

Abstract
Spinal muscular atrophy (SMA) is a common and often fatal neurodegenerative disease that primarily afflicts infants and young children. SMA is caused by abnormally low levels of the survival motor neuron (SMN) protein resulting from a combination of recessively inherited mutations in the SMN1 gene and the presence of an almost identical but partially functional copy gene, SMN2. Absence of the uniquely human SMN2 gene in SMA patients has never been reported because the SMN protein is indispensable for cell survival. Modeling SMA in animals therefore poses a challenge. This review describes the different strategies used to overcome this hurdle and model SMA in mice. We highlight new and emerging insights regarding SMA gained by studying the mice and illustrate how the animals serve as important tools to understand and eventually treat the human disease.
Keywords: Survival motor neuron, Spinal muscular atrophy, Mouse models, Neurodegeneration
Introduction
Proximal spinal muscular atrophy (SMA) is a common, frequently fatal, autosomal recessive neurodegenerative disease characterized by selective loss of the anterior horn cells of the spinal cord and concomitant skeletal muscle atrophy [1]. SMA is caused by homozygous mutations in the survival of motor neuron 1 gene (SMN1) and, consequently, a paucity of its translated product, the SMN protein [2]. Despite being the most frequent inherited cause of infant mortality in humans, SMA is less well recognized than other common neurodegenerative diseases, such as amyotrophic lateral sclerosis, Alzheimer’s disease, and Parkinson’s disease. Yet as a disease paradigm, SMA affords a distinct set of advantages in understanding the molecular basis of neurodegeneration. For instance, it is a monogenic rather than multifactorial disorder and thus is inherited according to simple mendelian principles. Sporadic cases of the disease are virtually unknown; therefore, the genetics of SMA are relatively simple. Second, it involves a defect in a ubiquitously expressed protein known to have a housekeeping role. Yet motor neurons are selectively vulnerable to SMN deficiency, akin to loss of specific neuronal populations in other, better-known neurodegenerative diseases caused by lesions in ubiquitously expressed genes. Understanding the biology of SMA, therefore, might shed light on the common properties of neurons that make them uniquely susceptible to genetic and environmental insults. Finally, SMA is a relatively frequent (~1:6400) disease condition, and although most genetic alterations in patients are SMN1 deletions, point mutations abound. These mutations not only will aid in defining important functional domains, thereby elucidating the role of the SMN protein in disease-relevant biochemical pathways, but also may inform our understanding of neurodegeneration in general.
Despite SMA’s relative obscurity among the lay public, much has been learned about it since the SMN1 gene was cloned. This is a result, in no small measure, of the analyses of a collection of excellent animal models of the disease. SMA model mice are arguably among the most faithful of the animal models in mimicking the human phenotype. This review not only attempts to bring the reader up to date with currently available model mice, but also serves to highlight the insights drawn from their study and the promise they and newer emerging models hold in more fully describing the mechanisms underlying SMA as a means to an eventual treatment for human patients.
The Molecular Genetics of SMA
SMA is caused by recessively inherited mutations in the SMN1 gene. However, patients always harbor one or more copies of an almost identical copy gene, SMN2 [3]. A C→T transition in exon 7 of the copy gene disrupts an exon splicing enhancer and/or creates an exon splicing silencer [4, 5]. As a consequence, SMN2 produces mostly an aberrantly spliced mRNA transcript lacking exon 7 (SMNΔ7) that is translated into an unstable and rapidly degraded protein. SMA patients therefore express vastly reduced levels of the SMN protein but may exhibit varying disease phenotypes, depending on SMN2 copy number [6, 7].
The SMN protein associates with numerous other molecules, interactions indicative of a multifunctional protein [1]. Yet the protein has been implicated unambiguously in just the one function—orchestrating the biogenesis of spliceosomal small nuclear ribonucleoprotein (snRNP) particles and pre-mRNA splicing [8–10]. Moreover, the precise pathway(s) linking SMN paucity to the SMA phenotype remain(s) poorly defined. One way to gain a better understanding of such pathways is through the use of animal models that can be genetically manipulated to study the biology of SMA. Several such models already exist. Here, we concentrate primarily on murine models.
Modeling SMA in Fish, Flies, and Worms
The existence of the SMN2 gene in humans alone, the presence of this gene in all affected individuals, and the absolute requirement of the SMN protein for cell survival posed an immediate challenge to modeling the disease in animals. This hurdle can partially be circumvented in fish, flies, and worms, in which there is a large maternal contribution of SMN to the developing zygote. Accordingly, several groups have attempted to model SMA in zebrafish (Danio rerio), Drosophila, and Caenorhabditis elegans [11–13]. Some of these models harbor mutations that spontaneously arose in their respective SMN genes. Others are based on technologies such as transposon insertion, RNA interference, and antisense morpholinos that were used to effect SMN depletion. Each has provided useful information and mimics certain aspects of the human disease phenotype. For instance, SMN knockdown in zebrafish causes motor axon pathfinding defects, and studies indicate that the protein functions cell-autonomously within motor neurons to ensure proper motor nerve outgrowth and neuromuscular junction (NMJ) formation [12]. Similarly, studies in Drosophila indicate that SMN depletion causes decreased NMJ boutons, whereas in worms, SMN mutations cause locomotor defects [11, 14]. However, some of the observations made in these models appear unique to the species involved, and it has been difficult to reconcile the different conclusions generated in different species. For example, model fish exhibit obvious motor nerve defects, whereas model worms fail to display any overt morphologic defect of the nervous system or loss of cholinergic neurons of the spinal cord. Mutant fish exhibit no defects in muscle organization, specification, or development, indicating a minimal effect of reduced SMN on muscle tissue. On the other hand, SMN depletion in fly muscle causes lethality, and rescue of a lethal phenotype in flies expressing ubiquitously low SMN requires restoration of protein to both nerves and muscles. In worms, it appears SMN acts primarily in nerves because muscle-specific restoration of the protein in mutants produces minimal rescue.
