

Abstract
Protein enzymes appear to use extensive packing and hydrogen-bonding interactions to precisely position catalytic groups within active sites. Due to their inherent backbone flexibility and limited side chain repertoire, RNA enzymes face additional challenges relative to proteins in precisely positioning substrates and catalytic groups. Here, we use the group I ribozyme to probe the existence, establishment, and functional consequences of an extended network of interactions in an RNA active site. The group I ribozyme catalyzes a site-specific attack of guanosine on an oligonucleotide substrate. We previously determined that the hydrogen bond between the exocyclic amino group of guanosine and the 2′-hydroxyl group at position A261 of the Tetrahymena group I ribozyme contributes to overall catalysis. We now use functional data, aided by double-mutant cycles, to probe this hydrogen bond in the individual reaction steps of the catalytic cycle. Our results indicate that this hydrogen bond is not formed upon guanosine binding to the ribozyme but instead forms at a later stage of the catalytic cycle. Formation of this hydrogen bond is correlated to other structural rearrangements in the ribozyme's active site that are promoted by docking of the oligonucleotide substrate into the ribozyme's active site, and disruption of this interaction has deleterious consequences for the chemical transformation within the ternary complex. These results, combined with earlier results, provide insight into the nature of the multiple conformational steps used by the Tetrahymena group I ribozyme to achieve its active structure and reveal an intricate, extended network of interactions that is used to establish catalytic interactions within this RNA's active site.
Enzymatic reactions use the same chemical groups and functionalities that can be found on simple molecule catalysts, but the enzymatic reaction take place in specialized pockets, referred to as active sites. The protein structure provides networks of packing and hydrogen bonding interactions to precisely position substrates and catalytic groups within the active site (1). The identification and functional characterization of these networks of interactions is a fundamental step in understanding of how enzymes provide their extraordinary rate acceleration and specificity.
Structural techniques, such as X-ray crystallography, are powerful tools to garner information about networks of interactions but alone are not sufficient to determine the role of specific interactions in catalysis. These limitations are exacerbated in case of RNA enzymes, because RNA molecules commonly adopt multiple folded conformations (2-7) and undergo conformational rearrangements upon ligand binding (8-17). This structural plasticity has been suggested to arise from the fundamental physical properties of RNA, which include a backbone with more degrees of freedom than the protein backbone, a limited number of side chains, all of similar shape and with hydrogen bond donors and acceptors, and a uniform negative charge along its backbone (18, 19).
Chemical biology probes, based on site-specific modifications of the RNA backbone and its nucleobases, are invaluable tools to test and extend models and proposals derived from structural approaches (5, 20-22). Further, as illustrated herein, functional studies can provide information about the conformational transitions and the connectivity of interactions that are used to establish active sites and precisely position substrates and active site groups (e.g., references 5, 23-26).
The group I ribozyme is an excellent system to identify and characterize the interactions needed for catalysis, as a wealth of structural and functional information allows investigation at unprecedented depth (27). This ribozyme uses an exogenous guanosine molecule to catalyze a nucleophilic attack on a specific phosphodiester bond of an oligonucleotide substrate in a reaction that mimics the first step of self-splicing of the intron (27, 28). The oligonucleotide substrate and the nucleophilic guanosine bind cooperatively, and one of the active site metal ions (commonly referred to as MC) has been proposed to mediate such coupling (24, 29). In addition, functional data suggest that a conformational change is involved in coupling (24, 29, 30). Whereas changes in the contacts made between the oligonucleotide substrate and the ribozyme during the course of the group I ribozyme reaction have been described and characterized (27), less is known about the changes associated with binding and reactivity of the guanosine nucleophile.
Previous functional (31) and structural (32-34) data identified a specific binding pocket for the guanosine nucleophile and defined interactions important for guanosine binding and positioning (31-40). In particular, recent work (36) has identified an important hydrogen bond between the exocyclic amino group of guanosine and the 2′-hydroxyl group of one of the most conserved residues in group I ribozymes (A261 using the Tetrahymena ribozyme numbering), as predicted by a recent structural model (41).
We have now investigated the role of this hydrogen bond in individual reaction steps of the Tetrahymena ribozyme catalytic cycle. We found that the hydrogen bond between the exocyclic amino group of guanosine and the 2′-hydroxyl group of residue A261 is not formed upon guanosine binding; rather, it is formed only at a later stage in the catalytic cycle. We show that formation of this hydrogen bond is involved in coupling between the two substrates. Our results refine and extend the model for the interactions made by the guanosine nucleophile, providing further insight into the dynamic nature of this RNA's catalytic cycle and the intricate network of interactions that constitute its active site.
Materials and Methods
Materials
Oligonucleotides were purchased from Dharmacon Inc. (Lafayette, CO) or IDT (Coralville, IA), or synthesized at the University of Chicago. 5′-32P-End-labeling of the oligonucleotide substrates for kinetic experiments was performed by standard methods (42). Oligonucleotides corresponding to nucleotides 260-274 of the ribozyme, containing a 5′-phosphoryl group and a 2′-OH or 2′-H group at position 261 were purified by anion exchange HPLC (0-200 mM NaCl over 3 min, then 200-500 mM NaCl over 30 min in a background of 20 mM Tris•HCl, pH 7.4, with a flow rate of 2 mL/min) using a DNAPAC PA-100 (9×250) column (Dionex, Austin, TX), and desalted by Sep-Pak (Waters, Milford, MA). AUCG and AUCI, two oligonucleotides containing the guanosine nucleophile with and without the exocyclic amino group, respectively, were used instead of G and I because their higher solubility, relative to their respective dissociation constants, more readily allows measurements of saturation (see Results). These oligonucleotides were purified by anion exchange HPLC (0-200 mM NaCl over 30 min in a background of 20 mM Tris•HCl, pH 7.4, flow rate = 2 mL/min) using the same column as above, and desalted by Sep-Pak.
Ribozyme Preparation
Wild type and variant ribozymes were constructed semi-synthetically using a single-step three-piece ligation (43). Constructs corresponding to nucleotides 22-259 and 275-409 of the Tetrahymena ribozyme were transcribed using a DNA template produced by PCR truncation of the plasmid-encoded ribozyme sequence, with excess GMP present in the transcription of the 3′-construct (275-409) to yield predominantly a 5′-monophosphate. The 5′-construct contained a 3′-flanking hammerhead cassette to ensure homogeneous 3′-ends; the terminal 2′,3′-cyclic phosphate was removed after gel purification by treatment with T4 polynucleotide kinase (44). The transcripts were ligated to the HPLC-purified 5′-phosphorylated synthetic oligonucleotides corresponding to nucleotides 260-274 via a single-step ligation with T4 DNA ligase and a DNA splint (complementary to nucleotides 239-295) to yield full-length ribozyme containing either a 2′-ribose (WT ribozyme) or a 2′-deoxyribose (A261H ribozyme) group at position A261. To improve yields, 30% glycerol was added to the ligation mixture, as previously reported (36). Yields were ∼10% in purified, fully ligated ribozyme. The wild type ribozyme was also prepared by in-vitro transcription as described previously (45). Measurement of dissociation rate constants (koff) for the wild type and A261H ribozymes suggested that the ligated ribozymes were ∼70% active, with ∼30% of the population that can bind substrates but that does not dock or react to products (see General Kinetic Methods and Measurement of Docking Equilibria below).
General Kinetic Methods
All cleavage reactions were single turnover, with ribozyme in excess of radiolabeled oligonucleotide substrate (*S), which was always present in trace quantities (<100 pM). Reactions were carried out at 30 °C in 45 mM NaHEPES/5 mM NaMOPS, pH 8.1, and 50 mM MgCl2. The pH of 8.1 was used to suppress a guanosine-independent reaction of the A261H ribozyme. Reaction mixtures containing 10 mM MgCl2 and 50 mM NaMOPS, pH 6.9, were pre-incubated at 50 °C for 30 min to renature the ribozyme (present at a 10-fold excess concentration compared to the final reaction conditions). The mixtures were allowed to equilibrate at room temperature for 10 min and then the ribozyme was aliquoted into different tubes to raise the pH and the MgCl2 concentration, to dilute the ribozyme concentration and to add additional reactants. Reactions were allowed to equilibrate at 30 °C for 5 min before the addition of *S. Reactions aliquots were removed at specific times to be quenched with two volumes of a solution containing 50 mM EDTA and 85% formamide. Radiolabeled oligonucleotides were separated by denaturing gel electrophoresis (7 M urea, 20% acrylamide) and quantitated using Phosphorimager analysis (GE Healthcare) with ImageQuant or TotalLab quantitation software. For slow reactions, rate constants were obtained from initial rates assuming endpoints of 97%. All other reactions were followed to completion, with endpoints of 97%. With ligated ribozymes, reactions in which reaction times were faster than equilibration between the active and inactive population of the ribozymes showed biphasic kinetics. In these cases, the decay time for the fast phase corresponded to the reaction's observed rate constant, and the slow decay time corresponded to the dissociation of the oligonucleotide substrate from the inactive ribozyme population and rebinding to the active ribozyme. Biphasic kinetics were not observed when transcribed ribozyme was used as a control. Observed rate constants were not corrected for the small population of inactive ribozymes because observed reaction rates for the ligated and the transcribed wild type ribozyme were within experimental errors of one another.
