

Abstract
The rate of porphyrin biosynthesis in mammals is controlled by the activity of the pyridoxal 5′-phosphate-dependent enzyme 5-aminolevulinate synthase (EC 2.3.1.37). Based on the postulate that turnover in this enzyme is controlled by conformational dynamics associated with a highly conserved active site loop, we constructed a variant library by targeting imperfectly conserved noncatalytic loop residues and examined the effects on product and porphyrin production. Functional loop variants of the enzyme were isolated via genetic complementation in Escherichia coli strain HU227. Colony porphyrin fluorescence varied widely when bacterial cells harboring the loop variants were grown on inductive media; this facilitated identification of clones encoding unusually active enzyme variants. Nine loop variants leading to high in vivo porphyrin production were purified and characterized kinetically. Steady state catalytic efficiencies for the two substrates were increased by up to 100-fold. Presteady state single turnover reaction data indicated that the second step of quinonoid intermediate decay, previously assigned as reaction rate-limiting, was specifically accelerated such that in three of the variants this step was no longer kinetically significant. Overall, our data support the postulate that the active site loop controls the rate of product and porphyrin production in vivo and suggest the possibility of an as yet undiscovered means of allosteric regulation.
Keywords: Diseases/Blood, Enzymes, Enzymes/Catalysis, Enzymes/Mechanisms, Enzymes/Pyridoxal Phosphate, Metabolism/Heme, Porphyrin
Introduction
Aminolevulinate (ALA)2 is the universal building block of tetrapyrolle biosynthesis (1). In nonplant eukaryotes and the α-subclass of purple bacteria, the production of ALA is catalyzed by the pyridoxal 5′-phosphate (PLP)-dependent enzyme 5-aminolevulinate synthase (ALAS) (EC 2.3.1.37), in a reaction involving the Claisen-like condensation of succinyl-coenzyme-A and glycine to yield CoA, carbon dioxide, and ALA (2). ALAS catalyzes the first committed step of tetrapyrrole biosynthesis in these organisms, which is also the rate-determining step of the pathway. Consequently, overexpression of ALAS in prokaryotic and eukaryotic cells results in accumulation of the photosensitizing heme precursor protoporphyrin IX (3). This property could potentially lead to novel applications of ALAS or ALAS variants in photodynamic therapy of tumors and other dysplasias (4).
ALAS is classified as a fold-type I PLP-dependent enzyme and, like the evolutionarily related l-amino acid transaminases (5), functions as a homodimer wherein a PLP cofactor is bound at each of the two active sites, which are recessed in clefts at the subunit interface (6, 7). X-ray crystal structures of ALAS from Rhodobacter capsulatus and the closely related enzyme 8-amino-7-oxononanoate synthase from Escherichia coli reveal an induced fit type mechanism wherein binding of substrates and product, respectively, trigger closure of an extended loop over the active site (6, 8) (Fig. 1). The inferred conformational dynamics of this loop are of interest because kinetic and crystallographic studies support the hypothesis that the rate of ALA (and hence protoporphyrin IX) production is controlled by opening of the active site loop coincident with product release (6, 9, 10). This mechanism, if correct, is notable in that the catalytic rate, which is 0.66/s at 37 °C, is slow in comparison with similar conformational motions measured in other enzymes (11), suggesting that the primary selective pressures for ALAS evolution are oriented away from speed.
FIGURE 1.

The position of the active site loop in the R. capsulatus ALAS crystal structure. In A, the ribbon representations of the three-dimensional structures of one monomer of holoenzymic (magenta) and succinyl-CoA-bound (green) ALAS from R. capsulatus are superimposed. Note the distinct positioning of the active site loop in the two structures (depicted in magenta in the ALAS holoenzyme structure and in teal in the succinyl-CoA-bound structure). In B, the active site loop in the closed conformation of ALAS (i.e. succinyl-CoA-bound structure) is perched above the catalytic cleft of the enzyme. Succinyl-CoA and the cofactor PLP are shown as sticks. The image was constructed using Pymol and Protein Data Bank entries 2BWN and 2BWO.
We posited that if loop dynamics were a primary determinant of the slow catalytic rate, then it should be straightforward to generate hyperactive ALAS variants by targeting nonconserved residues in the active site loop. Besides shedding light on the mechanism of ALA production, hyperactive ALASs might also find philanthropic utility in the generation of ALA for a variety of clinical and agricultural applications (4, 12–14). To test our hypothesis, we employed synthetic mixed base oligonucleotides to generate a library of ALAS loop variants, coupled with a simple screening strategy to isolate potentially hyperactive clones.
EXPERIMENTAL PROCEDURES
Materials
DEAE-Sephacel, β-mercaptoethanol, PLP, bovine serum albumin, succinyl-CoA, ALA-hydrochloride, α-keto-glutarate, α-ketoglutarate dehydrogenase, Bis-Tris, HEPES-free acid, MOPS, thiamin pyrophosphate, NAD+, and the bicinchoninic acid protein determination kit were purchased from Sigma-Aldrich. Ultrogel AcA 44 was from IBF Biotechnics. Glycerol, glycine, disodium ethylenediamine tetraacetic acid dihydrate, ammonium sulfate, ascorbic acid, and magnesium chloride hexahydrate were acquired from Fisher. SDS-PAGE reagents were acquired from Bio-Rad. PD-10 columns were from Amersham Biosciences. Restriction enzymes and polymerases were from New England Biolabs. Synthetic oligonucleotides were obtained from Integrated DNA Technologies.
