

Abstract
N139L substitution in D-channel of cytochrome oxidase from Rhodobacter sphaeroides results in a ∼15-fold decrease of turnover number and in loss of proton pumping. Time-resolved absorption and electrometric assays of the F→O transition in the N139L mutant oxidase result in 3 major findings. (1) Oxidation of the reduced enzyme by O2 shows ∼200-fold inhibition of the F→O step (k ∼ 2 s-1 at pH 8) which is not compatible with the enzyme turnover (∼30 s-1). Presumably, an abnormal intermediate Fdeprotonated is formed under these conditions, one proton-deficient relative to a normal F-state. In contrast, the F→O transition in N139L oxidase induced by single-electron photoreduction of intermediate F, generated by reaction of the oxidized enzyme with H2O2, decelerates to an extent compatible with enzyme turnover. (2) In the N139L, the protonic phase of Δψ-generation coupled to the flash-induced F→O transition greatly decreases in rate and magnitude and can be assigned to proton movement from E286 to the binuclear site, required for reduction of heme a3 from Fe4+=O2- to Fe3+-OH- state. Electrogenic reprotonation of E286 from the inner aqueous phase is missing from the F→O step in the mutant. (3) In the N139L, the KCN-insensitive rapid electrogenic phase may be actually composed of two components with lifetimes of ∼10 and ∼40 μs and the magnitude ratio of ∼3:2, respectively. The 10 μs phase matches vectorial electron transfer from CuA to heme a, whereas the 40 μs component is assigned to intraprotein proton displacement across ∼20% of the membrane dielectric thickness. This proton displacement might be triggered by rotation of the charged K362 side-chain coupled to heme a reduction. The two components of the rapid electrogenic phase have been resolved subsequently with other D-channel mutants as well as with cyanide-inhibited wild-type oxidase. The finding helps to reconcile the unusually high relative contribution of the microsecond electrogenic phase in the bacterial enzyme (∼ 30%) with the net electrogenicity of the F→O transition coupled to transmembrane transfer of 2 charges per electron.
The aerobic respiratory chains of mitochondria and many bacteria contain cytochrome c oxidase (COX)1 as the terminal oxygen-reactive enzyme (1-3). Two input centers of cytochrome oxidase, CuA and heme a, transfer electrons from the native donor, cytochrome c, to the oxygen-reducing site, formed by the high-spin iron of heme a3 and copper ion, CuB, inside the hydrophobic core of the protein, where oxygen is reduced to water (4, 5).
The highly exergonic reduction of dioxygen to water catalyzed by cytochrome c oxidase is coupled to the generation of a transmembrane difference of proton electrochemical potential, ΔμH+. The mechanism of generation of the ΔμH+ by COX consists of two processes. Firstly, as proposed originally by Mitchell (6), in the course of dioxygen reduction to water, the “chemical” protons are taken up electrogenically from the inner aqueous phase to the heme a3/CuB binuclear center where they combine with the electrons, coming from cytochrome c on the opposite side of the membrane. Secondly, it was discovered by Wikström (2, 7) that in addition to uptake of the chemical protons, a single proton is pumped electrogenically across the membrane for each electron transferred by cytochrome oxidase from cytochrome c to dioxygen, and a model was proposed (8, 9) which predicted involvement of 3 intraprotein proton channels in proton pumping by the enzyme.
Crystal structures of COX from different sources (4, 5, 10, 11) reveal 3 “pores” that could potentially serve as input protonic channels (K-, D- and H-channels) through which protons are taken up from the inner aqueous phase during catalytic turnover. The functional significance of the K- and D-proton channels has been well documented for prokaryotic enzymes (reviewed, (12, 13)) while evidence for the role of the H-channel has been obtained only for the mammalian oxidase (14-16).
Starting with the fully oxidized cytochrome oxidase, the catalytic redox cycle of the enzyme binuclear center can be depicted schematically as follows:
It is established that after O2 reacts with the reduced form of the binuclear center to form the “PM” intermediate, the next two electrons delivered to the binuclear center (the “PM” →F and F →O transitions) are accompanied by protons that are taken up exclusively via the D- channel (17-19). The proton transfer events in the first half of the reaction cycle (O → ….→ “PM”) are not yet fully understood, partly due to the existence of multiple forms of the oxidized state (20, 21). The K-channel is known to deliver the two chemical protons to the oxygen-reducing site, one accompanying each of the first two electrons required to reduce the fully oxidized heme a3/CuB metals in O (Fe3+/Cu2+) to the Fe2+/Cu+ state in R2. In addition, the K-channel may be involved in reprotonation of the active-site tyrosine (Y288 in Rhodobacter sphaeroides), which is likely to provide the chemical proton required for O-O bond cleavage during formation of the “PM” state (17, 22-26).
The proton-input channels of cytochrome oxidase have been studied extensively by biophysical techniques combined with site-directed mutagenesis (reviewed, (3, 12, 13)). Several theoretical investigations, including energy minimization and molecular dynamics simulations of proton transfer through the D-channel, have been reported recently (27-32). While mutations of many of the amino acid residues forming the “walls” of the D-channel do not affect functional activity of COX (33-35), several amino acid replacements in the lower part of the D channel result in a loss of proton pumping activity. Thus the D135N mutant of bo3-type quinol oxidase from Escherichia coli (36) and the homologous D132N and D132A mutants of cytochrome c oxidase from R. sphaeroides (37) were shown to be devoid of proton pumping while partly retaining electron transfer activity.
A landmark finding in the proton pumping studies - the N131D mutant of COX from Paracoccus denitrificans and the equivalent mutant N139D of the oxidase from R. sphaeroides, were reported not to pump protons but to retain full or even enhanced catalytic activity (35, 38). N139 is located near the entry of the D channel and is one of three asparagines (N139, N121 and N207, R. sphaeroides numbering) that form a constriction or neck in the channel. The N207D mutation has also been shown to decouple the proton pump without altering the oxidase activity of the enzyme (39).
The effect of decoupling the proton pump from electron transfer by a single amino acid residue substitution is remarkable, and the behaviour of the N139D mutant has been analyzed in considerable detail both experimentally (38, 40-42) and theoretically (30, 43). In our previous work (41), the kinetics of charge translocation across the membrane coupled to the F→O transition was examined by the one-electron photoreduction of the liposome-reconstituted N139D oxidase poised in the F state by reaction with H2O2. With the wild type bacterial oxidase, membrane-potential generation during the F→O transition is comprised of three phases (17, 41, 44), as observed earlier with the bovine enzyme (45, 46). A rapid phase (∼ 15 μs) is insensitive to KCN and is conventionally assigned to vectorial electron transfer from CuA to heme a. An intermediate phase (∼0.4 ms) and a slow phase (∼1.5 ms) are suppressed by KCN and are due to proton movements that accompany electron transfer from heme a to the heme a3/CuB binuclear center (17). The intermediate electrogenic phase was found to be missing in the non-pumping but catalytically active N139D mutant (41). Hence, this phase can be assigned to translocation of the “pumped” proton in the wild type oxidase. Accordingly, the slow protonic phase (∼1.5 ms in wild type enzyme) retained by the N139D mutant oxidase was attributed to transfer of the proton required for oxygen chemistry from the inner water phase to the binuclear center (41).
To get further insight into the critical role of the N139 residue in the proton pump, we have studied in this work the N139L mutant of the R. sphaeroides cytochrome c oxidase, in which a hydrophobic amino acid residue has been substituted in the same location. The N139L replacement results in about 15-fold deceleration of enzyme turnover, and the residual activity (∼7% at pH 6.5-8.5) is fully decoupled from proton pumping. The inhibition of turnover is found to be associated with inhibition of the F→O transition in the catalytic cycle as observed earlier for the non-pumping mutant D132(N,A) of the R. sphaeroides oxidase (17, 19) and its P. denitrificans homologue, D124N (47). Surprizingly, oxidation of the fully reduced N139L mutant oxidase by O2 reveals ∼200-fold inhibition of the F→O step (kv =1.8 s-1 at pH 8) which is not consistent with turnover rate at this pH (∼30 s-1). At the same time, the F→O transition induced in the mutant by single electron photoreduction of F is much faster, and its slowest phase agrees with the turnover rate.
The results of the time-resolved electrometric measurements of charge transfer across the membrane coupled to single-electron photoreduction of Compound F in liposome-reconstituted N139L mutant oxidase are consistent with inhibition of the proton uptake into the D-channel in the mutant, so that when protonated E286 donates the “chemical” proton to the binuclear site, its reprotonation from the inner aqueous phase is suppressed. Most interestingly, time-resolved electrometric studies of the generation of the membrane potential in the N139L oxidase, have revealed a new electrogenic phase (∼40 μs) within the KCN-insensitive microsecond part of the photoelectric response. Upon re-analysis of previous data, this second phase can also be discerned in the photoelectric response of the KCN-inhibited wild-type enzyme. The additional charge separation step in the KCN-insensitive electrogenic phase of the bacterial oxidases that is not present in the bovine oxidase, provides an explanation for ∼1.5-fold higher fractional contribution of the microsecond phase in the bacterial oxidase as compared to the bovine enzyme, a long-recognized discrepancy in the calibration of the voltage amplitudes for bacterial enzyme in relation to the observations with the bovine oxidase. The new KCN-insensitive electrogenic phase can be assigned provisionally to the K-channel dependent internal charge displacement coupled to the electron transfer from CuA to heme a or to the displacement of proton inside of the output proton path above the E286 residue.
Materials and Methods
Enzyme preparation and reconstitution into phospholipid vesicles
The his-tagged cytochrome oxidase was isolated from the R. sphaeroides cell membranes as described in (48).
Reconstitution of oxidase in proteoliposomes
Cytochrome oxidase vesicles (COVs) were made by the following method. Purified asolectin was suspended at 40 mg/mL in 2% cholate and 75 mM HEPES-KOH, pH 7.4, and sonicated until the solution was clear. Cytochrome oxidase (4 μM) was incubated on ice in 75 mM HEPES-KOH, pH 7.4, and 4% cholate for 1 h and then mixed with the sonicated asolectin to a final concentration of 2 μM oxidase and 20 mg/mL of lipid. COV formation was attained by stepwise addition of Bio-Beads which removes the cholate from the solution and the enzyme, leaving the protein incorporated into the vesicles. Then Bio-Beads were removed and the COV solution was dialyzed against 60mM KCl.
The steady state activity of the wild type cytochrome c oxidase and the N139L mutant enzyme was assayed amperometrically with a Clark-type oxygen electrode connected to an oxygen meter (Yellow Springs Instrument Co. Inc) in a buffer containing 50 mM potassium phosphate, pH 6.5, with 10 mM ascorbate, 0.5 mM TMPD (N,N,N′,N′-tetramethyl-p-phenylenediamine) and 45 μM cytochrome c as electron donor system. The reaction was started by the addition of cytochrome c oxidase to the solution. With the wild type enzyme, the final concentration of the enzyme was about 5 nM. Since the turnover of the N139L mutant is slow, about 30 nM oxidase was used in the assay.
