

Abstract
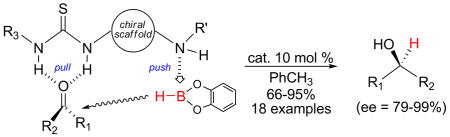
Prochiral ketones are reduced to enantioenriched, secondary alcohols using catecholborane and a family of air-stable, bifunctional thiourea-amine organocatalysts. Asymmetric induction is proposed to arise from the in situ complexation between the borane and chiral thiourea-amine organocatalyst resulting in a stereochemically biased boronate-amine complex. The hydride in the complex is endowed with enhanced nucleophilicity while the thiourea concomitantly embraces and activates the carbonyl.
The enantioselective reduction of prochiral ketones is a mainstay in the production of enantioenriched, secondary alcohols.1 As in other areas of chiral synthetic methodology, the trend has been away from stoichiometric reductants2 towards more economic and environmentally friendly catalytic processes3 and, in recent years, has embraced organocatalysis.4,5 One of the most prominent and frequently applied members of this latter category is the Corey-Bakshi-Shibata (CBS) catalyst, a chiral oxazaborolidine pioneered by Itsuno6 and further developed by Corey7 and other investigators.8 However, the sensitivity of oxazaborolidines to oxygen and moisture as well as the need in conjunction with a current project for a highly enantioselective reducing agent compatible with a challenging combination of highly sensitive functionality, prompted us to explore the utility of urea-/thiourea-based organocatalysts as an alternative to CBS oxazaborolidines.9,10
Whilst chiral ureas and thioureas have emerged as efficacious catalysts for a variety of nucleophilic conjugate additions11 and 1,2-carbonyl additions, e.g., hydrocyanation,12 Henry reaction,13 and Baylis-Hillman reaction,14 there are few examples of highly enantioselective hydride additions.15,16 However, the insights gained developing asymmetric oxy-Michael additions of boronic acids with α,β-unsaturated ketones17 revealed several unique attributes that we felt could be harnessed for enantioselective carbonyl reductions. Specifically, we envisioned that the union between a borane and a chiral thiourea-amine organocatalyst would result in a stereochemically biased boronate-amine complex.18 The hydride in the complex is endowed with enhanced nucleophilicity (the push) while the thiourea concomitantly embraces and activates the carbonyl (the pull) (Figure 1). As proof-of-concept, we developed of a family of robust, bifunctional thiourea-amine catalysts and describe herein their exploitation for the stereodefined reduction of prochiral ketones to enantioenriched, secondary alcohols.
Figure 1.

Proposed asymmetric catalysis
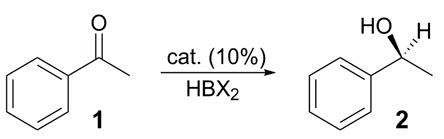
Despite its outstanding performance catalyzing the aforementioned oxy-Michael additions,17 thiourea catalyst A19 furnished (S)-(−)-1-phenylethanol (2) in poor yield and low enantioselectivity at room temperature in THF (Table 1, entry 1) using acetophenone (1) and BH3·THF as the model substrate and hydride source, respectively. Reasoning that the cinchona alkaloid moiety might be responsible, it was replaced with the simpler (R,R)-trans-N,N′-dimethylcyclohexane-1,2-diamine. The resultant monobasic catalyst B provided a modest improvement in yield and enantioselectivity, albeit delivering the R-enantiomer of 2 (entry 2). A survey of commercial boranes showed catecholborane delivered the best performance and that toluene was superior to other common solvents. In concert with the temperature dependency displayed by CBS catalysts,20 both yield and optical purity improved using this combination as the temperature was lowered to around −46 °C (entry 3), but then declined as the temperature was lowered still further (entry 4). Primary amine catalyst C (entry 5) was disappointing in all respects and was not further pursued. In contrast, the corresponding N-benzyl secondary amine catalyst D at −78 °C boosted the stereoselectivity upwards to 73% ee, albeit at the expense of yield (entry 6). Mindful of the preceding temperature dependency, catalyst D was evaluated over a wider temperature range (see Supporting Information). At −46 °C, the yield of 2 jumped to 88% and the enantioselectivity to 98% ee (entry 7); thereafter, the stereoselectivity slowly declined as the temperature was raised, e.g., 85% ee at −30 °C (entry 8). The biphasic behavior of the thiourea catalysts might be attributed to the slow breakdown of the catalyst-product complex below approximately −46 °C; presumably, the catalyst-product complex is functionally catalytic, but less enantioselective than the catalyst alone.20 Catalyst E, which differs from D by having an N-isobutyl substituent instead of an N-benzyl, showed a significant loss of enantioselectivity under otherwise identical reaction conditions (entry 9 vs. 7). This might be attributed to just steric differences, although alternative explanations, e.g., π-π bonding between the N-benzyl of D and the electron-rich catechol of the borane, warrant investigation. A comparison of catalyst F with D is also instructive. The former was prepared from a commercial, chiral acyclic-diamine, yet furnished results comparable to D (entry 10), indicating a wide latitude in the design of future catalysts.
