

Abstract
Background and Aims
Hepatocellular carcinoma is the third leading cause of cancer mortality worldwide and current chemotherapeutic interventions for this disease are largely ineffective. The retinoblastoma tumor suppressor (RB) is functionally inactivated at relatively high frequency in hepatocellular carcinoma and hepatoma cell lines. Here we interrogated the ability of CDK4/6-inhibition to inhibit hepatocyte proliferation and the impact of RB-status on this process.
Methods
Hepatoma cell lines and xenograft models harboring RB knockdown and mice harboring liver specific Rb deletion were utilized to define the role of RB function in response to CDK4/6 inhibition.
Results
Our study shows that CDK4/6-dependent, cell cycle progression in hepatoma cells was readily arrested by inhibition of CDK4/6 by PD-0332991 or p16ink4a irrespective of RB status. Interestingly, upon CDK4/6 inhibition, p107 protein stability was dramatically increased as a function of RB loss. This engagement of compensatory mechanisms was critical for cell cycle inhibition in the absence of RB, as both the E1A oncoprotein and overexpression of E2F proteins were capable of overcoming the impact of CDK4/6 inhibition. These findings were recapitulated in xenograft models. Furthermore, to determine how these findings relate to hepatocyte proliferation in vivo, mice were exposed to carbon tetrachloride to induce liver regeneration followed by treatment with PD-0332991. This treatment significantly inhibited hepatocyte proliferation. Strikingly, this facet of PD-0332991 function was retained even in RB-deficient livers.
Conclusions
These data demonstrate that CDK4/6 inhibition is a potent mediator of cytostasis, and RB loss can be readily compensated for in the context of both hepatoma cell lines and in liver tissue.
Keywords: Retinoblastoma tumor suppressor, HCC, CDK4/6 inhibitor
INTRODUCTION
Hepatocellular carcinoma (HCC) is the most common manifestation of primary human liver cancer and is the fifth most commonly diagnosed cancer worldwide, which results in greater than 500,000 annual deaths 1. The majority of HCC cases are diagnosed at relatively advanced stages and surgical resection is associated with a high rate of early recurrence 2. Non-resectable or metastatic HCC are associated with poor prognosis and systemic chemotherapeutic approaches provide minimal benefit 3. Therefore, there is a significant need to develop novel therapeutic approaches for the systemic therapy of this disease.
Many of the major genetic abnormalities associated with human HCC have been identified. Among the pathways frequently disrupted in HCC is the RB tumor suppressor pathway 4, 5. Classically, RB functions as a negative regulator of cell cycle progression 6, 7. In early G1 phase of the cell cycle, RB restrains cell cycle progression by forming transcriptional repressor complexes that function to inhibit the transactivation of E2F-responsive genes such as cyclin A and ribonucleotide reductase subunit 2 (RNR2), which are required for entry into S-phase 6–8. Importantly, the RB-related proteins p107/p130 also contribute to the regulation of these same genes 8, 9. In the presence of mitogenic signaling, RB and related proteins are inactivated by cyclin dependent kinases (cdk). The cooperative action of D-type cyclin/CDK4/6 and cyclin E/CDK2 relieve RB-mediated transcriptional repression. This allows unrestrained E2F activity and passage into S-phase 10–12. In contrast, anti-mitogenic signaling inhibits CDK activity, thus maintaining RB, p107 and p130 in their active/hypophosphorylated form, blocking cell cycle progression 10, 12. In HCC, RB is inactivated by several mechanisms, including direct mutation or loss of the gene itself, loss of p16INK4a expression and amplification of cyclin D1.
Given the central role of RB in mediating cell cycle inhibition and correct control of proliferation, therapeutic activation of this tumor suppressor has been explored via multiple mechanisms. RB plays a central role in the establishment of cell cycle arrest following exposure of cells to a variety of antimitogenic signals, including those elicited by traditional chemotherapeutic agents (e.g. cisplatin, and 5-fluorouracil) and specific targeted interventions 13–15. Prior studies have indicated that RB and related proteins are required for the cell cycle inhibitory response to CDK4/6 inhibition, such as achieved by antibody micro-injection or the ectopic expression of p16ink4a 16, 17. Herein, the highly specific CDK4/CDK6 inhibitor PD-332991 18, 19 was utilized to define the mechanisms through which RB-deficiency alone influences the response to CDK4/6-inhibition in HCC. The presented studies indicate that RB deficiency is not sufficient for the bypass of CDK4/6 inhibition. Rather, aberrations in the dependence for CDK4 phosphorylation of pocket proteins or coordinate disruption of pocket protein function is required to render HCC cell lines resistant to CDK4 inhibition.
MATERIALS AND METHODS
Cell culture
Human hepatocellular carcinoma cells Huh7, HepG2 and Hep3B were generous gifts from Drs. Craig Cameron (Pennsylvania State University), Jerry Lingrel (University of Cincinnati), and Alvaro Puga (University of Cincinnati) respectively. breast cancer cell lines: MCF7, BT-549; osteosarcoma cell lines: U2OS, SAOS2; and prostate cancer cell lines: PC3, DU-145 were obtained from ATCC. Cells were propagated by routine subculturing in DMEM containing 10% FCS supplemented with 100 U/ml penicillin/streptomycin and 2mM L-glutamine at 37°C and 5% CO2.
Plasmids
Retroviral vector pMSCV-LMP-miRB was used for the knockdown of RB and the pMSCV-LMP-miNS vector expressing a non-specific shRNA was utilized as a control. The pH2B-GFP expression plasmid, the pCycA-Luc and pCycA-CCRE reporter plasmids20, the E1A wild-type and E1A CXCDL mutant expression plasmids 21 have been previously described, and HA-ubiquitin expression plasmid was a kind gift from Andrew Aplin (Thomas Jefferson University).
Retroviral and adenoviral transductions
Huh7 and HepG2 cells were infected with miRB or miNS retroviruses and adenoviruses as described in detail in the supplemental materials and methods.
Drug Treatments
The specific CDK4/6 inhibitor PD-0332991 (PD) was generously provided by Pfizer 18, 19. Stock solutions of PD-0332991 and Roscovitine were prepared in DMSO.
BrdU labeling and bivariate flow cytometry
For cell cycle analyses, control or drug treated cells were incubated with BrdU for one hour before harvest. Cells were washed in PBS, fixed in cold 70% ethanol; and bivariate flow cytometry was utilized for dual analysis of BrdU incorporation and total DNA content as previously described 22.
Immunoblotting, RT-PCR, QRT-PCR and protein stability assay
Equal total protein was separated by SDS-PAGE. Proteins were detected by standard immunoblotting procedure using primary antibodies indicated in the figures. For determination of p107 protein stability, HepG2miNS and HepG2miRB cells were incubated with cycloheximide (CHX). Conditions and primers for RT-PCR and qRT-PCR are available on request.