Proton pumping measurements
Proton pumping was measured in a stirred cell as described (39). The cytochrome oxidase vesicles (COVs) in a 1.5 mL reaction mixture contains 60 mM KCl, 40 μM cytochrome c, 300 μM ascorbate, 10 μM valinomycin, and ∼0.4 μM liposome-reconstituted oxidase. After all of the components were added, except for ascorbate, the headspace of the cell was flushed with a constant stream of water-saturated argon for 30 minutes to remove the O2 from the sample. Ascorbate was then added. After equilibration, the reaction was initiated by injecting into the reaction mixture a small volume (10 μL) of air-saturated H2O equilibrated at 25° C. Calibration of the pH changes was performed by adding the same volume of a degassed solution of 1 mM HCl. The number of turnovers of the enzyme was calibrated based on calculating the amount of O2 in 10 μL of water at 25° C, assuming O2 concentration of 250 μM. Under these conditions, proton extrusion induced by O2 addition (at proton-pumping efficiency, H+/e- =1), as well as proton consumption observed in the presence of the uncoupler should equal the magnitude of Δ[H+] observed with HCl addition. The proton-pumping efficiency (H+/e-) of the oxidase was calculated as the ratio of pH decrease outside the vesicles induced by O2 addition to the pH change induced by addition of HCl.
Time-resolved measurement of electric-potential generation
Generation of the electric potential difference across the vesicle membrane was monitored by an electrometric technique (49), adapted for time-resolved experiments with COX (45). Details of the sample preparation and the method have been described in detail in the previous papers (41, 46).
Time-resolved spectrophotometric measurements in photoreduction experiments
Time-resolved absorption changes were followed by using a home-built spectrophotometer. The sample was placed in a rectangular semi-microcell with an optical pathway of 4 mm (Starna Cells). The monitoring light from a 100-watt halogen lamp was passed through a grating monochromator (Jobin-Yvon) and projected onto the cell in the sample holder at the 90° angle relative to the excitation laser beam (10 mm in diameter), passing through the entire sample volume vertically (downwards). After passing through the sample, the monitoring light was passed to the photomultiplier via a second grating monochromator and a dark-blue glass filter (OD540/OD450 = 7.5).
The reaction was initiated by a flash from a frequency-doubled neodymium YAG laser (Spectra Physics, Lab-170-10; λ=532 nm; 9 ns; 200-300 mJ). The photomultiplier signal (as well as the electrometric signal, see above) were digitized using a PC-installed Gage 8012A card (time scale, 2,000,000 points with programmed distribution; ordinate scale, 12-bit). The basic effective time resolution of the system was set to 0.1 μs for both electrometric and optical measurements. For the spectrophotometric measurements in the time window >10 μs, an RC filter was placed before the analog-to-digital converter to improve the signal-to-noise ratio. 15–36 traces spaced by 3–5 s were averaged for each absorption trace.
Routinely, the intraprotein electron transfer was measured by following of the reduced heme a absorption band (444 nm). In case of the short (microsecond) time-scale measurements, the signal at reference wavelength (470 nm) was subtracted from the 444 nm kinetics to remove the unspecific absorbance changes, caused by the influence of the laser beam on the photomultiplier.
Stopped-flow absorbance spectroscopy was performed using an Applied Photophysics DX 17MV rapid-mixing spectrophotometer operated in a diode array mode. The solution entering the flow cell is fully mixed within 1-2 milliseconds.
In the experiments shown, syringe A contained an anaerobic solution of 10 μM cytochrome c oxidase fully reduced by about 400 μM dithionite in a buffer containing 10 mM Tris-HCl, pH 8.0, 0.05% dodecylmaltoside and 100 μg/ml of catalase. Syringe B contained an O2-saturated (1 mM O2), 10 mM Tris-HCl buffer, pH 8.0, with 0.05% dodecylmaltoside. The solutions were mixed at a 1:1 ratio and the progress of the reaction was monitored at several different time sweeps.
Data analysis
Global analysis of the spectral/time surfaces was carried out using either MATLAB 6.1 (The MathWorks, South Natick, MA), with subroutines DISCRETE and SPLMOD (50) implemented as in (51) or Pro-Kinetisist version 1.5 program (Applied Photophysics). The experimental kinetics traces were fitted by programs Origin 7 and Discrete (52).
Results
Steady-state turnover of the enzyme and proton pumping
The cytochrome c oxidase activity of the N139L mutant (77 e-/s at pH 6.5), is less than 10 % that of the wild type enzyme (∼1100 e-/s at pH 6.5) but shows a very similar pH-dependence of the turnover rate (see Supporting Information (SI), Figure S1). For pH 8 at which all the time-resolved measurements were made, the turnover rate of the N139L mutant oxidase was about 30 e-/s as compared to ca. 500 s-1 in the wild type enzyme.
Both the wild type and N139L mutant oxidases were reconstituted into phospholipid vesicles. The cytochrome c oxidase activity of the wild type enzyme is stimulated about 10-fold by the uncouplers. However, the presence of the uncouplers has little influence on the steady state turnover of the vesicle-reconstituted N139L mutant enzyme. Rather, there is a small decrease of activity (∼20%). The “reverse respiratory control” has been previously observed with several non-pumping mutants of cytochrome c oxidase in which proton flux through the D-channel is inhibited (e.g., D132A (53, 54) and G204D (55)).
The vesicle-reconstituted oxidase was used to measure proton pumping using a pH-meter equipped with a fast-response glass electrode by adding 2.5 nmol of O2 (10 μl of air-saturated H2O) in the presence of excess reductant. As shown in Figure S2, protons are ejected from the vesicles with the wild type oxidase, while no proton pumping is observed with the N139L mutant oxidase.
Reaction of the fully reduced N139L mutant with O2 reveals inhibition of the F→O step
The N139L mutation inhibits dramatically oxidation of the fully reduced oxidase by oxygen (Figure 1). For the wild type enzyme, oxidation of the enzyme is essentially complete within a dead-time of the stopped-flow system, except for a tail of heme a oxidation in a small fraction of the enzyme (Figure 1A). In case of the N139L mutant, the oxidation process evolves for at least 10 s (Figure 1B). Global analysis of the spectra/time surface for N139L oxidation reveals 3 exponential processes with the rate constants of 8.5 s-1, 1.8 s-1 and 0.09 s-1. The difference spectra of the components that decay in these 3 phases are shown in Figure 2. The major component with a rate constant of 1.8 s-1 is rather close to a spectrum of the F→O transition observed for the wild type P. denitrificans oxidase (courtesy of Dr. M.Verkhovsky, University of Helsinki) as well as to the spectrum of the F→O transition in a N131V mutant of P. denitrificans oxidase (26).
Figure 1. N139L replacement decelerates oxidation of dithionite-reduced enzyme by oxygen.

(A) Wild type enzyme. (B) N139L mutant. The WT and mutant enzyme were reduced anaerobically by 400 μM dithionite and mixed with oxygen in a stopped-flow diode array spectrophotometer. Spectra of the reaction mixture were taken each 4 ms for WT and 5 ms for N139L. The panels show difference absorption spectra versus the final spectrum of the oxidized enzyme that was taken as a baseline. Only 1 of each 10 spectra is shown in the figure. Final concentrations after mixing: COX, 5 μM, oxygen 500 μM. For other conditions, see Experimental procedures.
Figure 2. Spectra of the kinetic phases resolved by global analysis for N139L oxidation.

The spectra/time surface recorded during the oxidation of the fully reduced N139L by oxygen could be fitted to a linear transition of the enzyme through 4 states: 1→2→3→4 (i.e., 3 exponential phases). The difference spectra (vs the final oxidized state) of the components which decay at the steps 1→2, 2→3 and 3→4 with the rate constants of 8.5, 1.8 and 0.09 s-1, respectively, are shown in the figure.
The F→O step during the oxidation of the fully reduced oxidase by O2 involves transfer of an electron shared by heme a and CuA to the ferryl-oxo (Fe4+=O2-) complex of heme a3, converting the latter to the ferric-hydroxy (Fe3+-OH-) state. The data in Figure 2 are consistent with such a process. Decay of the shoulder around 577 nm in the visible, together with the disappearance of the troughs at 413 nm and at 660 nm clearly reports the transition of heme a3 from the ferryl to the ferric state. Simultaneously, heme a is oxidized, as evidenced by intermediate position of the maximum in the Soret band at 442 nm (a band at 436 nm is expected for disappearance of the F580 state and a band at 448 nm for the oxidation of heme a) and by disappearance of the peak at ∼608 nm in the visible. Notably, the position of the maximum of the α-band is red-shifted by about 2 nm relative to the characteristic peak of heme a oxidation (∼606 nm). Therefore, it is possible that the difference spectrum includes some contribution from the decay of the “PR” to the F580 state.
The F→O transition of the N139L oxidase observed during oxidation of the fully reduced enzyme by oxygen at pH 8 shows an effective rate constant of about 2 s-1, which is about 200-times slower than is characteristic of the wild type enzyme (56). Presumably, the N139L mutation inhibits proton uptake required for the F→O transition.
Interpretation of the two minor components of the pre-steady state kinetics (8.5 s-1 and 0.09 s-1) is less obvious. The difference spectrum of the 8.5 s-1-phase shows a maximum at 435 nm with a trough at 414 nm in the Soret and a trough at ∼660 nm. These changes may be consistent with decay of heme a3 from a ferryl-oxo complex as in F580 or “PR” intermediates to the high-spin ferric state, but the absence of significant changes around 605 nm indicates that the electron is delivered to the binuclear site from CuA rather than from heme a. The very low-intensity changes in the ∼550-630 nm region (broad maxima at about 615-618 nm and ∼575 nm, minor minimum at ca. 600 nm) as well as a small but distinct trough at ∼448 nm may reflect superposition of the oppositely directed changes arising from reduction of heme a by CuA (maximum at ∼605 nm) and decay of a Ferryl-oxo form with a maximum at ∼608 nm (“PR” or modified F) in the small fraction of the enzyme. The slowest phase with k = 0.09 s-1 reflects a late process in which oxidation of heme a (decay of the maxima at 606 and 448 nm), is accompanied by reduction of the ferric/cupric binuclear site and formation of some oxygen intermediate (decay of the 660 nm charge–transfer band, and increase of the absorbance at 430 nm).
In conclusion, oxidation of the fully reduced oxidase by oxygen is drastically impaired by the N139L mutation, perhaps at several partial steps, of which inhibition of the F→O transition is the most evident. A similar conclusion has been arrived recently with respect to the homologous N131V mutant oxidase from P. denitrificans (26).
Single-electron photoreduction of the ferryl-oxo complex of the N139L oxidase
The impaired F→O step in the N139L mutant was further studied by an alternative approach involving single-electron photoreduction of Compound F to the O state (57). To this end the F580 state was prepared by treatment of the solubilized or liposome-reconstituted oxidase with H2O2 (58), and then using a photoreductant (RuBpy + aniline) to inject one electron into the enzyme. The electron initially reduces CuA, then heme a, and finally, moves to the heme a3/CuB center. This approach has been used earlier with the wild type and several mutant forms of cytochrome oxidase from R. sphaeroides (17, 41) and P.denitrificans (44, 59, 60). The oxidized N139L enzyme reacts with H2O2 to form compound F580 with normal spectral characteristics and with about the same efficiency as does the N139D mutant (41). About 65-70% of the enzyme is converted to the F state and, accordingly, in both N139D and N139L, 65-70% of the flash-reduced heme a undergoes rapid reoxidation. Flash-induced, single-electron reduction of the F state with the RuBpy/aniline system allows for time-resolved spectrophotometric and electrometric studies of the isolated F→O transition in the peroxide-reactive fraction of the enzyme.