Table 1.
Influence of select reaction parameters on yield and enantioselectivity.a
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | catalyst | borane (equiv) | solvent | temp (°C) | yieldb(%) | eec(%) | config |
| 1 | A | BH3·THF (0.7) | THF | 23 | 10 | 5 | S |
| 2 | B | BH3·THF (0.7) | THF | 23 | 40 | 13 | R |
| 3 | B | catechol borane (1.6) | toluene | −46 | 88 | 43 | R |
| 4 | B | catecholborane (1.6) | toluene | −78 | 65 | 27 | R |
| 5 | C | catecholborane (1.6) | toluene | −78 | 25 | 20 | S |
| 6 | D | catecholborane (1.6) | toluene | −78 | 24 | 73 | S |
| 7 | D | catecholborane (1.6) | toluene | −46 | 88 | 98 | S |
| 8 | D | catecholborane (1.6) | toluene | −30 | 88 | 85 | S |
| 9 | E | catecholborane (1.6) | toluene | −46 | 85 | 65 | S |
| 10 | F | catecholborane (1.6) | toluene | −46 | 83 | 96 | S |
Reaction conditions: catalyst (10 mol %), 24 h, argon atmosphere.
Isolated yield.
Measured by chiral HPLC.
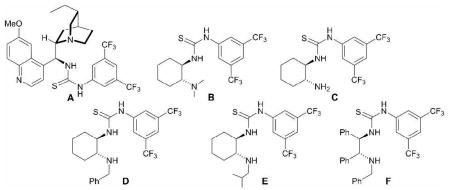
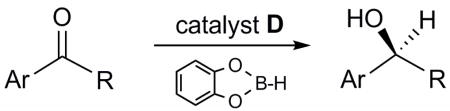
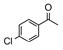
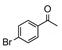
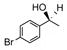
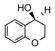
Catalyst D proved useful for the enantioselective reduction of a wide range of aryl ketones (Table 2). Simple phenyl alkyl ketones 3 and 5 were smoothly reduced with excellent stereocontrol to (S)-alcohols 4 (entry 1) and 6 (entry 2), respectively. Importantly, the presence of an ortho-substituent did not alter the level of enantioselectivity (entry 3, 7→8) nor did electron-withdrawing (entry 4) or electron-donating (entry 5) groups, although the latter did require a longer reaction time. Other functionality was also well tolerated including p-fluoro (entry 6), p-chloro (entry 7), and p-bromo (entry 8). The cyclic ketones 1-tetralone (19) and 4-chromanone (21) were likewise well behaved and furnished alcohols 20 (entry 9) and 22 (entry 10) in high yield and optical purity. Comparable results were obtained using 2-acetonaphthone (23, entry 11) and the heterocycle 2-acetylthiophene (25, entry 12).
Table 2.
