


Abstract
Here, we propose a framework for the design of synthetic protein networks from modular protein–protein or protein–peptide interactions and provide a starter toolkit of protein building blocks. Our proof of concept experiments outline a general work flow for part–based protein systems engineering. We streamlined the iterative BioBrick cloning protocol and assembled 25 synthetic multidomain proteins each from seven standardized DNA fragments. A systematic screen revealed two main factors controlling protein expression in Escherichia coli: obstruction of translation initiation by mRNA secondary structure or toxicity of individual domains. Eventually, 13 proteins were purified for further characterization. Starting from well-established biotechnological tools, two general–purpose interaction input and two readout devices were built and characterized in vitro. Constitutive interaction input was achieved with a pair of synthetic leucine zippers. The second interaction was drug-controlled utilizing the rapamycin-induced binding of FRB(T2098L) to FKBP12. The interaction kinetics of both devices were analyzed by surface plasmon resonance. Readout was based on Förster resonance energy transfer between fluorescent proteins and was quantified for various combinations of input and output devices. Our results demonstrate the feasibility of parts-based protein synthetic biology. Additionally, we identify future challenges and limitations of modular design along with approaches to address them.
INTRODUCTION
Cells rely on networks of interacting proteins for the fast integration and processing of information. Complementary changes in gene regulation require the time- and energy-consuming steps of transcription and translation. By comparison, protein regulatory networks are often already in place and merely need to be modified or re-arranged (1). Protein signaling is thus faster and, thanks to an amazing repertoire of protein functionalities, also much more versatile. Several recent studies have begun to re-route and manipulate the dynamics of natural protein networks (2–4). Nevertheless, major concepts of synthetic biology have been primarily tested on the level of gene (5) or RNA regulatory systems (6).
The engineering of systems from ‘standard biological parts’ (7–9) is one of these prevailing visions of synthetic biology. Parts are considered to be the smallest unit of engineering, typically encoded by a single DNA sequence and re-used for different aims in different contexts. Examples from the world of gene regulation include ribosomal binding sites (RBS), promoter regions, and genes for transcription factors or reporters. Standards are proposed that facilitate the physical exchange and assembly of such building blocks (10) and foster their refinement based on shared experiences (9). Parts are then combined into ‘devices’. Devices encapsulate a certain function with defined input and output. Basic examples could be a genetic toggle switch (11) or the AND gate within a RNA sensor (12). The notion of devices helps to abstract away from the underlying complexity and creates a higher level, functional rather than physical, unit of engineering. Definitions of parts and functional interfaces between devices can ultimately converge into full-fledged engineering frameworks. Such frameworks of functional composition outline standard boundaries between different categories of devices and set rules for their physical connection. Currently, frameworks have been proposed for the composition of transcription-based networks (7,13) as well as for the engineering of RNA regulatory systems (6).
We have recently outlined the contours of an engineering framework for protein synthetic biology (4). We propose to utilize the natural modularity of many proteins (14) and aim for the construction of synthetic protein circuits from protein interaction devices. In fact, modular interactions and synthetic co-recruitment have long been proven not only as a very effective way to interfere with cellular signaling (15), but have also been employed for metabolic engineering (16) or therapeutic targeting (17). Moreover, biochemists are routinely trimming or disassembling natural proteins into their globular domains or using fusions with various tags and reporters for detection, purification or life imaging. The question is now whether we can bundle this know-how into a synthetic biology framework of parts and devices and whether we will thus indeed be able to tame the intricate complexity of proteins and their networks. Some of the key requirements will be: (i) catalogs of reliable protein building blocks (parts); (ii) efficient DNA exchange formats and assembly methods for the composition of multi-domain constructs; (iii) streamlined protocols for protein production and characterization; (iv) well-characterized interaction input devices to trigger and control synthetic systems; (v) well-characterized interaction readout devices to follow their dynamics. Moreover, a rational design approach can build on the in vitro characterization of proteins and quantify their interactions in absolute biophysical measures.
Our proof of concept study elaborates on all five issues. We examine the feasibility of a prototypical work flow, beginning with the assembly of synthetic protein constructs from standard DNA parts, followed by expression and purification of proteins for biophysical characterization. We provide a starter toolkit for the design of protein interaction devices. We apply a streamlined BioBrick (18,19) assembly protocol to the construction of 25 synthetic two-domain fusion proteins from a set of 12 different globular domains as well as linker and purification tags. We evaluate the expression of these proteins in Escherichia coli and obtain soluble, purified samples for about half of them. The binding kinetics of two different interaction input devices is characterized by surface plasmon resonance (SPR) and we test the readout of interactions based on Förster resonance energy transfer (FRET). Domain swap experiments reveal factors limiting protein expression and shed light on the modularity and composability of simple input/output combinations.
All parts and constructs described in this article have been submitted to the Registry of Standard Biological Parts. Detailed protocols are maintained online on the OpenWetware community collaboration portal (http://www.openwetware.org). We aim to develop this small initial library of building blocks into a larger community-driven parts distribution for protein synthetic biology.
MATERIALS AND METHODS
Basic BioBrick protein parts
DNA coding for individual (basic) BioBrick-formatted protein parts was synthesized by GeneArt (Regensburg, Germany) and sequence-optimized for expression in E. coli. Parts were formatted as specified in BioBrick Foundation Request for Comments (BBF RFC) 25 (20). This standard proposal extends the classic BioBrick format [BBF RFC 10 (18)] by two restriction sites for in-frame protein fusions. Every part was thus preceded by the extended RFC 25 BioBrick prefix GAATTCGCGGCCGCTTCTAGATGGCCGGC (with restriction sites EcoRI, NotI, XbaI, NgoMIV) and followed by the extended BioBrick suffix ACCGGTTAATACTAGTAGCGGCCGCTGCAG (with restriction sites AgeI, SpeI, NotI, PstI). Codon optimization by GeneArt also removed any of these restriction sites from the part sequence itself. Every part is therefore compatible to standard BioBrick assembly according to BBF RFC 10 as well as to protein fusion BioBrick assembly according to BBF RFC 25. DNA constructs were delivered in GeneArt vector backbone pMA (Registry part number K157000).
Modification of BioBrick construction vectors
Construction vectors pSB1AC3, pSB1AK3 and pSB1AT3 were obtained from the Registry of Standard Biological Parts. The vector backbone without ccdB (part P1010) insert was linearized by PCR introducing the RFC 25 extensions including NgoMIV and AgeI restriction sites. The P1010 insert was amplified by PCR introducing 15-bp overlap with the modified ends of the vector backbone. The two overlapping PCR products were recombined using the In-Fusion kit (Clonetech). Three NgoMIV restriction sites within the Tc resistance cassette of pSB1AT3 were subsequently deleted using the QuickChange mutagenesis kit (Qiagen).
Streamlined BioBrick assembly
More detailed versions of these protocols are given at http://openwetware.org/wiki/Prbbbb:vector_pcr (vector backbone), http://openwetware.org/wiki/Prbbbb:fusion_assembly_v1 (assembly) and http://openwetware.org/wiki/PrbbBB:colony_pcr_v1 (colony screening).
A stock of 5× concentrated restriction mix A was prepared containing 1 U/µl EcoRI, 1 U/µl AgeI, 5× NEBuffer 1. Restriction mix B, 5× concentrated, contained 1.6 U/µl PstI, 1.7 U/µl NgoMIV, 5× NEBuffer 4, 5× BSA. A stock of 2× concentrated ligation mix contained 40 000 U/µl T4 DNA ligase in 2× T4 buffer. All plasmid DNA was normalized to a standard concentration of 50 ng/µl in water. Eight microliters of part A plasmid DNA was digested for 2 h with 2 µl restriction mix A (without further dilution). Eight microliters of part B plasmid DNA was digested for 2 h with 2 µl restriction mix B. Both restrictions were heat-inactivated for 20 min at 80°C. A stock of target vector backbone C was prepared by PCR from vectors pSB1AC3F, pSB1AK3F or pSB1AT3F using Phusion polymerase (Finnzymes) with primers BBa_J18910 and BBa_J18911, treated with DpnI, desalted, dephosphorylated with Antarctic phospatase, diluted to a standard concentration of 25 ng/µl and digested with restriction mix C containing EcoRI and PstI, followed by heat-inactivation. Four microliters of digest reaction A, 4 µl digest reaction B and 2 µl digest reaction C (vector) were mixed with 10 µl 2× ligation mix, incubated for 1 h at 16°C, heat-inactivated and 2 µl were used for transformation into 12 µl Top10 chemically competent cells (Invitrogen) according to the manufacturer's instruction. All restriction and ligation enzymes were purchased from New England Biolabs (NEB). Restrictions, ligations and transformation reactions were laid out on 96-well PCR plates as described in Figure 1 and incubated on Thermocycler heat blocks. Clones were selected by growth on agar plates containing either chloramphenicol, kanamycin, or tetracycline (depending on the target backbone).
Figure 1.

A streamlined BioBrick assembly protocol. The pairwise ligation of standardized parts was optimized for parallel processing on 96-well plates and thermocycler heat blocks. Plasmids with BioBrick-formatted parts are double-digested with restriction mix A or B, triple-ligated into a new vector backbone and transformed into competent cells. Transformants are screened by colony PCR and inoculated into deep-well plates for robotic or manual DNA miniprep. Plasmid DNA is diluted to a standard concentration and stored for further assembly rounds.
Colony PCRs were performed on 96-well PCR plates using AmpliTaq DNA Polymerase (Roche) and standard sequencing primers BBa_G00100, and BBa_G00101 with 30 cycles at 64°C annealing temperatures. PCR products were analyzed on agarose gels or on a Bioanalyzer (Agilent). For each transformation, four clones were screened and two positive clones were grown over night in square-bottom 96-deep-well plates using volumes of 1.5–1.8 ml 2xTY medium supplemented with appropriate antibiotic, sealed with gas-transmissible adhesive tape and incubated on a thermomixer (Eppendorf) at 37°C with rigorous shaking. Aliquots of cells were mixed with glycerol (20% final concentration) and stored at −80°C in 96-well plates. Remaining cells were harvested by centrifugation at optical densities (OD) around 1.5 and DNA isolated with a 96-well plate-based DNA miniprep kit (Millipore) and eluted with water (MilliQ). DNA concentrations were determined on a Nanodrop spectrophotometer (ThermoScientific) or on a plate reader (Tecan) using defined volumes in 384 square-well plates (UV-star, Greiner). DNA was diluted to 50 ng/µl in water and stored in 96-well plates at −20°C. Cell, DNA samples and sequencing results were tracked together with other part-related information in a custom-made database-backed web server, BrickIt, which we have made freely available under the GPL open source license for customization and local deployment (http://brickit.sf.net).
Protein expression screens and small-scale purification
A detailed version of the expression protocol is hosted at http://openwetware.org/wiki/Prbbbb:small_scale_expression_v1. Plasmids with the expression constructs were transformed into E. coli BL21(DE3) (Invitrogen) and spread on LB agar plates supplemented with 1% glucose, 100 µg/ml Ampicilin as well as a second antibiotic depending on the vector backbone (Kanamycin or Chloramphenicol). Agar plates were incubated for 12–14 h at 37°C. Two colonies each were picked into 1 ml LB medium containing the same antibiotics and 1% glucose and were incubated overnight shaking at 37°C in a 96-deep-well (square wells) plate with gas-transmissible sealing. Cell stocks were prepared with a final concentration of 10% glycerol and stored at −80°C.