Optical monitoring of the reoxidation kinetics of heme a
A striking difference has been found between the kinetics of the F→O step as observed during the oxidation and photoreduction experiments with the N139L mutant oxidase (Figure 3). The F→O transition induced by single electron photoreduction of Compound F (trace 2) is very much faster than the F→O step observed during the oxidation of the fully reduced enzyme (trace 1). The finding points to significant differences between the mechanisms of the F→O transition under these two sets of conditions.
Figure 3. Different kinetics of the F→O transition in N139L mutant oxidase as induced by single-electron photoreduction of Compound F (trace 2) and observed during oxidation of the fully reduced enzyme by O2 (trace 1).

Trace 1, oxidation of heme a followed at 608 minus 630 nm has been constructed from the dataset in Figure 1A. Trace 2, oxidation of heme a measured at 445 nm in the F→O transition triggered by single-electron photoreduction of Compound F (see Figure 4 and Table 1 for conditions and more details). The traces are given normalized to the magnitude of the absorption changes at each wavelength pair.
Nevertheless, inhibition of the F→O step in N139L mutant relative to the wild type enzyme is observed in the photoreduction experiments as well, but the effect is much less dramatic than in the case of the oxidation of the fully reduced enzyme by oxygen. Flash-induced reduction of heme a by RuBpy/aniline and its subsequent reoxidation by the binuclear site for N139L and the wild type oxidases are compared in Figure 4. The reduction of heme a proceeds with the same rate for the wild type and mutant enzymes (not shown), but reoxidation of heme a differs significantly. For the wild type oxidase, reoxidation of the flash-reduced heme a is virtually complete and reaches the plateau in ∼10 ms (Figure 4A). In the N139L mutant, the magnitude of the reoxidation is less (65-70%) and the process is slower. The incomplete reoxidation of heme a in the N139L oxidase (Figure 4A) is in agreement with the ∼70% conversion of heme a3 to the F state by the reaction with H2O2 as determined by absorption changes in the Soret band (ΔA436-414). A similar lower yield of peroxide-generated Compound F was previously reported for N139D and other mutants within the D channel (41). The reoxidation of heme a in both the wild type and N139L mutant oxidases is fully prevented by cyanide (not shown).
Figure 4. Reoxidation of photoreduced heme a during the F → O transition in the wild type and N139L mutant form of cytochrome oxidase.

The optical cell (see Experimental procedures) contained COX (10-20 μM) in 5 mM Tris-acetate, pH 8, and 0.05–0.1% dodecyl maltoside, with 40 μM RuBpy, 10 mM aniline and 2 mM H2O2. 16 traces have been averaged in each case. The absorption curves have been normalized: (A) by the magnitude of heme a photoreduction and (B) by the magnitude of heme a reoxidation.
In Figure 4B, the traces of the reoxidation of heme a from the N139L and wild type oxidases have been normalized by the amplitude of reoxidation of heme a. It is clear that the N139L mutation results in a significant decrease in the rate of electron transfer from heme a to the binuclear center. Results of the heme a oxidation kinetics fitting for N139L and wild type oxidase are shown in Table 1 (see also Figure S3). The deconvolution of the reoxidation of heme a in the N139L mutant gives three exponentials (Table 1). The slowest component with τ of 16-22 ms corresponds reasonably well to the turnover number of the N139L mutant (∼ 30 s-1 at pH 8; τ ∼ 33 ms) as well as to the rate of the electrogenic protonic phase associated with reoxidation of heme a (τ = 25-40 ms, Table 2). More detailed comparison of the optical and electrometric responses associated with heme a oxidation in the mutant oxidase is limited by relatively poor signal-to-noise ratio for both the absorption and the electrometric traces with N139L. The fast component of heme a reoxidation, with τ of 0.15-0.18 ms, is not observed with the wild type oxidase (Table 1, Figure S3), but a similar fast phase was found in the reoxidation kinetics of heme a of the N139D mutant oxidase (41).
TABLE 1.
Kinetics of heme a reoxidation in the wild type and N139L oxidases from R. sphaeroides
| Phase | Wild type | N139L | ||
|---|---|---|---|---|
| τ, ms | amplitude, % | τ, ms | amplitude, % | |
| 1 | 0.4 | 30-35 | 0.26-0.32 | 24-29 |
| 2 | 1.6 | 50-60 | 2.1-3.3 | 39-42 |
| 3 | 8-10 | 5-15 | 16-22 | 29-37 |
The data for N139L, have been obtained by deconvolution of the traces shown in Figure 4. For the wild type oxidase, a separate set of data with a better signal-to-noise ratio was used and deconvolution of heme a reoxidation to exponentials was performed by two different protocols (see Figure S3). The Table shows the results of the fitting procedure used in Figure S3,B (and see Figure S3,A and (41) for the other version).
TABLE 2.
Photoelectric responses of the wild type and N139D mutant forms of cytochrome c oxidase coupled to the F→O transition
| electrogenic phases | Wild type | N139L | Origin | ||||||
|---|---|---|---|---|---|---|---|---|---|
| this work | earlier papersd | this work | |||||||
| τ | contributiona, a. u. | τ | contributiona, a.u | τ | contribution, a.u. | ||||
| 1 | insensitive to KCN | 10 μs | 1.0 | 1.0 | single phase, 15-17 μs | 1.0 | 10 μs | 1.0 | electronic CuA→heme a |
| 2 | insensitive to KCN | 40 μs | 0.67 | 0.67 | 40 μs | 0.67 | protonic? | ||
| 3 | KCN-sensitive | 0.4 ms | 1.17b | 1.9c | 0.4 ms | 0.7b | 0.5-2.0 ms | 0.09b | protonic |
| 4 | KCN-sensitive | 1.6 ms | 2.84b | 2.2c | 1.6 ms | 1.7b | 25-40 ms | 0.30b | protonic |
| Total electrogenicity: arbitrary units | 5.7 | 3.4 | 2.1 | ||||||
| Total electrogenicity: elementary charges translocated across the membranee | 1.9 | 1.1 | 0.7 | ||||||
In all samples, the magnitude of the very first phase has been assigned a value of 1 arbitraty unit. For N139L, correction for 30% of non-reoxidizable heme a has been made.
apparent contributions of the two KCN-sensitive electrogenic protonic phases resolved by deconvolution as published in refs d;.
The true contributions recalculated from the apparent values (cf. Footnote b) taking into account serial sequence of the two protonic phases (64);
calculated from the electrogenicity value in arbitrary units assuming that the rapid electrogenic phase (CuA to heme a electron transfer = 1 arbitrary unit) corresponds to translocation of 1 elementary charge across ∼1/3 of the insulating dielectric layer.
Electrogenic processes coupled to the F→O transition
Time-resolved electrometric measurements using the single-electron photoinjection technique were performed to characterize the effect of the N139L mutation on membrane potential generation during the F→O transition. The data are illustrated by Figure 5 (see also Figures S4, S5) and are summarized in Table 2. Figure 5 compares the time-resolved electrogenic response for the N139L mutant with the results obtained previously for the wild type oxidase and the N139D mutant (17, 41). Each of the three oxidases exhibits a microsecond phase of voltage generation, and the data have been normalized to the magnitude of this phase.
Figure 5. Comparison of the electrogenic protonic phases in N139L, N139D and wild type cytochrome oxidase.
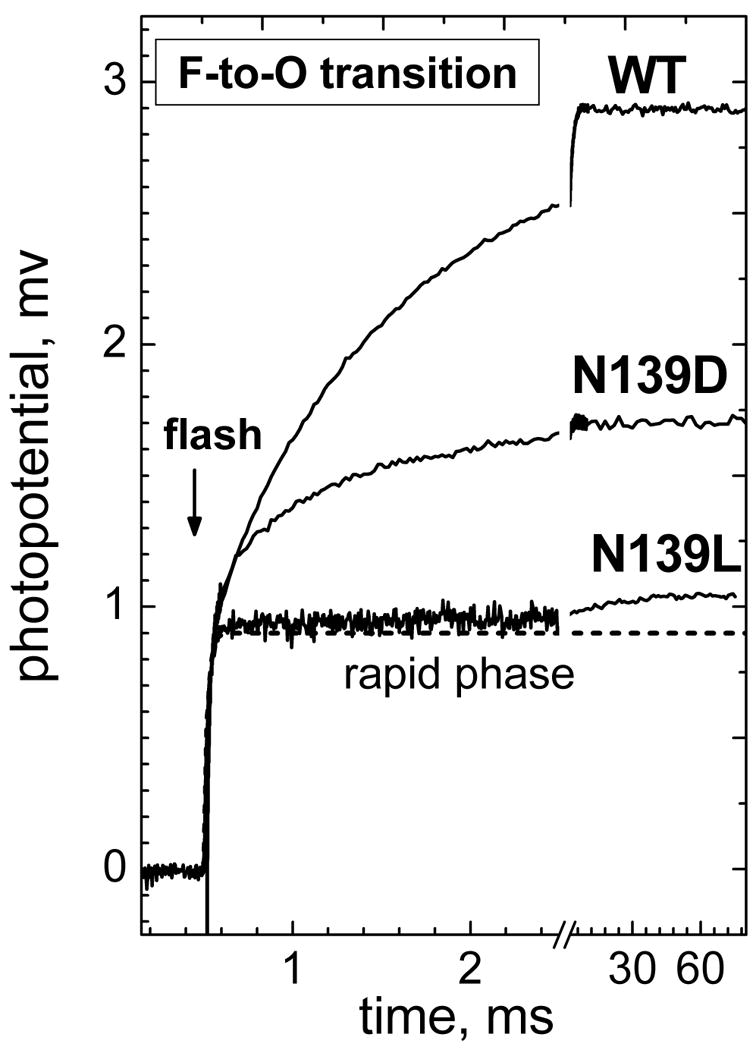
Experiments with collodion film-adhered liposomes with COX have been carried out in the buffer containing 5 mM Tris-acetate, pH 8, with 40 μM RuBpy and 10 mM aniline as the photoreducing system. 2 mM H2O2 was added to convert COX to the ferryl-oxo state prior to the flash. The traces have been normalized by the magnitude of the microsecond KCN-insensitive part of the response (indicated by a dashed line) and contribution of the membrane passive discharge has been subtracted from all the curves (cf. Figures S4, S5).
The KCN-sensitive phase of photoelectric response observed for N139L mutant is much smaller and slower than for the pumping wild type oxidase or for the non-pumping but active N139D mutant. In fact, the phase can be hardly resolved in N139L oxidase unless passive discharge of Δψ has been subtracted from the trace (Figures S4, S5). The amplitude of the KCN-sensitive electrogenic phase in the N139L oxidase constitutes only about 20% of the protonic electrogenic phase in the non-pumping N139D mutant. The phase is dominated (∼75%) by a slow component with τ of 25-40 ms, which is in reasonable agreement with the turnover number of the mutant enzyme under these conditions (∼30 s-1) and with the slowest component of the F→O transition measured optically (Table 1). There is also a minute faster phase with lifetime of ∼0.5-2 ms (∼25%).