Modeling SMA in Mice
The apparent inconsistencies noted earlier may very well derive from species-specific effects, but they leave important questions about the biology of SMA unanswered. Moreover, it may be argued that none of the previously described models harbors the SMN2 gene, which ensures constant low levels of the SMN protein in SMA patients and is a fundamental characteristic of the human disease. These factors, coupled with the advantages rodents offer in modeling human disease in a mammalian system, provided a compelling argument to model SMA in mice.
Before the identification of the SMN1 gene, a handful of mutant mice that spontaneously arose in various colonies were proposed as models of infantile/juvenile SMA. These included the “muscle deficient” (mdf), wobbler (wr), progressive motor neuronopathy (pmn), and “paralyśe” (par) mouse mutants. However, none of these is based on mutations in the SMN gene and—except for paralyśe, which has yet to be mapped—harbor lesions in unrelated loci [15–17].
The discovery that SMN1 gene mutations are the underlying cause of human SMA was followed shortly after by the identification of the murine orthologue Smn, of which only a single copy exists [18]. Homozygous Smn knockouts provided the first direct evidence that the SMN protein is indispensable for cell survival [19], a finding consistent with reports indicating that SMA patients always carry at least one copy of an SMN2 gene. However, the complete Smn knockout (Smn−/−) did not provide a useful model of SMA because the condition is lethal early in embryogenesis. In early 2000, two approaches were used to generate viable model mice. Frugier et al. [20] selectively inactivated murine Smn in nerves using a conditional (SmnF7) allele from which exon 7 is deleted. The resulting “neuronal” mutants suffered motor neuron loss and muscle atrophy and died of neuromuscular disease at around postnatal day 25 (P25). Because a cardinal feature of SMA is skeletal muscle atrophy, the investigators also sought to determine the effect of selectively abolishing protein expression in muscle tissue [21]. “Muscular” mutants displayed a prominent dystrophic phenotype, which could be modulated if SMN was knocked out in muscle satellite cells rather than differentiated myofibers. Nevertheless, these mice also died prematurely. In retrospect, these findings are hardly surprising given the essential requirement of SMN for cell viability and the specific strategy implemented, which causes abolition rather than reduction of the SMN protein, as seen in human SMA.
To mimic the genetics of human SMA in mice more accurately, two groups introduced the human SMN2 gene into animals lacking murine Smn [22, 23]. To maintain the aberrant splicing pattern of SMN2 and thus ensure low levels of functional protein from the gene, the investigators used a genomic fragment rather than cDNA to make the transgenic animals. One to two copies of the SMN2 transgene in SMN2;Smn−/− mice rescue embryonic lethality. Newborn mutants are indistinguishable from control littermates, but by P2 begin to develop a clear phenotype characterized by an inability to suckle, reduced size, and progressive weakness. Approximately 40% of the motor neurons in the spinal cord and brainstem of mutants are lost by 3 days of age, and the animals rarely live beyond P4. These animals and similar lines generated by Hsieh-Li et al. [23] represent what are arguably the first true murine models of severe, infantile human SMA. Incredibly, eight copies of the SMN2 transgene in SMN2;Smn−/− animals were found not only to rescue embryonic lethality, but also to completely ameliorate the SMA phenotype, supplying the first proof-of-concept study of the feasibility of modulating the SMN2 gene for therapeutic purposes [22]. Subsequent studies to test the in vivo function of SMNΔ7, the most commonly reported mutation in SMA patients, and SMN1A2G, a mild mutation, not only resulted in two additional models of SMA—a severe one and a mild one—but also provided important information about the two mutant isoforms [24, 25]. First, it was shown that SMNΔ7 is not deleterious by acting in a dominant negative manner, as was previously alleged [26]. Instead, it modestly attenuates the severe phenotype of SMN2;Smn−/− mice. Increasing SMNΔ7 expression does not mitigate disease further, likely because of a severely compromised ability to oligomerize. SMNΔ7 therefore is a severe mutation. On the other hand, expressing SMN1A2G in the severe SMA genetic background effects significant phenotypic rescue and results in a mildly affected animal. Importantly, neither SMNΔ7 nor SMN1A2G alone can rescue Smn−/− embryonic lethality, indicating that low levels of full-length SMN (FL-SMN) must be present to enable the formation of functional oligomeric complexes. The model mice described previously and those under development are summarized in Table 1.
Table 1.