Qualitative expression screen
A total of 2.5 ml 2xTY medium (with 1% glucose and both antibiotics) were inoculated with 25 µl from the same overnight culture and incubated shaking until an OD of ∼0.4–0.8. Protein expression was induced by adding IPTG to a final concentration of 0.5 mM. Samples were immediately split and two 1 ml aliquots of each were incubated on two different 96-deep-well plates for expression at 37°C and 20°C, respectively. Plates were sealed (gas-transmissible) and incubated on a thermomixer (Eppendorf) at 850 r.p.m. The 37°C cultures were harvested after 2.5 h by centrifugation for 10 min at 4000 r.p.m. at 4°C and pellets were subsequently frozen at −80°C. Cultures (20°C) were incubated over night and equally harvested and frozen. Cells were lysed by resuspension in 300 µl BugBuster buffer (Novagen), supplemented with 1 Tablet Complete mini protease inhibitor (Roche) per 10 ml lysis buffer. The lysis mix was incubated for 20 min shaking at room temperature. Cell debris was removed by 5 min centrifugation at 1500g at 4°C. The supernatant was transferred from the 96-well plate into 1.5 ml Eppendorf tubes and subjected to 40 min centrifugation at 20 000g and 4°C in order to split the soluble from the insoluble fraction. Ten microliters of the soluble fraction was denatured in sample buffer, separated by SDS–PAGE under reducing conditions and analyzed by western blotting with mouse monoclonal anti-polyHistidine antibody (Sigma) detected with peroxidase-conjugated sheep anti-mouse secondary antibody (Jackson ImmunoResearch, Baltimore, MD, USA) using Western Lighting ECL reagants (PerkinElmer). The insoluble fraction was redissolved in 50 µl 5× SDS–PAGE sample buffer, denatured and 5 µl were analyzed by SDS–PAGE and western blotting.
Quantitative purification screen
For each protein construct, we selected the clone with the highest soluble expression for a 4-ml scale purification screen. Starting cultures were inoculated overnight as before, 4 ml production cultures (2xTY, 1% glucose, two antibiotics) were inoculated 1:100 and incubated in 24-deep-well plates at 37°C. Most clones were inoculated on two different plates for expression at 37°C (all clones) and at 20°C (all but those with no expression at 20°C during the qualitative screen). Cultures were grown to OD of 0.4–0.8, switched to expression temperature, induced with IPTG at 0.5 mM and incubated shaking at 850 r.p.m. for 3 h (37°C) or 18 h (20°C). The final OD was measured, cells were harvested, frozen and lysed as described above (using 600 µl BugBuster buffer per sample) and split into soluble fraction and insoluble pellet. Pellets were re-dissolved in 500 µl solubilization buffer (50 mM HEPES, 0.5 M NaCl, 5 mM DTT, 1% Triton-X, 20 mM imidazole, 8 M urea, pH 7.4). Soluble and insoluble fractions were then subjected to Ni2+-affinity purification on HisMultiTrapHP 96-well plates (GE Healthcare) according to the manufacturer's instructions and eluted with elution buffer (20 mM sodium phosphate, 500 mM NaCl, 500 mM imidazole, pH 7.4). Eluates were probed by western blotting as before. Wash and eluate fractions were then analyzed on a 2100 BioAnalyzer capillary electrophoresis device (Agilent) using the Protein 80 kit according to the manufacturer's instructions. Protein concentrations were quantified by automatic integration of elution profiles with the BioAnalyzer software. Two clones each of 10 potentially toxic protein constructs (plus 2 controls) were subjected to another expression/purification screen after transformation into BL21(DE3)pLysE (Invitrogen) and growth in LB (rather than 2xTY) at 20°C.
RBS efficiency calculation
Each construct's leading 200 bp (80 bp before and 117 bp after the start codon) were submitted to the RBS calculator web server at http://voigtlab.ucsf.edu/software/. Each protein yield was normalized by the sample's final OD600 and mapped against the RBS score.
Large-scale protein purification
Each construct's best-performing clone was re-streaked from the cell stock onto LB plates supplemented with 1% glucose, ampicilin and kanamycin or chloramphenicol. Ten milliliters 2xTY (+1% glucose and the two appropriate antibiotics) were inoculated from a single fresh colony and incubated shaking over night at 37°C. Cells were pelleted by centrifugation, resuspended in 5 ml 2xTY and added to a 4 l flask with 1 l of culture medium (2xTY, 1% glucose, selection by either kanamycin or chloramphenicol). Production cultures were grown shaking at either 37°C or 20°C (depending on the previous screening result) to an OD of around 0.5 and then induced with 0.5 mM IPTG. Expression was stopped after 3 h (37°C) or after 17 h (20°C) and cells were harvested by centrifugation for 15 min at 6000g and 4°C, washed once in 15 ml PBS, weighed and stored at −80°C (20°C cultures were processed immediately). Pellets were resuspended in 5 ml binding buffer [25 mM Tris–Hcl, 20 mM imidazole, 0.5 M NaCl, 10% glycerol, pH 7.4, supplemented with ‘Complete’ protease inhibitor (Roche)] at 1 tablet per 50 ml for each gram of pellet and passed twice through a French press at about 10 000 psi pressure. The lysate was clarified by 30 min centrifugation at 50 000g and 4°C and then mixed with 5 ml Ni-NTA Agarose resin (Qiagen, washed twice in binding buffer) and incubated on a rotator for 30 min at 4°C. The resin was washed (1 min centrifugation at 2000g) first with 30 ml binding buffer, then twice with 30 ml washing buffer (25 mM Tris–Hcl, 40 mM imidazole, 0.5 M NaCl, 10% glycerol, pH 7.4) and protein was eluted by gravity flow with 10 ml elution buffer (as above but with 0.5 M imidazole) or by centrifugation (2 min at 2000g) with 2 × 5 ml of the same buffer. Eluates were filtered over a Miracloth (Calbiochem) to remove excess resin and passed into centrifugal spin concentrators (GE Healthcare) with 10 kD size exclusion limit. Samples were then exchanged into storage buffer (25 mM Tris–HCl pH 7.4, 150 mM NaCl, 10% glycerol, 1 mM DTT) by repeated concentration and dilution, and aliquots of about 3 mg/l were stored at −80°C.
His-tag removal
A 1 ml HISTrap HP column (GE Healthcare) was equilibrated in storage buffer and loaded with 1.2 ml protein (2–5 mg). The column was washed with 3–4 volumes buffer until the eluate remained colorless (all treated samples contained fluorescent protein). The column was loaded with 1.2 ml storage buffer containing 120 U GST-tagged PreScission protease (GE Healthcare) and incubated for 5 h at 4°C. It was then eluted through a 1 ml GSTrap HP column (GE Healthcare), equilibrated in the same buffer. Fractions were pooled by color and then concentrated and exchanged into HBS-P+ buffer (Biacore) using a 5 ml centrifugal spin concentrator with 10 kD exclusion limit (GE Healthcare).
SPR measurements
SPR experiments were performed on a Biacore T100 instrument (GE Healthcare) using HBS-P+ [10 mM HEPES, 0.15 M NaCl, 0.05% (v/v) polysorbate 20, 50 µM EDTA, pH 7.4] as running buffer. His-tagged proteins (ligand) were immobilized to channel 2 and 4, respectively, of a NTA sensor chip with 15 s injection at 10 µl/min. Dilution series of analyte protein were prepared in running buffer and injected over all channels at 70 µl/min flow. Following each binding, the chip surface was stripped with 60 s injection at 10 µl/min of regeneration buffer (10 mM HEPES, 0.35 M EDTA, 8 M urea, 0.5 M NaCl, pH 8.3) and re-loaded with 60 s at 10 µl/min of Nickel solution (500 µM NiCl2 in running buffer). The urea and salt supplements ensured the removal of any unspecifically bound ligand which we verified by reproducible base line and loading levels. Regular control injections of buffer-only as well as fixed analyte concentrations showed constant loading and binding curves over the course of each experimental series. Binding traces were analyzed with the manufacturer's evaluation software version 2.0.1. and fitted to a kinetic two-state 1:1 binding model. For each protein-loaded channel, we first subtracted the signal of its reference channel (i.e. 2–1or 4–3) and then the signal of a buffer-only injection.
Leucine zipper interaction
His-tagged Zip2∼mCitrine (P22) was diluted to 600 nM or 300 nM and immobilized to stable loading levels of about 1300 RU or 640 RU (response units). Dilution series of analyte protein Zip1∼mCherry (P21) without His-tag were prepared in running buffer starting with protein concentrations of 2 or 3 µM. The analyte was injected for 60 s at 70 µl/min and unbinding was monitored for 300 s.
FKBP12–FRB interaction
Running buffers were supplemented with 50 nM rapamycin. Under these conditions, we can assume a complete saturation of the FKBP12 receptor protein with rapamycin [KD = 0.2 nM (21)] that transforms the ternary protein–drug–protein interaction into a bi-molecular protein–protein binding (22). His-tagged FKBP12∼mCherry (P04) or His-tagged FKBP12∼Citrine (P03) were diluted to 0.15 µM with running buffer containing 0.2 µM rapamycin and immobilized to loading levels around 110 or 170 RU Following an additional 60 s of equilibration at normal flow, P03 and P04 showed a constant signal decline of about 0.5%/min and 2%/min, respectively. This decline was near-linear, reproducible and was corrected for by the subtraction of the buffer-only injection signal. Dilution series of analyte protein FRB∼Cherry (P10) without His-tag were prepared in running buffer with 50 nM rapamycin and starting from protein concentrations of 256 nM. The analyte was injected for 300 s at 70 µl/min and unbinding was monitored for 400 s.
FRET measurements
Measurements were performed on a plate reader (Tecan) in black flat-bottom 96-well plates. The donor protein was diluted to 0.75 µM in HBS-P + buffer (pH 7.4, see above). The acceptor protein was diluted to 1.5 µM in the same buffer. Three samples were then prepared for each protein pair: (i) 100 µl donor + 50 µl buffer, (ii) 100 µl buffer + 50 µl acceptor, (iii) 100 µl donor + 50 µl acceptor. Donor and acceptor were thus used at an equimolar concentration of 0.5 µM in 150 µl volumes. Each sample was prepared in six replicas on a single plate and was subjected to two different measurement regimes for the detection of either donor quenching or sensitized acceptor emission. Excitation and emission wavelengths for the four scenarios are listed in Table 1. The platereader's bandwidth required a gap of, at least, 30 nm between excitation and emission wavelength and excitation was therefore sometimes shifted away from the donor's absorption peak. In case of FKBP + FRB input interactions, measurements were performed before and after adding 2 µl 75 µM rapamycin to a final concentration of 1 µM. Extinction coefficients at different wavelenghts were determined on a Nanodrop spectrophotometer (Thermo Scientific) using two different constructs each for mCerulean (P05, P12), mCitrine (P19, P22), and mCherry (P04, P21).
Table 1.