Measurement of AUCG and AUCI Affinities
To determine the affinities of ribozyme complexes for AUCG and AUCI, the rate constant for reaction of *S was determined as a function of AUCG (or AUCI) concentration. Reactions with AUCI were followed up to 1 mM concentration. The ribozyme concentration was 50 nM to ensure that *S was fully bound to the ribozyme, as confirmed by native gels (>95% of *S was bound to the ribozymes). Because the dissociation rate constant of AUCG (or AUCI) from the ribozyme is faster than the rate constant for the chemical transformation (kc) for the substrates and conditions herein (reference 12), AUCG (or AUCI) affinity for the ribozyme can be determined by plotting the observed rate constant (kobs) for cleavage of *S as a function of AUCG (or AUCI) concentration and fitting it to Equation 1.
| (1) |
To determine AUCG (or AUCI) affinity in the closed complexes we used the -1d,rSA5 substrate to ensure that the chemical step was rate-limiting and that the observed (or ) equals [or ] (46). Measurement of the docking equilibria for the A261H ribozyme provided evidence that the -1d,rSA5 substrate reacts from the closed complex of this ribozyme (see Measurement of Docking Equilibria below). To determine the affinity of AUCG (or AUCI) in the open complex [ and , respectively], we used the oligonucleotide substrate -1r,dSA5, which favors the open complex (47).
Measurement of Docking Equilibria (Kdock)
Kdock can be determined from kinetic measurements of dissociation rate constants of S (koff) if Kdock > 1. To derive Kdock, we assumed that the second-order rate constant for the association of the oligonucleotide substrate S with the ribozyme (kon) is the same in the wild type and the A261H ribozymes and that the first-order rate constant for docking of bound S (kdock) is faster than koff. These assumptions are supported by the observations that association of S with the ribozyme involves only base-pairing interactions (48), which are the same in the wild type and in the A261H ribozymes, that kon has been shown to be unaffected by modifications of the wild type ribozyme (49-51), and that kdock is at least two orders of magnitude faster than koff in the wild type ribozyme (49). Following these assumptions, (see Scheme 1, in which the subscripts ‘o’ and ‘c’ represent the open and closed complexes, respectively).
SCHEME 1.
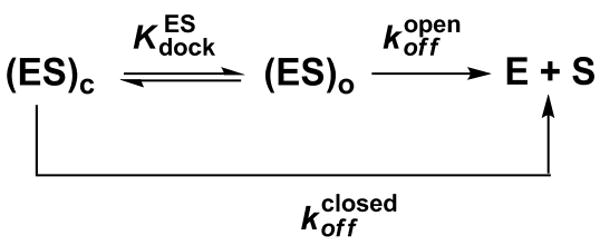
Dissociation rate constants (koff) of S from the E•S complexes of the wild type and A261H ribozymes were determined from pulse-chase gel-shift experiments (52, 53) carried out at pH 6.1 to minimize reactivity of the substrates. Briefly, trace *S was bound to saturating amounts of ribozyme (50-100 nM) and a large excess of unlabeled CCCUCdU (>10-fold over the ribozyme) was then added. At specified times aliquots were carefully loaded onto a running native gel in THEM buffer (33 mM Tris, 67 mM HEPES, 1 mM EDTA, 10 mM MgCl2). The remaining fraction of bound oligonucleotide was plotted against the time subsequent to addition of the chase. Control experiments confirmed full binding of *S prior to addition of the chase and the efficiency of the chase. Reactions of the ligated ribozymes showed biphasic decay of bound oligonucleotide, with a fast phase followed by a slower phase. The observed rate constant for the decay of the fast phase was the same as that of the open complex, an indication of a population of damaged ribozyme that cannot reach the closed conformation and cannot react. This interpretation is further supported by the biphasic kinetics observed in reactions of the ligated wild type ribozyme in which the observed rate constants for cleavage were comparable to, or faster than, the rate constant for the dissociation of *S from the damaged population, as noted above under General Kinetic Methods.
Docking equilibria in the absence of AUCG or AUCI were determined from the ratio of the dissociation rate constants (koff) of oligonucleotides -3m,-1d,rSA5, which predominantly binds the ribozyme in the open complex (49), and -1d,rSA5, which predominantly binds the ribozyme in the closed complex (49), according to Equation 2, which is derived from Scheme 1.
| (2) |
The docking equilibrium in the presence of AUCG for the A261H ribozyme was measured as described above, but with 150 μM AUCG present, which is saturating with respect to the ribozyme [see Table 2, ].
TABLE 2. Dissociation constants for AUCG and AUCI in the WT and A261H ribozymes (see Scheme 2).
| Ribozyme | Nucleophile | ||||||
|---|---|---|---|---|---|---|---|
| (μM) | Relative | (μM) | Relative | ||||
| WT | AUCG | 7.0 | (1.0) | 2.7 | (1.0) | ||
| AUCI | 380 | 54 | 360 | 130 | |||
| A261H | AUCG | 5.0 | 0.71 | 29 | 11 | ||
| AUCI | 180 | 26 | 390 | 140 | |||
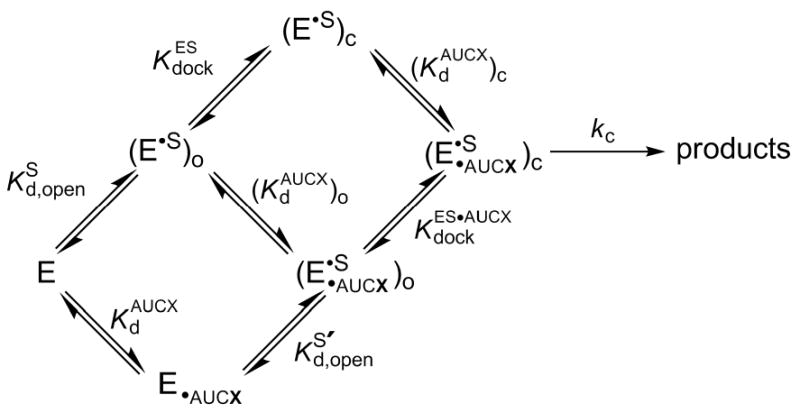
Because the oligonucleotide substrate -1d,rSA5 reacts from the ES•AUCG complex of the wild type ribozyme faster than it dissociates, the docking equilibrium in the presence of AUCG was determined from the thermodynamic cycle shown in Scheme 2, using the measured values of , , and according the Equation 3.
SCHEME 2.
| (3) |
Similarly, docking equilibria in the presence of AUCI for the wild type and A261H ribozymes were determined from the thermodynamic cycle shown in Scheme 2 and from the measured values of , , and according the Equation 4.
| (4) |
Results
The Tetrahymena group I ribozyme catalyzes a phosphoryl transfer reaction that mimics the first step of group I intron self-splicing (Figure 1 and Scheme 2) (27, 28). This reaction involves two substrates, an oligonucleotide (S) and an exogenous guanosine (G). S binds to the ribozyme by base-pairing (48), forming the ‘open complex’ (subscript ‘o’ in Scheme 2), and then docks into the ribozyme's active site (54, 55), forming tertiary interactions in the ‘closed complex’ (subscript ‘c’ in Scheme 2). G also binds to the ribozyme in a dedicated, highly conserved site (31-34), and the substrates can bind in either order. G binding and S docking are coupled, with G binding stronger when S is docked and vice versa (46). One of the catalytic metal ions, referred to as MC, has been implicated in this coupling by previous functional data (24). Within the bound complex, the 3′-OH of guanosine is deprotonated and attacks S at a specific phosphoryl group, transferring the phosphoryl group and its attached 3′-A tail (27).
FIGURE 1. Representation of the transition state of the reaction catalyzed by the Tetrahymena group I ribozyme.

The oligonucleotide substrate S (CCCUCUA) is shown on top, and the guanosine nucleophile is below. The metal ion interacting with the deprotonated 3′-OH of the guanosine nucleophile (MB, in grey) is shown as a separate metal ion from that interacting with the 2′-OH of the same residue, as inferred from functional data (39, 64); structural data suggest that the 3′-OH and the 2′-OH of the guanosine nucleophile interact with the same metal ion, MC (grey interaction with the 3′-oxygen of G; refs. (32, 61)). This unresolved issue does not impact the interpretation of the results herein. The hydrogen bond monitored in this work is highlighted by the red box.