For all three of the oxidase samples, the addition of KCN has no effect on the microsecond phase, but it eliminates the voltage generation in the millisecond region. Cyanide binds to ferric heme a3 and blocks electron transfer from heme a to heme a3. Therefore, the KCN-insensitive microsecond part of the photoelectric response is confined to the electron transfer from CuA to heme a and to any other charge transfer events that are coupled to this reaction, whereas the KCN-sensitive millisecond phase is due to proton movements associated with the electron transfer from heme a to heme a3.
As noted above, the F→O transition induced by single electron photoreduction of Compound F in N139L is much faster than the F→O decay observed during oxidation of the fully reduced mutant oxidase by oxygen (τ ∼ 0.5 s, Figures 1-3). Similar very slow F→O decays (τ of 0.4 – 0.6 s) have been observed at pH around 8 during the oxidation of the fully reduced D132N mutants (61), as well as in D124N and N139V mutants of P.denitrificans oxidase (26, 62). Under these conditions, the F→O decay is likely to be limited by slow electrogenic reprotonation of E286Rs (E278Pd) from the inner phase, as evidenced by the data obtained by FTIR (62) and electrometric measurements (47, 63).
In contrast, the photoreduction-induced F→O transition with the N139L mutant does not show evidence for electrogenic reprotonation of E286. Reprotonation of E286 from the N-phase with τ ∼0.5 s would give rise to Δψ generation simulated in Figure S5 by trace d (dotted line). Clearly, the curve is not compatible with the experimental data. Thus, the electrometric data provide further evidence for different mechanisms of the F→O transition as observed during the oxidation of the fully reduced oxidase by O2 (47, 62) and during single-electron photoreduction of Compound F.
Deconvolution of the microsecond phase of the electrogenic response of the N139L oxidase
In past studies, the initial KCN-insensitive microsecond phase of the electrogenic response observed in the flash-induced, single-electron redox transitions of cytochrome oxidase from either R. sphaeroides (17, 41, 64) or P. denitrificans (44, 59, 60, 65) was approximated routinely by a single exponential with τ of 10-20 μs (see also (66) for detailed analysis). This initial transient is followed by slower cyanide-sensitive protonic phases (Table 2). The very low contribution of the protonic phases in N139L oxidase makes it possible to analyze the kinetics of the initial microsecond phase with higher precision. Conventional fitting of the microsecond electrogenic phase in the N139L mutant to a single exponential gives a time constant of 15-17 μs, similar to the results obtained with the R. sphaeroides wild type oxidase and various mutant forms of the enzyme studied previously (17). However, the fit is very significantly improved by using a two-exponential deconvolution (Figures 6 A, B). The biphasic pattern of the response is retained in the cyanide-inhibited N139L oxidase (Figure 6 C, D), which excludes any contribution from the processes associated with electron transfer to the binuclear site. Moreover, a very similar biphasic pattern is evident upon re-analysis of the data for the cyanide-inhibited wild type oxidase (Figure 6 E, F).
Figure 6. Biphasic pattern of the microsecond electrogenic phase in N139L and wild-type oxidase.

For each of the enzyme samples, the adjacent panels compare deconvolution of the electrometric traces into one (A,C,E) or two (B,D,F) exponentials. Each panel shows an experimental trace, a fitted curve and their difference (plot of the residuals). Basic conditions, as in Figure 5. (A,B), N139L oxidase; (C,D), N139L oxidase in the presence of 0.5 mM KCN and 200 μM ferricyanide; (E,F), wild type oxidase in the presence of 0.5 mM KCN and 200 μM ferricyanide. Ferricyanide was added in case of the KCN-inhibited enzyme to ensure the oxidized state of heme a.
The optically monitored electron transfer from CuA to heme a in the wild type oxidases from R. sphaeroides (67) and P. denitrificans (65) has been firmly established to be monophasic with τ∼10μs. The same τ value of ∼10 μs is also observed in this work for the flash-induced reduction of heme a in the N139L mutant (Figure 7, circles). This time constant is noticeably less than the lifetime of 17 μs found for the microsecond electrogenic phase in a single-exponential fitting. Moreover, as shown in Figure 7, the kinetics of the microsecond electrogenic phase clearly deviates from the kinetics of reduction of heme a. It is noteworthy, that no such discrepancy between the absorption and electrometric curves associated with heme a reduction has been observed with bovine cytochrome oxidase (S.A. Siletsky, unpublished).
Figure 7. The kinetics of rapid phase of membrane potential generation does not match the kinetics of heme a reduction during single-electron photoreduction of Compound F in N139L oxidase.

Conditions for the absorption measurements, as in the Figures 4, S5; for the electrometric measurements, as in Figures 5,6. The amplitude of ΔΨ generation is 0.75 mV for the electrometric trace and the maximal value of ΔA445 for the absorption trace is 0.0067 O.D.
As shown above (Figure 6), a much better fit is obtained for the rapid, KCN-insensitive electrogenic phase with the N139L mutant as well as with the KCN-inhibited wild type enzyme using a two-exponential approximation. If the time constant for the first exponential is assigned a fixed value of 10 μs, the second exponential takes on a value of τ ∼35-40 μs, and the two components show relative apparent amplitudes of ca. 60% (10 μs) and 40% (40 μs). Hence, 60% of the charge transfer in the KCN-insensitive electrogenic phase matches the 10 μs vectorial electron transfer from CuA to heme a, and 40% of the charge transfer is delayed to about 40 μs. This is observed for both the N139L and wild type oxidases.
Resolution of the electrogenic responses of the wild type and N139L mutant oxidases into individual phases taking into account biphasic character of the microsecond, KCN-insensitive part of the response is summarized in Table 2 (the columns “this work”).
Discussion
Abolition of proton pumping
Replacement of N139 by either aspartate (38, 41), threonine (68), or leucine (this work) results in the loss of proton pumping. Similar loss of proton pumping is observed for other amino acid substitutions in the lower part of the D-channel, e.g. D132N and D132A (37), G204D (55), N207D (39) as well as N131(V,D,E,C) in the oxidase from P. denitrificans (26, 69). The unique explanation for the decoupling of the proton pump is not known and, in fact, it may not be the same for all of these mutations.
In those studies in which a non-ionizable residue was replaced by an ionizable one (e.g., N139D, N207D or G204D) or vise versa (D132N or D132A (70)), the analysis was focused on a possible shift of the pKa of E286 induced by a change in electrostatic interactions of E286 with the residues replaced (e.g., (42)) but computational work has questioned the possibility of any significant electrostatic interactions at such distances (30, 43, 71). In any case, the electrostatic explanation cannot be invoked to explain how the N139L replacement results in the loss of proton pumping.
A flawless operation of the proton pump machinery may depend critically on the exact timing of proton delivery to residue E286 via the D-channel, consistent with the models in which proton transfer from residue E286 to the so called “Pump Loading Site” (PLS), coupled to oxidation of heme a2+, must precede proton transfer from E286 to the oxygen reducing site (41, 60, 72). The decoupling mutations in the D-channel have been shown to alter the intrachannel water structure, the energetic profile of the proton trajectory along the D-channel and configuration of E286 (29, 30, 69, 73), and therefore should affect the kinetics of H+ transfer through the D-channel. Presumably, the delay of H+ delivery through the D-channel in the N139L mutant of the oxidase results in all protons ending up at the oxygen reducing site where they are consumed in the formation of H2O.
Inhibition of the catalytic cycle at the F→O step
The N139L mutation inhibits enzyme turnover about tenfold. As in the case of the D132N mutation (17, 19) or the D124N and N131V replacements in the P. denitrificans oxidase (26, 74), the inhibition of turnover involves primarily the F→O step of the catalytic cycle. Presumably the N139L mutation restricts proton transfer from D132 to the chain of waters resolved in the D-channel by crystal structure analysis (32, 75), starting just beyond the constricted “neck” consisting of three asparagines (N139, N121 and N207) in the wild type oxidase, and leading to E286.
A new important observation is that inhibition of the F→O step observed for oxidation of the fully reduced enzyme by O2 is much stronger than inhibition of the F→O transition induced by photoreduction of Compound F (Figure 3). Therefore, the mechanisms of the impaired F→O transitions in the N139L mutant oxidase may be different in the two types of experiments and are to be considered separately (Figure 8 A,B).
Figure 8. Different mechanisms of the F→O step observed with N139L oxidase during oxidation of the fully reduced enzyme and induced by photoreduction of Compound F.
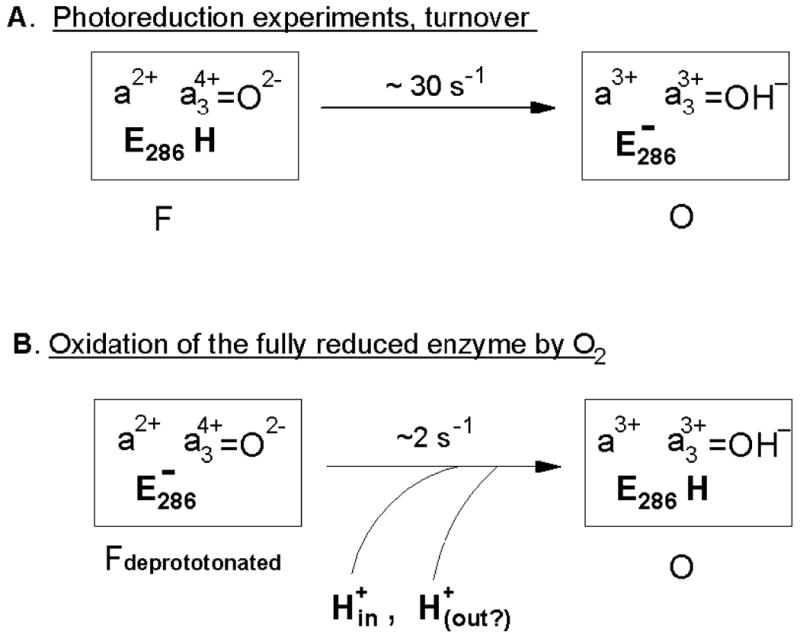
(A) In the photoreduction experiments and, presumably, during turnover, the inhibited F→O step is rate limited (ca. 30 s-1) by internal transfer of the chemical proton from the protonated E286-COOH to heme a3-bound oxene oxygen reduced to hydroxide state by an electron from ferrous heme a. The process is slower than in the wild type since E286-COO- anion formed at this step cannot be reprotonated rapidly in the mutant; accordingly, the driving force for protonation of the binuclear site is lower than in the case of proton delivery via E286 from the N-phase. (B) During oxidation of the fully reduced enzyme by oxygen, N139L mutant arrives at abnormal F state (Fdeprotonated) in which E286 is already deprotonated and cannot provide the chemical proton required for reduction of the oxene anion bound to heme a3. In this case, the step Fdeprotonated →O has to await for very slow (∼2 s-1) delivery of the external protons, that come possibly from the different sides of the membrane (see the text).