FRET measurement parameters
| FRET pair | Donor quenching | Acceptor emmission | ||
|---|---|---|---|---|
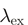 |
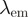 |
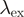 |
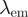 |
|
mCerulean  mCitrine mCitrine |
435 | 480 | 435 | 530 |
mCitrine  mCherry mCherry |
495 | 530 | 516 | 610 |
Excitation and emission wavelengths (in nm) used for different FRET pairs.
Data analysis
FRET efficiencies were determined with two complementary intensity measurements. (i) The decrease in donor emission (donor quenching) was quantified by comparing donor fluorescence intensities with and without acceptor protein after donor excitation. The donor-based FRET efficiency ED is then calculated as:
| (1) |
where 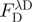 is the fluorescence intensity of the donor-only sample measured at the donor emission peak
is the fluorescence intensity of the donor-only sample measured at the donor emission peak 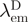 .
. 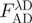 is the fluorescence of the combined donor + acceptor sample. Its raw value needs to be corrected for the acceptor-only fluorescence at the same wavelength:
is the fluorescence of the combined donor + acceptor sample. Its raw value needs to be corrected for the acceptor-only fluorescence at the same wavelength: 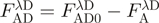 . Although in practice, this acceptor ‘bleed-through’ remained always negligible. Alternatively, (ii) the increase in sensitized acceptor emission was quantified by comparing acceptor fluorescence intensities with and without donor protein. Again, all samples were excited at, or close to the donor's maximum absorption but, this time, fluorescence intensities were recorded at the acceptor emission peak
. Although in practice, this acceptor ‘bleed-through’ remained always negligible. Alternatively, (ii) the increase in sensitized acceptor emission was quantified by comparing acceptor fluorescence intensities with and without donor protein. Again, all samples were excited at, or close to the donor's maximum absorption but, this time, fluorescence intensities were recorded at the acceptor emission peak 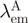 . The acceptor-based FRET efficiency EA is then calculated as:
. The acceptor-based FRET efficiency EA is then calculated as:
| (2) |
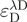 and
and 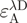 denote the donor and acceptor extinction coefficient at the same donor excitation wavelength (see below).
denote the donor and acceptor extinction coefficient at the same donor excitation wavelength (see below). 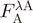 is the acceptor-only fluorescence measured at the acceptor emission maximum
is the acceptor-only fluorescence measured at the acceptor emission maximum 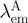 (after excitation at the donor absorbance peak).
(after excitation at the donor absorbance peak). 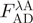 is the acceptor fluorescence of the combined donor + acceptor sample. It needs to be corrected for the donor-only contribution to the signal:
is the acceptor fluorescence of the combined donor + acceptor sample. It needs to be corrected for the donor-only contribution to the signal: 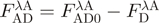 . Unlike in the donor quenching regime, both FRET pairs show a sizable emission overlap between donor and acceptor, and this correction is therefore important. Extinction coefficients are usually only published for a fluorophore's absorbance maximum (
. Unlike in the donor quenching regime, both FRET pairs show a sizable emission overlap between donor and acceptor, and this correction is therefore important. Extinction coefficients are usually only published for a fluorophore's absorbance maximum (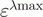 ). Off-peak coefficients like
). Off-peak coefficients like  were determined from the absorbance
were determined from the absorbance  normalized to the absorbance maximum
normalized to the absorbance maximum  :
:
| (3) |
and are given in Table 2.
Table 2.
Extinction coefficients in M−1cm−1
 |
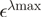 |
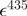 |
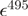 |
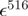 |
|
|---|---|---|---|---|---|
| mCerulean | 433 nm | 43 000b | |||
| mCitrine | 516 nm | 77 000b | 2400 | 33 000 | |
| mCherry | 587 nm | 72 000b | 9100 | 16 200 |
Exerimental extinction coefficients for different wavelenghts.
aAbsorbance maximum.
bTaken from ref. (23). The remaining values were measured here.
The measurements of donor-only, acceptor-only and donor + acceptor samples were individually averaged over six replicas each and the error of the final FRET efficiency was derived from standard error propagation of independent variables. We moreover performed an alternative analysis based on the fact that the FKBP:FRB interaction can be chemically ‘switched on’. Rather than using donor- or acceptor-only samples as reference for the FRET-off state, we directly compared donor or acceptor fluorescence before and after saturating the system with rapamycin. Donor-based FRET efficiency was then determined from a single sample, with the un-induced fluorescence replacing 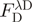 in Equation 1. Likewise, we replaced
in Equation 1. Likewise, we replaced 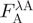 in the acceptor emission protocol (Equation 2). Nevertheless, while the donor quenching can now be measured on a single sample, the calculation of the acceptor-based FRET efficiency still requires a separate donor reference to correct for the donor signal ‘bleeding’ into the acceptor emission window.
in the acceptor emission protocol (Equation 2). Nevertheless, while the donor quenching can now be measured on a single sample, the calculation of the acceptor-based FRET efficiency still requires a separate donor reference to correct for the donor signal ‘bleeding’ into the acceptor emission window.
RESULTS
The BioBrick cloning format
The BioBrick standard (18,19) allows biological engineers to easily assemble any combination of parts by the iterative application of a single low-cost protocol. BioBrick-formatted DNA is wrapped by a standard prefix and a standard suffix sequence each containing a set of two restriction sites. The two inner restriction sites generate compatible cohesive ends which, after ligation, produce a mixed site that is not recognized by either restriction enzyme (18). This allows the iterative fusion of two BioBricks into a single new BioBrick, which can then be fused in a new round. The original BioBrick assembly format is documented as BioBrick Foundation Request For Comments number 10 or, in short, BBF RFC 10 (18). It is widely used, in particular, by student teams participating in the annual international Genetically Engineered Machine competition (iGEM) http://igem.org. A large and growing number of BioBrick-formatted DNA parts is available from the Registry of Standard Biological Parts (‘the registry’, in the following) http://partsregistry.org. Unfortunately, the original BBF RFC 10 standard was not designed for the fabrication of protein fusions. We thus adopted a modified format that extends the original prefix and suffix sequences by a new set of compatible inner restriction sites (AgeI and NgoMIV). The format is documented as BBF RFC 25 (20) and remains backward-compatible with the older standard. Finished fusion proteins can therefore be combined with the existing collection of parts using RFC 10 assembly.
BioBrick assembly is based on the repeated triple ligation of two parts A and B into a third vector backbone. A rotation between three different antibiotic resistancies allows to select for the target vector without the need to gel-purify A or B plasmid restrictions (3A assembly). The construction vectors pSB1AC3, pSB1AK3 and pSB1AT3 with three different antibiotic resistancies can be, in theory, used for both BBF RFC 10 and RFC 25 assembly reactions. However, three NgoMIV restriction sites in the tetracycline resistance cassette of pSB1AT3 render this plasmid incompatible with the BBF RFC 25 protocol. For consistency, we adapted all three vectors to BBF RFC 25. The modified construction vectors are denoted pSB1AC3F, pSB1AK3F and pSB1AT3F (F for fusion) and are available from the Registry under part numbers J18901, J18902 and J18903.
A collection of basic protein parts
Our initial collection of basic protein building blocks is listed in Table 3. All protein parts were codon optimized for expression in E. coli, adapted to the BBF RFC 25 (20) BioBrick format (18,19) and gene-synthesized from scratch.
Table 3.
Protein parts provided in this study
| IDa | Nickname | Description | Sizeb | Sourcec | Relatedd | References |
|---|---|---|---|---|---|---|
| J18912 | T7start | T7 promoter, RBS, start codon for expression in E. coli | 83 | pET3a | ||
| J18913 | T7stop | T7 terminator | 135 | pET3a | ||
| J18914 | FLAG | FLAG epitope tag (DYKDDDDK) | 24 | (26) | ||
| J18915 | 3xFLAG | 3-repeat FLAG epitope tag | 72 | (26) | ||
| J18909 | His6 | Hexahistidine affinity tag | 18 | K157011 | (24) | |
| J18916 | StrepII | StrepII affinity tag | 24 | pET52b | K157012 | (25) |
| J18917 | GST | Glutathione S-transferase tag | 687 | pGEX-2T | (27) | |
| J18918 | TEVsite | TEV protease cleavage site | 21 | K128002 | (28) | |
| J18919 | preSCsite | PreScission protease cleavage site | 24 | (29) | ||
| J18920 | 1xGS | 2 aa flexible Glycine–Serine linker | 6 | (42) | ||
| J18921 | 3xGS | 6 aa flexible Glycine–Serine linker | 12 | (42) | ||
| J18922 | 5xGS | 10 aa flexible Glycine–Serine linker | 30 | (42) | ||
| J18923 | ZipE34 | Engineered leucine zipper | 129 | (30,43) | ||
| J18924 | ZipR34 | Engineered leucine zipper | 129 | (30,43) | ||
| J18925 | FKBP12 | FKBP12 (FK506-binding protein) | 321 | (44,31) | ||
| J18926 | FRB | Engineered FKBP12-rapamycin-binding domain FRB(T2098L) | 279 | (44,45) | ||
| J18927 | LOV2 | Arabidopsis phototropin 1 LOV2 domain | 414 | (32) | ||
| I757011 | bla-frag1 | TEM-1 β-lactamase fragment 1 | 525 | iGEM 2007 | (36,37) | |
| I757012 | bla-frag1 | TEM-1 β-lactamase fragment 2 | 270 | iGEM 2007 | (36,37) | |
| J18928 | gLuc-frag1 | Gaussia luciferase fragment 1 | 276 | (38) | ||
| J18929 | gLuc-frag2 | Gaussia luciferase fragment 2 | 228 | (38) | ||
| J18930 | mCerulean | Engineered cyan fluorescent protein | 714 | (39) | ||
| J18931 | mCitrine | Engineered yellow fluorescent protein | 714 | (40) | ||
| J18932 | mCherry | Engineered red fluorescent protein | 705 | (41) |
aID in registry of standard biological parts.
bLength in base pairs.
cSource of amino acid sequence, if not literature.
dClosely related parts.
Our parts list includes the most popular tags for protein detection and affinity purification—Hexahistidine (24), StrepII (25), FLAG (26), and GST (27). These are complemented by recognition sites for TEV (28) and preScission (29) proteases as well as flexible Glycine-Serine linkers of different lengths. Our DNA assembly method allows to place these peptides in frame, in any order and at either ends or within a synthetic fusion protein. Taken together, our list therefore covers the fusion constructs that are most commonly used in modern protein purification protocols.
Another set of parts allows the triggering (input) of protein–protein interactions. ZipE34 and ZipR34 encode for a pair of leucine zippers that were rationally designed for constitutive, high-affinity heterodimeric binding (30). Alternatively, the drug-induced interaction between FKBP12 and FRB (31,22) is a widely used system that puts the co-recruitment of any two proteins under the control of rapamycin. Both interaction devices are characterized in dedicated sections below. A third interaction pair remains less established and here we only report the expression and purification of the LOV2 domain from Arabidopsis phototropin 1. According to one report (32), this domain should undergo a light-induced homodimerization.