Construction of the ALAS Active Site Loop Library
The design and experimental approach for construction of the library was based on previously described methods for shuffling mutagenesis (15–17). The targeted region corresponded to mALAS-2 amino acids Tyr422–Arg439 (supplemental Fig. S1). Codons for the 10 nonconserved positions of the loop were targeted for shuffling mutagenesis using synthetic oligonucleotides as delineated in supplemental Table S1 and described in the supplemental data. The ALAS active site library was generated by annealing three partially overlapping oligonucleotides (1 pmol each) and amplifying the annealed product with PCR. The reaction product was further amplified with another round of PCR using primers overlapping the 5′ and 3′ ends of the generated PCR product and extending to the restriction enzyme sites used in subsequent subcloning (i.e. XbaI and BamHI). To minimize the wild-type ALAS “background” during the screening of the library for functional ALAS variants, the library was subcloned into a mock ALAS expression vector. The mock vector contained the wild-type ALAS-encoding sequence from the pGF23 expression plasmid (21), with the exception of the region encoding the active site loop and flanked by the XbaI and BamHI restriction enzyme sites, which was replaced with a non-ALAS-related sequence. The primers, conditions for the annealing reaction and PCR, and mock vector used in this study are described in detail in the supplemental “Experimental Procedures.” The ligation reactions and DNA digestions with restriction endonucleases were according to standard protocols in molecular biology.
Screening of the ALAS Synthetic Library and Isolation of Functional ALAS Variants
Library screening and selection of functional variants was accomplished by reversing the ALA deficient phenotype of E. coli hemA− (HU227) (22, 23). HemA− cells can only survive if harboring a functional ALAS or when ALA (or hemin) is added to the medium (23). Electrocompetent E. coli HU227 cells were transformed with the library and plated onto LB/ampicillin medium without ALA to allow selection of the active ALAS variants as previously described (22) (supplemental Fig. S2). To score the total number of colonies produced and assess transformation efficiency, one-tenth of each transformation reaction was spread onto LB/ampicillin plates containing 10 μg/ml ALA. Functional ALAS clones (i.e. isolated from the ALA minus plates) were then picked and plated onto defined MOPS medium to induce protein overexpression (21) and screened for porphyrin overproduction using fluorescence microscopy. Briefly, the plates with the functional ALAS clones were examined with a Nikon Eclipse E1000 fluorescence microscope (Nikon, Tokyo) fitted with either a Nikon Triple Band filter set for excitation at 385–400 nm and emission at 450–465 nm for 4′,6-diamidino-2-phenylindole (DAPI) detection or a Nikon Cube BV2A excitation filter set for excitation at 400–440 nm and emission at 450–465 nm with a 610-nm long pass filter and a band pass filter at 550 nm ± 20 nm for porphyrin detection. Photographs were taken with a CCIR high performance COHU CCD camera, and the images were processed with Image software Genus 2.81 from Applied Imaging. The level of porphyrin fluorescence accumulated in the functional ALAS clones was compared with that of bacterial cells harboring wild-type ALAS and grown under the same experimental conditions (see Fig. 2). Colonies accumulating greater porphyrin fluorescence density than those harboring wild-type ALAS were individually grown overnight in LB/ampicillin medium in 96-well plates, and glycerol stocks generated from the cultures were submitted to the ICBR Genomics Core at the University of Florida for DNA sequencing of the corresponding plasmids.
FIGURE 2.

Bacterial cells harboring hyperactive ALAS variants accumulate greater porphyrin levels than those harboring wild-type ALAS. A, wild-type ALAS; B, hyperactive variant SS2. Panels 1, DAPI visualized cells. Panels 2, cells visualized for porphyrin (red) fluorescence. Panels 3, overlay of DAPI and red fluorescence visualized cells.
Overexpression, Purification, and Spectroscopic Analyses of ALAS Active Site Loop Variants
Overexpression was from the alkaline phosphatase (phoA) promoter, and the conditions for promoter induction were as previously described for mALAS-2 (21). However, induction of the F1, SS2, F10, and H1 variants was accomplished by growing the bacterial cells harboring the expression plasmids for these variants in MOPS medium supplemented with 10 mg/liter ascorbic acid for 30 h at 20 °C, because these conditions ameliorated the apparent toxicity observed with overexpression of the most active variants and produced better yields. Purification, storage, handling, and spectroscopic analysis of the mALAS-2 variants were conducted as described previously (9). Protein concentrations were determined by the bicinchoninic acid method using bovine serum albumin as the standard (21). Reported enzyme concentrations are based on the monomeric molecular mass of 56 kDa (21). Protein purity was assessed using SDS-PAGE.