To probe the interaction between the exocyclic amino group of guanosine and the 2′-hydroxyl group of residue A261 (highlighted in Figure 1 by a red box) throughout the catalytic cycle, we isolated individual reaction steps and determined double-mutant cycles (56, 57) for the individual reaction steps. Specifically, we asked whether replacement of the 2′-hydroxyl group at position A261 with a group no longer capable of making the proposed interaction had the same functional effect or a diminished effect in reactions with guanosine and with a guanosine analog (inosine) lacking the proposed partner of the 2′-hydroxyl group at position A261 (36). If the interaction between the exocyclic amino group of guanosine and the 2′-hydroxyl group of residue A261 contributed to a particular step in the catalytic cycle, we expected a diminished functional effect on binding or on the reaction rate upon the replacement of the 2′-hydroxyl group at position A261 when inosine was used as the nucleophile; in other words, we expected a negligible thermodynamic contribution from the interaction between the two monitored functional groups.
To carry out these measurements we used the wild type (WT) ribozyme and a ribozyme with a 2′-H group at position A261 (the A261H ribozyme) that cannot accept a hydrogen bond through this functional group. Because guanosine binds the open complex of the WT ribozyme with low affinity and because of the low solubility of G in aqueous solution (46, 58), we decided to use 5′-extensions that enhance affinity. In particular, the oligonucleotide of sequence AUCG binds tighter than guanosine, forming additional base-pairing and stacking interactions with the ribozyme, interactions referred to as P9.0 (59). Thus, we used AUCG and AUCI throughout our experiments for both the WT and the A261H ribozymes. Finally, we did not probe the effect of the hydrogen bond between the exocyclic amino group of AUCG and the 2′-hydroxyl group of residue A261 in the free ribozyme, because prior data have shown that guanosine affinity for the free ribozyme and for the open complex are identical (46), and there are no interactions made between S in the open complex and the ribozyme's active site (27); therefore guanosine (and AUCG) is expected to make the same interactions in the free ribozyme and in its open complex.
The hydrogen bond between the exocyclic amino group of AUCG and the 2′-hydroxyl group of residue A261 is not formed in the open complex
We first tested the effect of removal of the hydrogen bond between the exocyclic amino group of AUCG and the 2′-hydroxyl group of residue A261 on AUCG binding in the open complex. If this hydrogen bond were formed in the open complex, we expected AUCG to bind weaker to the A261H ribozyme compared to the WT ribozyme; further, we expected AUCI, which lacks the exocyclic amino group proposed to contact the 2′-hydroxyl group of residue A261, to bind with equal affinity to the WT ribozyme and to the A261H ribozyme.
In contrast to these expectations, AUCG binds with near equal affinity to the open complexes of the WT and A261H ribozymes [ and 5.0 μM for the WT and A261H ribozymes, respectively; Table 2], showing that replacement of the 2′-hydroxyl group of residue A261 with a 2′-H group has a negligible effect on AUCG binding. Consistent with this result, the affinities of AUCI for the WT and A261H ribozymes are within ∼2-fold [ and 380 μM for the WT and A261H ribozymes, respectively; Table 2]. The thermodynamic cycle in Figure 2A and the observed negligible interaction energy ( kcal/mol) strongly suggest that there is no functional communication between the 2′-hydroxyl group of residue A261 and the exocyclic amino group of AUCG in the open complex. Thus, the hydrogen bond between these two groups (36) is not formed in the open complex.
FIGURE 2. Double-mutant cycles for individual reaction steps.
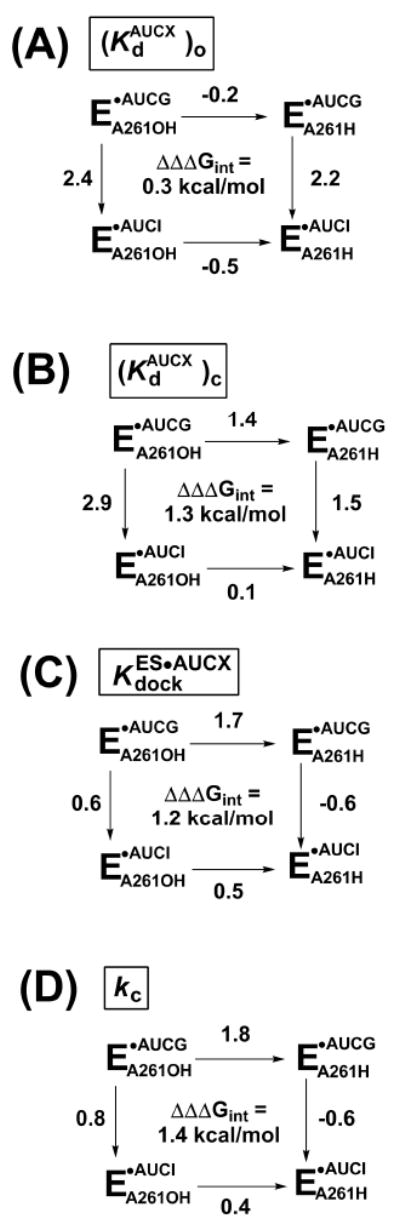
Reactions for WT (EA261OH) or 2′-deoxy A261 (EA261H) ribozyme with AUCG or AUCI. AUCX represents either AUCG or AUCI. The numbers next to each arrow represent the functional effect (ΔΔG in kcal/mol) of either replacing the 2′-hydroxyl group of residue A261 with a 2′-H residue (horizontal arrows) or ablating the exocyclic amine of AUCG, by use of AUCI as the nucleophile (vertical arrows). Values of ΔΔΔGint are calculated by subtracting the value on the right from the value on the right or, equivalently, the value on the bottom from the value on the top. The individual reaction steps (defined in Scheme 2) are as follows: (A) nucleophile binding to the open complex; (B) nucleophile binding to the closed complex; (C) docking of the oligonucleotide substrate S with bound nucleophile; and (D) the chemical step. ΔΔG values are from the rate and equilibrium constants of Tables 2, 3, and 4, and were rounded to a single decimal place to take in account the experimental errors.
The results summarized in Figure 2A also show that removal of the exocyclic amino group of AUCG has a dramatic effect on nucleophile binding: AUCI binds 54- and 36-fold weaker than AUCG in the WT and A261H ribozymes, respectively (Table 2), corresponding to ΔΔG values of 2.4 and 2.2 kcal/mol (vertical arrows in Figure 2A). This large effect on nucleophile binding is consistent with previous results for the WT ribozyme, which showed that inosine binds significantly weaker than guanosine (30, 35, 60). Because of the similar effect in the WT and A261H ribozymes upon removal of the exocyclic amino group of residue A261, our results suggest that the same contacts are formed between the exocyclic amino group of the guanosine nucleophile and the open complexes of the WT and A261H ribozymes. As noted above, these contacts do not involve the hydrogen bond with the 2′-hydroxyl group of residue A261. Presumably the N7 atom of G264 accepts a hydrogen bond from the exocyclic amino group of the guanosine nucleophile, as proposed by Michel et al. (31) and inferred from structural models of three different group I introns (32-34, 41, 61), as shown in Figure 1. We suggest that an additional non-productive contact is also made, as described below (see “Structural rearrangements between the open and closed complexes”).
The hydrogen bond between the exocyclic amino group of AUCG and the 2′-hydroxyl group of residue A261 is formed in the closed complex
Although the hydrogen bond between the exocyclic amino group of AUCG and the 2′-hydroxyl group of residue A261 is not formed in the open complex, prior functional data provided evidence that this hydrogen bond is formed in the transition state of the reaction (36). Therefore, this hydrogen bond can either be formed in the closed complex and maintained in the chemical step, or formed subsequently to the formation of the closed complex but prior to the chemical step. To distinguish between these possibilities we used, as above, a double-mutant cycle to evaluate the energetic consequences of replacement of the 2′-hydroxyl group of residue A261 with a 2′-H group on binding of the nucleophile (AUCG or AUCI), now applying the analysis to the closed complex.