We set out to detect protein–protein interactions by two different mechanisms: either through the complementation of enzyme fragments (33,34) (protein complementation assay, PCA) or through FRET between fluorescent proteins (35). Parts I757011 and I757012 encode for two fragments of an interaction reporter that would reconstitute TEM-1 β-lactamase activity (36). The two parts were designed by the Freiburg iGEM 2007 team based on an engineered variant of this enzyme (37). Another set of popular complementation systems converts protein–protein interactions into light signals. A recent implementation of this assay is based on the re-assembly of split luciferase from Gaussia princeps (38) and promised improved signal intensity and reversibility. Unfortunately, as we describe in further detail below, purification of both the split lactamase and the split luciferase fragments failed due to issues pertaining to folding and toxicity. We therefore focused our efforts on the FRET-based interaction readout and provide three engineered, monomeric variants of fluorescent proteins: mCerulean (cyan) (39), mCitrine (yellow) (40) and mCherry (red) (41). The three proteins can be combined into two different FRET pairs, where mCitrine either accepts FRET from mCerulean or acts as FRET donor for mCherry. Both scenarios are characterized below.
The parts list is completed by two cassettes for the initiation as well as termination of transcription and translation under the control of T7 polymerase. The start cassette includes a strong T7 promoter, E. coli RBS and start codon. The stop cassette begins with two consecutive stop codons (tagtga) followed by the T7 terminator. Both sequences were adapted from the prototypical expression vector pET3a (Novagen): we deleted an XbaI site, added stop codons and introduced the extended BioBrick prefix and suffix according to BBF RFC 25 (20). Initially, the RFC 25 BioBrick extension was only intended for protein-coding sequences. However, as a matter of convenience, the new parts J18912 and J18913 allow us to complete a full expression construct with the same protocol that is also used for the assembly of the protein coding region itself. The finished expression construct is self-contained. There is no need for further sub-cloning and expression is initiated directly from the assembled part in the standard BioBrick construction vector.
Complete sequence and additional information for all parts is available from the Registry under the IDs given in Table 3.
Construction of synthetic proteins from standard parts
We optimized the BioBrick assembly protocol for the parallel construction of composite fusion proteins from individual parts. Our modified work flow is outlined in Figure 1. Compared to the original protocol, we simplified liquid handling, reduced reagent volumes and adapted most of the process for the parallel processing of many assemblies on a 96-well plate format. Using this protocol, we have assembled expression constructs for 25 synthetic two-domain fusion proteins. The overall architecture of our constructs is described in Figure 2. Table 4 summarizes the detailed composition of all synthetic proteins. All constructs comprise seven BioBrick components and share the same architecture, with a 10 amino acid flexible linker separating two globular interaction input and readout domains that are followed by a preScission cleavage site and hexahistidine tag for affinity purification. Complete nucleotide sequences are given in the Supplementary Data and are available online from the Registry.
Figure 2.

Assembly of synthetic protein constructs from standard parts. Expression constructs for fusion proteins were built in several iterative steps using the pairwise BioBrick assembly protocol. Parts could be assembled into any order but here we adhered to a single domain architecture. Only positions of input and readout domain were occasionally swapped. All part boundaries are separated by the assembly ‘scar’ sequence Thr–Gly.
Table 4.
Synthetic proteins constructed in this study
| ID | Compositiona | Size | Registry |
|---|---|---|---|
| Domain1 – [GS]5 – domain2 ⋈H6 | (kD) | IDb | |
| P01 | FKBP ∼ Luciferase 1 | 25.2 | J18933 |
| P02 | FKBP ∼ Luciferase 2 | 23.5 | J18934 |
| P03 | FKBP ∼ mCitrine | 42.1 | J18935 |
| P04 | FKBP ∼ mCherry | 41.9 | J18936 |
| P05 | FKBP ∼ mCerulean | 42.1 | J18937 |
| P06 | FKBP ∼ β-lactamase 1 | 34.6 | J18938 |
| P07 | FKBP ∼ β-lactamase 2 | 24.9 | J18939 |
| P08 | FRB ∼ Luciferase 1 | 24.6 | J18940 |
| P09 | FRB ∼ Luciferase 2 | 22.9 | J18941 |
| P10 | FRB ∼ mCherry | 41.2 | J18942 |
| P11 | ZipE34 ∼ Luciferase 1 | 18.4 | J18943 |
| P12 | ZipE34 ∼ mCerulean | 35.2 | J18944 |
| P13 | ZipR34 ∼ Luciferase 2 | 16.7 | J18945 |
| P14 | ZipE34 ∼ β-lactamase 1 | 27.8 | J18946 |
| P15 | LOV2 ∼ Luciferase 1 | 29.5 | J18947 |
| P16 | LOV2 ∼ Luciferase 2 | 27.8 | J18948 |
| P17 | Luciferase 1 ∼ LOV2 | 29.5 | J18949 |
| P18 | mCitrine ∼ LOV2 | 46.5 | J18950 |
| P19 | FRB ∼ mCitrine | 40.7 | J18951 |
| P20 | FRB ∼ mCerulean | 40.6 | J18952 |
| P21 | ZipE34 ∼ mCherry | 35.0 | J18953 |
| P22 | ZipR34 ∼ mCitrine | 35.5 | J18954 |
| P23 | LOV2 ∼ mCitrine | 46.5 | J18955 |
| P24 | LOV2 ∼ mCerulean | 46.3 | J18956 |
| P25 | mCerulean ∼ LOV2 | 46.3 | J18957 |
aComposition of fusion proteins—all proteins follow the same layout of domain 1, GS linker, domain 2, pre-Scission restriction site ( ) and polyhistidine tag (
) and polyhistidine tag ( ); see text for details.
); see text for details.
bID in Registry of Standard Biological Parts (http://partsregistry.org); due to part assembly, all proteins start with the sequence MTG and end with TG. Proteins labeled in bold face were purified on large scale.
As shown schematically in Figure 2, six cloning reactions were needed for the assembly of a single fusion construct. Yet, several intermediate assemblies of two or three basic parts could be re-used for the construction of different targets and, under ideal conditions, the overall list should have required only about 70 steps. Not only due to intentional redundancies in our assembly pathways but also owing to the repetition of failed steps we, nevertheless, performed more than 150 cloning reactions in total. Single reactions typically yielded between 50% and 100% positive clones. By way of example, the success rate of each construct's final assembly round is given in Supplementary Table S1. Negative control reactions, lacking either part A or B, gave between zero and four colonies. The ligation-based process was not prone to point mutations and there was no need to sequence every intermediate product.
Occasional cloning failures often followed systematic patterns and we thus also document yields of less efficient assembly scenarios in Supplementary Tables S2 and S3. In particular, success rates were lower for the fusion of very short fragments or when parts were deployed directly from the gene synthesis vector pMA rather than from the standard pSB1 vector backbone. Counts of false positive transformants were moderately increased in the ‘mixed backbone scenario’ (pMA + pSB1, Supplementary Table S2), increased further if one of the parts was short and became, for unknown reasons, often unmanageable if both parts were excised from pMA (Supplementary Table S3). The assembly of some additional target constructs (combinations of luciferase or lactamase fragments 2 with leucine zipper domains) was given up after repeated failures to complete the fusion with T7start and T7stop. Other, more sporadic, assembly failures appeared unrelated to backbone or input parts and could manifest themselves in both lack of transformants or excessive false positive rates. Some were resolved by a simple repetition. Others indicated issues of quality control—sequencing of every single cloning intermediate would have been prohibitively expensive and errors could therefore occasionally propagate through different rounds of assemblies. Such effects were limited by redundancy: we archived two independent clones of each assembly step but could also bypass some bottlenecks on alternative assembly paths. All final constructs and only selected intermediate steps were verified by sequencing.
Protein expression efficiency
Our small-scale purification screen of all 25 synthetic protein constructs revealed widely varying expression levels. Detailed results are given in Table 5 and summarized in Figure 3A. Indeed, a similar variation could also be expected for the heterologous production of natural proteins. Two clones of each construct were tested at two different expression temperatures (37°C and 20°C). Here, we only report values for the best performing temperature and clone. In fact, protein yields can differ even between two clones grown under equal conditions and harboring the same construct. Overexpression of the heterologous protein puts a heavy burden on the cell. This leads to a high selective pressure and some host cell populations adapt better or worse or, occasionally, also escape protein production. For some of the more striking examples, Figure 3A includes cell densities and protein yields from the second clone grown under the same conditions: two clones of P19, grown in parallel at 37°C, gave similar protein yields. Yet, in one case, the protein remained fully soluble, whereas in the other case, virtually all of it was deposited in inclusion bodies. A similar pattern was observed for the expression of P15 in E. coli strain BL21-DE3-pLysE (see below) and the production of P19 differed by one order of magnitude between seemingly identical clones of the same strain.
Table 5.
Protein expression yields
| ID | Protein | BL21-DE3 37°Ca |
BL21-DE3 20°Cb |
DE3-pLysE 20°Cc |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| OD | Soluble (ng/µl) | Insol. | OD | Soluble (ng/µl) | Insol. | OD | Soluble (ng/µl) | Insol. | ||
| P01 | FKBP ∼ gLuc-frag 1 | 2.8 | 0 | 0 | 2.7 | 0 | 0 | |||
| P02 | FKBP ∼ gLuc-frag 2 | 0.8 | 0 | 0 | 2.5 | 0 | 0 | |||
| P03 | FKBP ∼ mCitrine | 3.6 | 50 | 275 | 8.3 | 184 | 0 | |||
| P04 | FKBP ∼ mCherry | 3.1 | 367 | n.d. | 8.4 | 597 | n.d. | 3.4 | 367 | 140 |
| P05 | FKBP ∼ mCerulean | 3.7 | 472 | 0 | 8.0 | 137 | 0 | |||
| P06 | FKBP ∼ bla-frag 1 | 1.5 | 0 | 0 | ||||||
| P07 | FKBP ∼ bla-frag 2 | 0.9 | 0 | 0 | ||||||
| P08 | FRB ∼ gLuc-frag 1 | 1.1 | 0 | 0 | 0.3 | 0 | 0 | |||
| P09 | FRB ∼ gLuc-frag 2 | 2.0 | 315 | 0 | 0.6 | 25 | 0 | |||
| P10 | FRB ∼ mCherry | 1.6 | 283 | 0 | 2.0 | 93 | n.d. | |||
| P11 | ZipE34 ∼ gLuc-frag 1 | 0.8 | 0 | 0 | 0.8 | 43 | 0 | |||
| P12 | ZipE34 ∼ mCerulean | 0.8 | 0 | 0 | 1.2 | 129 | 0 | 2.6 | 34 | 0 |
| P13 | ZipR34 ∼ gLuc-frag 2 | 0.2 | 0 | 0 | 2.0 | 0 | 0 | |||
| P14 | ZipE34 ∼ bla-frag 1 | 0.2 | 0 | 0 | 0.3 | 0 | 0 | |||
| P15 | LOV2 ∼ gLuc-frag 1 | 0.4 | 0 | 0 | 0.3 | 0 | 0 | 3.6 | 0 | 77 |
| P16 | LOV2 ∼ gLuc-frag 2 | 0.3 | 0 | 0 | 0.6 | 0 | 0 | |||
| P17 | gLuc-frag 1 ∼ LOV2 | 0.2 | 0 | 0 | 0.5 | 0 | 0 | 2.3 | 0 | 1084 |
| P18 | mCitrine ∼ LOV2 | 3.7 | 30 | 0 | 6.0 | 247 | 0 | 2.5 | 179 | 274 |
| P19 | FRB ∼ mCitrine | 4.2 | 0 | 650 | 8.2 | 0 | 0 | |||
| P20 | FRB ∼ mCerulean | 5.8 | 488 | 0 | ||||||
| P21 | ZipE34 ∼ mCherry | 2.7 | 384 | 0 | 3.7 | 278 | n.d. | |||
| P22 | ZipR34 ∼ mCitrine | 2.7 | 20 | 0 | 3.8 | 226 | 0 | |||
| P23 | LOV2 ∼ mCitrine | 3.2 | 0 | n.d. | 4.8 | 305 | 10 | |||
| P24 | LOV2 ∼ mCerulean | 1.2 | 0 | 0 | 3.8 | 226 | 0 | |||
| P25 | mCerulean ∼ LOV2 | 2.8 | 21 | 38 | 8.2 | 301 | 19 | |||
Protein yield from expression screens under different conditions. Protein constructs and IDs are the same as in Table 4. Cells were grown in 4 ml, proteins were purified on Ni affinity resin, probed by western blot and positive fractions quantified by capilary electrophoresis. Two clones each were screened under each condition and here we only report the result of the clone with the highest protein yield/cell. See text for details.
affinity resin, probed by western blot and positive fractions quantified by capilary electrophoresis. Two clones each were screened under each condition and here we only report the result of the clone with the highest protein yield/cell. See text for details.
aStandard screen in strain BL21-DE3 at 37°C.
bStandard screen in strain BL21-DE3 at 20°C.
cToxic screen in strain BL21-DE3-pLysE at 20°C. OD, cell density (OD ); soluble protein concentration in soluble fraction; insol protein concentration in insoluble fraction; n.d., a weak band was observed in the insoluble fraction but was not quantified.