Transfections and luciferase Assay
Transient transfections were performed using Fugene 6 transfection reagent (Roche). Promoter activity was analyzed by transient transfection of luciferase reporter constructs pCycA-Luc, pCycA-CCREmut-Luc. For all transfections, pCMV-β-galactosidase was co-transfected to normalize for transfection efficiency. Analysis of luciferase activity was performed as previously described 20.
Animals and animal treatments
Details on the PD-0332291 and CCl4 treatment in vivo are provided in the supplemental material and methods. The generation of the Rbf/f and Rbf/f; albcre+ mice has been previously described 23.
PCR genotyping, recombination, preparation of liver nuclei and protein analysis
DNA extraction and genotyping was carried out as previously described 23. Liver nuclei extraction, protein extraction and immunoblotting procedures were performed as previously described 23.
Histological and Immnunohistochemistry
For histological evaluation, liver tissue was embedded in paraffin and sectioned at 4µm. Slides were then deparaffinized and described in detail in the supplemental materials and methods.
RESULTS
CDK4/6 inhibitor causes cell cycle inhibition in hepatoma cells in an RB-independent fashion
To analyze the efficacy of CDK inhibition as a means to inhibit proliferation of HCC, the influence of PD0332991 (PD), a CDK4/6 specific inhibitor 18, 19, on HepG2 and Huh7 cell lines was evaluated. Initially, the impact of PD on cell cycle distribution and replication kinetics was determined by flow cytometry. As shown in Figure 1A, HepG2 and Huh7 cells respond effectively to PD, eliciting a G1/S cell cycle arrest. To define the underlying basis for the sensitivity, the expression and activity of multiple cell cycle regulatory proteins was evaluated. As expected, PD treatment lead to the dephosphorylation of RB and related pocket proteins p107 and p130 (Fig.1B). Furthermore, there was a significant reduction in the overall levels of p107 and corresponding increase in p130 levels concomitant with the changes in phosphorylation. Down-regulation of cyclin A (a consistent target of pocket protein-mediated transcriptional repression) was observed, while cyclin E levels were retained and there was a reduction in the phosphorylated active form of CDK2 (Fig.1C). These results are consistent with the ability of RB and pocket proteins to mediate downstream effects that result in a reduction in CDK2 activity and DNA replication.
Figure 1. CDK4/6 inhibitor causes cell cycle response in HCC cells.
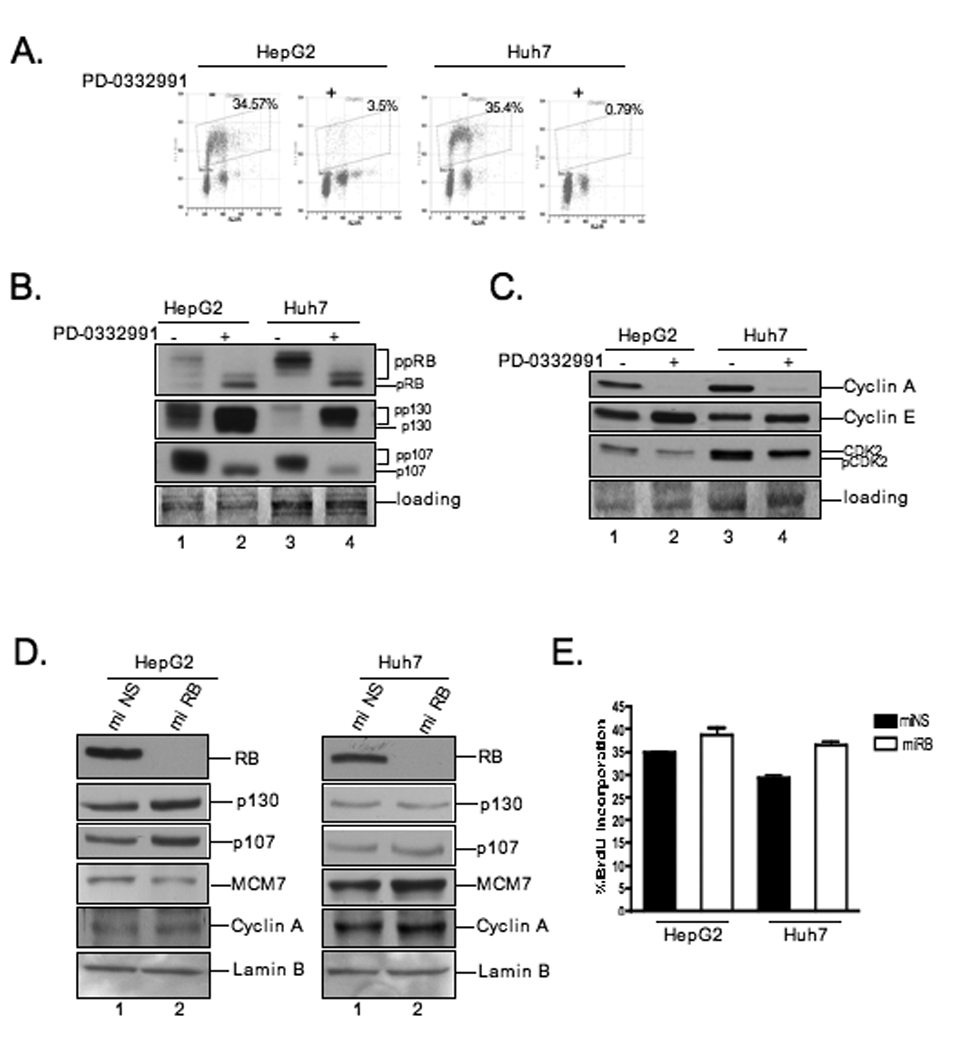
(A) Indicated cells were treated with 1µM PD-0332991 for 24 hours. Cells were pulsed with BrdU for one hour before harvest. BrdU incorporation was analyzed by bivariate flow cytometry. (B and C) Cell lysates were analyzed by immunoblot for indicated proteins. (D) Cells were infected with miRB or miNS retroviruses. Cells were harvested and lysates were analyzed by immunoblot for the indicated proteins. (E) Relative BrdU incorporation from at least three independent experiments; bars, SD.
To specifically define the influence of RB on the response of hepatoma cells to CDK4/6 inhibition, we utilized recombinant retroviruses to infect HepG2 and Huh7 cells and stably expressed synthetic micro RNA targeting RB (miRB) or a scrambled non-specific sequence (miNS). The efficiency of RB knockdown was evaluated by immunoblotting. Compared to control cells, HepG2 and Huh7 cells infected with the miRB producing retroviruses exhibited robust and stable reduction in RB protein level (Fig.1D, lanes:2). In this context, there were only modest alterations in the phosphorylation/abundance of pocket proteins p107 and p130. Similarly there was no evident change in protein levels of E2F responsive genes MCM7 and Cyclin A (Fig.1D). Analysis of DNA synthesis by BrdU incorporation revealed a modest, yet significant difference on DNA synthesis dependent on RB status (p=0.005) (Fig.1E). These findings suggested that the RB-pathway may be largely deregulated during the process of tumorigenesis, and thus deficiency of the RB protein would have minimal effects on theses parameters.