In contrast to the results obtained in the open complex, AUCG binds the closed complex of the WT ribozyme about 10-fold tighter than it binds the closed complex of the A261H ribozyme [Table 2, and 29 μM for the WT and A261H ribozymes, respectively]. This difference corresponds to ΔΔG = 1.4 kcal/mol (Figure 2B) and indicates that the A261H ribozyme is defective in AUCG binding. To determine whether the contact between the exocyclic amino group of AUCG and the 2′-hydroxyl group of residue A261 is responsible for this defect in binding, we established whether this effect is suppressed when AUCI is used as the nucleophile. Indeed, AUCI binds with the same affinity to the closed complexes of the WT and A261H ribozymes [Table 2, and 390 μM, respectively], providing experimental evidence for functional communication between the 2′-hydroxyl group of residue A261 and the exocyclic amino group of AUCG ( kcal/mol, Figure 2B). These results strongly suggest that the hydrogen bond between the exocyclic amino group of AUCG and the 2′-hydroxyl group of residue A261 is formed in the closed complex.
The A261H ribozyme binds AUCG 10-fold tighter than AUCI in this complex (Table 2, 29 and 390 μM, respectively, corresponding to a free energy difference of 1.5 kcal/mol, as shown in Figure 2B). This remaining energetic effect from removal of the exocyclic amino group of AUCG in the absence of the 2′-hydroxyl group of residue A261 suggests that additional interactions are made between the exocyclic amino group of AUCG and the A261H ribozyme. This observation is consistent with the prevailing model for guanosine binding shown in Figure 1, which proposes a hydrogen bond between the exocyclic amino group of (AUC)G and the N7 atom of residue G264 (31). As noted above, there is structural support for this proposal (32-34, 41, 61).
Structural rearrangements between the open and closed complexes
The WT ribozyme closed complex binds AUCG ∼130-fold stronger than AUCI (Table 2, 2.7 and 360 μM, respectively, corresponding to a ΔΔG = 2.9 kcal/mol, as shown in Figure 2B). This value is similar to the 54-fold tighter binding of AUCG, compared to AUCI, observed in the open complex of the WT ribozyme (Table 2, 7.0 and 380 μM, respectively, corresponding to a ΔΔG = 2.4 kcal/mol, as shown in Figure 2A). This similarity was not expected, as the open complex effect cannot reflect the loss of the same contacts as in the closed complex; the hydrogen bond between the exocyclic amino group of AUCG and the 2′-hydroxyl group of residue A261 is formed in the closed complex but not in the open complex.
Two models can account for the similar deleterious effect of removal of the exocyclic amino group of AUCG in the open and closed complexes despite the absence of the A261 2′-hydroxyl hydrogen bond in the open complex1. In one model (Figure 3A), the same number of hydrogen bonds are formed by the exocyclic amino group of AUCG and the ribozyme in the open complex and in the closed complex but one of these hydrogen bonds' partners is lost in going from the open to the closed complex. One candidate that may accept a hydrogen bond from the exocyclic amino group of AUCG in the open complex is the 2′-hydroxyl group of residue C262, as it is nearby (3.9 Å between the oxygen and the nitrogen atoms). This hydrogen bond would be replaced in the closed complex by that between the exocyclic amino group of AUCG and the 2′-hydroxyl group of residue A261. In the second model (Figure 3B), the hydrogen bond between the exocyclic amino group of AUCG and the N7 atom of G264 contributes more to AUCG binding, relative to AUCI, in the open complex than it does in the closed complex. This difference could arise from a change in the alignment of the hydrogen bond. We cannot distinguish between these two models, but regardless of this detail, the findings described above strongly suggest that the hydrogen bond between the exocyclic amino group of AUCG and the 2′-hydroxyl group of residue A261 is formed in the closed complex but not in the open complex, and indicate that rearrangements that involve the guanosine binding site occur in going from the open to the closed complex.
FIGURE 3. Models for the interactions formed by the exocyclic amino group of AUCG in the open and closed complexes of the WT ribozyme.
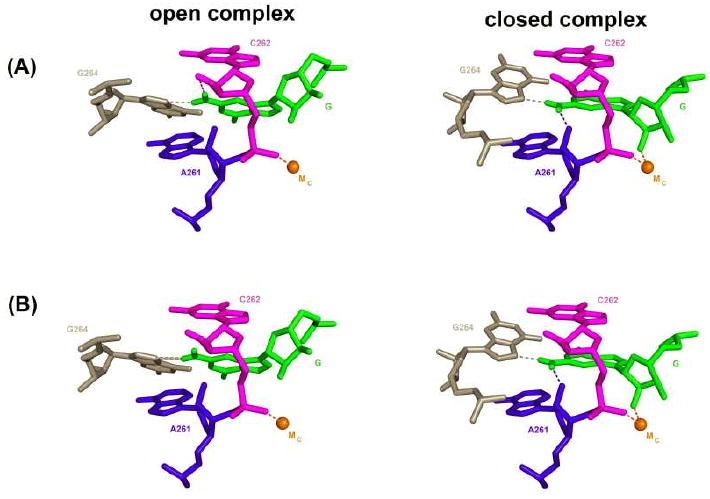
Representation of the closed complex is from the Azoarcus 3bo3 structural model (41); the model for the open complex is obtained by arbitrarily rotating the G nucleophile and G264. In model (A) the exocyclic amino group of AUCG forms two hydrogen bonds with the ribozyme (represented by dashed lines) in both the open and the closed complex, but one of these hydrogen bonds is different in the two complexes; the hydrogen bond between the exocyclic amino group of G and the 2′-hydroxyl group of residue C262, shown in the open complex, is purely speculative. In contrast, for model (B) only one hydrogen bond is formed with the exocyclic amino group of G264 in the open complex, and is represented by a thicker line. In both models the closed complex is the same with the exocyclic amino group of the G nucleophile making hydrogen bonds with the 2′-hydroxyl group of residue A261 and the N7 atom of G264. The contact with G264 is inferred from previous functional data (31) and suggested from structural data (32-34, 41, 61) but has not been experimentally tested. This and the following figures were generated with Pymol (DeLano, W.L. The PyMOL Molecular Graphics System (2002) on World Wide Web http://www.pymol.org)
Anticooperative binding of AUCG and docking of S in the A261H ribozyme suggests a network of interactions connecting the binding sites
We have presented data that strongly suggest that the hydrogen bond between the exocyclic amino group of AUCG and the 2′-hydroxyl group of residue A261 contributes to AUCG binding in the closed complex but not in the open complex. Because binding of AUCG is thermodynamically coupled to docking of the oligonucleotide substrate S (Scheme 2), internal consistency of the thermodynamic cycles requires an identical functional effect for this hydrogen bond in the docking step, as shown by the double-mutant cycle in Figure 2C. The hydrogen bond between the exocyclic amino group of AUCG and the 2′-hydroxyl group of residue A261 could be involved in stabilization of the docked conformation, and its removal could promote occupancy of alternative conformations that are unfavorable for docking.
As shown in Table 3, the docking equilibria for the WT and A261H ribozymes, in the absence of bound nucleophile, are identical within errors, implying that the 2′-deoxy modification in the A261H ribozyme is not deleterious for S docking in the absence of bound nucleophile. In the WT ribozyme binding of guanosine or guanosine analogs such as AUCG cooperatively enhances S docking (46), such that S docks in the ribozyme's core ∼3-fold tighter when AUCG is bound (Table 3, and ). The contact between the catalytic metal ion MC and the pro-SP oxygen atom of the transferred phosphoryl group (Figure 1) has been implicated in the thermodynamic coupling between guanosine binding and S-docking (24, 29). Unexpectedly, we found that in the A261H ribozyme docking of S is destabilized when AUCG is bound (Table 3, and ). This negative cooperativity suggests that the A261H and the WT ribozymes have structural differences that extend beyond the A261 2′-hydroxyl group contact with the exocyclic amino group of the guanosine nucleophile. It is possible that the native docked (closed) conformation is destabilized in the A261H ribozyme because of the close proximity of the polar hydrogen atom of the exocyclic amine of AUCG and the apolar hydrogen at the 2′-position of residue A261 (Figure 1). It is also possible that the 2′-hydroxyl group of residue A261 donates a hydrogen bond to a residue on the ribozyme, and removal of this group alters the ribozyme's conformation. Such alternative conformations could result in different positioning of catalytic metal ion MC, as this metal ion is liganded by the oxygen of the phosphoryl group 3′ of A261 (Figure 1 and reference 25). As noted above, MC is involved in coupling between S docking and G binding (24, 29).
TABLE 3. Docking equilibria in the absence and presence of AUCG or AUCI for the WT and A261H ribozymes (see Scheme 2).
| Ribozyme | Bound nucleophile | ||||||
|---|---|---|---|---|---|---|---|
| (unitless) | Relative | (unitless) | Relative | ||||
| WT | 18 | (1.0) | AUCG | 47* | (1.0) | ||
| AUCI | 18* | 2.6 | |||||
| A261H | 14 | 1.3 | AUCG | 3 | 16 | ||
| AUCI | 8* | 5.9 | |||||
= calculated from the thermodynamic framework of Scheme 2
Binding of AUCI has a different effect on S docking in the WT and A261H ribozymes (Table 3). In the WT ribozyme, binding of AUCI modestly destabilizes S docking, compared to binding of AUCG, whereas in the A261H ribozyme binding of AUCI modestly enhances S docking, compared to binding of AUCG. Again, although we cannot establish the structural details leading to these differences, subtle rearrangements within the active site may play a role.