Enantioselective reduction of aryl ketones.a
 | |||||
|---|---|---|---|---|---|
| entry | aryl ketone | alcohol | time (h) | yieldb(%) | eec(%) |
| 1 |
3 |
4 |
24 | 86 | 99 |
| 2 |
5 |
6 |
24 | 86 | 99 |
| 3 |
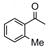 7 |
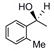 8 |
26 | 71 | 95 |
| 4 |
9 |
10 |
22 | 92 | 96 |
| 5 |
11 |
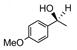 12 |
36 | 80 | 97 |
| 6 |
13 |
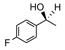 14 |
20 | 84 | 99 |
| 7 |
 15 |
16 |
22 | 94 | 99 |
| 8 |
 17 |
 18 |
22 | 95 | 99 |
| 9 |
19 |
20 |
24 | 86 | 99 |
| 10 |
21 |
 22 |
24 | 95 | 98 |
| 11 |
23 |
24 |
24 | 93 | 98 |
| 12 |
25 |
26 |
30 | 66 | 97 |
Reaction conditions: catalyst D (10 mol %), catecholborane (1.6 equiv), 4 Å molecular sieves, toluene, −46 °C, argon atmosphere.
Isolated yield.
Measured by chiral HPLC.
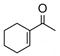
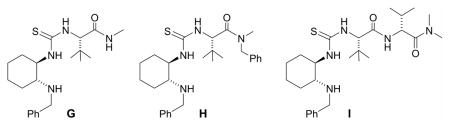
As an extension of our survey of structurally diverse prochiral carbonyls, α,β-unsaturated ketones 27, 29, and 31 were transformed in good yields and stereoselectivities to alcohols 28, 30, and 32, respectively, using catalyst D (Table 3, entries 1–3). The latter example deserves comment since it was obtained in appreciably better optical purity (97% ee) than that reported using the CBS catalyst (81% ee).21 Unsymmetrical dialkyl ketones, of course, were more challenging. While alcohol 34 was produced from ketone 33 in good yield using catalyst D, the chiral induction was quite modest (entry 4). Drawing inspiration from the recent work of Zuend and Jacobsen,22 we sought to improve catalytic performance with the introduction of an additional chiral center. Indeed, after extensive study of the structure-activity relationships of the catalyst scaffold (see Supporting Information for details), catalysts G and H were found to raise the stereoselectivity for the reduction of 33 to 63% ee (entry 5) and 79% ee (entry 6), respectively, validating this approach. Yet, catalyst I was less successful despite having a fourth chiral center. In the case of cycloalkyl alkyl ketone 35, catalysts D and H were comparable and furnished 36 with high enantioselectivity (entries 8 and 9).
Table 3.
Enantioselective reduction of alkenyl and alkyl ketones.a
| entry | ketone | alcohol | catalyst | yieldb(%) | %eec |
|---|---|---|---|---|---|
| 1 |
27 |
28 |
D | 78 | 90 |
| 2 |
29 |
30 |
D | 88 | 86 |
| 3 |
 31 |
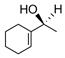 32 |
D | 82 | 97 |
| 4 |
33 |
34 |
D | 81 | 47 |
| 5 | 33 | 34 | G | 84 | 63 |
| 6 | 33 | 34 | H | 92 | 79 |
| 7 | 33 | 34 | I | 90 | 67 |
| 8 |
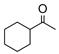 35 |
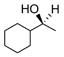 36 |
D | 60 | 89 |
| 9 | 35 | 36 | Hd | 68 | 91 |
Reaction conditions: catalyst (10 mol %), catecholborane (1.6 equiv), 4 Å molecular sieves, toluene, −46 °C, 24 h.
Isolated yield.
Measured by chiral HPLC.
36 h.
In summary, we describe a family of air-stable, bifunctional amino-thiourea cataysts for the enantioselective reduction of prochiral ketones using echolborane. Yields and % ee using aryl and α,β-unsaturated ketones rival or exceed those achievable using extant reagents. Promising results were also seen using unsymmetrical dialkyl ketones and a strategy for future catalyst optimization was demonstrated.
Supplementary Material
Acknowledgments
Financial support provided by the Robert A. Welch Foundation and NIH (GM31278, DK38226).
Footnotes
Supporting Information Available: Synthetic procedures and analytical data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Review: Itsuno S. Org React. 1998;52:395–576.Noyori R, Takeshi O, Sandoval CA, Muniz K. Asymmetric Synth. 2007:321–325.