To determine the consequence of RB deficiency on the response to CDK4/6 inhibition, HepG2 and Huh7 cells expressing miNS or miRB were exposed to the specific CDK4/6 inhibitor PD332991 for 24h. Cells were then pulse-labeled with BrdU for one hour before harvest. The influence of RB-deficiency on the cell cycle response to PD was then determined by bivariate flow cytometry. As expected, PD exposure induced cell cycle inhibition in both HepG2 and Huh7 cells expressing miNS. Strikingly, PD exposure also mediated potent cell cycle inhibition in the RB-deficient cells (Fig.2A). Similar results were observed when the CDK4/6-inhibitor p16ink4a was utilized to trigger cell cycle inhibition (Fig.2B). These data indicate that RB is not required for establishing a G1/S cell cycle arrest with CDK4/6 inhibition in these models.
Figure 2. RB-independent cell cycle arrest of hepatoma cells by CDK4/6 inhibition.

(A) HepG2 and Huh7 cells models were treated with 1µM PD-0332991 for 24 hours. BrdU incorporation was analyzed by bivariate flow cytometry, data are from at least three independent experiments; bars, SD. (B) Huh7 cells were infected with Ad-p16ink4a or Ad-GFP for 24 hours. BrdU incorporation was analyzed as in (A). (C) Hep3B cells were treated and analyzed as in (A). (D) Following treatment, cell lysates were analyzed by immunoblot for the indicated proteins. (E) Cells were treated as in (A). Cell lysates were analyzed by immunoblot for the indicated proteins. (F) Hep3B cells were treated with 40µM Roscovitine (RVT) for 24 hours. BrdU incorporation was analyzed as in (A). Cell lysates were immunoblotted for indicated proteins.
Spontaneously RB-deficient cells bypass CDK4-inhibition due to differential CDK utilization
The above finding was at odds with the relatively extensive literature associated with both pharmacological and protein inhibitors of CDK4/624. However, most of these studies have utilized cell lines that have been established in the absence of RB. To investigate the possibility of an intrinsic difference between these two models of RB loss, the RB-deficient Hep3B cell line was employed. Contrary to the findings with RB knockdown, the Hep3B cells failed to respond to PD-0332991 (Fig.2C). These results indicated that likely additional permutations present in Hep3B cells underlie the relative resistance to CDK4/6 inhibition. Comparing levels of cyclin D1, CDK4 and p16ink4a between the three different cell lines revealed that Hep3B cells express copious levels of p16ink4a protein and little cyclin D1 (Fig.2E). This basic principle was readily apparent in multiple established RB-deficient cancer cell lines, indicating that this finding is not just a peculiarity of Hep3b cells (Fig.S1), and suggests that such cells are proliferating in the absence of a functional CDK4/6-cyclin D complex. Consistent with this hypothesis, the phosphorylation status and levels of p130 and p107 were unchanged by treatment with PD-0332991 in Hep3B cells (Fig.2D). These findings suggested that Hep3B cells are particularly dependent on other CDK-complexes. Therefore, Hep3B cells were treated with Roscovitine, a CDK2/CDK1 inhibitor. Roscovitine inhibited cell cycle progression in Hep3B cells and mediated the repression of E2F-regulated target genes (Fig.2F). These data indicate that in Hep3B cells where CDK4/6 activity is minimal, other CDK complexes, which are sensitive to roscovitine, mediate cell cycle control. These results indicate the mechanism through which long-term loss of RB leads to an ultimate bypass of CDK4/6 inhibition. However, the exact mechanism for continued sensitivity with acute RB was unclear.
Compensation of RB loss by complex action of other pocket proteins
To investigate the basis of CDK4/6-inhibition mediated cell cycle arrest in RB-deficient hepatoma cell lines, the repression of E2F responsive genes was investigated. Interestingly, in the presence or absence of RB, there was significant down-regulation of the well-established E2F target genes cyclin A and RNR2 at the mRNA level (Fig.3A). This finding was further confirmed at the protein level (Fig.3B). To determine if this down regulation was due to transcriptional repression, we utilized reporter constructs encompassing the sites of RB-mediated transcriptional repression of the cyclin A promoter. In both HepG2 and Huh7 cells, repression of this reporter was observed in the presence of PD (Fig.3C). Furthermore, mutation of the cell cycle regulatory element (CCRE), containing the proximal E2F sites, in this promoter rendered these constructs unresponsive to PD, suggesting formation of E2F-repressor complexes is required for repression of the cyclin A promoter in these cells. However, lack of RB expression had no impact on the transcriptional repression in this setting. The findings indicate that factors other than RB are mediating the E2F-dependent down-stream effects to regulate cell cycle control.
Figure 3. G1/S phase arrest by transcriptional repression of RB/E2F target genes is independent of RB status after PD-0332991 treatment.