As indicated by the double mutant cycle of Figure 2C, there is energetic coupling between the 2′-hydroxyl group of residue A261 and the exocyclic amino group of AUCG in the docked conformation, with an interaction energy of 1.2 kcal/mol. But because structural models suggest no direct interactions between these functional groups and the docked oligonucleotide substrate, these results imply that the energetic connectivity between these two groups in the docked conformation arises indirectly via a network of interactions within the ribozyme's active site.
The ternary complex reacts slower for the A261H ribozyme than for the WT ribozyme
Finally, we asked whether the hydrogen bond between the exocyclic amino group of AUCG and the 2′-hydroxyl group of residue A261 affects the chemical step –i.e., reaction of ternary complex in the closed complex. Removal of the 2′-hydroxyl group of residue A261 had a large inhibitory effect of ∼20-fold on the chemical step, but removal of the exocyclic amine of AUCG had only a modest inhibitory effect of 4-fold. These results suggest that the monitored hydrogen bond does not have a large direct role in the stabilization of the chemical transition state, as it would be lost in both instances. Nevertheless, removal of both groups is deleterious to catalysis by ∼7-fold (Table 4). These results suggest that the interaction between the exocyclic amino group of AUCG and the 2′-hydroxyl group of residue A261, although remote from the side of the chemical transformation, nevertheless aids this transformation. Further, AUCI reacts slightly faster than AUCG in the A261H ribozyme, indicating a complex response to changes around the position occupied by the 2′-functional group of residue A261 (Table 4 and Figure 2D). As suggested in the previous section, these effects presumably arise from rearrangement within a precise, extensively tuned network of interactions that is used by the Tetrahymena group I ribozyme to establish its active conformation.
TABLE 4.
| Ribozyme | Nucleophile | kc | |
|---|---|---|---|
| (min-1) | Relative | ||
| WT | AUCG | 0.48 | (1.0) |
| AUCI | 0.13 | 3.7 | |
| A261H | AUCG | 0.023 | 21 |
| AUCI | 0.065 | 7.4 | |
Values of were > 1 for both the WT and A261H ribozymes (Table 3). Thus, both ribozymes reacted from the docked conformation.
Discussion
We have described results obtained by monitoring the hydrogen bond between the exocyclic amino group of AUCG and the 2′-hydroxyl group of residue A261 in the Tetrahymena group I ribozyme and obtaining double-mutant cycles for the individual steps of the reaction. We have shown that the monitored hydrogen bond is not formed in the open complex of the reaction but is formed when the oligonucleotide substrate docks into the active site. Further, we have shown that there is negative cooperativity in the A261H ribozyme between S docking and AUCG binding, suggesting that the lack of the interaction between the exocyclic amino group of AUCG and the 2′-moiety of residue A261 might lead to an alternative conformation that is suboptimal for docking. Finally, we have shown that there is an additional penalty in the transition state of the reaction for the removal of this hydrogen bond, suggesting that this interactions aids alignment for catalysis.
Because the residues involved in this hydrogen bond do not directly contact the docked oligonucleotide substrate or the atoms involved in the chemical transformation, these results strongly suggest a network of interactions that connects different regions of the ribozyme's active site. We now use the published structural models from multiple group I ribozymes crystals to place our functional results in a structural context.
A structural perspective for the observed functional effects
Structural models derived from the crystals of three different group I introns (32-34, 41, 61) show highly similar guanosine binding sites (Figure 4). The guanosine nucleophile forms a base triple with the G264-C311 base pair or the homologous base pair in the Azoarcus and Twort introns, and stacks between residues A261 and C262 (Tetrahymena ribozyme numbering is used throughout). These two bases have been proposed to be part of additional base triples (A261:A265-U310 and C262-G312:A263) that sit above and below the base triple formed with the guanosine nucleophile and presumably strengthen guanosine binding (34). The global similarity of the guanosine binding site holds despite the different constructs used in the different crystallizations, which likely resemble features of the closed complex in the case of the Twort and Azoarcus introns and of the open complex in the case of the Tetrahymena intron (62). Although the overall architecture is conserved, with root mean square deviations of the sugar phosphate atoms of <1 Å (62), overlaying the structural models of the guanosine binding site from different group I intron structures reveals differences in the atomic level models (Figure 4).
FIGURE 4. Superpositioning of structural models of the guanosine binding site from different group I introns crystal structures reveal subtle differences.
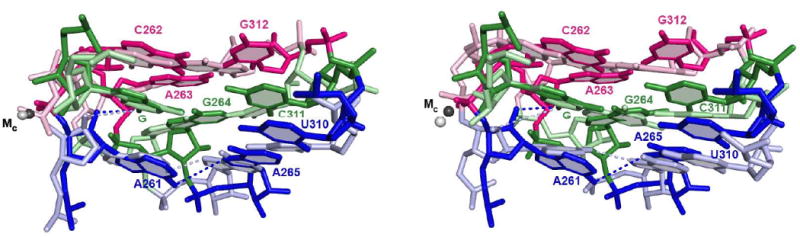
Model from the Azoarcus intron, PDB IS 3bo3 (41), is depicted in darker colors; models from the Tetrahymena intron (34) are in lighter colors and correspond to molecule C from the Tetrahymena ribozyme crystals (part A) and molecule A (part B). Numbering of residues is according to the Tetrahymena intron. Structures were superimposed by aligning the guanosine nucleophile (G). Nucleobases proposed to be part of the same base triple are depicted in the same color. Dashed lines connect the 2′-hydroxyl group of residue A261 and the exocyclic nitrogen atom of G, representing the hydrogen bond functionally detected and investigated herein, and the exocyclic nitrogen atom of residue A261 and the N7 atom of residue A265, representing a hydrogen bond proposed to stabilize the guanosine-binding site. Numerous other proposed hydrogen bonds within this site are omitted for clarity.
In Figure 4A, we overlay a structural model from the Azoarcus ribozyme structure (darker colors, PDB ID 3bo3, 3.3 Å resolution) and that derived from molecule C of the asymmetric unit of the Tetrahymena ribozyme crystal (lighter colors, PDB ID 1×8w, 3.8 Å resolution). These models show similar positioning of the nucleobases, but with differences in the positioning of some of the phosphoryl groups. In particular, the phosphoryl group of residue C262, which provides a ligand to the catalytic metal ion MC (37), adopts a different position in the two models. As mentioned before, MC is involved in transition state stabilization and in coupling between S docking and guanosine binding (24, 63, 64). In both structures the 2′-hydroxyl group of A261 points towards the exocyclic amino group of guanosine, in agreement with the hydrogen bond detected by functional data (36).2
The structural model from a different molecule of the asymmetric unit of the Tetrahymena ribozyme structure (molecule A) shows even more differences when compared that from Azoarcus molecule shown in Figure 4A (Figure 4B). There are more differences in base orientation, especially with respect to residues G264 and A261. Further, the 2′-hydroxyl group of A261 points away from the exocyclic amino group of guanosine, and the phosphoryl group of residue C262 is away from MC, with a distance between the pro-SP oxygen atom and MC of 4.3 Å.
The variations in these structural models may represent subtle but functionally important differences in the arrangement of residues around the guanosine nucleophile. Minimally they suggest that there are multiple potential stacking and hydrogen bond arrangements in and around the guanosine-binding site. Indeed, previous functional data strongly suggested rearrangements around the guanosine nucleophile during the catalytic cycle. First, binding of guanosine involves at least two steps, described as an encounter step and an accommodation step (12). Although the atomic-level details of these two steps have not been defined, it has been proposed that the absence of guanosine may cause a collapse of the base triples that would need to transiently open to allow access of guanosine (27). Second, coordination of the 2′-OH group of the guanosine nucleophile by the active site metal ion referred to as MC (Figure 1) occurs only in the closed complex (24). We have identified an additional conformational change in the guanosine binding process; the hydrogen bond between the exocyclic amino group of guanosine and the 2′-hydroxyl group of residue A261 (highlighted by the red box in Figure 1) is not formed in the open complex and forms subsequently in the closed complex (Figure 3).