Steady State and Presteady State Kinetic Characterization of ALAS Active Site Loop Variants
ALAS steady state activity of the ALAS active site loop variants was determined at 20 °C using a continuous spectrophotometric assay as described previously for wild-type ALAS (24). Activity data were acquired using a Shimadzu UV-visible 2100 dual beam spectrophotometer. The steady state kinetic parameters were determined by fitting the data to the Michaelis-Menten equation using nonlinear regression analysis software (SigmaPlot10; Systat). The steady state kinetic parameters of wild-type ALAS and the SS2 variant were also determined at 15, 25, 30, and 35 °C.
Rapid scanning stopped flow measurements were performed using a model RSM-100 stopped flow spectrophotometer (OLIS, Inc.). This instrument has a dead time of ∼2-ms and an observation chamber path length of 4 mm. Spectral scans covering a wavelength range of 300–570 nm were collected at a rate of 1000 scans/s and then averaged to 62 scans/s to reduce the time course data files to an appropriate size for global fit analyses. The temperature of the syringes and the stopped flow cell compartment was maintained at 20 °C by an external water bath. Presteady state kinetic reactions of the variant enzymes were examined under single turnover conditions, using final concentrations of 60 μm enzyme, 120 mm glycine, and 10 μm succinyl-CoA in 100 mm HEPES, pH 7.5, and 10% (v/v) glycerol as previously described (9). Single turnover data were evaluated using either a two- or three-kinetic step mechanism as described by Equations 1 and 2, respectively (9).
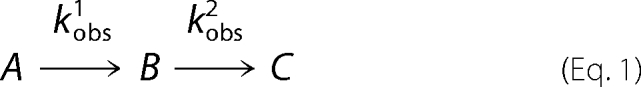 |
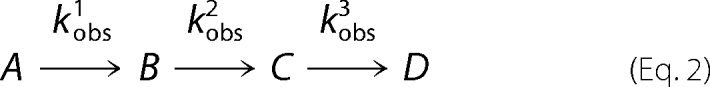 |
The spectral data were used to determine the observed rate constants via the Robust Global Fitting program. This approach utilizes the single value decomposition software provided by OLIS, Inc. as previously reported (9, 10). The quality of the calculated fits was judged by analysis of the residuals, and the simplest mechanism that described the experimental data was used.
Determination of the Dissociation Constants for the Binding of Glycine and ALA
The equilibrium dissociation constant (KD) for the binding of glycine to the SS2 variant was determined in 20 mm HEPES, pH 7.5, containing 10% glycerol at 20 °C and by titrating the SS2 variant (60 μm) with increasing concentrations of glycine (0.6–60 mm). The reaction was monitored by the increase in absorbance at 420 nm upon formation of the external aldimine between PLP and glycine (25).
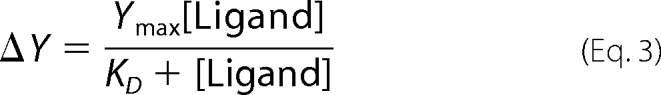 |
The KD value was determined by fitting the data to Equation 3, where ΔY is the absorbance increase at 420 nm, Ymax is the maximum increase in absorbance, and [Ligand] is the glycine concentration, using nonlinear regression analysis. Binding of ALA to the SS2 variant resulted in quenching of the fluorescence emission at 428 nm upon excitation at 330 nm, which reflected the formation of the ALA-external aldimine with the PLP cofactor. Thus to determine the dissociation constant for the binding of ALA to SS2, the change in fluorescence emission at 428 nm (λexc = 330 nm) was monitored upon titration of SS2 (60 μm) with increasing concentrations of ALA (0.5–128 mm). The changes in fluorescence at 428 nm were plotted as a function of ALA concentration, and the KD value was determined by fitting the data to Equation 3, using nonlinear regression analysis software. In this application of Equation 3, ΔY is the total change in fluorescence at 428 nm, Ymax is the initial fluorescence, and [Ligand] is the ALA concentration.
Determination of the Thermodynamic Activation Parameters
The temperature dependence of the steady state kinetic parameters of wild-type ALAS and the SS2 variant were examined as described above over the temperature range of 15–35 °C. The natural log of the calculated values for the turnover numbers (lnkcat) were plotted versus the inverse of temperature, and the data were fit to the Arrhenius equation.
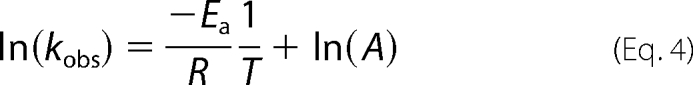 |
where Ea is the activation energy, R is the universal gas constant, T is the absolute temperature, and A is the frequency factor. The determined activation energies were then used to calculate the thermodynamic activation parameters, ΔH, ΔG, and ΔS, as previously described (10).
Data Analysis for the Definition of the Minimal Kinetic Mechanism of a Selected ALAS Active Site Loop Variant
The KinTekSim kinetic simulation software (26) was used to model the single wavelength kinetic traces at 510 nm for the reaction catalyzed by the SS2 variant and thus estimate forward and reverse rate constants. The interior rate constants were allowed to float, whereas the previously determined KD values for binding of glycine and ALA to the SS2 variant and wild-type ALAS were held constant (9).
