of PHF-1-positive cell number in the dorsal striatum of LRRK2-Wt mice (**p < 0.01; n = 3 per group). Quantitative data are expressed as mean ± SEM and were analyzed by one-way ANOVA with Bonferroni's post hoc analysis.
Despite increased brain kinase activity in LRRK2-G2019S mice, no sign of neuronal or other cell death was seen in any brain region, including those with high levels of exogenous LRRK2 or G2019S (e.g., cortex, striatum, and hippocampus; data not shown). Stereological cell counting revealed no differences in the number of DAergic neurons (TH-positive cells; TH+) in the SNc in LRRK2-Wt or LRRK2-G2019S versus littermate controls at 6 or 12 months (Fig. 6B; supplemental Fig. S4A, available at www.jneurosci.org as supplemental material). In addition, nigrostriatal terminals were not grossly altered at 6 or 12 months, with no difference in the optical density or appearance of TH+ staining in dorsal striatum among LRRK2-G2019S, LRRK2-Wt, and control mice (Fig. 6C; supplemental Fig. S4B, available at www.jneurosci.org as supplemental material). There was no apparent abnormality or loss of TH+ immunolabeling in SNc (including distal dendrites in the substantia nigra pars reticulata), internal capsule, or dorsal striatum even at 18–20 months in LRRK2-G2019S or LRRK2-Wt mice (data not shown). This may contribute to the lack of obvious motor impairment in LRRK2-G2019S mice at 12 months (Fig. 5). Importantly, this result further indicates that dysfunctional DAergic transmission caused by LRRK2-G2019S (Figs. 1, 3, 4) occurs without apparent degeneration of nigrostriatal terminals or cell bodies at this age.
In the absence of the overt loss of DAergic neurons or dystrophic terminals, we investigated other PD-associated pathological markers in these mice. Immunohistochemistry of LRRK2-G2019S and LRRK2-Wt brains up to 18 months showed no increases in levels or aggregation of α-synuclein or ubiquitin, which are found in Lewy bodies in the SNc in PD (data not shown). However, staining with anti-phospho-tau antibodies (PHF-1 against pS396/pS404 and CP13 against PS202/T205) showed significantly fewer phospho-tau-positive (+) cells in dorsal striatum of 18 month LRRK2-Wt, than in control or LRRK2-G2019S mice (Fig. 6D). This suggests that overexpression of LRRK2 may prevent the accumulation of phospho-tau in brain. Although no obvious change in the number of phospho-tau+ cells was observed in LRRK2-G2019S versus control mice, the significant difference between the two BAC lines suggests a contrasting, thereby inhibitory, effect of G2019S on the function of LRRK2 in regulating homeostatic phospho-tau levels.
Discussion
The establishment of two BAC transgenic strains expressing similar levels and patterns of Wt LRRK2 and G2019S mutation provides a unique opportunity to dissect the physiological function of LRRK2 and unravel the pathogenic role of LRRK2-G2019S mutation in PD. Our study provides the first in vivo evidence that overexpression of Wt LRRK2 increases striatal dopamine release and enhances motor performance. In contrast, overexpression of LRRK2 variant carrying the most common familial PD mutation G2019S fails to provide these benefits, indicated by an age-dependent decrease in DA content, impairment of evoked striatal DA release, and decreased DA uptake in LRRK2-G2019S mice. In addition, LRRK2 G2019S neither enhanced nor compromised motor function in mice up to 12 months. These results, therefore, suggest that LRRK2 regulates striatal DA transmission, thereby facilitating motor activity, and that G2019S mutation may cause an impairment of this function of native LRRK2.
An important finding in our study is that the age-dependent impairment in striatal DA release and loss of DA content in LRRK2-G2019S mice was not associated with obvious neuronal death or nigrostriatal terminal degeneration (by 18–20 months) or motor deficits (by 12 months) in LRRK2-G2019S mice. These data indicate that an initial consequence of G2019S mutation is a functional rather than physical denervation of the nigrostriatal DA pathway. Previous studies suggest that LRRK2 is localized at membrane structures, including synaptic vesicles (Hatano et al., 2007), and may regulate synaptic vesicle proteins and vesicle endocytosis and, thus, vesicle life cycle (Sakaguchi-Nakashima et al., 2007). Our data showing elevated DA release in LRRK2-Wt mice along with enhanced motor performance are consistent with this LRRK2 function. Conversely, decreased DA release and release sustainability in LRRK2-G2019S mice would be consistent with alteration of this function. Whether the progressive nature of DA impairment is a result of an age-dependent accumulation of LRRK2 protein (Li et al., 2007) or protein modification of LRRK2 over time remains to be established. Another potential function of LRRK2 is suggested by the observation that LRRK2-Wt overexpression decreases the number of phospho-tau (+) cells, suggesting a protective role of LRRK2 against tau-associated pathology. In contrast, this protection of LRRK2 is absent in G2019S mutant.