The F→O transition initiated by single-electron photoreduction of Compound F
The slowest step of electron transfer induced by single-electron photoreduction of F to O is characterized in N139L at pH 8 by τ ∼ 20 ms (Table 1). This result is in fair agreement with the ∼15-fold inhibition of the steady state cytochrome c oxidase activity of the N139L mutant but it is difficult to reconcile with the 200-300-fold inhibition of the F→O step observed during aerobic oxidation of the fully reduced enzyme. Conceivably, if the rate constant for the F→O step were 2 s-1 as found for oxidation of the fully reduced N139L enzyme by oxygen, it would not be possible for the enzyme to turn over with a rate of ∼30 s-1. The only straightforward inference at this point is that the behavior of the N139L enzyme in the F→O step in the photoreduction experiments is, in some critical way, more close to its behavior under steady state conditions, than oxidation of the fully reduced enzyme by oxygen.
Electrogenic phases observed with the N139L oxidase upon photoinjection of an electron into Compound F
The initial microsecond part of Δψ generation observed after photoinjection of a single electron into Compound F of cytochrome oxidase is associated with the e- transfer from CuA to heme a and is called the rapid electrogenic phase. In the bovine cytochrome c oxidase (45, 46) as well as in the wild type enzyme from R. sphaeroides (17, 41) or P. denitrificans (44, 60), the rapid phase is followed by two major electrogenic steps associated primarily with proton movements within the enzyme that accompany electron transfer from heme a to the heme a3/CuB active site. These two electrogenic steps are named the intermediate and slow electrogenic phases or protonic electrogenic phases I and II, respectively. Previous data-fitting protocols estimated the magnitude of the voltage generated during the slow phase to be about 3-fold larger than in the intermediate phase (17, 44, 45, 59). However, fitting of the data using a model of two consecutive, rather than two parallel protonic, electrogenic steps, shows that the intermediate and slow electrogenic phases have similar magnitudes (64, 66, 76). Hence, the intermediate and slow phases are likely to represent essentially the same proton transfer processes, namely, the two consecutive reprotonations of E286 via the D-channel (64, 76), with minor contributions from other coupled intraprotein charge movements. In each of the “PM”→F and F→O transitions, the first reprotonation occurs after the initially protonated E286-COOH donates its proton to the Pump Loading Site (protonic phase I). The second electrogenic phase (protonic phase II) corresponds essentially to reprotonation of E286, following transfer of the proton required for the oxygen chemistry from E286-COOH to the oxygen-reducing site.
In the N139D mutant, proton pumping is absent. Accordingly, only one of the two protonic electrogenic phases is observed, corresponding to the transfer of the “chemical” proton from the N-phase to the oxygen reducing site (41). The total voltage generated during the F→O transition in this mutant is, as expected, about half of the magnitude observed with the wild type oxidase (41) (Figure 5).
With the N139L mutant, the extent of the electrogenic proton movement is further dramatically decreased when the equivalent experiment is performed (Figure 5, Table 2). It is clear that the magnitudes of both of the major KCN-sensitive electrogenic proton transfer phases are suppressed in the mutant and, accordingly, that neither of the two reprotonations of E286 from the N-phase take place in the F→O transition of the mutant enzyme.
Origin of the residual electrogenic protonic phase in the N139L oxidase upon electron photoinjection
The very slow F→O transition during oxidation of the fully reduced D132N mutant by O2 (ca. 2 s-1, (61)) is coupled to the uptake of protons (70) and to an electrogenic phase with a large magnitude ((63, 77). Similar observations have been made for oxidation of the fully reduced D124N mutant oxidase from P. denitrificans (47, 62) Hence, the severe inhibition of the F→O step under these conditions can be naturally explained by the slow H+ uptake from the N-side to reprotonate E286 (or E278Pd). The same explanation may apply to the strongly inhibited F→O transition during oxidation of the fully reduced N139L studied in this work (Figure 8 B).
In contrast, the very low magnitude of the protonic electrogenic phase rules out such an explanation in case of the F→O transition induced in the N139L mutant by the single-electron photoreduction of Compound F (Figure 5). The magnitude of the residual protonic electrogenic phase accompanying the photoinduced F→O transition with the N139L mutant corresponds to about 20% of the protonic phase observed with the decoupled N139D mutant, i.e. to less than 15% of charge transfer across the entire membrane. Such a magnitude is in agreement with the electrogenicity expected for proton transfer from E286 to the binuclear site (66). Therefore, we tentatively assign the residual electrogenic protonic phase with τ of 25-40 ms, observed in the single electron photoinjection-induced F→O transition of the N139L mutant, primarily to the transfer of the “chemical” proton from the protonated E286 to the Fe4+=O2- species of heme a3 (Figures 8A, 10). In the simplest case, E286 will remain deprotonated following this proton transfer (Figure 8A), but it is possible that E286 could be partly or completely reprotonated by adjacent intrachannel proton donors, e.g. Zundel cation (30, 31, 78), should such a proton donor exist (32).
Figure 10. Electrogenic reactions coupled to single-electron photoreduction of Compound F to O in N139L mutant of cytochrome c oxidase.
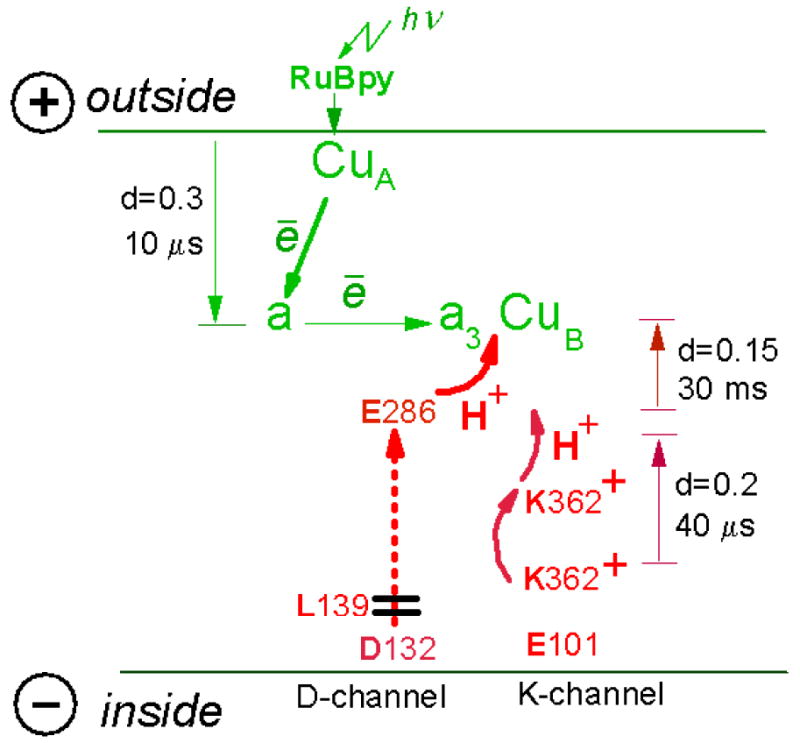
There are 3 electrogenic phases associated with single-electron photoreduction of compound F of N131L oxidase. First, there is 10 μs vectorial electron transfer from CuA to heme a across ca. 0.3 of the membrane dielectric thickness. Second, there is a residual KCN-sensitive protonic phase with τ∼20-30 ms matching roughly the slowest phase of electron transfer from heme a to the oxygen-reducing site; the phase corresponds to elementary charge transfer across ca. 0.15 of the membrane dielectric and is assigned to transfer of the “chemical” H+ from the protonated E286 to heme a34+=O2- intermediate. Third, there is a newly-resolved KCN-insensitive electrogenic phase with τ of ca. 40 μs and with a magnitude corresponding to ca. 2/3 of the 10 μs CuA → heme a electron transfer phase, or to ∼20% of transmembrane charge transfer. This phase is discerned in both pumping and non-pumping mutants of R.sphaeroides oxidase and is tentatively proposed to be triggered by displacement of the charged side chain of K362 in the K-channel (see the text for other possibilities). The magnitude of the phase corresponds to elementary charge transfer across ca. 0.2 of the membrane dielectric thickness; therefore it cannot be fully accounted for by K362 movement and should include some other charge(s) displacements triggered by K362 rotation (see the text).
Hence, in the photoreduction experiments and, supposedly, under steady-state turnover conditions, the N139L mutant oxidase is likely to reach the O state with a single proton deficiency (presumably, deprotonated E286) (Figure 8A). Refill of this deficiency (reprotonation of E286) under multiple turnover conditions may take place upon addition of the next electron to the oxidized enzyme, i.e. in the O→E transition of the next turnover. In this case it can involve the K-channel that is known to service proton delivery associated with the O →E transition (24, 59, 79). Alternatively, the missing proton may be taken up from the P-phase as proposed originally for D132A mutant by East-Lancing group (12, 53) to explain the “inversed respiratory control” in the liposome-reconstituted mutant enzyme. Actually, the two processes may compete with each other depending on availability of electrons and protons. It is interesting that in either case, the N139L mutant oxidase should be able to turn over under steady-state conditions without participation of its blocked D-channel at all.
Inhibition of the F→O step during oxidation of the fully reduced N139L
Oxidation of the fully reduced N139L mutant oxidase by oxygen is inhibited at the F→O step to a much greater extent than is the F→O transition induced by single-electron reduction of Compound F (Figure 3, Figure 8A,B). A likely scenario for the impaired oxidation of the fully reduced N139L oxidase by oxygen, taking into account relevant data on other oxidase mutants with the blocked D-channel (26, 47, 61, 62, 70, 77, 80), is illustrated by Figure 9. After several steps, an abnormal proton-deficient form of intermediate F is formed (Fdeprotonated) in which E286 is deprotonated, and the reaction halts because there is no internal H+ available for conversion of ferryl-oxo intermediate of heme a3 (a34+ =O2-) to the oxidized ferric-hydroxy state (a33+ - OH−) (Figure 8A). In contrast, in the F580 state of N139L obtained by treatment of the mutant enzyme by H2O2 or formed during the steady-state turnover, E286 is presumed to be protonated, so that the intermediate is ready for the F580→O step (Figure 8A).
Figure 9. Proposed mechanism for impaired oxidation of the fully reduced N139L oxidase by oxygen.