Genetic mouse models of spinal muscular atrophy
| Study | Genotypea | Phenotype | JAX stock no. | Notesa |
|---|---|---|---|---|
| Schrank et al. [19] | Smn+/− | Normal | 6214b | Homozygous knockouts (Smn−/−) are embryonic lethal |
| Monani et al. [22] | SMN2+/+; Smn−/− | Very severe SMA phenotype; maximum survival ~P8 | 5024b | Homozygous mutants harbor 2 copies of human SMN2 (~35-kB genomic fragment) |
| Le et al. [24] | SMN2+/+;Δ7+/+;Smn−/− | Severe phenotype; maximum survival ~P23 | 5025b | Demonstrates that SMNΔ7 is capable of mitigating SMA phenotype |
| Monani et al. [25] | SMN2+/+; A2G+/−;Smn−/− | Mild phenotype; mean survival ~8 mo | 5026b | Phenotype variable on different strain backgrounds. Mice hemizygous for both transgenes are more severely affected. |
| Hsieh-Li et al. [23] | SMN2+/−; Smn−/− | Mild phenotype accompanied by severe necrosis of extremities | 5058b | Hemizygous mutants harbor ~2 copies of human SMN2 (~115-kB genomic fragment) |
| Bowerman et al. [60] | SMN2B/− | Intermediate phenotype; mean survival ~P30 | N/A | 2B allele harbors a 3-bp substitution in the Smn exon 7 ESE |
| Frugier et al. [20] | NSE-Cre; SmnF7/Δ7 | Intermediate phenotype; mean survival ~P25 | 6146b (SmnF7 mice) | NSE-Cre effects abolition of SMN protein in neurons |
| Cifuentes-Diaz et al. [21] | HSA-Cre; SmnF7/Δ7 | Intermediate phenotype; mean survival ~P30 | See above | HSA-Cre effects abolition of SMN protein in myofibers |
| JAX | SmnmSmn1–6, SMN2 7,8/+ | Normal | 8453c | Engineered allele is hybrid for mSmn exons 1–6 and human SMN2 exons 7–8 |
| JAX | Smn1SMN2; mSmn1–6SMN2 7,8/+ | Characterization of homozygotes is incomplete | 8714c | Engineered allele harbors 1 copy of human SMN2 and 1 mSmnEx1–6SMN2Ex7,8 hybrid gene |
| JAX | Smn3SMN2; mSmn1–6SMN2 7,8/+ | Characterization of homozygotes is incomplete | 9378c | Engineered allele harbors 3 copies of human SMN2 and 1 mSmnEx1–6SMN2Ex7,8 hybrid gene |
mSmn and Smn refer to murine Smn
Available
Under development
ESE exon splicing enhancer, JAX Jackson Laboratories, N/A not applicable, P postnatal day, SMA spinal muscular atrophy
New and Emerging Insights From Model Mice
In the decade since the first genetic mouse models of SMA were reported, the animals have been widely disseminated within the scientific community and have significantly enhanced our understanding of the biology of the disease and of ways to treat it. In the following section, we highlight new insights into human SMA based on the study of these important biological tools and describe how they have contributed to preclinical development.
Natural History of SMA
An apparent conundrum from early observations in severe mice was the onset of profound muscle weakness before motor neuron loss in the spinal cord [24]. Studies to ascertain whether this might be explained by initial defects that appear distally rather than centrally within the spinal cord revealed striking and profound abnormalities at the NMJs [27•, 28•]. In severe mice, these abnormalities appear as early as P2 and are characterized by abnormal neurofilament infiltrates in the presynapse, poor terminal arborization, and impaired maturation of the acetylcholine receptor clusters (Fig. 1). These perturbations are reflected in neurotransmission defects in severe as well as mildly affected model mice. Importantly, similar NMJ defects were detected in human patients [27•]. However, it is not clear whether the defects are a direct consequence of reduced SMN protein or a byproduct of the disease. Nevertheless, the findings reveal a novel aspect of SMA pathology and raise the possibility that an intervention at the NMJ may be an option in treating the disease. Such an option may not be viable if defects of the neuromuscular system appear in utero. Studies in severely affected SMA infants demonstrated that the motor unit is functionally normal during the first few weeks of life, following which a catastrophic loss of motor unit numbers is observed [29]. Conversely, in mutant zebrafish embryos, neurodevelopmental defects are obvious. To determine whether embryonic defects truly characterize severe SMA, the motor neuronal marker HLXB9::GFP was introduced into SMN2;Smn−/− mice and a systematic analysis of the neuromuscular system was undertaken [30]. These studies indicate that the gross morphology of the motor nerves is normal but that fully half of all intercostal NMJs lack synaptic input. It is puzzling that the latter observation is consistent with neither the normal motor unit number estimates in human patients nor independent observations we (S. Kariya and U. Monani, unpublished finding) and others [28•] have made in severely affected mice. The disparate observations in mice might be attributed to strain-related differences and will require a more thorough analysis in congenic strains.
Fig. 1.