RESULTS
Construction of the Active Site Loop Library and Screening of Functional ALAS Variants
A multiple sequence alignment of 65 different ALAS proteins was compiled with ClustalW software to determine the naturally occurring amino acid diversity present at specific positions in the active site loop (supplemental Fig. S1). This diversity was incorporated into mixed base oligonucleotide primers that were utilized to construct a library of theoretically over 108 active site loop variants.
A simple and effective two-step library screening strategy was developed for isolation of colonies potentially harboring hyperactive ALAS variants (supplemental Fig. S2). First, the library was transformed into the ALA auxotrophic E. coli strain HU227 to eliminate nonactive and very poorly active library variants (23). Next, the intensity of porphyrin fluorescence following induction of ALAS expression was used as a presumptive indicator of relative activity. The porphyrin fluorescence in individual colonies differed substantially, as exemplified in Fig. 2, where bacterial cells harboring a hyperactive ALAS variant are compared with cells harboring the wild-type ALAS expression plasmid (21). Colonies were visualized with a DAPI filter in panels A1 and B1, by red fluorescence in panels A2 and B2, and as overlays in panels A3 and B3. A decrease in colony diameter was also observed to be a general feature of bacterial cells harboring hyperactive ALAS variants, presumably because of oxidative toxicity associated with overproduction of ALA (27). Thus porphyrin fluorescence intensity proved to be a reliable indicator of ALAS activity.
Qualitative Analysis of the Isolated Active Site Loop Variants
1820 of 3800 screened colonies rescued growth of HU227 E. coli cells. Plasmid DNA from 194 colonies producing high porphyrin concentrations was sequenced in the region encompassing the active site loop and confirmed the integrity of the library (data not shown). Nine independent ALAS variants from bacterial cells producing exceptionally high porphyrin concentrations were purified and characterized. Sequence data for this subset indicated that functional mutations were present at eight of the 10 positions targeted, the exceptions being Glu436 and Leu438 (Table 1).
TABLE 1.
Amino acids substitutions in hyperactive mALAS-2 variants
Targeted positions are indicated in italics. Amino acids observed in other ALASs are indicated with bold type, while those underlined are not observed in nature.
| Wild-type ALAS | Y422 | V | Q | A | I | N | Y | P | T | V | P | R | G | E | E | L | L | R439 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Single variants | ||||||||||||||||||
| A8 | T | |||||||||||||||||
| D8 | Q | |||||||||||||||||
| G7 | H | |||||||||||||||||
| Quadruple variant | ||||||||||||||||||
| F1 | K | K | Q | Q | ||||||||||||||
| Penta variant | ||||||||||||||||||
| F10 | I | Q | N | T | N | |||||||||||||
| Hexa variants | ||||||||||||||||||
| A4 | I | P | C | R | K | N | ||||||||||||
| F3 | G | H | H | N | K | K | ||||||||||||
| H1 | N | N | I | E | K | K | ||||||||||||
| Hepta variant | ||||||||||||||||||
| SS2 | L | R | E | I | N | Q | K | |||||||||||
Steady State and Presteady State Kinetic Analyses of the Active Site Loop Variants
Steady state kinetic characterizations were carried out at 20 °C and are summarized in Table 2. Hyperactivity was arbitrarily defined as at least a 10-fold increase in catalytic efficiency toward one or both substrates, and each of the purified variant enzymes met this criterion. The catalytic efficiencies for succinyl-CoA were increased up to 100-fold, whereas those for glycine were increased in proportion to increases in kcat, because the Km values for glycine were either unchanged or slightly elevated.
TABLE 2.
Kinetic parameters for the reactions of hyperactive ALAS enzymes
Enzymatic reactions were monitored at 20 °C. WT, wild type; NA, not applicable.
| Enzyme | Steady state parameters |
Presteady state parameters |
||||||
|---|---|---|---|---|---|---|---|---|
| kcat | KmGly | KmSCoA | kcat/KmGly | kcat/KmSCoA | Qfa | Qd1b | Qd2c | |
| s−1 | mm | μm | mm−1 · s−1 | μm−1 · s−1 | s−1 | s−1 | s−1 | |
| WT | 0.02 ± 0.01 | 14 ± 2.0 | 11 ± 1.0 | 1.1 × 10−3 | 1.8 × 10−3 | 0.8 | 0.53 | 0.01 |
| SS2 | 0.31 ± 0.06 | 12 ± 1.2 | 3.0 ± 0.3 | 0.02 | 0.10 | 16 | 1.7 | NA |
| A4 | 0.16 ± 0.01 | 14 ± 1.1 | 2.3 ± 0.4 | 0.01 | 0.07 | 7.1 | 1.7 | 0.07 |
| F3 | 0.20 ± 0.01 | 13 ± 1.1 | 2.6 ± 0.4 | 0.02 | 0.08 | 7.7 | 1.1 | 0.17 |
| H1 | 0.23 ± 0.14 | 13 ± 1.4 | 2.0 ± 0.3 | 0.02 | 0.11 | 10 | 1.5 | NA |
| F10 | 0.17 ± 0.01 | 16 ± 1.8 | 1.1 ± 0.7 | 0.01 | 0.15 | 3.7 | 0.98 | 0.15 |
| F1 | 0.20 ± 0.02 | 16 ± 1.2 | 2.9 ± 0.4 | 0.01 | 0.07 | 4.0 | 1.1 | NA |
| A8 | 0.07 ± 0.01 | 25 ± 3.7 | 2.3 ± 0.4 | 2.8 × 10−3 | 0.03 | 4.8 | 0.81 | 0.12 |
| D8 | 0.03 ± 0.01 | 24 ± 3.1 | 1.7 ± 0.7 | 1.3 × 10−3 | 0.18 | 5.4 | 0.44 | 0.02 |
| G7 | 0.07 ± 0.01 | 15 ± 1.7 | 1.5 ± 0.1 | 4.6 × 10−3 | 0.05 | 4.4 | 0.87 | 0.12 |
a Rate for quinonoid intermediate formation.