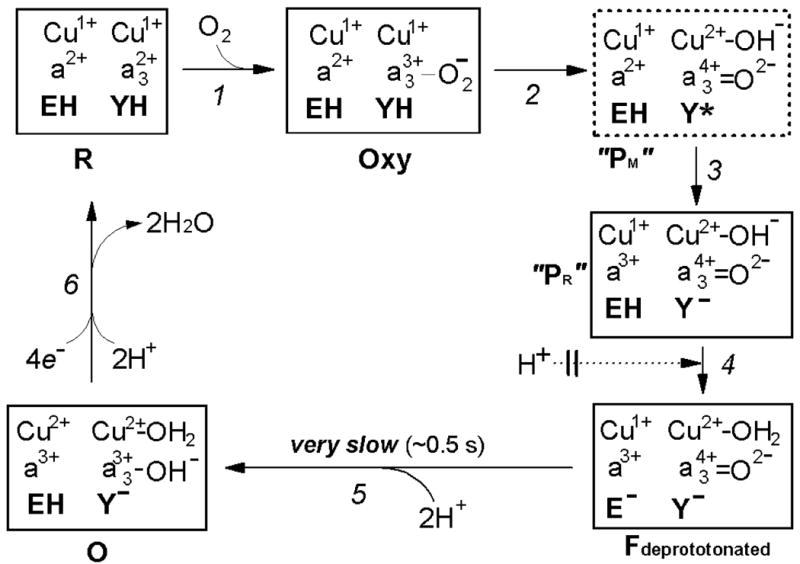
EH and YH denote the protonated glutamic acid E286 and tyrosine Y288, respectively. The transition of the oxycomplex of the fully reduced oxidase (Fe2+-O2 CuB1+) to the initial ferryl-oxene state (Fe4+=O2- Cub2+-OH-) during the reductive splitting of the O-O bond requires extraction of at least one proton from the protein by oxygen. The proton is most likely donated initially by Y288-OH, oxidized to the neutral radical Y288-O• at the O-O-bond breaking step (80, 89-91). In concordance with recent views (84, 85), we assume that the O-O-bond splitting mechanism may be essentially the same for oxidation of the fully reduced and the mixed-valence enzyme, and in either case it results initially in formation of a peroxidase Compound I-type intermediate of the binuclear site (Fe4+=O2- state of heme a3 + amino acid radical, Y288-O•). In this intermediate of cytochrome oxidase, the O-O bond is already broken (92), but it is still labeled here as “Peroxy” state (“PM”) solely to avoid discrepancy with the outdated traditional nomenclature in the literature (see (17, 22) for more consistent possible nomenclature of the oxidase compounds). Insofar, this intermediate has not been directly observed during the oxidation of the fully reduced COX and therefore is shown in a dashed line box. When the reaction starts from the fully reduced state of the oxidase with both CuA and heme a initially reduced, the neutral Y288• radical in the putative “PM” intermediate is rapidly reduced by heme a to tyrosine anion, Y, forming a peroxidase Compound II-type intermediate denoted as “PR” in cytochrome oxidase literature. This designation, although widely used, is misleading because the oxygen-reducing site in “PR” contains 1 reducing equivalent more than “PM” and, actually, its redox state corresponds to the next intermediate F580 analogous to Compound II of peroxidases. For the reasons not yet understood, the “PR” intermediate retains a strong absorption band of heme a3 at 607 nm, typical of the Compound I-type intermediate of cytochrome oxidase (“PM”), and hence may be denoted as F607. In the wild type enzyme, the F607 (“PR”) intermediate converts to the “normal” F580 state with a maximum at ∼580 nm upon H+ uptake by the enzyme. The proton converting F607 (“PR”) to F580 is believed to be provided by E286-COOH that is then rapidly reprotonated via the D-channel. Where exactly the proton goes, has not been established yet with certainity. Originally, it was believed that the proton goes to the Y288 anion. However, according to the recent data (26), Y288 is likely to remain deprotonated in F580 at pH >8. Alternatively, the H+ may be accepted by CuB-bound hydroxide (cf. ref. (85)) as depicted provisionally in Figure 9, or by some other group (e.g. water) in the surroundings of the oxygen reducing site. In the N139L or D132(N,A) mutants, in which the entrance to D-channel is blocked, the “chemical” proton can still be extracted from E286-COOH, allowing for transition to the F580 state, but subsequent rapid reprotonation of E286 is no longer possible. This conclusion is corroborated by the fact that there is no proton uptake coupled to the “PR”→F580 transition in the D132A mutant oxidase of R. sphaeroides (70); moreover, deprotonated state of E286 (E278Pd) has been directly demonstrated for D124N mutant oxidase from P. denitrificans by ATR-FTIR spectroscopy (62). Accordingly, cytochrome oxidase gets stuck at a specific intermediate denoted as Fdeprotonated (Figures 8, 9) that is 1 proton deficient relative to the normal F state. At this unusual state, the proton deficiency is shown to reside on E286 anion, but actually it may be shared among E286 –COOH/E286-COO-and [Y288-OH/Y288-O- ---CuB-H2O/OH-] proton acceptors. Subsequent reduction of Fdeprotonated to the oxidized state by the fourth electron stored within the equilibrating heme a/CuA pair is rate limited (k ∼ 2 s-1) by delivery of at least one, but probably two protons required to protonate the reduced oxygen at heme a3 and reprotonate E286. Only one of the 2 protons is delivered electrogenically from the N-phase (see the text).
The reductive conversion of Compound Fdeprotonated to the O state (Fe3+-OH−) requires two more protons – one to form the heme a33+ -bound hydroxide and the other to regenerate the protonated form of E286. Uptake of 2 protons coupled to the very slow F→O transition in D132A mutant oxidase was indeed demonstrated in (70). However, the magnitude of the slow electrogenic phase coupled to the F→O transition in either membrane-reconstituted D132N oxidase ((63, 77) and personal communication of P.Brzezinki) or in a homologous D124N mutant enzyme from P.denitrificans (74) and personal communication from M.Verkhovsky) corresponds to electrogenic uptake of only 1 proton. Therefore, if the data on the D132N (D124Pd) mutants apply to N139L, only one of the two protons required for the Fdeprotonated →O transition in N139L is delivered from the N-phase. The second proton may reach the active site from the P-phase (Figures 8B, 9; cf. refs. (53, 81)).
What is the reason for the extra proton deficiency during oxidation of the fully reduced N139L enzyme as compared to the photoreduction experiments or turnover conditions?
Comparing the scenarios, we conclude that during the oxidation of the fully reduced oxidase in which D-channel is blocked near the N-side orifice, the enzyme arrives at the Fdeprotonated state with 1 proton less then in case of the F580 state obtained with H2O2 treatment, or under turnover conditions. The photoreduction experiments and the experiments under multiple turnover conditions have in common the fact that intermediate F580 of cytochrome oxidase is formed by passing through the “PM” state as a major relatively long-lived intermediate, rather than through the “PR” state as in the case of oxidation of the fully reduced enzyme.
It is proposed that on a way from ferric-peroxy state (Compound 0) to intermediate F580 (Compound II) via the compound “PM” (Compound I), the oxygen-reducing center is able to recruit additional proton(s) relative to the reaction trajectory via Compound “PR”. The exra proton(s) could be borrowed from internal water/hydronium molecules around Y288 (25, 82, 83) to refill the H+ vacancy created in the binuclear site during the splitting of the O-O bond with subsequent reprotonation via the K-channel (17, 22) that should occur before closing of the K-channel triggered by scission of the O-O bond and formation of Compound I (“PM”) (24). The lifetime of the open state of the K-channel after scission of the O-O bond may be related to the lifetime of cytochrome oxidase Compound I (“PM”) but in any case, the channel closes before Compound II-type intermediate (“PR” or F580 state) is formed (17, 19). During the oxidation of the fully reduced COX (Figure 9), the Compound I intermediate (“PM”) is very short-lived (84, 85), so that the ferrous-oxy complex of COX (“Compound A”) proceeds in effect directly to Compound II-type intermediate “PR”. Accordingly, the K-channel closes untimely, before reprotonation of the group(s) participating in the proton-dependent scission of the O-O bond takes place.
Origin of a novel KCN-insensitive microsecond electrogenic phase. Rapid internal charge transfer may accompany electron transfer from CuA to heme a
The rapid (microsecond) phase of the electrogenic response with the N139L mutant can be analyzed more accurately than is usually possible due to the very low contribution of subsequent protonic phases. This circumstance has revealed that the microsecond phase in N139L appears to be a biphasic process. A very similar biphasic pattern was confirmed in several other cases, including the KCN-inhibited wild type enzyme (Figure 6 E, F, Table 2) and E286Q mutant (data not included). At the same time no evidence for biphasic pattern of the rapid electrogenic phase could be found for bovine oxidase (S.A. Siletsky, unpublished data; ref. (64)).
It is clear that both components of the rapid electrogenic phase must be associated with electron transfer from CuA to heme a. However, the electron transfer process itself, measured optically, is monophasic, as reported from several laboratories (refs. (65, 67), Figure 7 in this work). The first of the two microsecond phases discerned in the electrometric response is, therefore, assigned to charge separation resulting directly from the vectorial electron transfer from CuA to heme a with τ of ∼10 μs (Figure 10). The second microsecond phase with τ of about 40 μs, may then originate from internal proton or other charge transfer process triggered by or coupled to the reduction of heme a. The amplitude of the second KCN-insensitive electrogenic phase corresponds to a charge translocation across about 20% of the membrane dielectric thickness.
The finding of an additional component to the rapid electrogenic phase in the bacterial oxidase can explain why in the wild type oxidases from either R. sphaeroides (17, 41) or P. denitrificans (44) the contribution of the rapid electrogenic phase to the overall response is consistently about 30%, whereas it is close to 20% with the bovine enzyme (45, 46). It also resolves a controversy pointed out at Sigrid Juselius Foundation International Symposium “Currents of Life” in Helsinki (2000) and considered later on in (60). If the entire microsecond phase in the bacterial oxidases corresponds to electron transfer from CuA to heme a across 1/3 (or even 40% (66)), of the membrane dielectric, than the overall charge translocation coupled to the F→O step attains a value close to 1 per electron (Table 2, column “earlier works”), which is much less than 2 charges per electron expected for the transition. However, if it is only the 10 μs component of the rapid electrogenic phase that corresponds to the vectorial e- transfer from CuA to heme a, then the overall electrogenicity of the F→O transition in the wild type enzyme becomes very close to translocation of 2 charges per electron across the membrane (Table 2, column “this work”).
The physical process underlying the 40 μs electrogenic phase remains to be identified. There are at least 4 reasonable specific possibilities:
intraprotein proton displacement within the D-channel “below” E286 (e.g., polarization of the water wire inside the channel (17);
charge displacement along the proton conducting water chain from E286 to the “pump loading site”, e.g., polarization of this water chain coupled to heme a reduction, as proposed in (86);
proton release from the cluster [−OH-Mg2- (II-E254)-H3O+] to the P-phase triggered by heme a reduction by CuA, as proposed recently by M. Sharpe et al. (78, 87) and corroborated by molecular dynamics simulations in (88);
Explanation (1) and to a lesser extent (2), may be compromised by the fact that all of the mutants in the D-channel for which the data of sufficient quality are available to us (D132N, E286D, E286Q, N139D, N139L) reveal the same biphasic pattern of the rapid phase. Nevertheless, it is worthy noting that the magnitude and rate constant of the putative 40 μs phase are not that different from those of the ∼70 μs electrogenic phase resolved during oxidation of the fully reduced P.denitrificans oxidase and assigned to E278 → “pump loading site” proton transfer coupled to “A” →“PR” transition (74).
Possibility (3) based on a proposal by M. A. Sharpe et al (78, 87) supported by a recent work (88) would be in ideal agreement with the size of the effect (40% of the rapid phase) as well as with the insensitivity of the phase to heme a3 binding with cyanide. However, the proposed release of H+ from the Mg2+ -(II-Glu254) -bound water to the P-phase coupled to heme a reduction is expected to occur in both bovine and bacterial oxidases (87, 88), whereas the additional component of the rapid electrogenic phase appears to be absent from the bovine oxidase.
Internal charge displacement within the K-channel coupled to reduction of heme a has been demonstrated experimentally (68), hence possibility (4) can be invoked to explain the 40 μs component of the rapid electrogenic phase (Figure 10). This charge displacement would likely include rotation of the charged K362 side chain in the direction of the binuclear site. It is noted that such a rotation can account for only part of the magnitude of the 40μs phase, however this event could trigger additional proton displacement(s) around the binuclear site through electrostatic interactions with the Y-288/CuB domain (85). The lack of discernible H/D-solvent kinetic isotope effect of the rapid electrogenic phase (41) would be in excellent agreement with K362 side chain movement limiting the rate of the process. Therefore, we assume provisionally, that (i) the novel 40 μs component of the rapid electrogenic phase in the bacterial oxidase is associated with electrogenic swinging of K362 charged side-chain within the K-channel, and that (ii) this process does not take place during the rapid electrogenic phase in the bovine oxidase.