An intricate network of interactions within the Tetrahymena group I ribozyme active site
The hydrogen bond between the exocyclic amino group of guanosine and the 2′-hydroxyl group of residue A261 connects several residues implicated in guanosine binding, contributing to an intricate network of interactions that may have implications for the selective recognition of guanosine and overall transition state stabilization within the group I ribozyme active site (Figure 5). This hydrogen bond (shown with a dashed blue line) directly connects the ribozyme with the guanosine nucleophile (shown in green), acting as part of the guanosine-binding site. Further, it establishes an additional connection between the sugar and the base moieties of the guanosine nucleophile (in addition to the covalent bond between them), through a network that comprises MC (shown as an orange sphere), the phosphoryl group between residues A261 and C262 (in blue), and the aforementioned sugar moiety of residue A261 (also blue). In addition, the guanosine nucleophile appears to stack upon residue A261 (Figure 4), and the base of residue A261 also appears to stack with residue A306 (Figure 5); A306 provides an additional ligand to MC through its pro-RP oxygen (references 32, 65, and MF, Jihee Lee and DH, unpublished results). Intriguingly, the other non-bridging phosphoryl oxygen of residue A306, the pro-SP oxygen, provides a ligand to catalytic metal ion MA (32, 50, 65), which contacts the oligonucleotide substrate S (Figure 1 and Figure 5). Stacking of A261 may affect positioning of the phosphoryl group of A306, and thus MA and MC position, providing an additional means to connect the two substrates and their interacting groups within the ribozyme's active site.
FIGURE 5. Network of interactions in the group I ribozyme active site.

Residues are numbered according to the Tetrahymena intron and shown for the Azoarcus 3bo3 structural model (41). This structure contains the products of the reactions carried out herein, with the reactive phosphoryl group and the A residue transferred on the 3′-oxygen of the guanosine nucleophile. Different regions of the ribozyme are colored differently. The two active site metal ions present in the model are shown as orange and yellow spheres and correspond to MC and MA in Figure 1, respectively. Dashed lines represent metal ion contacts or hydrogen bonds and are color coded according to their location. Atoms involved in these contacts are represented by spheres, blue for nitrogen and red for oxygen; hydrogen atoms are now shown. The hydrogen bond between the 2′-hydroxyl group of residue A261 and the exocyclic amino group of G, investigated herein, is represented by the dashed blue line. Stacking of the nucleobases of residues C262, A261, A306 and the G nucleophile is suggested from the X-ray structures (32-34, 41, 61). Additional base triples and stacking interactions are omitted for clarity (see Figure 4).
Roles of conformational changes in the group I ribozyme
As shown in the Results, the hydrogen bond between the exocyclic amine of AUCG and the 2′-hydroxyl group of residue A261 is not formed in the open complex, but forms in the closed complex. We proposed that one of the hydrogen atoms of the exocyclic amino group of AUCG changes its partner as the oligonucleotide substrate S docks into the ribozyme's active site (Figure 3A). A conformational change that establishes the communication between the oligonucleotide substrate and the guanosine nucleophile was also proposed by McConnell and Cech (30) and by Shan and Herschlag (24, 29), and their proposals are consistent with our results. Likewise, our results are consistent with the observations of Karbstein and Herschlag (12), who determined that guanosine binding to the closed complex of the Tetrahymena group I ribozyme is orders of magnitude slower than the diffusion limit and proposed that the free ribozyme adopts an alternative local structure in the guanosine-binding site that must be disrupted to allow G binding.
But what is the possible role of this conformational change? As discussed by Karbstein and Herschlag (12), the conformational change that occurs at the guanosine binding site can increase specificity for the terminal guanosine (G414), which is attached to the intron and used in the second step of self-splicing (27). G414 has two additional helices at its 3′- and 5′-positions that allow it to bind faster than free G. In this kinetic model for specificity, binding of G alone is slower than diffusion, but G414 can bind faster and thus react faster that an incorrect G in the second step of self-splicing, because of the additional helices that hold G414 near to its binding site and give it multiple chances to bind.
Another possible role of the conformational change is to reconcile the advantage from having interactions on multiple sides of a substrate with the requirements to allow substrates and products to enter and leave the active site (66, 67). Functional data indicate that there are numerous interactions between the ribozyme and the guanosine nucleophile, and this guanosine is nearly fully sequestered from solution (Figure 6). Thus, a conformational change may be required to allow guanosine to enter and leave this site; the species formed after guanosine attack, with its attached 3′-sequence, may be particularly difficult to remove from the active site without a conformational change.
FIGURE 6. Space-filling representation of the environment around the guanosine nucleophile.
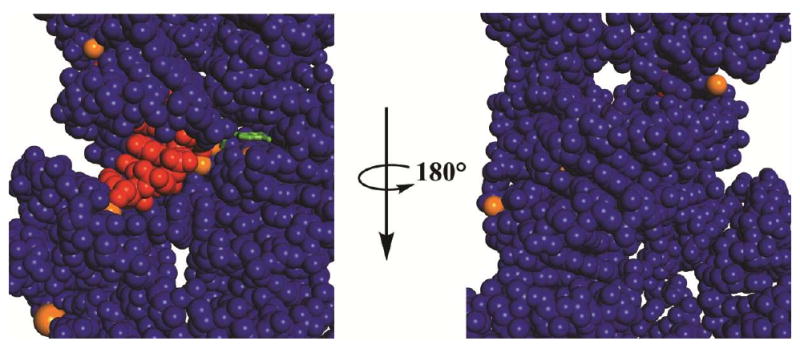
The guanosine nucleophile is in green, the oligonucleotide substrate in red, metal ions in orange, and the ribozyme in blue. The guanosine nucleophile is shown as a stick representation and all other atoms as space filling. Only the central portion of the Azoarcus structural model is shown (3bo3, ref. 41).
Conclusions
We have shown that the guanosine-binding site of the Tetrahymena group I ribozyme undergoes a conformational change that is coupled to docking of the oligonucleotide substrate into the catalytic core. This conformational change involves the formation of a hydrogen bond between the exocyclic amino group of the guanosine nucleophile and the 2′-hydroxyl group of residue A261.
The results presented herein highlight the importance of the 2′-hydroxyl group of residue A261 in the Tetrahymena group I ribozyme, the connectivity between groups and interactions within the active site, and the enhancement of this connectivity in the ternary complex prior to the chemical transformation. These observations expand the picture, both structural and functional, of an extensive and intricate network of interactions used by this RNA enzyme to achieve its extraordinary catalysis and specificity. Future functional studies should reveal additional aspects of this network of interaction and additional connections and networks within and surrounding this complex, highly cooperative active site.
Table 1. Oligonucleotide substrates used in this work.
Letters represent RNA bases, unless otherwise specified; ‘d’ represents a 2′-deoxy substitution; ‘m’ represents a 2′-methoxy substitution.
| Oligonucleotide | Abbreviation |
|---|---|
| d(CCCUC)UA5 | -1r,dSA5 |
| CCCUCd(U)A5 | -1d,rSA5 |
| CCCm(U)Cd(U)A5 | -3m,-1d,rSA5 |
| CCCUCdU | -1d,rP |
Acknowledgments
We thank Tara Benz-Moy for helpful discussion, Max Greenfeld, Jonathan Lassila, Rob Spitale, and other members of the Herschlag and Piccirilli labs for comments on the manuscript, and Dr. Qing Dai for oligonucleotide synthesis.
Footnotes
In the models depicted in Figure 2, we have drawn MC and the A261 and C262 residues with the same positioning in the open and closed complex, and the guanosine nucleophile and the G264 residue changing their positioning. This is an oversimplified representation presented to aid visualization. It is likely that all of these groups rearrange somewhat within the ribozyme's structure in the transition from the open to the closed complex.
The exocyclic amine of A261 does not have the proper geometry in either structure to donate a hydrogen bond to A265 in the complexes of Figure 4A (dashed lines) and is significantly out of the plane described by the A265-U310 base-pair. This last observation suggests that the proposed A261:A265-U310 base triple might not be functionally relevant or, alternatively, that the ribozyme further rearranges to reach the native conformation. For the structural model of the Tetrahymena ribozyme in Figure 4B the exocyclic amine of A261 is in the same plane of the A265-U310 base-pair, compared to the out-of-plane arrangement in the Azoarcus structure, and is only 3.0 Å from the N7 atom of A265, in contrast to the out-of-plane arrangement and to the 4.0 Å distance between these groups in the Azoarcus structure.
This work was supported by a grant from the NIH (GM 49243) to D.H. and by a grant from the Howard Hughes Medical Institute to J.A.P.