b Rate for first step of quinonoid intermediate decay.
c Rate for second step of quinonoid intermediate decay.
Single turnover reactions were utilized to determine the kinetics of quinonoid intermediate formation and decay (Table 2). In the wild-type enzyme, single turnovers occur in three steps (supplemental Fig. S3), which have been assigned to 1) decarboxylation of a PLP-bound α-amino-β-ketoadipate intermediate to form a PLP-ALA quinonoid intermediate, 2) protonation of the quinonoid intermediate to form the bound product, and 3) opening of the active site loop coincident with product released (10). Quinonoid intermediate formation for the A4 and F3 hexa-variants were increased by 9-fold (7.1 and 7.7 s−1, respectively) compared with wild-type ALAS (0.8 s−1). Additionally, the rates corresponding to the first step of quinonoid intermediate decay in these variants (1.7 and 1.1 s−1) were also increased over that of wild-type ALAS (0.53 s−1). Similarly, the second step of quinonoid intermediate decay, a step hypothesized to include opening of the active site loop and ALA dissociation, was markedly increased in these variants, with values of 0.07 and 0.17 s−1, rates 7- and 17-fold higher when compared with 0.01 s−1 for wild-type ALAS. These data support the increased catalytic efficiency observed from the experiments performed in the steady state. The singly mutated variants A8, D8, and G7 also formed the quinonoid intermediate at least 4-fold faster when compared with wild-type ALAS (4.8, 5.4, and 4.4 s−1, respectively; Fig. 3, B–D). However, the first rate of biphasic quinonoid intermediate decay was similar to that of wild-type ALAS. In the remaining three variants, the lifetime of the transient quinonoid intermediate was dramatically different (Figs. 3, G and H, and 4A). Instead of proceeding by a mechanism similar to wild-type ALAS, these enzymes appear to condense the biphasic rate of decay into a single step. Consequently, the data are fitted to a two-step sequential mechanism. SS2, H1, and F1 each form the quinonoid intermediate faster than wild-type ALAS. Most notably, SS2 does so at a rate that is 20-fold faster (16 s−1 versus 0.8 s−1 for wild-type ALAS) (Fig. 4A). The rate associated with quinonoid intermediate decay was also enhanced in these variants. Because the SS2 variant exhibited the largest enhancement in the turnover number and distinct kinetic properties from those of the parent, wild-type ALAS, we decided to focus on the definition of the kinetic mechanism of this variant.
FIGURE 3.

The single turnover reactions of isolated hyperactive ALAS variants. The presteady state kinetic parameters were determined under single turnover conditions (60 μm enzyme-glycine complex and 10 μm succinyl-CoA) at 20 °C by monitoring absorbance changes at 510 nm. The variant-catalyzed reactions A4, D8, G7, A8, F3, and F10 (A–F, respectively) resemble that of the wild-type enzyme in that quinonoid lifetime is observed as a single kinetic step assigned to quinonoid intermediate formation and a two-step kinetic process assigned to its decay (9). The reaction kinetics of the F1 and H1 variants (G and H, respectively) are markedly altered in that the second of the two kinetic steps assigned to quinonoid intermediate decay is not observed.
FIGURE 4.

The SS2 variant-catalyzed reaction. Equilibrium and presteady state kinetic parameters for the SS2 variant were determined at 20 °C by monitoring changes in absorbance at 510 nm associated with quinonoid intermediate lifetime, under single turnover conditions (A); monitoring the change in absorbance at 420 nm (internal aldimine) (B); and monitoring the change in fluorescence emission at 428 nm upon excitation at 330 nm following the addition of ALA (C).