Supplementary Material
Acknowledgments
We are grateful to Dr. M.Verkhovsky from Helsinki University and Dr. P.Brzezinski from University of Stockholm for information of the details of their studies on D124N and D132N mutants of cytochrome oxidase.
†This research was supported by the National Institutes of Health HL16101 (to RBG); the U.S. Civilian Research and Development Foundation Cooperative Grants Program #2836 (to RBG and AAK); the Russian Fund for Basic Research Grants 04-4-48856, 06-04-48608 and 09-04-00140 (to SAS), and the Howard Hughes Medical Institute International Scholar Award 55005615 (AAK).
Footnotes
Abbreviations: COX - cytochrome c oxidase; COVs - cytochrome oxidase vesicles; WT, wild type; O, R2, R4 - the oxidized, 2e- reduced and 4e- reduced forms of COX; PM, PR, F - ferryl-oxo intermediates of heme a3 in the COX catalytic cycle, corresponding to Compounds I (PM) and II (PR, F) of peroxidases. P-phase, N-phase, the positively and negatively charged aqueous phases, separated by the coupling membrane. RuBpy, tris-bipyridyl complex of ruthenium (II).
Supporting Information Available. It contains experimental Figures S1-S5 with pertinent comments. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ferguson-Miller S, Babcock GT. Heme/copper terminal oxidases. Chem Rev. 1996;7:2889–2907. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 2.Wikström M. Cytochrome c oxidase: 25 years of the elusive proton pump. Biochim Biophys Acta. 2004;1655:241–247. doi: 10.1016/j.bbabio.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 3.Belevich I, Verkhovsky MI. Molecular mechanism of proton translocation by cytochrome c oxidase. Antioxidants and Redox Signaling. 2008;10:1–29. doi: 10.1089/ars.2007.1705. [DOI] [PubMed] [Google Scholar]
- 4.Iwata S, Ostermeier C, Ludwig B, Michel H. Structure at 2.8 Å resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature. 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 5.Tsukihara T, Aoyama H, Yamashita E, Takashi T, Yamaguichi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 Å. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 6.Mitchell P. Chemiosmotic coupling and energy transduction. Glynn Research Ltd.; Bodmin: 1968. [Google Scholar]
- 7.Wikström M. Proton pump coupled to cytochrome c oxidase in mitochondria. Nature. 1977;266:271–273. doi: 10.1038/266271a0. [DOI] [PubMed] [Google Scholar]
- 8.Konstantinov AA. Role of protons in the operational mechanism of the 3rd conjugation point of the respiratory chain of mitochondria: cytochrome oxidase as an electron-proton generator of the membrane potential. Doklady Akad Nauk SSSR. 1977;237:713–716. [PubMed] [Google Scholar]
- 9.Artzatbanov VY, Konstantinov AA, Skulachev VP. Involvement of intramitochondrial protons in redox reactions of cytochrome a. FEBS Lett. 1978;87:180–185. doi: 10.1016/0014-5793(78)80327-5. [DOI] [PubMed] [Google Scholar]
- 10.Soulimane T, Buse G, Bourenkov GB, Bartunik HD, Huber R, Than ME. Structure and mechanism of the aberrant ba3-cytochrome c oxidase from Thermus thermophilus. EMBO J. 2000;19:1766–1776. doi: 10.1093/emboj/19.8.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Svensson-Ek M, Abramson J, Larsson G, Törnroth S, Brzezinski P, Iwata S. The x-ray crystal structures of wild-type and EQ(I-286) mutant cytochrome c oxidases from Rhodobacter sphaeroides. J Mol Biol. 2002;321:329–339. doi: 10.1016/s0022-2836(02)00619-8. [DOI] [PubMed] [Google Scholar]
- 12.Hosler JP, Ferguson-Miller S, Mills DA. Energy transduction: Proton transfer through the respiratory complexes. Annu Rev Biochem. 2006;75:165–187. doi: 10.1146/annurev.biochem.75.062003.101730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brzezinski P, Gennis RB. Cytochrome c oxidase: Exciting progress and remaining mysteries. J Bioenerg Biomembr. 2008;40:521–531. doi: 10.1007/s10863-008-9181-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee HM, Das TK, Rousseau DL, Mills D, Fergusson-Miller S, Gennis R. Mutations in the putative H-channel in the cytochrome c oxidase from Rhodobacter sphaeroides show that this channel is not important for proton conduction but reveals modulation of the properties of heme a. Biochemistry. 2000;39:2989–2996. doi: 10.1021/bi9924821. [DOI] [PubMed] [Google Scholar]
- 15.Tsukihara T, Shimokata K, Katayama Y, Shimada H, Muramoto K, Aoyama H, Mochizuki M, Shinzawa-Itoh K, Yamashita E, Yao M, Ishimura Y, Yoshikawa S. The low-spin heme of cytochrome c oxidase as the driving element of the proton-pumping process. Proc Natl Acad Sci USA. 2003;100:15304–15309. doi: 10.1073/pnas.2635097100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimokata K, Katayama Y, Murayama H, Suematsu M, Tsukihara T, Muramoto K, Aoyama H, Yoshikawa S, Shimada H. The proton pumping pathway of bovine heart cytochrome c oxidase. Proc Natl Acad Sci U S A. 2007;104:4200–4205. doi: 10.1073/pnas.0611627104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Konstantinov AA, Siletsky S, Mitchell D, Kaulen A, Gennis RB. The roles of the two proton input channels in cytochrome c oxidase from Rhodobacter sphaeroides probed by the effects of site-directed mutations on time resolved electrogenic intraprotein proton transfer. Proc Natl Acad Sci USA. 1997;94:9085–9090. doi: 10.1073/pnas.94.17.9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vygodina TV, Pecoraro C, Mitchell D, Gennis R, Konstantinov AA. The mechanism of inhibition of electron transfer by amino acid replacement K362M in a proton channel of Rhodobacter sphaeroides cytochrome c oxidase. Biochemistry. 1998;37:3053–3061. doi: 10.1021/bi971876u. [DOI] [PubMed] [Google Scholar]
- 19.Smirnova I, Adelroth P, Gennis RB, Brzezinski P. Aspartate-132 in cytochrome c oxidase from Rhodobacter sphaeroides is involved in a two-step proton transfer during oxo-ferryl formation. Biochemistry. 1999;38:6826–6833. doi: 10.1021/bi982865j. [DOI] [PubMed] [Google Scholar]
- 20.Moody AJ. “As prepared” forms of fully oxidised haem/Cu terminal oxidases. Biochim Biophys Acta. 1996;1276:6–20. doi: 10.1016/0005-2728(96)00035-7. [DOI] [PubMed] [Google Scholar]
- 21.Bloch D, Belevich I, Jasaitis A, Ribacka C, Puustinen A, Verkhovsky MI, Wikström M. The catalytic cycle of cytochrome c oxidase is not the sum of its two halves. Proc Natl Acad Sci USA. 2004;101:529–533. doi: 10.1073/pnas.0306036101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Konstantinov A. Cytochrome c oxidase as a proton-pumping peroxidase: Reaction cycle and electrogenic mechanism. J Bioenerg Biomembr. 1998;30:121–130. doi: 10.1023/a:1020571930850. [DOI] [PubMed] [Google Scholar]
- 23.Blomberg M, Siegbahn PEM, Babcock GT, Wikström M. O-O bond splitting mechanism in cytochrome oxidase. J Inorg Chem. 2000;80:1238–1243. doi: 10.1016/s0162-0134(00)00080-5. [DOI] [PubMed] [Google Scholar]
- 24.Pecoraro C, Gennis RB, Vygodina TV, Konstantinov AA. Role of the K-channel in the pH-dependence of the reaction of cytochrome c oxidase with hydrogen peroxide. Biochemistry. 2001;40:9695–9708. doi: 10.1021/bi010115v. [DOI] [PubMed] [Google Scholar]
- 25.Yoshioka S, Kawai H, Yamaguchi K. Theoretical study of role of H2O molecule on initial stage of reduction of O2 molecule in active site of cytochrome c oxidase. Chemical Physics Letters. 2003;374:45–52. [Google Scholar]
- 26.Gorbikova EA, Wikstrom M, Verkhovsky MI. The protonation state of the cross-linked tyrosine during the catalytic cycle of cytochrome c oxidase. J Biol Chem. 2008;283:34907–34912. doi: 10.1074/jbc.M803511200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cukier RI. Theory and simulation of proton-coupled electron transfer, hydrogen-atom transfer, and proton translocation in proteins. Biochim Biophys Acta. 2004;1655:37–44. doi: 10.1016/j.bbabio.2003.06.011. [DOI] [PubMed] [Google Scholar]
- 28.Wikström M, Ribacka C, Molin M, Laakkonen L, Verkhovsky MI, Puustinen A. Gating of proton and water transfer in the respiratory enzyme cytochrome c oxidase. Proc Natl Acad Sci USA. 2005;102:10478–10481. doi: 10.1073/pnas.0502873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olsson MH, Warshel A. Monte Carlo simulations of proton pumps: On the working principles of the biological valve that controls proton pumping in cytochrome c oxidase. Proc Natl Acad Sci U S A. 2006;103:6500–5. doi: 10.1073/pnas.0510860103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu J, Voth GA. Free energy profiles for H+ conduction in the D-pathway of cytochrome c oxidase: A study of the wild type and N98D mutant enzymes. Biochim Biophys Acta. 2006;1757:852–9. doi: 10.1016/j.bbabio.2006.05.028. [DOI] [PubMed] [Google Scholar]
- 31.Xu J, Sharpe MA, Qin L, Ferguson-Miller S, Voth GA. Storage of an excess proton in the hydrogen-bonded network of the D-pathway of cytochrome c oxidase: Identification of a protonated water cluster. J Am Chem Soc. 2007;129:2910–2913. doi: 10.1021/ja067360s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henry RM, Yu CH, Rodinger T, Pomes R. Functional hydration and conformational gating of proton uptake in cytochrome c oxidase. J Mol Biol. 2009;387:1165–1185. doi: 10.1016/j.jmb.2009.02.042. [DOI] [PubMed] [Google Scholar]
- 33.Mitchell DM, Fetter JR, Mills DA, Adelroth P, Pressler MA, Kim Y, Aasa R, Brzezinski P, Malmstrom BG, Alben JO, Babcock GT, Ferguson-Miller S, Gennis RB. Site-directed mutagenesis of residues lining a putative proton transfer pathway in cytochrome c oxidase from Rhodobacter sphaeroides. Biochemistry. 1996;35:13089–13093. doi: 10.1021/bi961416l. [DOI] [PubMed] [Google Scholar]