); soluble protein concentration in soluble fraction; insol protein concentration in insoluble fraction; n.d., a weak band was observed in the insoluble fraction but was not quantified.
Figure 3.

Protein expression screens. (A) Absolute protein concentrations and cell densities (green) after expression and purification under standard conditions (left) or under conditions optimized for toxic proteins (right). Concentrations in soluble and insoluble fractions are stacked on top of each other. Detailed data are listed in Table 5 and here we only show the clone and condition with highest yield/OD. (B) Control of protein expression by RBS obstruction. Total protein yields from (A) were normalized to cell density (OD) and are mapped against the RBS calculator score (51) reporting on the formation of mRNA secondary structure. Proteins with no or very low expression are denoted by open symbols and were not considered for the linear regression. The protein ID corresponding to each data point is shown in Supplementary Figure S1. The RBS score (here divided by 1000) considers only a sequence window surrounding the translation initiation site and is thus identical for constructs that share the same leading domain. Some constructs with zero expression therefore collapsed into the same data point.
There were 12 constructs for which the standard screen did not turn up any detectable protein in any clone at either temperature. The majority of the latter also showed sharply decreased growth rates (e.g. P13–P17) indicating that these synthetic proteins were toxic for their expression host. The 12th failure, P18, showed signs of plasmid loss and we could retrieve a well-expressing clone in a later test (not shown). Toxicity issues were not occurring at random—essentially, expression failed for all but one design containing either the split β-lactamase or luciferase fragments. Expression worked, to varying degrees, for every other protein.
We tried to rescue the production of nine split luciferase reporters and subjected them to a second screen optimized for the expression of toxic proteins. Conditions were modified to reduce the leaky expression of the lac-repressed T7 polymerase before the actual induction of protein production with IPTG. We switched growth from rich to normal LB medium in order to reduce premature lac activation due to yeast galactosides (46). Moreover, we transferred these constructs into a modified expression host, in which background expression of T7 polymerase is counteracted by T7 lysozyme (47). As another ‘stress avoiding’ measure (46), the new screen was performed at 20°C temperature both before and after induction. P04 and P12 were included as controls. The modified conditions led to moderate production of several additional proteins (Figure 3). However, they did not provide us with a matching pair for the in vitro characterization of the luciferase interaction readout device. Overall, our standard expression screen generated synthetic protein for 14 out of our 25 constructs, and the modified screen turned up some, albeit typically low, quantities for an additional three from the putatively toxic list. The three rescued proteins (P11, P15 and P17) represent different combinations of luciferase fragment 1 with either the LOV2 or ZipE34 domain.
Yields of the successfully overexpressed proteins still display considerable variation–from close to zero to over 1000 ng/µl in the elution volume. Indeed, a large part of this variation can be explained in terms of mRNA secondary structure formation limiting the rate of translation initiation (48–50). Figure 3B correlates protein yields from the two expression screens (normalized to cell count) with a recently developed score for translation initiation efficiency (51). The score of RBS calculator is based on only the sequence surrounding the start codon and the values for each leading domain are given in Table 6. The calculation incorporates several binding terms for the interaction between ribosomal and messenger RNA. In our case, the affected sequences are all constant and our constructs merely differ in the coding region following the first three codons. The formation of intra-mRNA secondary structure remains therefore the only term of relevance. Taken together, issues of toxicity and mRNA secondary structure appeared to be the two major factors controlling protein production.
Table 6.
RBS calculator scores
| Leading parts | Score |
|---|---|
| T7start - FKBP | 6691 |
| T7start - FRB | 9590 |
| T7start - ZipE34 | 10 031 |
| T7start - ZipR34 | 3899 |
| T7start - LOV2 | 2600 |
| T7start - gLucFrag1 | 60 698 |
| T7start - mCitrine | 17 215 |
| T7start - mCerulean | 17 215 |
The score predicts the efficiency of intiation of translation based on mRNA secondary structure formation near the translation intiation site. See ‘Materials and Methods’ section for details.
Interaction input device—leucine zipper
Leucine zipper pairs offer arguably the most simple and best understood protein–protein interaction partners available so far (52). Here, we have chosen two short (43 aa) but tightly interacting helices that form a parallel heterodimeric bundle. ZipE34 and ZipR34 are the pair with highest affinity and selectivity among a larger set of rationally designed variants (30). They are derived from the B-Zip domain of the chicken transcription factor VBP and were published along with several mutants of varying affinities as constructs B-EE34 (I) and A-RR34. The same pair, as well as two of the lower affinity variants, have already been utilized for the synthetic recruitment of negative or positive modulators to a yeast protein signaling scaffold (43). Based on thermal denaturation experiments, the peptides were reported to interact with a dissociation constant of 6.1 nM.
We sought to characterize this interaction more directly and subjected two of our ZipR34 and ZipE34 containing proteins to SPR experiments. The His tag of protein P21 (ZipE34∼mCherry) was removed by proteolysis with preScission protease. Unmodified P22 (ZipR34∼mCitrine) was then in turn immobilized via its remaining His tag on the NTA sensor surface and exposed to varying concentrations of its binding partner P21. Binding and unbinding traces of one experiment are shown in Figure 4. Data were fit to a kinetic 1:1 binding model implying a simple two state reaction. Averaging over three experiments, we determined an association rate 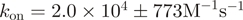 and dissociation rate
and dissociation rate 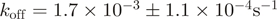 corresponding to an observed equilibrium dissociation constant
corresponding to an observed equilibrium dissociation constant 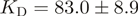 nM. The two-state model fitted the data well (average
nM. The two-state model fitted the data well (average 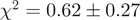 ), except at the highest analyte concentrations above 2 µM, where we observed systematic deviations (data not shown). These were excluded from the analysis, and as such we note that the simple two-state assumption may not be accurate for high local concentrations of the two binding partners. We did not detect any homodimeric binding between either ZipE34 or ZipR34 even at the highest analyte concentrations of 3 µM (data not shown).
), except at the highest analyte concentrations above 2 µM, where we observed systematic deviations (data not shown). These were excluded from the analysis, and as such we note that the simple two-state assumption may not be accurate for high local concentrations of the two binding partners. We did not detect any homodimeric binding between either ZipE34 or ZipR34 even at the highest analyte concentrations of 3 µM (data not shown).
Figure 4.

Binding kinetics of interaction input devices. (A) Constitutive interaction between Leucine zippers E34 and R34 (proteins P21 and P22) at analyte concentrations from 22 to 1500 nM. (B) Rapamycin-induced interaction between FKBP12 and FRB(T2098L) (proteins P10 and P04) at analyte concentrations from 0.25 to 32 nM. SPR traces of binding and unbinding were fitted to a 1:1 interaction model (red line).
Interaction input device—FKBP12:FRB (T2098L)
The rapamycin-induced interaction between FKBP12 (FK506 binding protein) and FRB (FKBP–Rapamycin binding domain) can be considered a textbook example of a re-usable interaction device (31) [see also references in our review (4)]. However, rapamycin is a potent immunosuppressor with antibiotic properties and can be, depending on cellular context, toxic or cytostatic (53). We replaced the wild type FRB from human mTOR by a FRB variant with mutation T2098L (54). This single mutation allows to trigger the FKBP12:FRB interaction with a non-toxic rapamycin derivative, AP21967, and is supposed to have little effect on rapamycin binding (54). At convenience, the interaction can thus be stimulated with either compound. Kinetics and equilibrium constants of the wt ternary FKBP:rapamycin:FRB interaction have already been characterized (22). However, quantitative data for the FKBP:rapamycin:FRB(T2098L) system have so far not been available.
As before, we used the His-tag of one binding partner, now P04 FKBP∼mCherry, for immobilization onto the NTA sensor surface, proteolyically removed the His-tag from the binding partner, P10 FRB∼mCherry, and analyzed their interaction by SPR. Following the same strategy as Banaszynski and colleagues (22), we saturated the system flow with 50 nM rapamycin. At this concentration, we can assume the complete saturation of the immobilized FKBP with rapamycin [KD=0.2 nM (21,22)], while the interaction between FRB and rapamycin remains negligible [wt KD=26 µM (22)].
Experimental traces of binding and unbinding are shown in Figure 4A. The kinetic fit to a 1:1 binding model gives a dissociation constant KD = 0.24 ± 0.03 nM (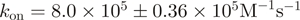 and
and 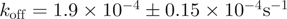 ). These values are averaged over four experiments at two different chip loading levels. Notably, steady state was not reached even at very high analyte concentrations, where the 1:1 binding model became increasingly inadequate. Inline with expectations, the FKBP:FRB(T2098L) interaction was strictly depending on rapamycin. Even 1 µM analyte concentrations gave no detectable binding signal in the absence of the drug (data not shown).
). These values are averaged over four experiments at two different chip loading levels. Notably, steady state was not reached even at very high analyte concentrations, where the 1:1 binding model became increasingly inadequate. Inline with expectations, the FKBP:FRB(T2098L) interaction was strictly depending on rapamycin. Even 1 µM analyte concentrations gave no detectable binding signal in the absence of the drug (data not shown).
Interaction readout devices—FRET
Our list of protein parts (Table 3) contains three fluorescent proteins that can be combined into two different FRET pairs for real-time detection of protein–protein interactions: cyan protein mCerulean (39) had been specifically engineered for FRET applications and acts as FRET donor for yellow fluorescent protein mCitrine (40). In the alternative setting, mCitrine is used as FRET donor for the red acceptor protein mCherry (41). In our engineering framework, each of the two pairs is considered an ‘interaction readout device’. Each device converts the input interaction between two attached domains or peptides into a measurable FRET signal. Our fusion constructs allowed us to characterize every FRET pair in combination with different input interactions. All in all, we determined FRET efficiencies for six different pairs of synthetic proteins. These experiments are easily performed on a standard plate reader instrument and an in-depth description of measurements and analysis can be found in the ‘Meterials and Methods’ section.