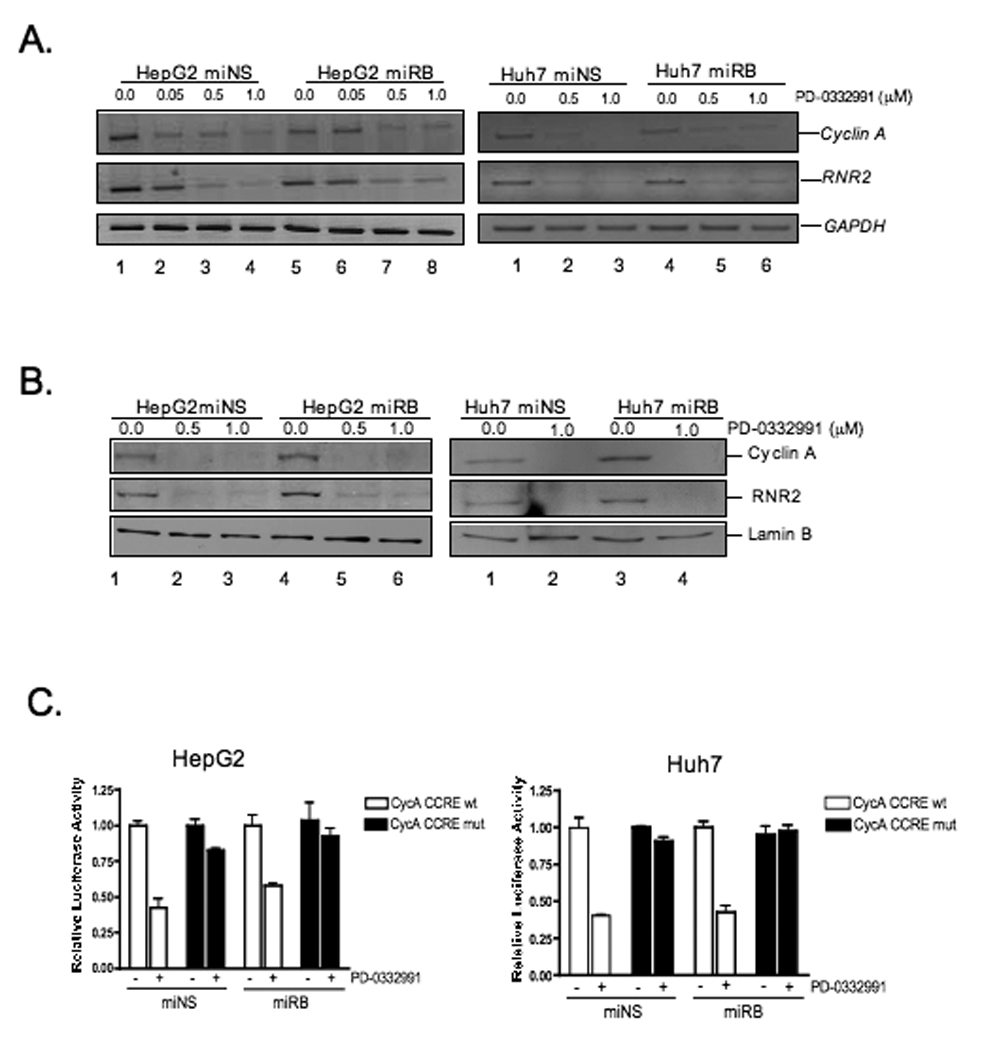
(A) Cells were treated with increasing concentrations of PD-0332991 for 24 hours. Reverse transcription-PCR was done targeting the indicated genes. (B,C) Cells were treated as in (A) and cell lysates were analyzed by immunoblot for the indicated proteins. (D) Cells were transfected with luciferase reporter constructs pCycA-Luc, pCycA-CCREmut-Luc and CMV-ß-galactosidase expression plasmid. After transfection, cells were treated as in (A) and luciferase activity was measured. Average relative luciferase activities from at least three independent experiments; bars, SD.
To investigate the mechanism of CDK4/6-mediated transcriptional repression in the absence of RB, the levels of p107 and p130 were determined. Following exposure to PD, levels of p130 protein increased in cells both RB-proficient and RB-deficient (Fig.4A). This finding suggested that variances in p130 levels are not responsible for the observed repression. However, in the case of p107, there is a precipitous reduction in protein levels following PD exposure. This effect is abrogated in the absence of RB (Fig.4A). These findings suggest that maintenance of p107 expression could be responsible for the observed effects on transcriptional repression in the absence of RB.
Figure 4. p107 protein levels and promoter occupancy is differentially regulated in RB-deficient cells exposed to PD-0332991.
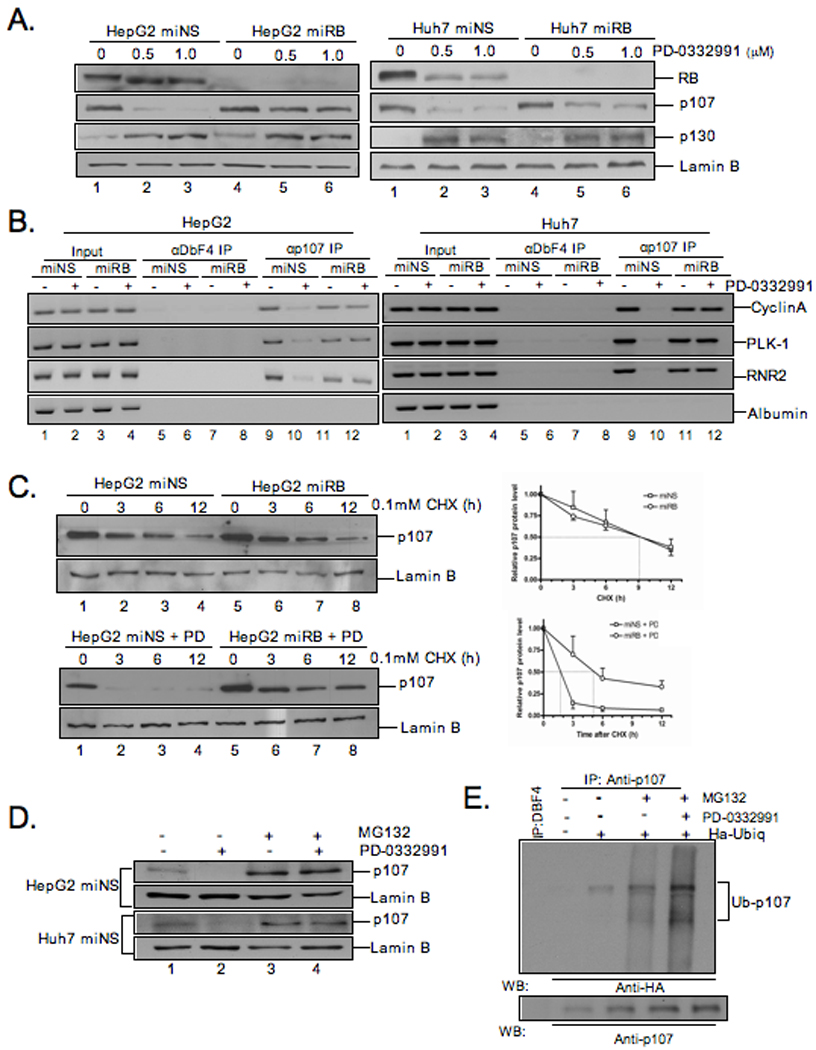
(A) Cells treated with PD-0332991 were harvested and lysates were analyzed by immunoblot for indicated proteins. (B) Chromatin was isolated from cells treated as in (A). CHIP assays were performed using antibodies for p107 and Dbf4 (negative control). Input and immunoprecipitated DNA were amplified by PCR with primers for the promoters of the indicated genes. (C) HepG2miNS/miRB cells were treated with 0.1mM cycloheximide for twelve hours. Lysates were analyzed by immunoblot for indicated proteins. Rate of p107 protein levels were plotted relative to p107 protein levels in untreated cells; bars, SD. Cells were treated with 1µM PD-0332991 for 24h. Twelve hours later, CHX (0.1mM) was added. Cells were harvested and analyzed as in (A). (D) Cells where treated with 1µM PD-0332991 and 1µM MG132 for 24h. Cells were harvested and lysates were analyzed by immunoblot for the indicated proteins. (E) HepG2 cells were transfected with HA-tagged ubiquitin and treated as in (D). Protein extracts were immunoprecipitated with anti-p107 and analyzed by immnublot for the indicated proteins.