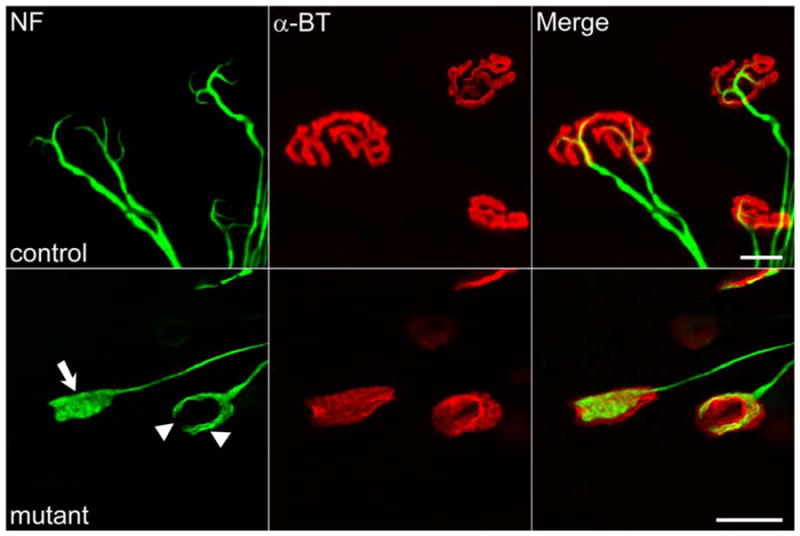
Neuromuscular junction (NMJ) defects in spinal muscular atrophy as seen in model mice. NMJs are characterized by abnormal infiltrates of neurofilament (NF) protein in nerve terminals (arrow), poor terminal arborization (arrowheads), and relatively immature, plaque-like acetylcholine receptor clusters. NMJs from wild-type littermates are shown for comparison. α-BT—α-bungarotoxin. Scale bar=20 μm
Cellular Site of Action of the SMN Protein
Motor neuron degeneration and muscle atrophy are widely accepted hallmarks of human SMA. However, the cellular site of action of the SMN protein has yet to be clearly established. One possibility is that SMN functions cell-autonomously in the motor neurons and that muscle atrophy is a secondary effect. Equally plausible is that SMN functions independently in muscle and perhaps in other cell types to ensure the proper functioning of the neuromuscular system. As noted earlier, the different possibilities have been investigated in non-murine model systems, but the results have varied. To answer the question using severely affected SMN2;Smn−/− model mice, Gavrilina et al. [31•] expressed SMN in motor neurons using a prion (PrP)-SMN cDNA transgene and in skeletal muscle fibers using a human skeletal actin (HSA)-SMN construct. This important study concluded that muscle-specific SMN expression fails to effect rescue whereas high levels of protein in neurons are sufficient to ameliorate the neuromuscular phenotype. The implication is that the primary cellular site of action of the SMN protein is the motor neuron. However, it is important to consider two caveats of this particular study. First, the PrP-SMN transgene in “rescued” SMA mice is characteristically leaky and is expressed not just in all cells of the nervous system, but also at modest but significant levels in muscle tissue. Therefore, it is not clear precisely which neuronal populations require wild-type levels of SMN and whether protein expression in muscle and/or even glia contributes to the eventual rescue. Second, the HSA element used in the study to drive muscle-specific SMN expression does so in myofibers and not myoblasts. If SMN is required in muscle progenitors rather than differentiated myofibers, HSA-SMN transgenes should not be expected to mitigate the SMA phenotype. Despite these caveats, the study is an important first step in resolving an important problem in SMA biology and has spurred complementary research involving distinct and improved transgenes to more precisely address questions pertaining to the cellular site of action of SMN.
SMN Functions and the SMA Phenotype
One of the most vexing problems in the study of neurodegenerative diseases is to construct precise biochemical pathways between the housekeeping function attributed to a causative gene and the unique cellular pathology that characterizes the disorder. SMA is no different. Although there is little doubt that SMN functions in the biogenesis of spliceosomal snRNPs, it is not clear how perturbations in this pathway cause selective motor neuron loss. One possibility is that a distinct function is disrupted. This notion is plausible considering the myriad purported interactions of the SMN protein, but the evidence is circumstantial at best. In an attempt to uncover disease-relevant functions of the SMN protein and dissociate them from those that do not directly affect the SMA phenotype, researchers have taken advantage of point mutations and tested their effects on motor neuronal phenotypes identified in model systems [32]. One such mutation, A111G, is reported to cause severe SMA [33] and is located in the tudor domain (responsible for spliceosomal Sm protein binding) of the protein. However, mutant molecules do oligomerize and are capable of interacting with the Sm proteins [34]. The ability of the mutant molecule to mediate Sm core assembly in in vitro assays, combined with the fact that it results in severe SMA, raised the possibility that a non–snRNP-related function is disrupted in the human disease and underlies the motor neuronal phenotype. To determine its effect specifically on a motor neuronal phenotype, Carrel et al. [32] tested the ability of the mutation to rescue axon outgrowth defects in model fish. Initial findings suggested that it fails to rescue such defects, lending credence to the existence of a novel, non–snRNP-related SMN function that underlies the neuro-muscular phenotype of SMA. However, subsequent experiments indicated that at sufficiently high concentrations, the mutant molecule did rescue the axonal phenotype. Furthermore, when the protein was expressed in severe (SMN2;Smn−/−) mice, it not only assembled Sm proteins onto U small nuclear RNAs (U-snRNAs) in an in vitro assembly assay, but also significantly mitigated the SMA phenotype [35••]. Interestingly, a line of transgenic mice expressing low levels of the mutant protein were not rescued, nor were spinal cord extracts from the animals able to assemble snRNPs efficiently. These findings, coupled with a previous report from the authors demonstrating correspondingly greater assembly activity in mild (SMN2;SMN1A2G;Smn−/−) versus severe (SMN2;Smn−/−) model mice [36••], seem to suggest there is a perfect correlation between SMN’s function in snRNP biogenesis and the SMA phenotype. Thus, one might argue against a novel function of SMN in SMA and propose that snRNP assembly defects truly explain the SMA phenotype. However, a close examination of the data reveals that the correlation is limited to widely differing phenotypes—for example, severe versus mild—in which there underlies a clearly discernible difference in protein levels. It does not apply to instances in which differences in protein level are subtle and phenotypes relatively similar. Indeed, differences in snRNP-related function in two different lines of severe model mice (SMN2;SMNΔ7;Smn−/− and SMN2; Smn−/−) are indistinguishable [36••]. Yet, phenotype as measured by survival is significant (mean survival in days, 13.3±0.3 for SMN2;SMNΔ7;Smn−/− and 5.2±0.2 for SMN2;Smn−/−). Although it is possible that the assays currently being used are simply not sensitive enough to detect putative differences in snRNP function between the two different severe lines, it is equally likely that at very low levels of protein, two or more SMN functions are hierarchically disrupted depending, for instance, on the relative affinities with which the relevant complexes normally form within a motor neuron. Further studies are required to assess SMN function(s).
Model Mice as Tools in Therapeutics Development
In addition to hastening our understanding of the basic biology of SMA, model mice constitute an important tool in the quest for an effective treatment for patients. Here we describe some of the more promising options based on recent studies in mice.