The lack of nigrostriatal degeneration in LRRK2-G2019S mice was initially surprising because of previous in vitro studies showing that increased kinase activity in LRRK2-G2019S was correlated with enhanced neurotoxicity (Smith et al., 2006; West et al., 2007). Despite significantly higher kinase activity of brain LRRK2-G2019S protein than its Wt counterpart, our LRRK2-G2019S mice showed no evidence of the neurodegeneration characteristic of PD. This offers further support for the notion that LRRK2-G2019S mutation can impair LRRK2 neuronal function without activating cell death pathways at early disease stages. Consistent with this, many carriers of PD-related mutations of LRRK2 do not develop PD symptoms. Indeed, an international human genetics study of PD involving >1000 subjects showed that the penetrance of the G2019S mutation is only 28% by age of 59, increasing to 51% by age 69 (Healy et al., 2008). Thus, the LRRK2-mediated pathogenic pathway may be particularly susceptible to modification by other genetic or cellular mechanisms. Interestingly, although expression of mutant LRRK2 in Drosophila consistently produces neurodegeneration, coexpression of another PD-related protein parkin can prevent this (Ng et al., 2009).
Two recent LRRK2 mouse models expressing R1441G/C mutation also provide evidence for impaired DA transmission (Li et al., 2009; Tong et al., 2009). However, neither previous study examined nigrostriatal DA release and uptake dynamics, the effect of normal LRRK2 overexpression, or the link between LRRK2 kinase/GTPase activity to neurotoxicity, as here. Our study further indicates that LRRK2-Wt overexpression decreases the number of phospho-tau (+) cells, suggesting a protective role of LRRK2 against tau-associated pathology that is not observed with the G2019S mutation. Interestingly, although both previous studies examined the R1441C/G mutation, which is a relatively rare mutation in PD, those mice exhibited several dramatic differences in pathology and behavior, some of which also differ from our findings in LRRK2-G2019S mice. Factors contributing to these phenotypic differences may include mutation examined (G2019S vs R1441G/C), source species (murine vs human), genetic background (C57BL/6J vs FVB), and transgene expression levels.
Together, these data support previous evidence that alteration of a single PD-related gene, whether autosomal dominant (α-synuclein) or autosomal recessive (DJ-1, Parkin, and Pink1), in mouse models is unlikely to recapitulate the full spectrum of PD symptoms. Finally, our study, along with others (Li et al., 2009), reveals a causal role for the LRRK2 mutant in the early pathogenesis of PD: impairment of striatal DA transmission. Recent PET imaging in presymptomatic individuals carrying the LRRK2 familial mutation suggests dysfunction of striatal DA transmission as an early event in disease progression in PD (Adams et al., 2005; Nandhagopal et al., 2008). Moreover, other genetic mouse models for PD also exhibit aberrant DA transmission in the absence of nigrostriatal degeneration (Abeliovich et al., 2000; Goldberg et al., 2003; Chen et al., 2005; Kitada et al., 2007; Zhou et al., 2008). Thus the emerging evidence, including the present findings from LRRK2 BAC transgenic mice, suggests a common mechanism for PD pathogenesis whereby familial PD mutations impair striatal DA transmission before the degeneration of DAergic neurons.
Footnotes
This work was supported by United States National Institutes of Health–National Institute of Neurological Disorders and Stroke Grants NS061152 (Z.Y.), NS060809 (Z.Y.), RNS055683A (Z.Y.), NS036362 (M.E.R.), and NS028642 (C.N.); the Michael J. Fox Foundation (Z.Y.); the Bachmann-Strauss Dystonia & Parkinson Foundation (Z.Y.); and the Dean's Office of Mount Sinai School of Medicine (Mouse and Rat Phenotyping Shared Resource Facility). We thank Ruth Walker and Sarah Funderburk for critical comments on the manuscript and Joanne Moy and Doug Williams for assistance in behavioral testing. We thank Dr. Huaibin Cai for the gift of LRRK2 KO tissues.