To determine if the altered levels of p107 were contributing the transcriptional epression of E2F-target genes in the absence of RB, the association of p107 with target gene promoters was evaluated by chromatin immunoprecipitation. In hepatoma cells, p107 protein is present at the cyclin A, RNR2, and Plk1 promoters but not the E2F-independent albumin promoter (Figure.4B). However, treatment with PD results in a substantial reduction in p107 from promoter elements. This effect was particularly dependent on the presence of RB, as p107 was maintained on the promoters in those cells lacking RB when treated with PD. Together, these data suggest that differential regulation of p107 levels is compensating for the lack of RB, and this effect is specific to conditions facilitating transcriptional repression and cell cycle inhibition in response to CDK4/6 inhibition.
To determine the mechanism of differential p107 regulation, we initially evaluated p107 mRNA levels. In both RB-proficient and deficient cells exposed to PD, there was a modest yet reproducible reduction in p107 RNA levels (Fig.S2). These analyses suggest that the differential p107 regulation is not due to differences in mRNA levels. To determine if RB-status was modifying p107 protein stability, the half-life of p107 was determined utilizing the protein synthesis inhibitor cycloheximide. Interestingly, basal p107 half-life was not significantly altered by RB-deficiency (Fig.4C). In contrast, following PD exposure, p107 half-life was rapidly attenuated in RB-proficient cells (Fig.4C lanes:2,3,4). However, p107 remained relatively stable in the presence of PD in RB-deficient cells (Fig.4C lanes:6,7,8). To further analyze the mechanism of p107 regulation we treated RB-proficient cells with PD and exposed them to the proteosomal inhibitor MG132. Under these conditions, p107 protein levels are effectively maintained (Fig.4D). These data suggest that the enhanced turnover of p107 is proteasome dependent. To define the role of ubiquitylation of p107 in this process, HepG2 cells were transfected with HA-ubiquitin and then treated with vehicle or PD-0332991 in the presence of MG132. The incorporation of HA-ubiquitin in the stabililzed p107 protein was determined by immunoprecipitation of p107 followed by immunoblotting with HA. These data showed p107 ubiquitylation in cells treated with PD-0332991 and MG132 showing that p107 is degraded by the proteosome following ubiquitination (Fig.4E).
E2F-mediated repression is required for CDK4/6 inhibition to trigger arrest
The combined analyses describe a regulatory circuit, wherein the upregulation of p130 and/or maintenance of p107 expression with CDK4/6 inhibition are eliciting the downstream effects of PD to facilitate transcriptional repression in the absence of RB. To specifically determine whether such pocket proteins were critical for mediating cell cycle inhibition in the absence of RB, we employed adenovirus E1A to inactivate both p107 and p130 25. Under this condition, cell cycle progression was actively restored (p=0.03) (Fig.5A). This effect was specifically dependent on interaction of E1A with pocket proteins, as the CXDL mutant of E1A was incapable of facilitating this effect (p=0.05) (Fig.5A). Second, we utilized E2F2 over-expression as a means to reverse transcriptional repression manifested in the absence of RB. In this context, flow cytometric analysis demonstrated that in Ad-GFP-infected HepG2miRB and Huh7miRB cells, PD significantly attenuated cell cycle progression (Fig.5B). In contrast, cells ectopically expressing E2F2 continued to incorporate BrdU at the same levels as untreated cells, indicating that inhibition of E2F activity is required for cell cycle arrest in HepG2miRB and Huh7miRB cells exposed to PD. In this context, the RB-independent repression of target genes that occurs with PD treatment was reversed with expression of E2F2 (Fig.5C). Together, these data demonstrate a requirement for the repression of E2F activity for the inhibition of cell cycle progression in response to PD in hepatoma cells that have lost RB.
Figure 5. E1A or E2F overexpression reverses cell cycle arrest in RB cells exposed to PD-0332991.

(A) Cells were co-transfected with H2B-GFP expression plasmid and indicated E1A expression plasmid, and treated with 1µM PD-0332991 for 24h. The percentage of BrdU-positive cells was determined from three independent experiments; bars, SD. (B) Cells were infected with adenoviruses harboring either GFP (vector) or E2F2. Cells were treated with 1µM PD-0332991 for 24 hours. Cells were pulsed with BrdU one hour before harvest. Cell cycle was analyzed by bivariate flow cytometry. (C) Infected cells treated 1µM PD-0332991 were harvested and analyzed by immunoblot for indicated proteins.
RB-independent cell cycle arrest in vivo upon treatment with CDK4 inhibitor
To determine the impact of PD in tumors we employed xenograft models. Specifically, RB-proficient and deficient Huh7 tumors were developed following inoculation in nude mice. After tumors reached 100 mm3, the mice were treated by gavage with 150 mg/kg of PD or with lactate buffer as a control. Treatment proceeded for three days, at which point the mice received an intraperitoneal injection of BrdU and were sacrificed. Analyses of BrdU incorporation in tumor sections demonstrated that PD treatment was highly effective at suppressing proliferation of tumors (Fig.6A). Strikingly, this occurred irrespective of RB-status. In this context, analysis of p107 protein levels demonstrated its retention specifically in the RB-deficient tumors upon PD treatment (Fig.6B). Together these data illustrate that CDK4/6 inhibition can have profound impact on proliferation in tumors irrespective of RB-status.
Figure 6. PD-0332991 treatment is highly effective in xenograft models irrespective of RB status.

(A) Huh7-miNS/miRB xenografts where treated with 150mg/kg PD-0332991. Nude mice where injected with BrdU 1 hour prior to sacrifice. Sectioned tumors were immunohistochemically stained and scored for BrdU. Relative BrdU incorporation from at least three independent experiments; bars, SD. Representative images of BrdU staining at 10x magnification. (B) Tumor samples were harvested and lysates were analyzed by immunoblot for the indicated proteins.