Equilibrium Constants for the SS2 Variant
The equilibrium dissociation constants for the binding of glycine and ALA to the SS2 variant at pH 7.5 and 20 °C were resolved to generate a model of the kinetic mechanism for this variant (Fig. 4, B and C). Schiff base formation between the cofactor of the SS2 variant and glycine was monitored spectroscopically at 420 nm. Although the ratio of the absorbance of the bands at 330 and 420 nm (i.e. Abs330 nm/Abs420 nm) for the SS2 holoenzyme indicated a modest decrease in relation to that of wild-type ALAS (data not shown), the absorbance values at 420 nm, corresponding to the ALAS-glycine external aldimine, were dependent on ligand concentration. The glycine concentration-dependent Abs420 nm data were fit to a standard hyperbola (Equation 3), and the KD for glycine was found to be 4.12 ± 0.57 mm. This value is 50% lower than that of the wild-type enzyme. The increased affinity of SS2 for glycine suggests that the active site lid mutations facilitate substrate binding, a finding coincident with increased catalytic efficiency. The dissociation constant for ALA, as measured in the SS2 variant, was determined by monitoring the change in fluorescence emitted at 428 nm upon excitation at 330 nm. To determine the KD for the product, the enzyme was reacted with increasing concentrations of ALA. The dissociation constant for ALA was determined to be 1.82 ± 0.19 mm, a value 62% higher compared with that of wild-type ALAS. This value is consistent with an increased rate of product release.
Thermodynamic Properties of Wild-type ALAS and the SS2 Variant
The dependence of the turnover number (kcat) on temperature was characterized over the range 288–308 K for both wild-type ALAS and the SS2 variant (Fig. 5). The kcat values were calculated using the same experimental method, and data treatment was as described for the reported steady state kinetic parameters. The turnover numbers for each enzyme were used to construct an Arrhenius plot, from which the thermodynamic activation parameters were derived (supplemental Table S2). The Arrhenius plot of the experimental data showed a linear dependence in the temperature range for both enzymes. An activation energy (Ea) of 48 kcal/mol was calculated for the wild-type enzyme, whereas for the SS2 enzyme, Ea was determined to be 18 kcal/mol, a value 80% lower compared with that of wild-type ALAS. This suggests that the SS2-catalyzed reaction may decrease the energy barrier for enzyme motions that are coupled to the reaction. Different slope values were calculated from the linear relationship between the kcat values for both enzymes and temperature. The slope value associated with wild-type ALAS was −24000, compared with −9200 for the SS2 variant. These two dissimilar values suggest that the SS2-catalyzed reaction proceeds by an alternate reaction pathway, where chemistry and lid mobility determines the rate-limiting step (Fig. 6).
FIGURE 5.

The thermal dependence of the SS2 variant-catalyzed reaction. kcat values were calculated by performing steady state kinetics at different temperatures. A, an Arrhenius plot depicting the difference between the wild-type (●) and the SS2 variant-catalyzed (○) reactions at 288, 293, 298, 303, and 308 K. The error bars are plotted over and partially obscured by the data points.
FIGURE 6.

The simulated kinetic mechanisms of the wild-type ALAS-catalyzed (A) and SS2 variant-catalyzed (B) reactions. Transition from an internal aldimine between the PLP cofactor and lysine 313 to an external aldimine with glycine is the first step. Binding of succinyl-CoA occurs next, which is followed by formation of the quinonoid intermediate and protonation of the quinonoid intermediate to yield an aldimine bound molecule of ALA. Finally, the release of ALA completes the reaction. E, wild-type ALAS or SS2 variant; G, glycine; EG, enzyme-glycine complex; SCoA, succinyl-CoA; EGSCoA, ALAS-glycine-succinyl-CoA complex; EQ, observable quinonoid intermediate; EALA, ALAS-ALA external aldimine; EALA1, ALAS-ALA external aldimine with active site loop closed; EALA2, ALAS-ALA external aldimine with active site loop open. (The rate and dissociation constants for the wild-type ALAS-catalyzed reaction were previously reported (10).)
DISCUSSION
A growing body of experimental evidence supports the theory that enzyme dynamics are not only integral to but also often synchronous with key kinetic steps during biocatalysis (28, 29). Product release is rate-determining for many enzymatic reactions, and in several cases where this step has been more closely examined, a conformational change of the enzyme has been discovered to be the key kinetic barrier (30–33). If this reaction mechanism were true for ALAS, it should be possible to increase the relatively slow rate of reaction by targeting noncatalytic residues in the conformationally mobile active site loop region.
Screening a small subset of the active site loop library resulted in isolation of nine different hyperactive ALASs. Turnover numbers and catalytic efficiencies were increased for each of the purified variant enzymes (Table 2). The catalytic efficiencies for succinyl-CoA were enhanced to a greater extent than those for glycine, consistent with the role of the active site loop in binding this substrate. The variety of sequence alterations leading to hyperactivity suggests that ALAS is not evolving toward catalytic perfection and probably has evolved toward a highly controlled rate of ALA production consistent with its role in the control of porphyrin biosynthesis. It also suggests the possibility of one or more as yet undiscovered allosteric effectors or post-translational modifications that modulate ALAS activity in vivo.
The accelerations observed for the presteady state rates associated with intermediate decarboxylation (quinonoid intermediate formation) and protonation of the ALA bound quinonoid intermediate (first step of quinonoid intermediate decay) indicate that nonconserved loop residues can have effects not only on the rate of product release but also on multiple chemical steps in the catalytic cycle. It is thus reasonable to posit that loop dynamics are attuned with multiple steps in the reaction cycle.