Pharmacologic Studies
Proof-of-principle experiments in transgenic mice demonstrating the feasibility of modulating the SMN2 gene to derive therapeutic benefit prompted numerous in vitro screening studies to identify small molecules capable of upregulating the gene. Among the most promising class of compounds to emerge were the histone deacetylase inhibitors (HDACi). Numerous molecules of this class of compounds subsequently were tested in model mice and shown to mitigate the disease. For instance, sodium butyrate was shown in early studies to extend survival by approximately 50% in severe mice [37]. However, experiments to determine the effect of the molecule on SMN protein and on cellular pathology were scant, and doses administered to the mutants varied widely. Subsequently, the authors tested the effect of a second HDACi, valproic acid, which is US Food and Drug Administration approved for certain applications in humans. Although this study used mild rather than severe model mice, treated animals were found to express increased SMN protein, exhibit reduced neuromuscular pathology, and perform better in motor function assays [38]. Unfortunately, valproic acid provides very limited benefit in SMA patients [39]. Perhaps the most exciting report so far involves a third HDACi, trichostatin A (TSA), which was administered in combination with aggressive nutritional supplementation to mutants. In the absence of nutritional support, TSA produced a 19% increase in median survival of severe SMA model mice. In combination with nutritional support, survival was increased by about 170% [40]. One important finding from the TSA studies is that treatment must be initiated early in the course of the disease to afford maximum benefit. Other molecules that may be promising based on results using SMA model mice include aminoglycosides, which function by forcing translational readthrough [41, 42]; indoprofen [43]; and a novel C5-quinazoline derivative found to target the DcpS enzyme [44]. However, none of these small molecules on its own enhanced survival in severe model mice by more than 30%, emphasizing the challenges of upregulating SMN in whole organisms.
Although small molecules have distinct advantages as therapeutic agents for neurodegenerative diseases, they are by no means the only option. Accordingly, SMA model mice are being used to test and develop a range of therapeutic molecules. Considering the aberrant splicing pattern of the SMN2 gene, a particularly promising class of molecules includes those that redirect the copy gene to transcribe increased levels of FL-SMN. Two studies describing such molecules used antisense oligonucleotides that bind to their cognate sequences in intron 7 of the SMN2 gene and block a putative intronic splicing silencer [45, 46]. Delivery of the antisense oligonucleotides systemically or into the cerebrospinal fluid of SMN2 transgenic mice resulted in vastly increased levels of FL-SMN transcript. In the latter study, spinal cord tissue of treated SMA model mice also expressed increased protein and animals performed better in motor function assays. In a similar study, bifunctional RNAs that simultaneously block splicing suppressor elements and recruit splicing factors were tested in severe model mice and shown to modestly mitigate the disease phenotype [47]. These studies clearly illustrate how SMA model mice have been used to test therapeutic molecules.
Genetic Studies
Restoring SMN to the tissues of affected individuals is an obvious therapeutic option. However, interfering with or enhancing non–SMN-related genetic pathways that are incidentally affected in the disease, but eventually contribute to the overall phenotype, also could inform our understanding of the disorder and point to alternative therapies. Accordingly, several genes known to have general effects on the neuromuscular system have been targeted in SMA model mice. Overexpressing the anti-apoptotic protein Bcl-xL or inactivating the proapoptotic protein Bax is reported to mitigate the disease phenotype [48, 49]. However, the results should be interpreted with caution. Bcl-xL enhanced survival in relatively long-lived mild mice but failed to do so in severely affected mutants [48]. Bax, on the other hand, affected the disease phenotype based on a shift in disease severity in mixed-strain background SMA model mice [49]. However, such shifts in disease severity may very well be the result of strain background differences, which routinely confound cross-breeding studies. To test the effect of perturbing a second pathway known to affect muscle development, researchers disrupted myostatin signaling by either genetically over-expressing or systemically administering the antagonist follistatin to SMN2;SMNΔ7;Smn−/− mice [50, 51]. Genetically inhibiting myostatin signaling failed to provide therapeutic benefit, whereas administering recombinant follistatin enhanced mean survival by around 30% by preventing early death rather than by extending lifespan. Intriguingly, the benefit described in the latter study was observed in only one of three cohorts of treated mice, which failed to show improvement in motor function [51]. In experiments to determine the effect of a third pathway known to retard axonal die-back and neuromuscular synapse loss, WldS protein [52] was overexpressed in SMA model mice [53, 54]. Reduced SMN did not abrogate the protective effect of the WldS protein, but overexpressing the latter in model mice did not mitigate the SMA phenotype [54].
One interesting outcome of these genetic studies is the profound variation observed in the SMA phenotype. This variation, likely the result of strain-background differences, may significantly confound interpretation of the results of a genetic cross but also is indicative of modifier loci that significantly alter the disease process. Such loci have been reported in flies [55]. Identifying similar genes in SMA model mice may be yet another way to exploit these important biological tools to understand the human disease.
Gene and Cell Therapy Studies
SMA model mice also are proving useful in optimizing gene and cell replacement protocols for the human disease. Of particular note is a recent report demonstrating considerable therapeutic benefit after transplantation of “primed” neural stem cells into severe SMA model mice [56]. Not only was mean survival increased by approximately 39%, but motor function and body weight, both of which are affected in mutants, were increased as well. Interestingly, fewer than 2% of transplanted cells eventually established themselves in the spinal cord parenchyma and expressed the motor neuronal marker HLXB9::GFP. Fewer still extended axons into the ventral root, suggesting that therapeutic benefit likely is gained not from improved motor connectivity but through trophic support supplied by transplanted cells to existing host tissue.