Overall, we determined four FRET efficiency values for each protein pair involving FKBP and FRB—we followed either donor or acceptor fluorescence while using either the external or internal (no rapamycin) reference. Only two FRET efficiencies each (donor quenching or acceptor emission) were determined for the connection of leucine zipper input to mCerulean mCitrine or mCitrine
mCitrine or mCitrine mCherry output. We have constructed fusion proteins for all six combinations of, on the one hand, FKBP or FRB binding domain and, on the other hand, mCerulean, mCitrine or mCherry readout domain. This allowed us to also perform ‘role swapping’ experiments, where the FRET donor domain was either attached to the FKBP or to the FRB part of the interaction input.
mCherry output. We have constructed fusion proteins for all six combinations of, on the one hand, FKBP or FRB binding domain and, on the other hand, mCerulean, mCitrine or mCherry readout domain. This allowed us to also perform ‘role swapping’ experiments, where the FRET donor domain was either attached to the FKBP or to the FRB part of the interaction input.
FRET efficiencies for the six different pairs of interacting proteins are shown in Table 7. The two different ways of determining the FRET-off state—either with separate control samples or by measuring in absence of rapamycin—were typically equivalent within or close to error margins. More importantly, the role-swapped FRET efficiencies were in good agreement for both the mCerulean mCitrine and the mCitrine
mCitrine and the mCitrine mCherry pair, at least, when connected to the FKBP:FRB input. That means, swapping donor and acceptor domain from one to the other half of the interaction input pair did not affect the observed FRET efficiency. Note that this comparison is more significant as it has encompassed a much larger range of possible experimental error. Ideally, for a given system, both donor- and acceptor-based FRET measurements should yield identical FRET efficiencies. This is indeed what was seen for the two combinations of FKBP:FRB input with mCerulean
mCherry pair, at least, when connected to the FKBP:FRB input. That means, swapping donor and acceptor domain from one to the other half of the interaction input pair did not affect the observed FRET efficiency. Note that this comparison is more significant as it has encompassed a much larger range of possible experimental error. Ideally, for a given system, both donor- and acceptor-based FRET measurements should yield identical FRET efficiencies. This is indeed what was seen for the two combinations of FKBP:FRB input with mCerulean mCitrine output. However, the mCitrine
mCitrine output. However, the mCitrine mCherry pair showed a marked difference between acceptor- and donor-based measurements. Gross experimental error could be ruled out as both values were perfectly reproduced between the domain-swapped experiments with different protein constructs. The discrepancy indicates a partial inactivation of the donor (mCitrine) domain. The misfolding or photoinactivation of some donor fluorophores would not affect the donor-based measurement of FRET efficiencies as every intact donor would still be paired up with an intact acceptor and the signal is normalized to the intact donor population. However, defective donor domains would, indeed, dilute the FRET signal measured on the acceptor side (55), which is normalized to the intensity of (intact) acceptor molecules. Conversely, a certain fraction of defective acceptor would invert this effect and lead to a relative decrease of the donor quenching. The absence of such a decrease from our mCerulean
mCherry pair showed a marked difference between acceptor- and donor-based measurements. Gross experimental error could be ruled out as both values were perfectly reproduced between the domain-swapped experiments with different protein constructs. The discrepancy indicates a partial inactivation of the donor (mCitrine) domain. The misfolding or photoinactivation of some donor fluorophores would not affect the donor-based measurement of FRET efficiencies as every intact donor would still be paired up with an intact acceptor and the signal is normalized to the intact donor population. However, defective donor domains would, indeed, dilute the FRET signal measured on the acceptor side (55), which is normalized to the intensity of (intact) acceptor molecules. Conversely, a certain fraction of defective acceptor would invert this effect and lead to a relative decrease of the donor quenching. The absence of such a decrease from our mCerulean mCitrine pairs would, actually, suggest an approximately equivalent degree of mCerulean inactivation. Both protein domains are closely related (96% sequence identity) and similar maturation efficiencies would therefore not be surprising.
mCitrine pairs would, actually, suggest an approximately equivalent degree of mCerulean inactivation. Both protein domains are closely related (96% sequence identity) and similar maturation efficiencies would therefore not be surprising.
Table 7.
FRET efficiencies for different combinations of interaction input and output devices
| Protein pair |
FRET method |
|||
|---|---|---|---|---|
|
Donor quenchinga |
Acceptor emmissionb |
|||
| Parallelc | ±rap.d | Parallelc | ±rap.d | |
FKBP∼mCerulean  FRB∼mCitrine FRB∼mCitrine |
0.23 ± 0.010 | 0.21 ± 0.007 | 0.20 ± 0.004 | 0.19 ± 0.015 |
FRB∼mCerulean  FKBP∼mCitrine FKBP∼mCitrine |
0.19 ± 0.004 | 0.18 ± 0.004 | 0.18 ± 0.002 | 0.17 ± 0.008 |
FKBP∼mCitrine  FRB∼mCherry FRB∼mCherry |
0.28 ± 0.006 | 0.25 ± 0.003 | 0.10 ± 0.010 | 0.10 ± 0.012 |
FRB∼mCitrine  FKBP∼mCherry FKBP∼mCherry |
0.29 ± 0.008 | 0.25 ± 0.004 | 0.09 ± 0.022 | 0.12 ± 0.022 |
Zip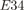 ∼mCerulean ∼mCerulean  Zip Zip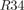 ∼mCitrine ∼mCitrine |
0.001 ± 3e-5 | 0.060 ± 0.006 | ||
Zip ∼mCitrine ∼mCitrine  Zip Zip ∼mCherry ∼mCherry |
0.017 ± 4e-4 | 0.021 ± 0.015 | ||
aMeasuring quenching of donor fluorescence.
bMeasuring sensitized acceptor emission.
cComparing independent samples with and without donor (external control).
dComparing fluorescence before and after adding rapamycin (internal control).
Surprisingly, we observed only very low resonance transfer when we connected our two readout devices to the constitutive leucine zipper interaction. Furthermore, the efficiencies from the different setups do not give a consistent picture, owing, probably, to the fact that below efficiencies of 0.05, the underlying fluorescent changes were minor compared to the strong basal emissions of donor and acceptor.
DISCUSSION
A protein interaction device framework
Natural signaling proteins are often composed of multiple domains that integrate signals by cooperative binding and unbinding with partner domains of other proteins (1). Not only the individual domains but also their interactions are recurring in numerous variations of the same design patterns across different networks. This natural modularity of interactions, rather than of mere proteins, is the departure point for our concept of protein interaction devices. We propose to ‘refacture’ protein–protein or protein–peptide interactions into functional devices for the construction of protein circuits. On a functional level, devices would communicate by the formation or release of protein interactions, leading to co-recruitment or changes in the local concentrations of attached domains. On a physical level, they would be connected through protein fusions.
We explored strategies and methods that may become critical for the routine design of protein systems. Key issues are, in our opinion, the composition of synthetic proteins from modular parts, the biophysical characterization of protein devices, as well as their physical connection and interplay.
From parts to DNA—design and assembly of protein parts
As a proof of concept, we have assembled 25 synthetic proteins, each from five protein parts and flanked by two non-coding parts for the regulation of expression and translation. We set out from a small catalog of 24 basic building blocks. The design of this initial parts list could tap into a large body of protein engineering literature. In fact, variants of most parts had already been used and re-used in various heterologous fusion proteins. Classic cloning strategies are ill-suited for the recombination of proteins from multiple unrelated domains and linker segments. Moreover, also current gene synthesis work flows are not adapted to this task. Our final constructs are long but, actually, mere recombinations of a small set of unique sequences. Nevertheless, this redundancy would not translate to cost savings as all would have to be synthesized de novo. We thus adopted a hybrid approach combining the gene synthesis of all basic parts with their assembly by a standardized iterative cloning method (BioBrick assembly) (18). The original BioBrick format does not allow for protein fusions and we therefore applied a modified, backwards-compatible method (BBF RFC 25) optimized for the assembly of protein fusion constructs. (20). Consequently, not only all our protein parts but also final constructs remain physically compatible with a large collection of building blocks in the Registry of Standard Biological Parts and could be integrated into larger genetic circuits. On the downside, despite our further streamlined protocol, cloning remained a time- and cost-consuming bottleneck of the whole process. Faster, easily accessible and more efficient methods for the assembly of DNA from recurring fragments are still wanting.
From DNA to proteins—production of synthetic proteins
A single established protocol for expression and purification was sufficient for the production of soluble protein from many synthetic constructs (14 out of 25). Three more could be recovered with a simple set of routine modifications. It seems feasible to compile a small set of consensus screens and protocols (56) covering the purification of most synthetic proteins. In fact, thanks to the synthetic layout of our constructs we could pinpoint two major factors explaining much of the variation in expression. First, all the failing constructs contained one of the artificially split protein domains. Second, the expression levels of most remaining proteins appeared to be controlled by RNA secondary structure interactions between coding sequence and RBS. Inadvertently, our data complement results of several recent reports (49–51) on this correlation between the obstruction of ribosomal binding and protein expression levels. While our data set is much more sparse, it does add quality to the relation. Previous work examined variants of a single reporter protein in combination with different RBS sequences. Here, we show the inverse experiment. While the RBS remained constant, very different domains were swapped into the N-terminal position and tested in combination with several unrelated C-terminal domains.
Notably, RBS obstruction has been known for some time (48) and is accounted for by the codon optimization algorithms of most gene synthesis providers. However, our part sequences were, quite intentionally, optimized outside their final context and are supposed to be re-usable in different positions and with different expression systems. Here, we are reaching some obvious limit of a purely parts-centric sequence design. Similar side effects may have to be accounted for at various levels. Nevertheless, in this case, there is a technical fix (49): a short N-terminal part can be codon-optimized for a given RBS and should uncouple the remaining protein sequence from interference with translation initiation.
RBS obstruction was unrelated to the outright expression failure of several protein constructs. Issues of mis-folding and aggregation are the most likely reasons for the toxicity of split protein domains. A case in point is luciferase fragment 1 (gLuc-frag1). Constructs containing this domain did not express under standard conditions but production of several proteins containing this domain, for example P15 and P17, could be rescued with the protocol optimized for toxic proteins. Yet, as shown in Figure 3, the highest growth rates were obtained for clones that channeled the product into inclusion bodies where it could not interfere with the cellular machinery. Furthermore, Gaussia princeps luciferase contains five disulfide bonds and even the intact enzyme is a difficult expression target (57). Complementation systems based on firefly (58) or Renilla (59) luciferase may offer a more robust alternative. Experiences about expression levels and solubility of proteins are of very practical importance but remain often qualitative and hidden from literature. The development of appropriate measurement standards may be a worthy challenge for protein synthetic biologists.
From proteins to systems—device characterization
Despite considerable efforts (9,60,61), most standard biological parts currently available are merely standardized with respect to cloning and remain otherwise uncharacterized. In particular, gene regulatory devices are often sensitive to their cellular context and activities can only be measured in relative terms. Measurement kits with reference standards have been proposed as a solution (61). In contrast, we can characterize proteins and their interactions in vitro and assign absolute numbers to many of their properties. Here, we have characterized two protein–protein interactions, which we consider prototypical ‘interaction input devices’, and combined them with two different FRET pairs that we consider ‘interaction readout devices’. Molecular interactions are commonly characterized by their dissociation constant KD. Yet, the kinetics of an interaction can often be much more important and accurate modeling of system dynamics requires on- and off-rates rather than equilibrium binding constants (62,63).