References
- Abeliovich A, Schmitz Y, Fariñas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- Adams JR, van Netten H, Schulzer M, Mak E, Mckenzie J, Strongosky A, Sossi V, Ruth TJ, Lee CS, Farrer M, Gasser T, Uitti RJ, Calne DB, Wszolek ZK, Stoessl AJ. PET in LRRK2 mutations: comparison to sporadic Parkinson's disease and evidence for presymptomatic compensation. Brain. 2005;128:2777–2785. doi: 10.1093/brain/awh607. [DOI] [PubMed] [Google Scholar]
- Avshalumov MV, Chen BT, Marshall SP, Peña DM, Rice ME. Glutamate-dependent inhibition of dopamine release in striatum is mediated by a new diffusible messenger, H2O2. J Neurosci. 2003;23:2744–2750. doi: 10.1523/JNEUROSCI.23-07-02744.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avshalumov MV, Patel JC, Rice ME. AMPA receptor-dependent H2O2 generation in striatal medium spiny neurons but not dopamine axons: one source of a retrograde signal that can inhibit dopamine release. J Neurophysiol. 2008;100:1590–1601. doi: 10.1152/jn.90548.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosgraaf L, Van Haastert PJ. Roc, a Ras/GTPase domain in complex proteins. Biochim Biophys Acta. 2003;1643:5–10. doi: 10.1016/j.bbamcr.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Chen BT, Moran KA, Avshalumov MV, Rice ME. Limited regulation of somatodendritic dopamine release by voltage-sensitive Ca channels contrasted with strong regulation of axonal dopamine release. J Neurochem. 2006;96:645–655. doi: 10.1111/j.1471-4159.2005.03519.x. [DOI] [PubMed] [Google Scholar]
- Chen L, Cagniard B, Mathews T, Jones S, Koh HC, Ding Y, Carvey PM, Ling Z, Kang UJ, Zhuang X. Age-dependent motor deficits and dopaminergic dysfunction in DJ-1 null mice. J Biol Chem. 2005;280:21418–21426. doi: 10.1074/jbc.M413955200. [DOI] [PubMed] [Google Scholar]
- Fleming SM, Salcedo J, Fernagut PO, Rockenstein E, Masliah E, Levine MS, Chesselet MF. Early and progressive sensorimotor anomalies in mice overexpressing wild-type human alpha-synuclein. J Neurosci. 2004;24:9434–9440. doi: 10.1523/JNEUROSCI.3080-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloeckner CJ, Kinkl N, Schumacher A, Braun RJ, O'Neill E, Meitinger T, Kolch W, Prokisch H, Ueffing M. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet. 2006;15:223–232. doi: 10.1093/hmg/ddi439. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem. 2003;278:43628–43635. doi: 10.1074/jbc.M308947200. [DOI] [PubMed] [Google Scholar]
- Greggio E, Cookson MR. Leucine-rich repeat kinase 2 mutations and Parkinson's disease: three questions. ASN Neuro. 2009;1:e00002. doi: 10.1042/AN20090007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K, Cookson MR. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Guo L, Gandhi PN, Wang W, Petersen RB, Wilson-Delfosse AL, Chen SG. The Parkinson's disease-associated protein, leucine-rich repeat kinase 2 (LRRK2), is an authentic GTPase that stimulates kinase activity. Exp Cell Res. 2007;313:3658–3670. doi: 10.1016/j.yexcr.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatano T, Kubo S, Imai S, Maeda M, Ishikawa K, Mizuno Y, Hattori N. Leucine-rich repeat kinase 2 associates with lipid rafts. Hum Mol Genet. 2007;16:678–690. doi: 10.1093/hmg/ddm013. [DOI] [PubMed] [Google Scholar]
- Healy DG, Falchi M, O'Sullivan SS, Bonifati V, Durr A, Bressman S, Brice A, Aasly J, Zabetian CP, Goldwurm S, Ferreira JJ, Tolosa E, Kay DM, Klein C, Williams DR, Marras C, Lang AE, Wszolek ZK, Berciano J, Schapira AH, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet Neurol. 2008;7:583–590. doi: 10.1016/S1474-4422(08)70117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintz N. BAC to the future: the use of bac transgenic mice for neuroscience research. Nat Rev Neurosci. 2001;2:861–870. doi: 10.1038/35104049. [DOI] [PubMed] [Google Scholar]