The most logical interpretation of the absence of a second step of quinonoid intermediate decay in the reactions of the SS2, H1, and F1 variants is that this step has become faster than the first step of quinonoid intermediate decay and is thus kinetically insignificant. This would imply a fundamental change in the nature of the rate-determining step of the reaction cycle in these variants, such that opening of the active site loop coincident with ALA release is no longer the slowest step. The steady state temperature dependence studies with the SS2 variant support this conclusion, because the Q10 for the catalytic rate constant is reduced from seven to two. From the single turnover rates it can be inferred that in the SS2 variant-catalyzed reaction product release has been accelerated by at least 170-fold (Qd2SS2 must be ≥Qd1SS2, which is in turn 170-fold greater than Qd2wt).
The nature of the amino acid changes observed in the isolated hyperactive variants can in most cases be logically interpreted in terms of enzyme structure and function. For instance, a previous study found that the kcat value increased 20 and 100% for the R433L and R433K mALAS-2 variants, respectively (34). The R. capsulatus crystal structures subsequently revealed that this loop residue comes within bifurcated hydrogen bonding distance of a conserved acidic residue (Asp81 in mALAS-2) in the closed conformation. Disruption of this electrostatic interaction would be expected to destabilize the closed conformation and provide a rationale for the observed increase in activity, and changes at this position were observed in two-thirds of the hyperactive variants isolated.
In general, the hyperactive loop variants harbor mutations that increase both hydrophilicity and basicity. These changes presumably destabilize and increase the “floppiness” of the loop by increasing solubility while eliminating hydrophobic and electrostatic interactions that would otherwise stabilize the loop in the closed conformation. Asparagine and glutamine would be expected to occur at an average rate of 12% at the codons potentially coding for these residues, based on the synthetic oligonucleotides used to prepare the library (supplemental Table S1), yet they are observed at twice this rate in the isolated hyperactive loop variants (Table 1). Similarly, lysine is expected to randomly occur, on average, 7% of the time at the positions that include codons for it but is observed in 17% of the hyperactive variant positions potentially coding for it. The strongest selective pressure was observed for wild-type residues, however, as exemplified by a 6-fold greater occurrence of proline at position 432 than would be expected based on the synthetic oligonucleotides used to prepare the library.
In conclusion, the isolation and characterization of a variety of stable hyperactive ALAS variants lead us to propose that the loop is an important determinant in ALAS activity and may be a hotspot for generation of hyperactive variants. These and other ALA-overproducing ALAS variants may be useful in multiple clinical and agricultural applications. Accumulation of ALA and the consequent build-up of the protoporphyrin IX photosensitizer has long been recognized a powerful vehicle in the photodynamic therapy of tumors and other dysplasias (18, 35). Similarly, ALA can be used as a photodynamic, selective, and biodegradable herbicide or insecticide (19). In tumor photodynamic therapy, the porphyrin drug is targeted to and accumulated in the tumor and irradiated to elicit the desired pharmacological response (e.g. apoptosis) (20). The hyperactive ALAS variants described in this study, which are characterized by enhanced rates of turnover, could potentially add another dimension to photodynamic therapy. Delivery of the DNA encoding the hyperactive ALAS variants to the specific tumor tissue would lead to in vivo localized accumulation of the porphyrin photosensitizer drug, thus increasing therapy specificity and effectiveness. Although the data presented here provide circumstantial support for the hypothesis that ALAS activity is primarily controlled by loop dynamics, this is still unproven and warrants further investigation. The rates of loop motion may be too slow for analysis using current NMR-based approaches (32), however; future studies using a Förster resonance energy transfer-based labeling approach may be most appropriate.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants DK63191 and GM080270 (to G. C. F.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental text, Tables S1 and S2, and Figs. S1–S3.
- ALA
- 5-aminolevulinate
- ALAS
- 5-aminolevulinate synthase
- mALAS-2
- murine erythroid ALAS
- MOPS
- 4-morpholinopropanesulfonic acid
- PLP
- pyridoxal 5′-phosphate
- SS2
- synthetically shuffled variant 2
- DAPI
- 4′,6-diamidino-2-phenylindole.