We therefore subjected both a constitutive leucine zipper and the drug-inducible interaction between FKBP12 and modified FRB to SPR analysis. The measurements confirmed that both pairs functioned correctly also in the context of fusion proteins. The ‘always on’ ZipE34:ZipR34 interaction was indeed exclusively heterodimeric. The FKBP12:FRB binding strictly depended on its inducer, rapamycin and switched from practically zero background to a high-affinity interaction. As of yet, no kinetic parameters have been determined for neither system, although a dissociation constant was published for ZipE34:ZipR34 (30) and SPR data were available for the interaction between FKBP12 and wild type FRB (22).
The kinetically determined affinity of the leucine zipper (KD = 83 nM) was one order of magnitude lower than the earlier value from thermal unfolding (KD = 6.1 nM). However, discrepancies were to be expected. The tethering to a much larger globular domain (mCherry) likely slows diffusion of the free molecule. The fused domain may also exert mechanical forces that counteract binding or accelerate unbinding. In fact, this could serve as another illustration of ‘side effects’ between parts. Single-domain controls and further experiments with other synthetic fusion constructs would be needed to delineate and properly account for these effects.
In contrast, the affinity for the rapamycin-induced binding of FKBP12 to the modified FRB(T2098L) appears higher than the affinity published (22) for the unmodified system (KD = 0.24 nM versus 12 nM). A moderate decrease in on-rate is more than compensated by a two orders of magnitude lower off rate. The T2098L mutation had been designed to accommodate the non-toxic ‘rapalog’ AP21967 and was supposed to have no or only minor influence on rapamycin binding. In fact, more qualitative data, based on a reporter gene assay, suggested a slightly detrimental effect. On the other hand, rapamycin is not a natural ligand of this system and affinity-increasing mutations seem therefore plausible.
Aside from the possibility of triggering synthetic interaction systems, we also need means to follow their dynamics. FRET assays can report molecular interactions with high time and spatial resolution inside or outside of cells without the requirement of co-factors. Unlike protein complementation methods, modern FRET pairs do not create interactions themselves and the signal is therefore reversible. On the other hand, FRET lacks the signal amplification that is afforded by enzymatic or genetic interaction reporters. The signal is strongly distance dependent and can be, for this reason, very sensitive to the steric arrangement of the two FRET probes within a complex. We have tested the interaction readout with two FRET pairs by connecting them to the constitutive leucine zipper and the inducible FKBP12:FRB interaction.
Despite rather long flexible linkers between the interaction input module and the readout pair, FKBP12:FRB input was converted into good and consistent FRET efficiencies. There was an excellent and quantitative agreement between domain swapping experiments, that means, donor or acceptor could be equally connected to either of the interaction partners. This perfect agreement of FRET efficiencies, in fact, indicates the ‘correct functioning’ (42) of the long (14 aa including assembly scars) and very flexible linker that uncoupled the dynamics of binding and reporter domain and ensured the orientational averaging of acceptor and donor fluorophores (64). The comparison of acceptor- and donor-based measurements across different domain-swapped pairs indicated a fraction of ‘dark’ or inactive mCitrine domains which seems to equally apply to mCerulean but not (or less) to mCherry. YFP, the common ancestor of mCitrine and mCerulean is indeed known for poor maturation rates (55) and, probably also depending on expression conditions, some of this problem appears to persist. Nevertheless, while there is room for improvement, the input/output system presented here may be quite suitable as a general purpose FRET reference.
On the other hand, disappointing FRET signals from the coupling to the leucine zipper remind of the challenges in FRET probe design in particular and parts-based protein composition in general. The interaction itself was independently verified by SPR. Compared to the FKBP12:FRB system, fusion to the leucine zipper should lower rather than increase the distance between FRET probes. The zipper's lower affinity, compared to the FKBP:FRB device, should have had little influence—measurements were taken at concentrations well above the system's KD (albeit not at full saturation of the bound state). Also, the interaction dynamics (koff∼10−2s−1) seem far too slow for affecting nanosecond time scale FRET processes. Issues may therefore have arisen from a less favorable orientation of the fluorophores (64) or from end-fraying of the leucine zipper (65), perhaps aggravated by the fusion with the heavy GFP domains. Notably, this result would not have been easily predicted from our deterministic measurements on the system's individual components. Structure-based dynamic modeling may help to make the connection of interaction input and output devices more predictable (66,67). Yet, for the time being, experimental characterization remains a key requirement.
CONCLUSION
We here explored the feasibility of a parts- and device-based approach to protein synthetic biology. Our model work flow started with the design of re-usable parts capturing basic protein functionalities, followed by their assembly into modular fusion proteins, the expression and purification of these proteins and, last not least, the in vitro constitution and biophysical characterization of simple interaction devices. Modular protein–protein interactions hold promise as the functional building blocks of larger systems. Their characterization and refacturing into protein interaction devices could, ultimately, become the basis of a versatile engineering framework. We here provide tested building blocks and protocols for the construction and rewiring of reference interactions as well as their dynamic detection. Our proof of concept experiments clearly demonstrate the feasibility of parts-based protein design. They, nevertheless, also revealed typical stumbling blocks: DNA assembly remains a technical bottleneck, whereas protein expression may be more amenable to rational optimization than perhaps expected. Input and output interactions could indeed be rewired in different combinations. However, their exact interplay was not always easily predicted from the characterization of the individual components. Rational engineering thus needs to be guided by additional rules and refined modeling tools. In fact, simple interaction systems like the ones constructed here, could provide a perfect test bed for exploring the connection rules of both synthetic and natural signaling modules. The comparison of synthetic constructs shed light on FRET probe design and delineated factors controlling protein expression. Thus, systematic domain swapping among modular and idealized synthetic proteins also proves a promising strategy for the study of more basic questions.
ACCESSION NUMBER
Sequences are deposited at the Registry of Standard Biological Parts under IDs: J18909 and J18912 to J18957.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Human Frontiers Science Program (LT-fellowship R.G.); European Union project PROSPECTS (to L.S.); Juan de la Cierva fellowship of the Spanish ministry of Science and Education (to A.M.S.) in part. Funding for open access charge: PROSPECTS.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The robotic DNA miniprep protocol was implemented together with Raul Gomez and Anja Leimpek from the CRG robotic platform. The extended BioBrick format BBF RFC 25 was originally developed for the Freiburg iGem 2007 team by Katja M. Arndt and Kristian M. Müller. Plasmids containing parts I757011 and I757012 were kindly provided by Janina Speck and Kristian M. Müller. We thank Timo Zimmermann for the discussion of FRET experiments and Kiana Toufighi for critical reading of the article.
REFERENCES
- 1.Gibson TJ. Cell regulation: determined to signal discrete cooperation. Trends Biochem. Sci. 2009;34:471–482. doi: 10.1016/j.tibs.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 2.Pryciak PM. Designing new cellular signaling pathways. Chem. Biol. 2009;16:249–254. doi: 10.1016/j.chembiol.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeke A, Lukács M, Lim WA, Reményi A. Scaffolds: interaction platforms for cellular signalling circuits. Trends Cell Biol. 2009 doi: 10.1016/j.tcb.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grünberg R, Serrano L. Strategies for protein synthetic biology. Nucleic Acid Res. 2010 doi: 10.1093/nar/gkq139. doi:10.1093/nar/gkq139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McDaniel R, Weiss R. Advances in synthetic biology: on the path from prototypes to applications. Curr. Opin. Biotechnol. 2005;16:476–483. doi: 10.1016/j.copbio.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 6.Win MN, Liang JC, Smolke CD. Frameworks for programming biological function through RNA parts and devices. Chem. Biol. 2009;16:298–310. doi: 10.1016/j.chembiol.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Endy D. Foundations for engineering biology. Nature. 2005;438:449–453. doi: 10.1038/nature04342. [DOI] [PubMed] [Google Scholar]
- 8.Andrianantoandro E, Basu S, Karig DK, Weiss R. Synthetic biology: new engineering rules for an emerging discipline. Mol. Syst. Biol. 2006;2 doi: 10.1038/msb4100073. 2006.0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Canton B, Labno A, Endy D. Refinement and standardization of synthetic biological parts and devices. Nat. Biotechnol. 2008;26:787–793. doi: 10.1038/nbt1413. [DOI] [PubMed] [Google Scholar]
- 10.Arkin A. Setting the standard in synthetic biology. Nat. Biotechnol. 2008;26:771–774. doi: 10.1038/nbt0708-771. [DOI] [PubMed] [Google Scholar]
- 11.Gardner TS, Cantor CR, Collins JJ. Construction of a genetic toggle switch in Escherichia coli. Nature. 2000;403:339–342. doi: 10.1038/35002131. [DOI] [PubMed] [Google Scholar]
- 12.Win MN, Smolke CD. Higher-order cellular information processing with synthetic RNA devices. Science. 2008;322:456–460. doi: 10.1126/science.1160311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marchisio M, Stelling J. Computational design of synthetic gene circuits with composable parts. Bioinformatics. 2008;24:1903–1910. doi: 10.1093/bioinformatics/btn330. [DOI] [PubMed] [Google Scholar]
- 14.Pawson T, Nash P. Assembly of cell regulatory systems through protein interaction domains. Science. 2003;300:445–452. doi: 10.1126/science.1083653. [DOI] [PubMed] [Google Scholar]
- 15.Crabtree GR, Schreiber SL. Three-part inventions: intracellular signaling and induced proximity. Trends Biochem. Sci. 1996;21:418–422. doi: 10.1016/s0968-0004(96)20027-1. [DOI] [PubMed] [Google Scholar]
- 16.Dueber JE, Wu GC, Malmirchegini GR, Moon TS, Petzold CJ, Ullal AV, Prather KLJ, Keasling JD. Synthetic protein scaffolds provide modular control over metabolic flux. Nat. Biotech. 2009;27:753–759. doi: 10.1038/nbt.1557. [DOI] [PubMed] [Google Scholar]