- Imai Y, Gehrke S, Wang HQ, Takahashi R, Hasegawa K, Oota E, Lu B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008;27:2432–2443. doi: 10.1038/emboj.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito G, Okai T, Fujino G, Takeda K, Ichijo H, Katada T, Iwatsubo T. GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson's disease. Biochemistry. 2007;46:1380–1388. doi: 10.1021/bi061960m. [DOI] [PubMed] [Google Scholar]
- Jaleel M, Nichols RJ, Deak M, Campbell DG, Gillardon F, Knebel A, Alessi DR. LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson's disease mutants affect kinase activity. Biochem J. 2007;405:307–317. doi: 10.1042/BJ20070209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John CE, Jones SR. Voltammetric characterization of the effect of monoamine uptake inhibitors and releasers on dopamine and serotonin uptake in mouse caudate-putamen and substantia nigra slices. Neuropharmacology. 2007;52:1596–1605. doi: 10.1016/j.neuropharm.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Jaber M, Giros B, Wightman RM, Caron MG. Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proc Natl Acad Sci U S A. 1998;95:4029–4034. doi: 10.1073/pnas.95.7.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy RT, Jones SR, Wightman RM. Dynamic observation of dopamine autoreceptor effects in rat striatal slices. J Neurochem. 1992;59:449–455. doi: 10.1111/j.1471-4159.1992.tb09391.x. [DOI] [PubMed] [Google Scholar]
- Khan HA. Analytical characterization of a sensitive radioassay for tyrosine hydroxylase activity in rodent striatum. Neurochem Res. 2004;29:1467–1472. doi: 10.1023/b:nere.0000029557.45306.4d. [DOI] [PubMed] [Google Scholar]
- Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G, Bonsi P, Zhang C, Pothos EN, Shen J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci U S A. 2007;104:11441–11446. doi: 10.1073/pnas.0702717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SB, Kim W, Lee S, Chung J. Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem Biophys Res Commun. 2007;358:534–539. doi: 10.1016/j.bbrc.2007.04.156. [DOI] [PubMed] [Google Scholar]
- Li X, Tan YC, Poulose S, Olanow CW, Huang XY, Yue Z. Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson's disease R1441C/G mutants. J Neurochem. 2007;103:238–247. doi: 10.1111/j.1471-4159.2007.04743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liu W, Oo TF, Wang L, Tang Y, Jackson-Lewis V, Zhou C, Geghman K, Bogdanov M, Przedborski S, Beal MF, Burke RE, Li C. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson's disease. Nat Neurosci. 2009;12:826–828. doi: 10.1038/nn.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limberger N, Trout SJ, Kruk ZL, Starke K. “Real time” measurement of endogenous dopamine release during short trains of pulses in slices of rat neostriatum and nucleus accumbens: role of autoinhibition. Naunyn Schmiedebergs Arch Pharmacol. 1991;344:623–629. doi: 10.1007/BF00174745. [DOI] [PubMed] [Google Scholar]
- Liu Z, Wang X, Yu Y, Li X, Wang T, Jiang H, Ren Q, Jiao Y, Sawa A, Moran T, Ross CA, Montell C, Smith WW. A Drosophila model for LRRK2-linked parkinsonism. Proc Natl Acad Sci U S A. 2008;105:2693–2698. doi: 10.1073/pnas.0708452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52:587–593. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Nandhagopal R, Mak E, Schulzer M, McKenzie J, McCormick S, Sossi V, Ruth TJ, Strongosky A, Farrer MJ, Wszolek ZK, Stoessl AJ. Progression of dopaminergic dysfunction in a LRRK2 kindred: a multitracer PET study. Neurology. 2008;71:1790–1795. doi: 10.1212/01.wnl.0000335973.66333.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CH, Mok SZ, Koh C, Ouyang X, Fivaz ML, Tan EK, Dawson VL, Dawson TM, Yu F, Lim KL. Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. J Neurosci. 2009;29:11257–11262. doi: 10.1523/JNEUROSCI.2375-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C. Interaction between diffusion and Michaelis-Menten uptake of dopamine after iontophoresis in striatum. Biophys J. 1995;68:1699–1715. doi: 10.1016/S0006-3495(95)80348-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, van der Brug M, López de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Martí-Massó JF, Pérez-Tur J, Wood NW, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Patel J, Trout SJ, Kruk ZL. Regional differences in evoked dopamine efflux in brain slices of rat anterior and posterior caudate putamen. Naunyn Schmiedebergs Arch Pharmacol. 1992;346:267–276. doi: 10.1007/BF00173539. [DOI] [PubMed] [Google Scholar]