- 2.Lin G-Q, Li Y-M, Chan ASC. Principles and Applications of Asymmetric Synthesis. Wiley-Interscience; New York, NY: 2001. pp. 355–367. [Google Scholar]
- 3.(a) Berkessel A, Gröger H. Asymmetric Organocatalysis. Wiley-VCH Verlag GmbH; Weinheim, Germany: 2005. pp. 314–322. [Google Scholar]; (b) Cho BT. Chem Soc Rev. 2009;38:443–452. doi: 10.1039/b811341f. [DOI] [PubMed] [Google Scholar]
- 4.Kagan HB. Review. In: Dalko PI, editor. Enantioselective Organocatalysis. Wiley-VCH; Weinheim, Germany: 2007. pp. 391–401. [Google Scholar]
- 5.Alternative methodology for enantioselective ketone reductions: Wu X, Xiao J. J Chem Soc, Chem Commun. 2007:2449–2466. doi: 10.1039/b618340a.Nakamura K, Matsuda T. Curr Org Chem. 2006;10:1217–1246.Zhou L, Wang Z, Wei S, Sun J. Chem Commun. 2007:2977–2979. doi: 10.1039/b703307a.
- 6.Itsuno S, Ito K, Hirao A, Nakahama S. J Chem Soc, Chem Commun. 1983;8:469–470. [Google Scholar]
- 7.Corey EJ, Shibata S, Bakshi RK. J Org Chem. 1988;53:2861–2863. [Google Scholar]
- 8.Review: Corey EJ, Helal CJ. Angew Chem Int Ed. 1998;37:1986–2012. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z.
- 9.Li Derun, Falck John R. 2009253919. US Pat Appl. 2009 CAN 151:425227, AN 2009:1231134.
- 10.Recent example using borane-amine complex: Zhou Y, Wang YW, Dou W, Zhang D, Liu WS. Chirality. 2009;21:657–662. doi: 10.1002/chir.20652.
- 11.Doyle AG, Jacobsen EN. Chem Rev. 2007;107:5713–5743. doi: 10.1021/cr068373r. [DOI] [PubMed] [Google Scholar]
- 12.Sigman MS, Jacobsen EN. J Am Chem Soc. 1998;120:4901–4902. [Google Scholar]
- 13.Shibasaki M, Groger H, Kanai M. In: Comprehensive Asymmetric Catalysis. Supp 1. Jacobsen EN, Pfaltz A, Yamamoto H, editors. Chapter 29.3 Springer; Berlin, Germany: 2003. [Google Scholar]
- 14.Masson G, Housseman C, Zhu J. Angew Chem Int Ed. 2007;46:4614–4628. doi: 10.1002/anie.200604366. [DOI] [PubMed] [Google Scholar]
- 15.Procuranti B, Connon SJ. J Chem Soc, Chem Commun. 2007:1421–1423. doi: 10.1039/b618792g. [DOI] [PubMed] [Google Scholar]
- 16.Thioureas with metals: Touchard F, Fache F, Lemaire M. Tetrahedron: Asymmetry. 1997;8:3319–3326.Bernard M, Delbecq F, Fache F, Sautet P, Lemaire M. Eur J Org Chem. 2001:1589–1596.
- 17.Li DR, Murugan A, Falck JR. J Am Chem Soc. 2008;130:46–48. doi: 10.1021/ja076802c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.For similar chiral Lewis Base catalyzed silane additions, see: Kocovsk P, Malkov AV. In: Enantioselective Organocatalysis. Dalko PI, editor. Wiley-VCH; Weinheim, Germany: 2007. pp. 255–286.Malkov AV, Stewart-Liddon AJP, Ramirez-Lopez P, Bendova L, Haigh D, Kocovsk P. Angew Chem, Int Ed. 2006;45:1432–1435. doi: 10.1002/anie.200503941.Denmark SE, Fu J. J Am Chem Soc. 2000;122:12021–12022.
- 19.Vakulya B, Varga S, Csámpai A, Soos T. Org Lett. 2005;7:1967–1969. doi: 10.1021/ol050431s. [DOI] [PubMed] [Google Scholar]
- 20.Stone GB. Tetrahedron: Asymmetry. 1994;5:465–472. [Google Scholar]
- 21.Corey EJ, Bakshi RK. Tetrahedron Lett. 1990;31:611–614. [Google Scholar]
- 22.Zuend SJ, Jacobsen EN. J Am Chem Soc. 2007;129:15872–15883. doi: 10.1021/ja0735352. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.