To directly challenge these findings in a genetic mouse model, we utilized previously characterized mouse strains Rbf/f and Rbf/f; albcre+ 23. These mice harbor a liver-specific deletion of the Rb locus. This deletion event was clearly observed by genomic PCR (Fig.7A). In this model, the adult mouse liver is quiescent irrespective of RB-status. In order to induce hepatocyte proliferation, CCl4 was administered to 8-week-old mice. CCl4 administration causes liver damage that drives the liver into a regenerative process to replace the liver mass. At 48 hours after CCl4 administration, DNA synthesis was analyzed by demonstrating an increase in BrdU incorporation (p=0.02) (Fig.7B). These findings suggested that in the context of liver regeneration, there was compensation associated with RB loss. In agreement with this hypothesis, we detected elevated levels of p130 present in quiescent RB-deficient livers, while elevated levels of p107 were observed in proliferative RB-deficient livers (Fig.7C). In these quiescent livers, there were undetectable levels of E2F regulated gene products (MCM7 and Cyclin A) at the protein level, suggesting that elevated p130 contributes to the maintenance of quiescence in this model. To specifically decipher the impact of CDK4/6 inhibition on liver regeneration, 8 week-old mice were challenged with CCl4 and treated with 5 doses of PD by gavage. Animals were sacrificed 48 hours after CCl4 treatment. CCl4 potently induced liver damage resulting in large areas of necrosis proximal to the portal vein (Fig.8D, note arrows). PD treatment completely abrogated the proliferative response triggered by CCl4 administration (p=0.004) (Fig.7E). Strikingly, this effect was independent of RB status (p=0.003) as determined by BrdU incorporation (Fig.7E). Thus, CDK4/6-inhibition is a potent means of suppressing hepatocyte proliferation in vivo, in a fashion that is not intrinsically dependent on the RB tumor suppressor.
Figure 7. Cell cycle arrest independent of RB status in vivo after PD-0332991 treatment.
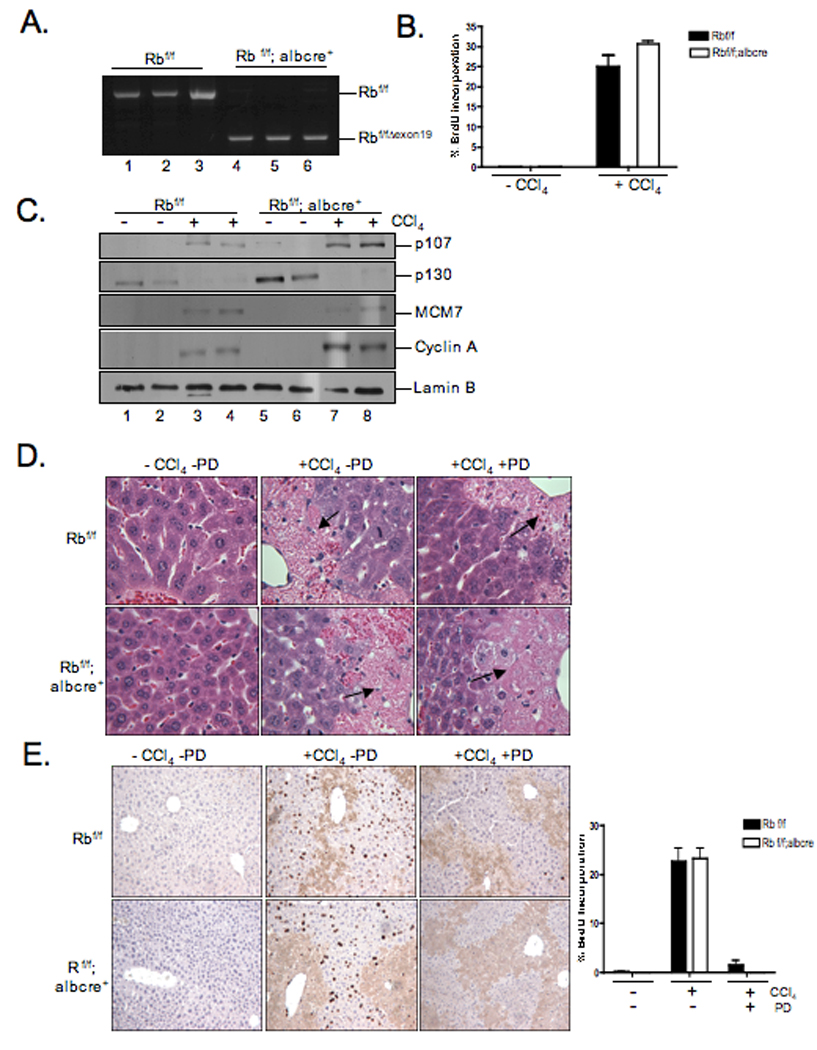
(A) Cre-mediated recombination of the floxed Rb locus (RbexonΔ19) was detected by PCR analysis of genomic DNA. (B, E) Mice were injected with CCl4 by i.p. 24 hours later treated with five doses of 150mg/kg PD-0332991. Mice where injected with BrdU 1 hour prior to sacrifice. BrdU incorporation was detected and scored. Percentage of BrdU incorporation from at least three independent experiments; bars, SD. Representative images of BrdU staining are shown at 10X magnification. (C) Liver tissue samples were harvested and lysates were analyzed by immunoblot for indicated proteins. (D) Liver tissue sections were stained with H&E. Representative images were taken of necrotic areas of the livers at 10X magnification.
DISCUSSION
Hepatocellular carcinoma is the fifth most commonly diagnosed cancer worldwide. One lesion that occurs in this tumor type is disruption of the RB-tumor suppressor pathway 4, 5. Reactivation of RB function via inhibition of CDK4/6 activity is a highly efficient means to inhibit proliferation of HCC cell lines in culture and liver tissue in vivo. According to previous work, RB loss is associated with the resistance to CDK4/6 inhibitors. In this study, we evaluated the functional impact of RB-deficiency on the response to CDK4/6 inhibition in the context of hepatocellular carcinoma. Surprisingly, RB alone is not sufficient for abrogation of response to inhibition of CDK4/6 in vitro and in vivo, rather there is a redundancy between pocket proteins that is particularly evident in this context.
It is clear that there is a major need for developing novel, more effective treatment of HCC. Currently, non-resectable HCC has a five-year survival rate of less than 10%. Thus, new models of therapeutic intervention are critical for effectively treating the disease. Here, we evaluated the efficacy of inhibiting CDK4/6 in the treatment of HCC. Most HCC tumors harbor some permutation of the CDK4/cyclinD1-p16ink4a-RB pathway, and as such, suppression of CDK4/6 function may represent an important means to target the oncogenic addictions present in HCC. In keeping with this supposition, we observed that the PD compound was very effective at limiting the proliferation of HCC cell lines and inhibiting proliferation in xenograft tumors. One of the key limitations in effectively treating HCC with therapeutic agents is the strong mechanism for detoxification in the liver. Nevertheless, we observed that CDK4/6 inhibition was an exceedingly potent mechanism to suppress proliferation in regenerating livers. This finding indicates that physiologically it is possible to attain sufficient concentrations of PD in a functional liver, and that PD has the capacity to inhibit proliferation. Although this finding is from normal liver tissue, the proliferative capacity of regenerating liver exceeds many tumors. Importantly, it is well appreciated that CDK4/6 activity is dispensable for the proliferation of mouse cells/tissues. However, our findings would strongly argue that pharmacological inhibition of CDK4/6 is distinct from genetic deletion in controlling the proliferative capacity of tissue. Thus, CDK4/6 inhibition could represent an effective means to limit tumorigenic proliferation or other hyper-proliferative conditions in the liver.