- Patel JC, Witkovsky P, Avshalumov MV, Rice ME. Mobilization of calcium from intracellular stores facilitates somatodendritic dopamine release. J Neurosci. 2009;29:6568–6579. doi: 10.1523/JNEUROSCI.0181-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips PE, Hancock PJ, Stamford JA. Time window of autoreceptor-mediated inhibition of limbic and striatal dopamine release. Synapse. 2002;44:15–22. doi: 10.1002/syn.10049. [DOI] [PubMed] [Google Scholar]
- Sakaguchi-Nakashima A, Meir JY, Jin Y, Matsumoto K, Hisamoto N. LRK-1, a C. elegans PARK8-related kinase, regulates axonal-dendritic polarity of SV proteins. Curr Biol. 2007;17:592–598. doi: 10.1016/j.cub.2007.01.074. [DOI] [PubMed] [Google Scholar]
- Schmitz Y, Lee CJ, Schmauss C, Gonon F, Sulzer D. Amphetamine distorts stimulation-dependent dopamine overflow: effects on D2 autoreceptors, transporters, and synaptic vesicle stores. J Neurosci. 2001;21:5916–5924. doi: 10.1523/JNEUROSCI.21-16-05916.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönfuss D, Reum T, Olshausen P, Fischer T, Morgenstern R. Modelling constant potential amperometry for investigations of dopaminergic neurotransmission kinetics in vivo. J Neurosci Methods. 2001;112:163–172. doi: 10.1016/s0165-0270(01)00465-4. [DOI] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Moore DJ, Liang Y, West AB, Dawson VL, Dawson TM, Ross CA. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc Natl Acad Sci U S A. 2005;102:18676–18681. doi: 10.1073/pnas.0508052102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci. 2006;9:1231–1233. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- Tong Y, Pisani A, Martella G, Karouani M, Yamaguchi H, Pothos EN, Shen J. R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc Natl Acad Sci U S A. 2009;106:14622–14627. doi: 10.1073/pnas.0906334106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Tang B, Zhao G, Pan Q, Xia K, Bodmer R, Zhang Z. Dispensable role of Drosophila ortholog of LRRK2 kinase activity in survival of dopaminergic neurons. Mol Neurodegener. 2008;3:3. doi: 10.1186/1750-1326-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet. 2007;16:223–232. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- Wolozin B, Saha S, Guillily M, Ferree A, Riley M. Investigating convergent actions of genes linked to familial Parkinson's disease. Neurodegener Dis. 2008;5:182–185. doi: 10.1159/000113697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Reith ME, Wightman RM, Kawagoe KT, Garris PA. Determination of release and uptake parameters from electrically evoked dopamine dynamics measured by real-time voltammetry. J Neurosci Methods. 2001;112:119–133. doi: 10.1016/s0165-0270(01)00459-9. [DOI] [PubMed] [Google Scholar]
- Zhou W, Milder JB, Freed CR. Transgenic mice overexpressing tyrosine-to-cysteine mutant human alpha-synuclein: a progressive neurodegenerative model of diffuse Lewy body disease. J Biol Chem. 2008;283:9863–9870. doi: 10.1074/jbc.M710232200. [DOI] [PubMed] [Google Scholar]
- Zigmond MJ, Abercrombie ED, Berger TW, Grace AA, Stricker EM. Compensations after lesions of central dopaminergic neurons: some clinical and basic implications. Trends Neurosci. 1990;13:290–296. doi: 10.1016/0166-2236(90)90112-n. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Müller-Myhsok B, Dickson DW, Meitinger T, Strom TM, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]