An even more promising avenue for future SMA therapies has emerged from recent gene replacement studies in SMA model mice. Three independent groups used slightly different versions of adeno-associated virus (AAV) to deliver SMN to severely affected model mice. Although AAV6 injections into muscle tissue of neonatal mice had no effect [57], administering AAV8 and AAV9 into the nervous system (cerebral ventricles) and systemically (temporal vein), respectively, brought about a remarkable reversal of the disease phenotype [58, 59]. These studies provide proof of principle that gene replacement strategies are both effective and promising.
Conclusions and Future Directions
The studies highlighted in this review leave no doubt that the SMA model mice we and others generated almost a decade ago have proven their utility from both basic science and clinical standpoints. Yet, numerous questions remain, some of which may be addressed using the currently available model mice whereas others will require a newer, more sophisticated generation of engineered lines. For instance, it is not yet certain when during the course of the disease SMN must be restored to produce full phenotypic rescue. Conversely, it is far from clear whether a requirement for the protein applies equally to all periods of postnatal development or whether there are developmentally vulnerable and/or refractory periods. These questions will require model mice in which the protein can be temporally induced. Similarly, and notwithstanding the findings of Workman et al. [35••], the function of SMN in SMA, particularly the severe form of the disease, remains to be fully defined. If, as some suggest, the protein truly has a unique function in axons, it may be identified by engineering mice in which SMN is restricted to certain subcellular compartments of neurons. Attempts to answer these questions using current and newer generations of model mice surely will move us a step closer to better understanding the disease and designing effective treatments for human patients.
Acknowledgments
We are grateful to Drs. D. C. De Vivo, S. Przedborski, and C. E. Henderson for advice and to S. Patruni for critically reading this manuscript. Work in the laboratory is funded by National Institute of Neurological Disorders and Stroke (NINDS) R01 NS057482, NINDS P01 NS055923, US Department of Defense (DoD) W81XWH-09-1-0245, DoD W81XWH-08-1-0009, the Motor Neuron Center, Columbia University, and the SMA Foundation, which also was instrumental in coordinating the generation of the newer model mice.
Footnotes
Disclosure No potential conflicts of interest relevant to this article were reported.
Contributor Information
Gyu-Hwan Park, Email: ghp2105@columbia.edu.
Shingo Kariya, Email: sk2656@columbia.edu.
Umrao R. Monani, Email: um2105@columbia.edu.
References
Papers of particular interest, published recently, have been highlighted as:
•Of importance
••Of major importance
- 1.Monani UR. Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron specific disease. Neuron. 2005;48:885–895. doi: 10.1016/j.neuron.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 3.Monani UR, Lorson CL, Parsons DW, et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999;8:1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- 4.Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet. 2002;30:377–384. doi: 10.1038/ng854. [DOI] [PubMed] [Google Scholar]
- 5.Kashima T, Manley JL. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet. 2003;34:460–463. doi: 10.1038/ng1207. [DOI] [PubMed] [Google Scholar]
- 6.Coovert DD, Le TT, McAndrew PE, et al. The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet. 1997;6:1205–1214. doi: 10.1093/hmg/6.8.1205. [DOI] [PubMed] [Google Scholar]
- 7.Feldkotter M, Schwarzer V, Wirth R, et al. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. 2003;70:358–368. doi: 10.1086/338627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Q, Fischer U, Wang F, Dreyfuss G. The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell. 1997;90:1013–1021. doi: 10.1016/s0092-8674(00)80367-0. [DOI] [PubMed] [Google Scholar]
- 9.Fischer U, Liu Q, Dreyfuss G. The SMN-SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell. 1997;90:1023–1029. doi: 10.1016/s0092-8674(00)80368-2. [DOI] [PubMed] [Google Scholar]
- 10.Pellizzoni L, Kataoka N, Charroux B, Dreyfuss G. A novel function for SMN, the spinal muscular atrophy disease gene product, in pre-mRNA splicing. Cell. 1998;95:615–624. doi: 10.1016/s0092-8674(00)81632-3. [DOI] [PubMed] [Google Scholar]
- 11.Chan YB, Miguel-Aliaga I, Franks C, et al. Neuromuscular defects in a Drosophila survival motor neuron gene mutant. Hum Mol Genet. 2003;12:1367–1376. doi: 10.1093/hmg/ddg157. [DOI] [PubMed] [Google Scholar]
- 12.McWhorter ML, Monani UR, Burghes AH, Beattie CE. Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J Cell Biol. 2003;162:919–931. doi: 10.1083/jcb.200303168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miguel-Aliaga I, Culetto E, Walker DS, et al. The Caenorhabditis elegans orthologue of the human gene responsible for spinal muscular atrophy is a maternal product critical for germline maturation and embryonic viability. Hum Mol Genet. 1999;8:2133–2143. doi: 10.1093/hmg/8.12.2133. [DOI] [PubMed] [Google Scholar]
- 14.Briese M, Esmaeili B, Fraboulet S, et al. Deletion of smn-1, the Caenorhabditis elegans ortholog of the spinal muscular atrophy gene, results in locomotor dysfunction and reduced lifespan. Hum Mol Genet. 2009;18:97–104. doi: 10.1093/hmg/ddn320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmitt-John T, Drepper C, Mussmann A, et al. Mutation of Vps54 causes motor neuron disease and defective spermiogenesis in the wobbler mouse. Nat Genet. 2005;37:1213–1215. doi: 10.1038/ng1661. [DOI] [PubMed] [Google Scholar]