It has been recognized, through numerous studies, that deficiency in RB is associated with a resistance to CDK4/6 inhibition 17. Consistent with this phenomenon is the finding that pharmacological inhibitors such as PD332991 fail to effect RB-negative tumor cell lines 19. Interestingly, the basis through which RB status is important for this resistance to CDK4/6 inhibition has not been rigorously interrogated. In the current study, we recapitulated many of these prior findings, in that the Rb-deficient hepatoma cell line, Hep3B, is resistant to the inhibition of cell cycle progression by PD-332991. However, analyses of cell cycle control in this cell line revealed three important points. First, signaling down-stream of RB to inactivate CDK2 function was not achieved. Second, other pocket proteins present in Hep3B cells were not affected by PD-332991. Third, and most importantly, there are exceedingly high levels of p16ink4a and minimal levels of cyclin D1. These findings are consistent with the concept that CDK4/6 activity is not required for cell cycle regulation in such RB negative cells instead the cell cycle is controlled by CDK2. Consistent with this model, CDK2 inhibition by Roscovitine inhibits cell cycle progression in Hep3B cells and mediates repression of classic E2F-regulated target genes. Such general findings were also observed in a number of other tumor cell models, including prostate, osteosarcoma and breast cancer. Overall, these data raise the critical question of whether the loss of RB or the aberrant cell cycle control circuitry observed in established RB-deficient tumors cell lines is the underlying mechanism that causes the resistance to CDK4/6 inhibition.
To specifically interrogate the functional requirement for RB in the response to CDK4/6 inhibition, two cell lines (HepG2 and Huh7) with RB knockdown and mouse livers with RB deletion were employed. These approaches have been extensively validated and are known to compromise RB function, although the response to such acute RB-deficiency is context dependent 15, 26. In HCC cell lines, Rb-deficiency had little impact on cell cycle progression or deregulation of E2F/RB responsive genes. Most importantly, the knockdown of RB did not obviate the acute response to CDK4/6-inhibition, as profound cell cycle inhibition occurred in the presence of PD-332991 and p16ink4a overexpression. These findings challenge the dogma that RB is required for the response to CDK4 inhibition, and rather suggest that the re-wiring of the cell cycle machinery, as selected for in RB-deficient cells, can be responsible for resistance. Interestingly, RB-deficient cells exhibited significant repression of E2F-target genes following CDK4/6 inhibition, suggesting that the potential involvement of p107/p130 pocket proteins compensate for RB loss 7, 27. It is well established that p107 expression is elevated in systems where RB is lost 23, 28. In contrast, we have detected little effect on the basal expression of p107 and p130 following RB knockdown. Rather, under conditions of CDK4/6 inhibition, p107 levels were dramatically elevated due to altered protein stability. Importantly, the attenuation of E2F-activity by p107 plays an important function in the control of cell cycle progression in the absence of RB, as reagents that compromise pocket protein mediated E2F-repression were highly effective at bypassing the impact of PD-332991 on cell cycle control as seen by expression of the E1A oncoprotein. Thus, pocket proteins are similarly important in controlling the sensitivity to CDK4/6 inhibition through the inhibition of the E2F transcriptional program, involving compensatory mechanisms. Our data shows that the compensatory mechanism seen in the HCC cell lines can also be observed in vivo with genetic deletion of RB. In this context, compensation is clearly apparent in the RB-deficient liver due to increased p107/p130 levels that maintain quiescence 29. Using an induced liver regeneration model, CDK4/6 inhibition had a profound effect on cell cycle in RB-deficient hepatocytes. In fact, there was no difference in the overall sensitivity to the effects of PD in this model. These findings strongly suggest that the dependence on RB for CDK4/6 inhibition is not universal, and that distinct tissues harbor the ability for compensation of RB loss with such therapy.
Our combined data suggests that in the context of HCC, PD-0332991 could prove beneficial. Furthermore, the studies described herein suggest that there are two critical determinants for sensitivity to CDK4/6 inhibition. The first would involve the selection of RB-deficient tumor cells for a CDK4/6-independent cell cycle. It has been previously postulated that RB loss obviates the requirement for CDK4, and thus selection occurs as a perceived compensatory event to limit cell cycle 17. However, a secondary explanation is that induction of p16ink4a in response to oncogenic stresses supports a selection for RB loss during tumor progression 30. Our studies on CDK4/6-inhibition would support the latter mechanism. However, irrespective of its etiology, such cell cycles intrinsically lack the target for PD and are therefore resistant. Thus, the studies described suggest that detection of high-levels of p16ink4a in rapidly proliferating cells or low levels of Cyclin D1 represent a specific marker of cell cycle deregulation that will not respond to CDK4/6 inhibition. The second determinant for sensitivity to CDK4/6 inhibition would involve deregulation of pocket-protein status and control of E2F activity. In this context, the combined cohort of pocket proteins is inter-related and can define the ultimate response to CDK4/6-inhibition. Thus, it will be critical to consider the interplay and deregulation of these factors as determinants of sensitivity to agents such as PD-33299 in a clinical setting.
Supplementary Material
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Potential conflict of interest: Nothing to report.
Funding source: The National Institute of Health (NIH).