References
- 1.Jencks WP. Economics of enzyme catalysis. Cold Spring Harb Symp Quant Biol. 1987;52:65–73. doi: 10.1101/sqb.1987.052.01.010. [DOI] [PubMed] [Google Scholar]
- 2.Ditzler MA, Aleman EA, Rueda D, Walter NG. Focus on function: single molecule RNA enzymology. Biopolymers. 2007;87:302–316. doi: 10.1002/bip.20819. [DOI] [PubMed] [Google Scholar]
- 3.Johnson TH, Tijerina P, Chadee AB, Herschlag D, Russell R. Structural specificity conferred by a group I RNA peripheral element. Proc Natl Acad Sci USA. 2005;102:10176–10181. doi: 10.1073/pnas.0501498102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xie Z, Srividya N, Sosnick TR, Pan T, Scherer NF. Single-molecule studies highlight conformational heterogeneity in the early folding steps of a large ribozyme. Proc Natl Acad Sci USA. 2004;101:534–539. doi: 10.1073/pnas.2636333100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blount KF, Uhlenbeck OC. The structure-function dilemma of the hammerhead ribozyme. Annu Rev Biophys Biomol Struct. 2005;34:415–440. doi: 10.1146/annurev.biophys.34.122004.184428. [DOI] [PubMed] [Google Scholar]
- 6.Herschlag D. RNA chaperones and the RNA folding problem. J Biol Chem. 1995;270:20871–20874. doi: 10.1074/jbc.270.36.20871. [DOI] [PubMed] [Google Scholar]
- 7.Nelson JA, Uhlenbeck OC. Hammerhead redux: Does the new structure fit the old biochemical data? RNA. 2007;14:605–6015. doi: 10.1261/rna.912608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aboul-ela F, Karn J, Varani G. Structure of HIV-1 TAR RNA in the absence of ligands reveals a novel conformation of the trinucleotide bulge. Nucleic Acids Res. 1996;24:3974–3981. doi: 10.1093/nar/24.20.3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brodsky AS, Williamson JR. Solution structure of the HIV-2 TAR-argininamide complex. J Mo l Biol. 1997;267:624–639. doi: 10.1006/jmbi.1996.0879. [DOI] [PubMed] [Google Scholar]
- 10.Famulok M, Hartig JS, Mayer G. Functional aptamers and aptazymes in biotechnology, diagnostics, and therapy. Chem Rev. 2007;107:3715–3743. doi: 10.1021/cr0306743. [DOI] [PubMed] [Google Scholar]
- 11.Puglisi JD, Tan R, Calnan BJ, Frankel AD, Williamson JR. Conformation of the TAR RNA-arginine complex by NMR spectroscopy. Science. 1992;257:76–80. doi: 10.1126/science.1621097. [DOI] [PubMed] [Google Scholar]
- 12.Karbstein K, Herschlag D. Extraordinary slow binding of guanosine to the Tetrahymena group I ribozyme: Implications for RNA preorganization and function. Proc Natl Acad Sci U S A. 2003;100:2300–2305. doi: 10.1073/pnas.252749799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Korostelev A, Ermolenko DN, Noller HF. Structural dynamics of the ribosome. Curr Opin Chem Biol. 2008;12:674–683. doi: 10.1016/j.cbpa.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Montange RK, Batey RT. Riboswitches: emerging themes in RNA structure and function. Annu Rev Biophys. 2008;37:117–133. doi: 10.1146/annurev.biophys.37.032807.130000. [DOI] [PubMed] [Google Scholar]
- 15.Simon AE, Gehrke L. RNA conformational changes in the life cycles of RNA viruses, viroids, and virus-associated RNAs. Biochim Biophys Acta. 2009;1789:571–583. doi: 10.1016/j.bbagrm.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leulliot N, Varani G. Current topics in RNA-protein recognition: control of specificity and biological function through induced fit and conformational capture. Biochemistry. 2001;40:7947–7956. doi: 10.1021/bi010680y. [DOI] [PubMed] [Google Scholar]
- 17.Williamson JR. Induced fit in RNA-protein recognition. Nat Struct Biol. 2000;7:834–837. doi: 10.1038/79575. [DOI] [PubMed] [Google Scholar]
- 18.Narlikar GJ, Herschlag D. Mechanistic Aspects of Enzymatic Catalysis: Lessons from Comparison of RNA and Protein Enzymes. Annu Rev Biochem. 1997;66:19–59. doi: 10.1146/annurev.biochem.66.1.19. [DOI] [PubMed] [Google Scholar]
- 19.Sigler PB. An analysis of the structure of tRNA. Annu Rev Biophys Bioeng. 1975;4:477–527. doi: 10.1146/annurev.bb.04.060175.002401. [DOI] [PubMed] [Google Scholar]
- 20.Christian EL. Identification and Characterization of Metal Ion Binding by Thiophilic Metal Ion Rescue. In: Hartmann RK, editor. Handbook of RNA Biochemistry. Wiley-VCH Verlag GmbH & Co; Weinheim: 2005. [Google Scholar]
- 21.Das SR, Fong R, Piccirilli JA. Nucleotide analogues to investigate RNA structure and function. Curr Opin Chem Biol. 2005;9:585–593. doi: 10.1016/j.cbpa.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 22.Grasby JA, Gait MJ. Synthetic oligoribonucleotides carrying site-specific modifications for RNA structure-function analysis. Biochimie. 1994;76:1223–1234. doi: 10.1016/0300-9084(94)90053-1. [DOI] [PubMed] [Google Scholar]
- 23.Das SR, Piccirilli JA. General acid catalysis by the hepatitis delta virus ribozyme. Nat Chem Biol. 2005;1:45–52. doi: 10.1038/nchembio703. [DOI] [PubMed] [Google Scholar]
- 24.Shan S, Herschlag D. Probing the role of metal ions in RNA catalysis: Kinetic and thermodynamic characterization of a metal ion interaction with the 2′-moiety of the guanosine nucleophile in the Tetrahymena group I ribozyme. Biochemistry. 1999;38:10958–10975. doi: 10.1021/bi990388e. [DOI] [PubMed] [Google Scholar]
- 25.Strobel SA, Ortoleva-Donnelly L. A hydrogen-bonding triad stabilizes the chemical transition state of a group I ribozyme. Chem Biol. 1999;6:153–165. doi: 10.1016/S1074-5521(99)89007-3. [DOI] [PubMed] [Google Scholar]
- 26.Wang S, Karbstein K, Peracchi A, Beigelman L, Herschlag D. Identification of the hammerhead ribozyme metal ion binding site responsible for rescue of the deleterious effect of a cleavage site phosphorothioate. Biochemistry. 1999;38:14363–14378. doi: 10.1021/bi9913202. [DOI] [PubMed] [Google Scholar]
- 27.Hougland JL, Piccirilli JA, Forconi M, Lee J, Herschlag D. How the group I intron works: A case study of RNA structure and function. In: Gesteland RF, Cech TR, Atkins JF, editors. The RNA World. 3. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2006. pp. 133–206. [Google Scholar]
- 28.Zaug AJ, Cech TR. The intervening sequence RNA of Tetrahymena is an enzyme. Science. 1986;231:470–475. doi: 10.1126/science.3941911. [DOI] [PubMed] [Google Scholar]
- 29.Shan S, Herschlag D. Dissection of a metal-ion-mediated conformational change in the Tetrahymena ribozyme catalysis. RNA. 2002;8:861–872. doi: 10.1017/s1355838202020216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McConnell TS, Cech TR. A positive entropy change for guanosine binding and for the chemical step in the Tetrahymena ribozyme reaction. Biochemistry. 1995;34:4056–4067. doi: 10.1021/bi00012a024. [DOI] [PubMed] [Google Scholar]
- 31.Michel F, Hanna M, Green R, Bartel DP, Szostak JW. The guanosine binding site of the Tetrahymena ribozyme. Nature. 1989;342:391–395. doi: 10.1038/342391a0. [DOI] [PubMed] [Google Scholar]
- 32.Adams PL, Stahley MR, Kosek AB, Wang J, Strobel SA. Crystal structure of a self-splicing group I intron with both exons. Nature. 2004;430:45–50. doi: 10.1038/nature02642. [DOI] [PubMed] [Google Scholar]
- 33.Golden BL, Kim H, Chase E. Crystal structure of a phage Twort group I ribozyme-product complex. Nat Struct Mol Biol. 2005;12:82–89. doi: 10.1038/nsmb868. [DOI] [PubMed] [Google Scholar]
- 34.Guo F, Gooding AR, Cech TR. Structure of the Tetrahymena ribozyme: base triple sandwich and metal ion at the active site. Mol Cell. 2005;16:351–362. doi: 10.1016/j.molcel.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 35.Bass BL, Cech TR. Specific interaction between the self-splicing RNA of Tetrahymena and its guanosine substrate: implications for biological catalysis by RNA. Nature. 1984;308:820–826. doi: 10.1038/308820a0. [DOI] [PubMed] [Google Scholar]