- 16.Schmidt WM, Kraus C, Höger H, et al. Mutation in the Scyl1 gene encoding amino-terminal kinase-like protein causes a recessive form of spinocerebellar neurodegeneration. EMBO Rep. 2007;8:691–697. doi: 10.1038/sj.embor.7401001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin N, Jaubert J, Gounon P, et al. A missense mutation in Tbce causes progressive motor neuronopathy in mice. Nat Genet. 2002;32:443–447. doi: 10.1038/ng1016. [DOI] [PubMed] [Google Scholar]
- 18.DiDonato CJ, Chen XN, Noya D, et al. Cloning, characterization, and copy number of the murine survival motor neuron gene: homolog of the spinal muscular atrophy-determining gene. Gen Res. 1997;7:339–352. doi: 10.1101/gr.7.4.339. [DOI] [PubMed] [Google Scholar]
- 19.Schrank B, Gotz R, Gunnersen JM, et al. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci U S A. 1997;94:9920–9925. doi: 10.1073/pnas.94.18.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frugier T, Tiziano FD, Cifuentes-Diaz C, et al. Nuclear targeting defect of SMN lacking the C-terminus in a mouse model of spinal muscular atrophy. Hum Mol Genet. 2000;9:849–858. doi: 10.1093/hmg/9.5.849. [DOI] [PubMed] [Google Scholar]
- 21.Cifuentes-Diaz C, Frugier T, Tiziano FD, et al. Deletion of murine SMN exon 7 directed to skeletal muscle leads to severe muscular dystrophy. J Cell Biol. 2001;152:1107–1114. doi: 10.1083/jcb.152.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monani UR, Sendtner M, Coovert DD, et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn (−/−) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet. 2000;9:333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- 23.Hsieh-Li HM, Chang JG, Jong YJ, et al. A mouse model for spinal muscular atrophy. Nat Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- 24.Le TT, Pham LT, Butthbach ME, et al. SMN Delta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- 25.Monani UR, Pastore MT, Gavrilina TO, et al. A transgene carrying an A2G missense mutation in the SMN gene modulates phenotypic severity in mice with severe (type I) spinal muscular atrophy. J Cell Biol. 2003;160:41–52. doi: 10.1083/jcb.200208079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kerr DA, Nery JP, Traystman RJ, et al. Survival motor neuron protein modulates neuron-specific apoptosis. Proc Natl Acad Sci U S A. 2000;97:13312–13317. doi: 10.1073/pnas.230364197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27•.Kariya S, Park G-H, Maeno-Hikichi Y, et al. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17:2552–2569. doi: 10.1093/hmg/ddn156. This study describes a previously unappreciated hallmark of human SMA—profound defects of the distal motor unit—using severe as well as mildly affected model mice. Both pre- and postsynaptic specializations were shown to display abnormalities. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28•.Kong L, Wang X, Choe DW, et al. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J Neurosci. 2009;29:842–851. doi: 10.1523/JNEUROSCI.4434-08.2009. This study independently reported distal defects of the motor unit in severe SMA model mice and showed for the first time that synaptic vesicle release may be perturbed in the human disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bromberg MB, Swoboda KJ. Motor unit number estimation in infants and children with spinal muscular atrophy. Muscle Nerve. 2002;25:445–447. doi: 10.1002/mus.10050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McGovern VL, Gavrilina TO, Beattie CE, Burghes AH. Embryonic motor axon development in the severe SMA mouse. Hum Mol Genet. 2008;18:2900–2909. doi: 10.1093/hmg/ddn189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31•.Gavrilina TO, McGovern VL, Workman E, et al. Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect. Hum Mol Genet. 2008;17:2900–2909. doi: 10.1093/hmg/ddm379. This report is the first to attempt to define which tissues require wild-type levels of SMN to rescue the SMA phenotype in model mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carrel TL, McWhorter ML, Workman E, et al. Survival motor neuron function in motor axons is independent of functions required for small nuclear ribonucleoprotein biogenesis. J Neurosci. 2006;26:11014–11020. doi: 10.1523/JNEUROSCI.1637-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun Y, Grimmler M, Schwarzer V, et al. Molecular and functional analysis of intragenic SMN1 mutations in patients with spinal muscular atrophy. Human Mutat. 2005;25:64–71. doi: 10.1002/humu.20111. [DOI] [PubMed] [Google Scholar]
- 34.Shpargel KB, Matera AG. Gemin proteins are required for efficient assembly of Sm-class ribonucleoproteins. Proc Natl Acad Sci U S A. 2005;102:17372–17377. doi: 10.1073/pnas.0508947102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35••.Workman E, Saieva L, Carrel TL, et al. A SMN missense mutation complements SMN2 restoring snRNPs and rescuing SMA mice. Hum Mol Genet. 2009;18:2215–2229. doi: 10.1093/hmg/ddp157. This study provides important data suggesting that SMN’s role in snRNP biogenesis is inextricably intertwined with the SMA phenotype. Rescue of snRNP assembly and restoration of minor spliceosome complexes, even if effected by mutant molecules, correlate with disease severity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36••.Gabanella F, Butchbach ME, Saieva L, et al. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS One. 2007;2:e921. doi: 10.1371/journal.pone.0000921. These experiments showed for the first time that SMN deficiency selectively affects a subset of spliceosomal snRNPs and that there is a remarkable correlation between assembly of snRNP complexes and disease phenotype. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang JG, Hsieh-Li HM, Jong YJ, et al. Treatment of spinal muscular atrophy by sodium butyrate. Proc Natl Acad Sci U S A. 2001;98:9808–9813. doi: 10.1073/pnas.171105098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsai LK, Tsai MS, Ting CH, Li H. Multiple therapeutic effects of valproic acid in spinal muscular atrophy model mice. J Mol Med. 2008;86:1243–1254. doi: 10.1007/s00109-008-0388-1. [DOI] [PubMed] [Google Scholar]