REFERENCES
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J.Clin. 2005;55:74. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.Yang Z, Wu X, Tsui TY, Hou Y, Luk JM, Fan ST. Long-term liver allograft survival induced by combined treatment with rAAV-hCTLA4Ig gene transfer and low-dose FK506. Transplantation. 2003;75:303–308. doi: 10.1097/01.TP.0000046938.50680.C4. [DOI] [PubMed] [Google Scholar]
- 3.Zhu AX. Systemic therapy of advanced hepatocellular carcinoma: how hopeful should we be? Oncologist. 2006;11:790–800. doi: 10.1634/theoncologist.11-7-790. [DOI] [PubMed] [Google Scholar]
- 4.Laurent-Puig P, Zucman-Rossi J. Genetics of hepatocellular tumors. Oncogene. 2006;25:3778–3786. doi: 10.1038/sj.onc.1209547. [DOI] [PubMed] [Google Scholar]
- 5.Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet. 2002;31:339–346. doi: 10.1038/ng0802-339. [DOI] [PubMed] [Google Scholar]
- 6.Knudsen ES, Knudsen KE. Retinoblastoma tumor suppressor: where cancer meets the cell cycle. Exp Biol Med (Maywood) 2006;231:1271–1281. doi: 10.1177/153537020623100713. [DOI] [PubMed] [Google Scholar]
- 7.Cobrinik D. Pocket proteins and cell cycle control. Oncogene. 2005;24:2796–2809. doi: 10.1038/sj.onc.1208619. [DOI] [PubMed] [Google Scholar]
- 8.Blais A, Dynlacht BD. E2F-associated chromatin modifiers and cell cycle control. Curr Opin Cell Biol. 2007;19:658–662. doi: 10.1016/j.ceb.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dannenberg JH, te Riele HP. The retinoblastoma gene family in cell cycle regulation and suppression of tumorigenesis. Results Probl Cell Differ. 2006;42:183–225. doi: 10.1007/400_002. [DOI] [PubMed] [Google Scholar]
- 10.Mittnacht S. Control of pRB phosphorylation. Curr Opin Genet Dev. 1998;8:21–27. doi: 10.1016/s0959-437x(98)80057-9. [DOI] [PubMed] [Google Scholar]
- 11.Harbour JW, Dean DC. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev. 2000;14:2393–2409. doi: 10.1101/gad.813200. [DOI] [PubMed] [Google Scholar]
- 12.Wang JY, Knudsen ES, Welch PJ. The retinoblastoma tumor suppressor protein. Adv Cancer Res. 1994;64:25–85. doi: 10.1016/s0065-230x(08)60834-9. [DOI] [PubMed] [Google Scholar]
- 13.Mayhew CN, Perkin LM, Zhang X, Sage J, Jacks T, Knudsen ES. Discrete signaling pathways participate in RB-dependent responses to chemotherapeutic agents. Oncogene. 2004;23:4107–4120. doi: 10.1038/sj.onc.1207503. [DOI] [PubMed] [Google Scholar]
- 14.Knudsen KE, Booth D, Naderi S, Sever-Chroneos Z, Fribourg AF, Hunton IC, Feramisco JR, Wang JY, Knudsen ES. RB-dependent S-phase response to DNA damage. Mol Cell Biol. 2000;20:7751–7763. doi: 10.1128/mcb.20.20.7751-7763.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bosco EE, Wang Y, Xu H, Zilfou JT, Knudsen KE, Aronow BJ, Lowe SW, Knudsen ES. The retinoblastoma tumor suppressor modifies the therapeutic response of breast cancer. J Clin Invest. 2007;117:218–228. doi: 10.1172/JCI28803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lukas J, Parry D, Aagaard L, Mann DJ, Bartkova J, Strauss M, Peters G, Bartek J. Retinoblastoma-protein-dependent cell-cycle inhibition by the tumour suppressor p16. Nature. 1995;375:503–506. doi: 10.1038/375503a0. [DOI] [PubMed] [Google Scholar]
- 17.Lukas J, Muller H, Bartkova J, Spitkovsky D, Kjerulff AA, Jansen-Durr P, Strauss M, Bartek J. DNA tumor virus oncoproteins and retinoblastoma gene mutations share the ability to relieve the cell's requirement for cyclin D1 function in G1. J Cell Biol. 1994;125:625–638. doi: 10.1083/jcb.125.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Toogood PL, Harvey PJ, Repine JT, Sheehan DJ, VanderWel SN, Zhou H, Keller PR, McNamara DJ, Sherry D, Zhu T, Brodfuehrer J, Choi C, Barvian MR, Fry DW. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J Med Chem. 2005;48:2388–2406. doi: 10.1021/jm049354h. [DOI] [PubMed] [Google Scholar]
- 19.Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, Toogood PL. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3:1427–1438. [PubMed] [Google Scholar]
- 20.Knudsen KE, Fribourg AF, Strobeck MW, Blanchard JM, Knudsen ES. Cyclin A is a functional target of retinoblastoma tumor suppressor protein-mediated cell cycle arrest. J Biol Chem. 1999;274:27632–27641. doi: 10.1074/jbc.274.39.27632. [DOI] [PubMed] [Google Scholar]
- 21.Moran E. A region of SV40 large T antigen can substitute for a transforming domain of the adenovirus E1A products. Nature. 1988;334:168–170. doi: 10.1038/334168a0. [DOI] [PubMed] [Google Scholar]
- 22.Knudsen ES, Buckmaster C, Chen TT, Feramisco JR, Wang JY. Inhibition of DNA synthesis by RB: effects on G1/S transition and S-phase progression. Genes Dev. 1998;12:2278–2292. doi: 10.1101/gad.12.15.2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mayhew CN, Bosco EE, Fox SR, Okaya T, Tarapore P, Schwemberger SJ, Babcock GF, Lentsch AB, Fukasawa K, Knudsen ES. Liver-specific pRB loss results in ectopic cell cycle entry and aberrant ploidy. Cancer Research. 2005 doi: 10.1158/0008-5472.CAN-04-4221. In press. [DOI] [PubMed] [Google Scholar]
- 24.VanderWel SN, Harvey PJ, McNamara DJ, Repine JT, Keller PR, Quin J, 3rd, Booth RJ, Elliott WL, Dobrusin EM, Fry DW, Toogood PL. Pyrido[2,3-d]pyrimidin-7-ones as specific inhibitors of cyclin-dependent kinase 4. J Med Chem. 2005;48:2371–2387. doi: 10.1021/jm049355+. [DOI] [PubMed] [Google Scholar]
- 25.Parreno M, Garriga J, Limon A, Mayol X, Beck GR, Jr, Moran E, Grana X. E1A blocks hyperphosphorylation of p130 and p107 without affecting the phosphorylation status of the retinoblastoma protein. J Virol. 2000;74:3166–3176. doi: 10.1128/jvi.74.7.3166-3176.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zagorski WA, Knudsen ES, Reed MF. Retinoblastoma deficiency increases chemosensitivity in lung cancer. Cancer Res. 2007;67:8264–8273. doi: 10.1158/0008-5472.CAN-06-4753. [DOI] [PubMed] [Google Scholar]
- 27.Sage J, Mulligan GJ, Attardi LD, Miller A, Chen S, Williams B, Theodorou E, Jacks T. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev. 2000;14:3037–3050. doi: 10.1101/gad.843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruiz S, Santos M, Segrelles C, Leis H, Jorcano JL, Berns A, Paramio JM, Vooijs M. Unique and overlapping functions of pRb and p107 in the control of proliferation and differentiation in epidermis. Development. 2004;131:2737–2748. doi: 10.1242/dev.01148. [DOI] [PubMed] [Google Scholar]
- 29.Mayhew CN, Bosco EE, Fox SR, Okaya T, Tarapore P, Schwemberger SJ, Babcock GF, Lentsch AB, Fukasawa K, Knudsen ES. Liver-specific pRB loss results in ectopic cell cycle entry and aberrant ploidy. Cancer Res. 2005;65:4568. doi: 10.1158/0008-5472.CAN-04-4221. [DOI] [PubMed] [Google Scholar]
- 30.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.