- 36.Forconi M, Sengupta RN, Liu MC, Sartorelli AC, Piccirilli JA, Herschlag D. Structure and function converge to identify a hydrogen bond in a group I ribozyme active site. Angew Chem Int Ed Engl. 2009;48:7171–7175. doi: 10.1002/anie.200903006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hougland JL, Kravchuk AV, Herschlag D, Piccirilli JA. Functional identification of catalytic metal ion binding sites within RNA. PLoS Biol. 2005;3:1536–1548. doi: 10.1371/journal.pbio.0030277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hougland JL, Sengupta RN, Dai Q, Deb SK, Piccirilli JA. The 2′-hydroxyl group of the guanosine nucleophile donates a functionally important hydrogen bond in the tetrahymena ribozyme reaction. Biochemistry. 2008;47:7684–7694. doi: 10.1021/bi8000648. [DOI] [PubMed] [Google Scholar]
- 39.Shan S, Kravchuk AV, Piccirilli JA, Herschlag D. Defining the catalytic metal ions interactions in the Tetrahymena ribozyme reaction. Biochemistry. 2001;40:5161–5171. doi: 10.1021/bi002887h. [DOI] [PubMed] [Google Scholar]
- 40.Sjogren AJ, Petterson E, Sjoberg BM, Stromberg R. Metal Ion interaction with cosubstrate in self-splicing group I introns. Nucleic Acids Res. 1997;25:648–653. doi: 10.1093/nar/25.3.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lipchock SV, Strobel SA. A relaxed active site after exon ligation by the group I intron. Proc Natl Acad Sci USA. 2008;105:5699–5704. doi: 10.1073/pnas.0712016105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zaug AJ, Cech TR. The Tetrahymena intervening sequence ribonucleic acid enzyme is a phosphotransferase and an acid phosphatase. Biochemistry. 1986;25:4478–4482. doi: 10.1021/bi00364a002. [DOI] [PubMed] [Google Scholar]
- 43.Moore MJ, Sharp PA. Site-specific modification of pre-mRNA - The 2′-hydroxyl groups at the splice sites. Science. 1992;256:992–997. doi: 10.1126/science.1589782. [DOI] [PubMed] [Google Scholar]
- 44.Morl M, Lizano E, Willkomm DK, Hartmann RK. Production of RNAs with homogeneous 5′ and 3′ ends. In: Hartmann RK, editor. Handbook of RNA Biochemistry. Wiley-VCH Verlag GmbH & Co; Weinheim: 2005. pp. 22–35. [Google Scholar]
- 45.Zaug AJ, Grosshans CA, Cech TR. Sequence-specific endoribonuclease activity of the Tetrahymena Ribozyme- Enhanced cleavage of certain oligonucleotide substrates that form mismatched ribozyme substrate complexes. Biochemistry. 1988;27:8924–8931. doi: 10.1021/bi00425a008. [DOI] [PubMed] [Google Scholar]
- 46.McConnell TS, Cech TR, Herschlag D. Guanosine binding to the Tetrahymena ribozyme: Thermodynamic coupling with oligonucleotide binding. Proc Natl Acad Sci U S A. 1993;90:8362–8366. doi: 10.1073/pnas.90.18.8362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herschlag D, Eckstein F, Cech TR. Contributions of 2′-hydroxyl groups of the RNA substrate to binding and catalysis in the Tetrahymena ribozyme. An energetic picture of an active site composed of RNA. Biochemistry. 1993;1993:8299–8311. doi: 10.1021/bi00083a034. [DOI] [PubMed] [Google Scholar]
- 48.Herschlag D, Cech TR. Catalysis of RNA cleavage by the Tetrahymena Thermophila ribozyme. 1. Kinetic description of the reaction of an RNA substrate complementary to the active site. Biochemistry. 1990;29:10159–10171. doi: 10.1021/bi00496a003. [DOI] [PubMed] [Google Scholar]
- 49.Bartley LE, Zhuang X, Das R, Chu S, Herschlag D. Exploration of the transition state for tertiary structure formation between an RNA helix and a large structured RNA. J Mol Biol. 2003;328:1011–1026. doi: 10.1016/s0022-2836(03)00272-9. [DOI] [PubMed] [Google Scholar]
- 50.Forconi M, Lee J, Lee JK, Piccirilli JA, Herschlag D. Functional identification of ligands for a catalytic metal ion in group I introns. Biochemistry. 2008;47:6883–6894. doi: 10.1021/bi800519a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Forconi M, Piccirilli JA, Herschlag D. Modulation of individual steps in group I intron catalysis by a peripheral metal ion. RNA. 2007;13:1656–1667. doi: 10.1261/rna.632007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karbstein K, Carroll KS, Herschlag D. Probing the Tetrahymena group I ribozyme reaction in both directions. Biochemistry. 2002;41:11171–11183. doi: 10.1021/bi0202631. [DOI] [PubMed] [Google Scholar]
- 53.Mei R, Herschlag D. Mechanistic investigations of a ribozyme derived from the Tetrahymena group I intron: Insights into catalysis and the second step of self-splicing. Biochemistry. 1996;35:5796–5809. doi: 10.1021/bi9527653. [DOI] [PubMed] [Google Scholar]
- 54.Bevilacqua PC, Turner DH. Comparison of binding of mixed ribose deoxyribose analogs of CUCU to a ribozyme and to GGAGAA by equilibrium dialysis: Evidence for ribozyme specific interactions with 2′-OH groups. Biochemistry. 1991;30:10632–10640. doi: 10.1021/bi00108a005. [DOI] [PubMed] [Google Scholar]
- 55.Herschlag D. Evidence for processivity and two-step binding of the RNA substrate from studies of J1/2 mutants of the Tetrahymena ribozyme. Biochemistry. 1992;31:1386–1399. doi: 10.1021/bi00120a015. [DOI] [PubMed] [Google Scholar]
- 56.Fersht A. Structure and mechanism in protein science. W.H. Freeman and Company; New York: 1999. [Google Scholar]
- 57.Horovitz A. Double-mutant cycles: a powerful tool for analyzing protein structure and function. Fold Des. 1996;1:R121–126. doi: 10.1016/S1359-0278(96)00056-9. [DOI] [PubMed] [Google Scholar]
- 58.Pitha PM, Huang WM, Ts'o PO. Physicochemical basis of the recognition process in nucleic acid interactions, IV. Costacking as the cause of mispairing and intercalation in nucleic acid interactions. Proc Natl Acad Sci U S A. 1968;61:332–339. doi: 10.1073/pnas.61.1.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moran S, Kierzek R, Turner DH. Binding of guanosine and 3′ splice site analogues to a group I ribozyme: Interactions with functional groups of guanosine and with additional nucleotides. Biochemistry. 1993;32:5247–5256. doi: 10.1021/bi00070a037. [DOI] [PubMed] [Google Scholar]
- 60.Bao P, Wu QJ, Yin P, Jiang Y, Wang X, Xie MH, Sun T, Huang L, Mo DD, Zhang Y. Coordination of two sequential ester-transfer reactions: exogenous guanosine binding promotes the subsequent omegaG binding to a group I intron. Nucleic Acids Res. 2008;36:6934–6943. doi: 10.1093/nar/gkn824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stahley MR, Strobel SA. Structural evidence for a two-metal-ion mechanism of group I intron splicing. Science. 2005;309:1587–1590. doi: 10.1126/science.1114994. [DOI] [PubMed] [Google Scholar]
- 62.Vincens Q, Cech TR. Atomic level architecture of group I introns revealed. Trends Biochem Sci. 2006;31:41–51. doi: 10.1016/j.tibs.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 63.Shan S, Narlikar GJ, Herschlag D. Protonated 2′-aminoguanosine as a probe of the electrostatic environment of the active site of the Tetrahymena group I ribozyme. Biochemistry. 1999;38:10976–10988. doi: 10.1021/bi9903897. [DOI] [PubMed] [Google Scholar]
- 64.Shan S, Yoshida A, Piccirilli JA, Herschlag D. Three metal ions at the active site of the Tetrahymena group I ribozyme. Proc Natl Acad Sci USA. 1999;96:12299–12304. doi: 10.1073/pnas.96.22.12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stahley MR, Strobel SA. Structural metals in the group I intron: A ribozyme with a multiple metal ion core. J Mol Biol. 2007;372:89–102. doi: 10.1016/j.jmb.2007.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Herschlag D. The Role of Induced Fit and Conformational-Changes of Enzymes in Specificity and Catalysis. Bioorganic Chemistry. 1988;16:62–96. [Google Scholar]
- 67.Wolfenden R. Enzyme catalysis: conflicting requirements of substrate access and transition state affinity. Mol Cell Biochem. 1974;3:207–211. doi: 10.1007/BF01686645. [DOI] [PubMed] [Google Scholar]