- 17.Cironi P, Swinburne IA, Silver PA. Enhancement of cell type specificity by quantitative modulation of a chimeric ligand. The J. Biol. Chem. 2008;283:8469–8476. doi: 10.1074/jbc.M708502200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knight T. 2003. [(19 Februry 2010, date last acessed)]. Idempotent vector design for standard assembly of biobricks. http://dspace.mit.edu/handle/1721.1/21168?show=full. [Google Scholar]
- 19.Shetty R, Endy D, Knight T. Engineering BioBrick vectors from BioBrick parts. J. Biol. Eng. 2008;2:5. doi: 10.1186/1754-1611-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grünberg R, Arndt K, Müller K. 2009. [(19 Februry 2010, date last acessed)]. BBF RFC 25: Fusion protein (Freiburg) biobrick assembly standard. http://dspace.mit.edu/handle/1721.1/45140. [Google Scholar]
- 21.Bierer BE, Mattila PS, Standaert RF, Herzenberg LA, Burakoff SJ, Crabtree G, Schreiber SL. Two distinct signal transmission pathways in t lymphocytes are inhibited by complexes formed between an immunophilin and either FK506 or rapamycin. Proc. Natl Acad. Sci. USA. 1990;87:9231–9235. doi: 10.1073/pnas.87.23.9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banaszynski LA, Liu CW, Wandless TJ. Characterization of the FKBP.rapamycin.FRB ternary complex. J. Am. Chem. Soc. 2005;127:4715–4721. doi: 10.1021/ja043277y. [DOI] [PubMed] [Google Scholar]
- 23.Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat. Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- 24.Hengen P. Purification of His-Tag fusion proteins from escherichia coli. Trends Biochem. Sci. 1995;20:285–286. doi: 10.1016/s0968-0004(00)89045-3. [DOI] [PubMed] [Google Scholar]
- 25.Schmidt TG, Koepke J, Frank R, Skerra A. Molecular interaction between the strep-tag affinity peptide and its cognate target, streptavidin. J. Mol. Biol. 1996;255:753–766. doi: 10.1006/jmbi.1996.0061. [DOI] [PubMed] [Google Scholar]
- 26.Hopp TP, Prickett KS, Price VL, Libby RT, March CJ, Cerretti DP, Urdal DL, Colo PJ. A short polypeptide marker sequence useful for recombinant protein identification and purification. Nat. Biotechnal. 1988;6:1204–1210. [Google Scholar]
- 27.Walker J, Crowley P, Moreman AD, Barrett J. Biochemical properties of cloned glutathione s-transferases from schistosoma mansoni and schistosoma japonicum. Mol. Biochem. Parasitol. 1993;61:255–264. doi: 10.1016/0166-6851(93)90071-5. [DOI] [PubMed] [Google Scholar]
- 28.Dougherty WG, Cary SM, Parks TD. Molecular genetic analysis of a plant virus polyprotein cleavage site: a model. Virology. 1989;171:356–364. doi: 10.1016/0042-6822(89)90603-x. [DOI] [PubMed] [Google Scholar]
- 29.Walker PA, Leong LE, Ng PW, Tan SH, Waller S, Murphy D, Porter AG. Efficient and rapid affinity purification of proteins using recombinant fusion proteases. Bio technology. 1994;12:601–605. doi: 10.1038/nbt0694-601. [DOI] [PubMed] [Google Scholar]
- 30.Acharya A, Ruvinov SB, Gal J, Moll JR, Vinson C. A heterodimerizing leucine zipper coiled coil system for examining the specificity of a position interactions: amino acids i, v, l, n, a and k. Biochemistry. 2002;41:14122–14131. doi: 10.1021/bi020486r. [DOI] [PubMed] [Google Scholar]
- 31.Belshaw PJ, Ho SN, Crabtree GR, Schreiber SL. Controlling protein association and subcellular localization with a synthetic ligand that induces heterodimerization of proteins. Proc. Natl Acad. Sci. USA. 1996;93:4604–4607. doi: 10.1073/pnas.93.10.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakasone Y, Eitoku T, Matsuoka D, Tokutomi S, Terazima M. Kinetic measurement of transient dimerization and dissociation reactions of arabidopsis phototropin 1 LOV2 domain. Biophys. J. 2006;91:645–653. doi: 10.1529/biophysj.106.084772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kerppola TK. Complementary methods for studies of protein interactions in living cells. Nat. Methods. 2006;3:969–971. doi: 10.1038/nmeth1206-969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Remy I, Michnick SW. Application of protein-fragment complementation assays in cell biology. BioTechniques. 2007;42:137–145. doi: 10.2144/000112396. [DOI] [PubMed] [Google Scholar]
- 35.Piston DW, Kremers G. Fluorescent protein FRET: the good, the bad and the ugly. Trends Biochem. Sci. 2007;32:407–414. doi: 10.1016/j.tibs.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 36.Galarneau A, Primeau M, Trudeau L, Michnick SW. (beta)-Lactamase protein fragment complementation assays as in vivo and in vitro sensors of protein-protein interactions. Nat. Biotechnol. 2002;20:619–622. doi: 10.1038/nbt0602-619. [DOI] [PubMed] [Google Scholar]
- 37.Hecky J, Mason JM, Arndt KM, Mller KM. A general method of terminal truncation, evolution and re-elongation to generate enzymes of enhanced stability. Methods Mol. Biol. 2007;352:275–304. doi: 10.1385/1-59745-187-8:275. [DOI] [PubMed] [Google Scholar]
- 38.Remy I, Michnick SW. A highly sensitive protein-protein interaction assay based on gaussia luciferase. Nat. Methods. 2006;3:977–979. doi: 10.1038/nmeth979. [DOI] [PubMed] [Google Scholar]
- 39.Rizzo MA, Springer GH, Granada B, Piston DW. An improved cyan fluorescent protein variant useful for FRET. Nat. Biotechnol. 2004;22:445–449. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]
- 40.Griesbeck O, Baird GS, Campbell RE, Zacharias DA, Tsien RY. Reducing the environmental sensitivity of yellow fluorescent protein. mechanism and applications. J. Biol. Chemi. 2001;276:29188–29194. doi: 10.1074/jbc.M102815200. [DOI] [PubMed] [Google Scholar]
- 41.Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 42.Neuweiler H, Lllmann M, Doose S, Sauer M. Dynamics of unfolded polypeptide chains in crowded environment studied by fluorescence correlation spectroscopy. J. Mol. Biol. 2007;365:856–869. doi: 10.1016/j.jmb.2006.10.021. [DOI] [PubMed] [Google Scholar]
- 43.Bashor CJ, Helman NC, Yan S, Lim WA. Using engineered scaffold interactions to reshape MAP kinase pathway signaling dynamics. Science. 2008;319:1539–1543. doi: 10.1126/science.1151153. [DOI] [PubMed] [Google Scholar]
- 44.Chen J, Zheng XF, Brown EJ, Schreiber SL. Identification of an 11-kDa FKBP12-rapamycin-binding domain within the 289-kDa FKBP12-rapamycin-associated protein and characterization of a critical serine residue. Proc. Natl Acad. Sci. USA. 1995;92:4947–4951. doi: 10.1073/pnas.92.11.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liberles SD, Diver ST, Austin DJ, Schreiber SL. Inducible gene expression and protein translocation using nontoxic ligands identified by a mammalian three-hybrid screen. Proc. Natl Acad. Sci. USA. 1997;94:7825–7830. doi: 10.1073/pnas.94.15.7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sevastsyanovich Y, Alfasi S, Overton T, Hall R, Jones J, Hewitt C, Cole J. Exploitation of GFP fusion proteins and stress avoidance as a generic strategy for the production of high-quality recombinant proteins. FEMS Microbiol. Lett. 2009 doi: 10.1111/j.1574-6968.2009.01738.x. [Epub ahead of print; 27 July 2009] [DOI] [PubMed] [Google Scholar]
- 47.Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. Use of t7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 48.deSmit MH, vanDuin J. Secondary structure of the ribosome binding site determines translational efficiency: a quantitative analysis. Proc. Natl Acad. Sci. USA. 1990;87:7668–7672. doi: 10.1073/pnas.87.19.7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kudla G, Murray AW, Tollervey D, Plotkin JB. Coding-sequence determinants of gene expression in escherichia coli. Science. 2009;324:255–258. doi: 10.1126/science.1170160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seo SW, Yang J, Jung GY. Quantitative correlation between mRNA secondary structure around the region downstream of the initiation codon and translational efficiency in Escherichia coli. Biotechnol. Bioeng. 2009;104:611–616. doi: 10.1002/bit.22431. [DOI] [PubMed] [Google Scholar]
- 51.Salis HM, Mirsky EA, Voigt CA. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 2009 doi: 10.1038/nbt.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mason JM, Arndt KM. Coiled coil domains: stability, specificity and biological implications. Chembiochem. 2004;5:170–176. doi: 10.1002/cbic.200300781. [DOI] [PubMed] [Google Scholar]
- 53.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 54.Bayle JH, Grimley JS, Stankunas K, Gestwicki JE, Wandless TJ, Crabtree GR. Rapamycin analogs with differential binding specificity permit orthogonal control of protein activity. Chem. Biol. 2006;13:99–107. doi: 10.1016/j.chembiol.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 55.Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- 56.Gräslund S, Nordlund P, Weigelt J, Bray J, Gileadi O, Knapp S, Oppermann U, Arrowsmith C, Hui R, Ming J, et al. Protein production and purification. Nat. Methods. 2008;5:135–146. doi: 10.1038/nmeth.f.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goerke AR, Loening AM, Gambhir SS, Swartz JR. Cell-free metabolic engineering promotes high-level production of bioactive gaussia princeps luciferase. Metab. Eng. 2008;10:187–200. doi: 10.1016/j.ymben.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 58.Luker KE, Smith MCP, Luker GD, Gammon ST, Piwnica-Worms H, Piwnica-Worms D. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc. Natl Acad. Sci. USA. 2004;101:12288–12293. doi: 10.1073/pnas.0404041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Paulmurugan R, Gambhir SS. Monitoring protein-protein interactions using split synthetic renilla luciferase protein-fragment-assisted complementation. Anal. Chem. 2003;75:1584–1589. doi: 10.1021/ac020731c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Anderson JC, Clarke EJ, Arkin AP, Voigt CA. Environmentally controlled invasion of cancer cells by engineered bacteria. J. Mol. Biol. 2006;355:619–627. doi: 10.1016/j.jmb.2005.10.076. [DOI] [PubMed] [Google Scholar]
- 61.Kelly J, Rubin A, Davis J, Ajo-Franklin C, Cumbers J, Czar M, deMora K, Glieberman A, Monie D, Endy D. Measuring the activity of BioBrick promoters using an in vivo reference standard. J. Biol. Eng. 2009;3:4. doi: 10.1186/1754-1611-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van der Sloot AM, Kiel C, Serrano L, Stricher F. Protein design in biological networks: from manipulating the input to modifying the output. Protein Eng. Des. Sel. 2009;22:537–542. doi: 10.1093/protein/gzp032. [DOI] [PubMed] [Google Scholar]
- 63.Kiel C, Serrano L. Cell type-specific importance of ras-c-raf complex association rate constants for MAPK signaling. Sci. Signal. 2009;2:ra38. doi: 10.1126/scisignal.2000397. [DOI] [PubMed] [Google Scholar]
- 64.van derMeer B. Kappa-squared: from nuisance to new sense. Rev. Mol. Biotechnol. 2002;82:181–196. doi: 10.1016/s1389-0352(01)00037-x. [DOI] [PubMed] [Google Scholar]
- 65.Holtzer ME, Lovett EG, d'Avignon DA, Holtzer A. Thermal unfolding in a GCN4-like leucine zipper: 13C alpha NMR chemical shifts and local unfolding curves. Biophys. J. 1997;73:1031–1041. doi: 10.1016/S0006-3495(97)78136-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Evers TH, vanDongen EMWM, Faesen AC, Meijer EW, Merkx M. Quantitative understanding of the energy transfer between fluorescent proteins connected via flexible peptide linkers. Biochemistry. 2006;45:13183–13192. doi: 10.1021/bi061288t. [DOI] [PubMed] [Google Scholar]
- 67.Pham E, Chiang J, Li I, Shum W, Truong K. A computational tool for designing FRET protein biosensors by rigid-body sampling of their conformational space. Structure. 2007;15:515–523. doi: 10.1016/j.str.2007.03.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.