REFERENCES
- 1.Jordan P. M. (1991) Biosynthesis of Tetrapyrroles, Elsevier, Amsterdam [Google Scholar]
- 2.Lendrihas T., Zhang J., Hunter G. A., Ferreira G. C. (2009) Protein Sci. 18, 1847–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whatley S. D., Ducamp S., Gouya L., Grandchamp B., Beaumont C., Badminton M. N., Elder G. H., Holme S. A., Anstey A. V., Parker M., Corrigall A. V., Meissner P. N., Hift R. J., Marsden J. T., Ma Y., Mieli-Vergani G., Deybach J. C., Puy H. (2008) Am. J. Hum. Genet. 83, 408–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gagnebin J., Brunori M., Otter M., Juillerat-Jeanneret L., Monnier P., Iggo R. (1999) Gene Ther. 6, 1742–1750 [DOI] [PubMed] [Google Scholar]
- 5.Eliot A. C., Kirsch J. F. (2004) Annu. Rev. Biochem. 73, 383–415 [DOI] [PubMed] [Google Scholar]
- 6.Astner I., Schulze J. O., van den Heuvel J., Jahn D., Schubert W. D., Heinz D. W. (2005) EMBO J. 24, 3166–3177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan D., Ferreira G. C. (1996) Biochemistry 35, 8934–8941 [DOI] [PubMed] [Google Scholar]
- 8.Webster S. P., Alexeev D., Campopiano D. J., Watt R. M., Alexeeva M., Sawyer L., Baxter R. L. (2000) Biochemistry 39, 516–528 [DOI] [PubMed] [Google Scholar]
- 9.Hunter G. A., Ferreira G. C. (1999) J. Biol. Chem. 274, 12222–12228 [DOI] [PubMed] [Google Scholar]
- 10.Hunter G. A., Zhang J., Ferreira G. C. (2007) J. Biol. Chem. 282, 23025–23035 [DOI] [PubMed] [Google Scholar]
- 11.Benkovic S. J., Hammes-Schiffer S. (2003) Science 301, 1196–1202 [DOI] [PubMed] [Google Scholar]
- 12.Sasaki K., Watanabe M., Tanaka T., Tanaka T. (2002) Appl. Microbiol. Biotechnol. 58, 23–29 [DOI] [PubMed] [Google Scholar]
- 13.Fukuda H., Casas A., Batlle A. (2005) Int. J. Biochem. Cell Biol. 37, 272–276 [DOI] [PubMed] [Google Scholar]
- 14.Kelty C. J., Brown N. J., Reed M. W., Ackroyd R. (2002) Photochem. Photobiol. Sci. 1, 158–168 [DOI] [PubMed] [Google Scholar]
- 15.Ness J. E., Kim S., Gottman A., Pak R., Krebber A., Borchert T. V., Govindarajan S., Mundorff E. C., Minshull J. (2002) Nat. Biotechnol. 20, 1251–1255 [DOI] [PubMed] [Google Scholar]
- 16.Ostermeier M. (2003) Trends Biotechnol. 21, 244–247 [DOI] [PubMed] [Google Scholar]
- 17.Zha D., Eipper A., Reetz M. T. (2003) ChemBioChem 4, 34–39 [DOI] [PubMed] [Google Scholar]
- 18.Ziolkowski P., Osiecka B. J., Oremek G., Siewinski M., Symonowicz K., Saleh Y., Bronowicz A. (2004) Exp. Ther. Oncol. 4, 121–129 [PubMed] [Google Scholar]
- 19.Wang L. J., Jiang W. B., Huang B. J. (2004) Physiol. Plant 121, 258–264 [DOI] [PubMed] [Google Scholar]
- 20.Dolmans D. E., Fukumura D., Jain R. K. (2003) Nat. Rev. Cancer 3, 380–387 [DOI] [PubMed] [Google Scholar]
- 21.Ferreira G. C., Dailey H. A. (1993) J. Biol. Chem. 268, 584–590 [PubMed] [Google Scholar]
- 22.Gong J., Ferreira G. C. (1995) Biochemistry 34, 1678–1685 [DOI] [PubMed] [Google Scholar]
- 23.Li J., Deslouches B., Cosloy S. D., Russell C. S. (2003) Biochim. Biophys. Acta 1626, 102–105 [DOI] [PubMed] [Google Scholar]
- 24.Hunter G. A., Ferreira G. C. (1995) Anal. Biochem. 226, 221–224 [DOI] [PubMed] [Google Scholar]
- 25.Nandi D. L. (1978) J. Biol. Chem. 253, 8872–8877 [PubMed] [Google Scholar]
- 26.Barshop B. A., Wrenn R. F., Frieden C. (1983) Anal. Biochem. 130, 134–145 [DOI] [PubMed] [Google Scholar]
- 27.Hunter G. A., Rivera E., Ferreira G. C. (2005) Arch. Biochem. Biophys. 437, 128–137 [DOI] [PubMed] [Google Scholar]
- 28.Eisenmesser E. Z., Millet O., Labeikovsky W., Korzhnev D. M., Wolf-Watz M., Bosco D. A., Skalicky J. J., Kay L. E., Kern D. (2005) Nature 438, 117–121 [DOI] [PubMed] [Google Scholar]
- 29.Benkovic S. J., Hammes G. G., Hammes-Schiffer S. (2008) Biochemistry 47, 3317–3321 [DOI] [PubMed] [Google Scholar]
- 30.Hanson J. A., Duderstadt K., Watkins L. P., Bhattacharyya S., Brokaw J., Chu J. W., Yang H. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 18055–18060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rozovsky S., McDermott A. E. (2001) J. Mol. Biol. 310, 259–270 [DOI] [PubMed] [Google Scholar]
- 32.Boehr D. D., Dyson H. J., Wright P. E. (2006) Chem. Rev. 106, 3055–3079 [DOI] [PubMed] [Google Scholar]
- 33.Raber M. L., Freeman M. F., Townsend C. A. (2009) J. Biol. Chem. 284, 207–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan D., Harrison T., Hunter G. A., Ferreira G. C. (1998) Biochemistry 37, 1478–1484 [DOI] [PubMed] [Google Scholar]
- 35.Hua Z., Gibson S. L., Foster T. H., Hilf R. (1995) Cancer Res. 55, 1723–1731 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.