- 39.Swoboda KJ, Scott CB, Reyna SP, et al. Phase II open label study of valproic acid in spinal muscular atrophy. PLoS One. 2009;4:e5268. doi: 10.1371/journal.pone.0005268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Narver HL, Kong L, Burnett BG, et al. Sustained improvement of spinal muscular atrophy mice treated with trichostatin A plus nutrition. Ann Neurol. 2008;64:465–470. doi: 10.1002/ana.21449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mattis VB, Ebert AD, Fosso MY, et al. Delivery of a readthrough inducing compound, TC007, lessens the severity of a spinal muscular atrophy animal model. Hum Mol Genet. 2009;18:3906–3913. doi: 10.1093/hmg/ddp333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heier CR, DiDonato CJ. Translational readthrough by the aminoglycoside geneticin (G418) modulates SMN stability in vitro and improves motor function in SMA mice in vivo. Hum Mol Genet. 2009;18:1310–1322. doi: 10.1093/hmg/ddp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lunn MR, Root DE, Martino AM, et al. Indoprofen upregulates the survival motor neuron protein through a cyclooxygenase-independent mechanism. Chem Biol. 2004;11:1489–1493. doi: 10.1016/j.chembiol.2004.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Butchbach ME, Singh J, Thornorsteinsdóttir M, et al. Effects of 2,4-diaminoquinazoline derivatives on SMN expression and phenotype in a mouse model for spinal muscular atrophy. Hum Mol Genet. 2009 Nov 25; doi: 10.1093/hmg/ddp510. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hua Y, Vickers TA, Okunola HL, et al. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82:834–848. doi: 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williams JH, Schray RC, Patterson CA, et al. Oligonucleotide-mediated survival of motor neuron protein expression in CNS improves phenotype in a mouse model of spinal muscular atrophy. J Neurosci. 2009;29:7633–7638. doi: 10.1523/JNEUROSCI.0950-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baughan TD, Dickson A, Osman EY, Lorson CL. Delivery of bifunctional RNAs that target an intronic repressor and increase SMN levels in an animal model of spinal muscular atrophy. Hum Mol Genet. 2009;18:1600–1611. doi: 10.1093/hmg/ddp076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsai LK, Tsai MS, Ting CH, et al. Restoring Bcl-x(L) levels benefits a mouse model of spinal muscular atrophy. Neurobiol Dis. 2008;3:361–367. doi: 10.1016/j.nbd.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 49.Tsai MS, Chiu YT, Wang SH, et al. Abolishing Bax-dependent apoptosis shows beneficial effects on spinal muscular atrophy model mice. Molec Ther. 2006;13:1149–1155. doi: 10.1016/j.ymthe.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 50.Sumner CJ, Wee CD, Warsing LC, et al. Inhibition of myostatin does not ameliorate disease features of severe spinal muscular atrophy mice. Hum Mol Genet. 2009;18:3145–3152. doi: 10.1093/hmg/ddp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rose FF, Jr, Mattis VB, Rindt H, Lorson CL. Delivery of recombinant follistatin lessens disease severity in a mouse model of spinal muscular atrophy. Hum Mol Genet. 2009;18:997–1005. doi: 10.1093/hmg/ddn426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mack TG, Reiner M, Beirowski B, et al. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- 53.Rose FF, Jr, Meehan PW, Coady TH, et al. The Wallerian degeneration slow (Wld(s)) gene does not attenuate disease in a mouse model of spinal muscular atrophy. Biochem Biophys Res Commun. 2008;375:119–123. doi: 10.1016/j.bbrc.2008.07.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kariya S, Mauricio R, Dai Y, Monani UR. The neuroprotective factor Wld(s) fails to mitigate distal axonal and neuromuscular junction (NMJ) defects in mouse models of spinal muscular atrophy. Neurosci Lett. 2009;449:246–251. doi: 10.1016/j.neulet.2008.10.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang HC, Dimlich DN, Yokokura T, et al. Modeling spinal muscular atrophy in Drosophila. PLoS One. 2008;3:e3209. doi: 10.1371/journal.pone.0003209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Corti S, Nizzardo M, Nardini M, et al. Neural stem cell transplantation can ameliorate the phenotype of a mouse model of spinal muscular atrophy. J Clin Invest. 2008;118:3316–3330. doi: 10.1172/JCI35432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Setola V, Towne C, Kieran D, et al. AAV-mediated gene therapy for SMA: in vitro and in vivo effects of FL-SMN and a-SMN [abstract]. Presented at the 13th Annual Spinal Muscular Atrophy Research Group Meeting; Cincinnati, OH. June 18–20, 2009. [Google Scholar]
- 58.Passini M, Bu J, Roskelley EM, et al. CNS delivery of human SMN1 by adeno-associated virus is highly efficacious in a mouse model of spinal muscular atrophy type I [abstract]. Presented at the 13th Annual Spinal Muscular Atrophy Research Group Meeting; Cincinnati, OH. June 18–20, 2009. [Google Scholar]
- 59.Foust K, Kaspar BK. The kinetics of scAAV9 expression in the spinal cord after neonatal intravenous injection [abstract]. Presented at the 13th Annual Spinal Muscular Atrophy Research Group Meeting; Cincinnati, OH. June 18–20, 2009. [Google Scholar]
- 60.Bowerman M, Anderson CL, Beauvais A, et al. SMN, profilin IIa and plastin 3: a link between the deregulation of actin dynamics and SMA pathogenesis. Mol Cell Neurosci. 2009;42:66–74. doi: 10.1016/j.mcn.2009.05.009. [DOI] [PubMed] [Google Scholar]