- 34.Pfitzner U, Odenwald A, Ostermann T, Weingard L, Ludwig B, Richter OM. Cytochrome c oxidase (heme aa3) from Paracoccus denitrificans: Analysis of mutations in putative proton channels of subunit I. J Bioenerg Biomembr. 1998;30:89–97. doi: 10.1023/a:1020515713103. [DOI] [PubMed] [Google Scholar]
- 35.Pfitzner U, Hoffmeier K, Harrenga A, Kannt A, Michel H, Bamberg E, Richter OM, Ludwig B. Tracing the D-pathway in reconstituted site-directed mutants of cytochrome c oxidase from Paracoccus denitrificans. Biochemistry. 2000;39:6756–6762. doi: 10.1021/bi992235x. [DOI] [PubMed] [Google Scholar]
- 36.Thomas JW, Puustinen A, Alben JO, Gennis RB, Wikström M. Substitution of asparagine for aspartate-135 in subunit I of the cytochrome bo ubiquinol oxidase of Escherichia coli eliminates proton-pumping activity. Biochemistry. 1993;32:10923–10928. doi: 10.1021/bi00091a048. [DOI] [PubMed] [Google Scholar]
- 37.Fetter JR, Qian J, Shapleigh J, Thomas JW, Garcia-Horsman A, Schmidt E, Hosler J, Babcock GT, Gennis RB, Ferguson-Miller S. Possible proton relay pathways in cytochrome c oxidase. Proc Natl Acad Sci USA. 1995;92:1604–1608. doi: 10.1073/pnas.92.5.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pawate AS, Morgan J, Namslauer A, Mills D, Brzezinski P, Ferguson-Miller S, Gennis RB. A mutation in subunit I of cytochrome oxidase from Rhodobacter sphaeroides results in an increase in steady-state activity but completely eliminates proton pumping. Biochemistry. 2002;41:13417–13423. doi: 10.1021/bi026582+. [DOI] [PubMed] [Google Scholar]
- 39.Han D, Namslauer A, Pawate AS, Morgan JE, Nagy S, Vakkasoglu AS, Brzezinski P, Gennis RB. Replacing Asn207 by aspartate at the neck of the D channel in the aa3-type cytochrome c oxidase from Rhodobacter sphaeroides results in decoupling the proton pump. Biochemistry. 2006;45:14064–14074. doi: 10.1021/bi061465q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Namslauer A, Pawate A, Gennis R, Brzezinski P. Redox-coupled proton translocation in biological systems: Proton shuttling in cytochrome c oxidase. Proc Natl Acad Sci USA. 2003;100:15543–15547. doi: 10.1073/pnas.2432106100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Siletsky SA, Pawate AS, Weiss K, Gennis RB, Konstantinov AA. Transmembrane charge separation during the ferryl-oxo-> oxidized transition in a non-pumping mutant of cytochrome c oxidase. J Biol Chem. 2004;279:52558–52565. doi: 10.1074/jbc.M407549200. [DOI] [PubMed] [Google Scholar]
- 42.Branden G, Pawate AS, Gennis RB, Brzezinski P. Controlled uncoupling and recoupling of proton pumping in cytochrome c oxidase. Proc Natl Acad Sci U S A. 2006;103:317–322. doi: 10.1073/pnas.0507734103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Olsson MHM, Sharma PK, Warshel A. Simulating redox coupled proton transfer in cytochrome c oxidase: Looking for the proton bottleneck. FEBS Lett. 2005;579:2026–2034. doi: 10.1016/j.febslet.2005.02.051. [DOI] [PubMed] [Google Scholar]
- 44.Ruitenberg M, Kannt A, Bamberg E, Ludwig B, Michel H, Fendler K. Single-electron reduction of the oxidized state is coupled to proton uptake via the K pathway in Paracoccus denitrificans cytochrome c oxidase. Proc Natl Acad Sci USA. 2000;97:4632–4636. doi: 10.1073/pnas.080079097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zaslavsky D, Kaulen A, Smirnova IA, Vygodina TV, Konstantinov AA. Flash-induced membrane potential generation by cytochrome c oxidase. FEBS Lett. 1993;336:389–393. doi: 10.1016/0014-5793(93)80843-j. [DOI] [PubMed] [Google Scholar]
- 46.Siletsky S, Kaulen AD, Konstantinov AA. Resolution of electrogenic steps coupled to conversion of cytochrome c oxidase from the peroxy to the ferryl-oxo state. Biochemistry. 1999;38:4853–4861. doi: 10.1021/bi982614a. [DOI] [PubMed] [Google Scholar]
- 47.Belevich I, Verkhovsky MI, Wikstrom M. Proton-coupled electron transfer drives the proton pump of cytochrome c oxidase. Nature. 2006;440:829–832. doi: 10.1038/nature04619. [DOI] [PubMed] [Google Scholar]
- 48.Mitchell DM, Gennis RB. Rapid purification of wildtype and mutant cytochrome c oxidase from Rhodobacter sphaeroides by Ni2+-NTA affinity chromatography. FEBS Lett. 1995;368:148–150. doi: 10.1016/0014-5793(95)00626-k. [DOI] [PubMed] [Google Scholar]
- 49.Drachev LA, Jasaitis AA, Kaulen AD, Kondrashin AA, Liberman EA, Nemecek IB, Ostroumov SA, Semenov A, Skulachev VP. Direct measurement of electric current generation by cytochrome oxidase, H+-ATPase and bacteriorhodopsin. Nature. 1974;249:321–324. doi: 10.1038/249321a0. [DOI] [PubMed] [Google Scholar]
- 50.Provincer SW, Vogel RH. Regularization techniques for inverse problems in molecular biology. Prog Sci Comp. 1983;2:304–319. [Google Scholar]
- 51.Morgan JE, Verkhovsky MI, Puustinen A, Wikström M. Identification of a “peroxy” intermediate in cytochrome bo3 of Escherichia coli. Biochemistry. 1995;34:15633–15637. doi: 10.1021/bi00048a005. [DOI] [PubMed] [Google Scholar]
- 52.Provencher SW. A Fourier method for the analysis of exponential decay curves. Bio phys J. 1976;16:27–41. doi: 10.1016/S0006-3495(76)85660-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fetter JR, Sharpe MA, Qian J, Mills DA, Ferguson-Miller S, Nicholls P. Fatty acids stimulate activity and restore respiratory control in a proton channel mutant of cytochrome c oxidase. FEBS Lett. 1996;393:155–160. doi: 10.1016/0014-5793(96)00874-5. [DOI] [PubMed] [Google Scholar]
- 54.Mills DA, Tan Z, Ferguson-Miller S, Hosler J. A role for subunit III in proton update into the D pathway and possible proton exit pathway in Rhodobacter sphaeroides cytochrome c oxidase. Biochemistry. 2003;42:7410–7417. doi: 10.1021/bi0341307. [DOI] [PubMed] [Google Scholar]
- 55.Han D, Morgan JE, Gennis RB. G204D, a mutation that blocks the proton-conducting D-channel of the aa3-type cytochrome c oxidase from Rhodobacter sphaeroides. Biochemistry. 2005;44:12767–74. doi: 10.1021/bi051141m. [DOI] [PubMed] [Google Scholar]
- 56.Branden G, Gennis RB, Brzezinski P. Transmembrane proton translocation by cytochrome c oxidase. Biochim Biophys Acta. 2006;1757:1052–1063. doi: 10.1016/j.bbabio.2006.05.020. [DOI] [PubMed] [Google Scholar]
- 57.Nilsson T. Photoinduced electron transfer from tris(2,2′-bipyridyl)ruthenium to cytochrome c oxidase. Proc Natl Acad Sci USA. 1992;89:6497–6501. doi: 10.1073/pnas.89.14.6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vygodina TV, Konstantinov AA. H2O2-induced conversion of cytochrome c oxidase peroxy complex to oxoferryl state. Ann NY Acad Sci. 1988;550:124–138. doi: 10.1111/j.1749-6632.1988.tb35329.x. [DOI] [PubMed] [Google Scholar]
- 59.Ruitenberg M, Kannt A, Bamberg E, Fendler K, Michel H. Reduction of cytochrome c oxidase by a second electron leads to proton translocation. Nature. 2002;417:99–102. doi: 10.1038/417099a. [DOI] [PubMed] [Google Scholar]
- 60.Verkhovsky MI, Belevich I, Bloch DA, Wikström M. Elementary steps of proton translocation in the catalytic cycle of cytochrome oxidase. Biochim Biophys Acta. 2006;1757:401–407. doi: 10.1016/j.bbabio.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 61.Salomonsson L, Branden G, Brzezinski P. Deuterium isotope effect of proton pumping in cytochrome c oxidase. Biochim Biophys Acta. 2008;1777:343–350. doi: 10.1016/j.bbabio.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 62.Gorbikova EA, Belevich NP, Wikström M, Verkhovsky MI. Time-resolved ATR-FTIR spectroscopy of the oxygen reaction in the D124N mutant of cytochrome c oxidase from Paracoccus denitrificans. Biochemistry. 2007;46:13141–13148. doi: 10.1021/bi701614w. [DOI] [PubMed] [Google Scholar]
- 63.Lepp H, Brzezinski P. Internal charge transfer in cytochrome c oxidase at a limited proton supply: Proton pumping ceases at high pH. Biochim Biophys Acta. 2009;1790:552–557. doi: 10.1016/j.bbagen.2009.03.023. [DOI] [PubMed] [Google Scholar]
- 64.Siletsky SA, Han D, Brand S, Morgan JE, Fabian M, Geren L, Millett F, Durham B, Konstantinov AA, Gennis RB. Single-electron photoreduction of the Pm intermediate of cytochrome c oxidase. Biochim Biophys Acta. 2006;1757:1122–1132. doi: 10.1016/j.bbabio.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 65.Belevich I, Bloch DA, Wikstrom M, Verkhovsky MI. Exploring the proton pump mechanism of cytochrome c oxidase in real time. Proc Natl Acad Sci U S A. 2007;104:2685–2690. doi: 10.1073/pnas.0608794104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sugitani R, Medvedev ES, Stuchebrukhov AA. Theoretical and computational analysis of the membrane potential generated by cytochrome c oxidase upon single electron injection into the enzyme. Biochim Biophys Acta. 2008;1777:1129–39. doi: 10.1016/j.bbabio.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zaslavsky D, Sadoski RC, Wang K, Durham B, Gennis RB, Millett F. Single electron reduction of cytochrome c oxidase compound F: Resolution of partial steps by transient spectroscopy. Biochemistry. 1998;37:14910–14916. doi: 10.1021/bi981490z. [DOI] [PubMed] [Google Scholar]
- 68.Lepp H, Salomonsson L, Zhu JP, Gennis RB, Brzezinski P. Impaired proton pumping in cytochrome c oxidase upon structural alteration of the D pathway. Biochim Biophys Acta. 2008;1777:897–903. doi: 10.1016/j.bbabio.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Durr KL, Koepke J, Hellwig P, Muller H, Angerer H, Peng G, Olkhova E, Richter OM, Ludwig B, Michel H. A D-pathway mutation decouples the Paracoccus denitrificans cytochrome c oxidase by altering the side-chain orientation of a distant conserved glutamate. J Mol Biol. 2008;384:865–877. doi: 10.1016/j.jmb.2008.09.074. [DOI] [PubMed] [Google Scholar]
- 70.Adelroth P, Hosler J. Surface proton donors for the D-pathway of cytochrome c oxidase in the absence of subunit III. Biochemistry. 2006;45:8308–8318. doi: 10.1021/bi0605843. [DOI] [PubMed] [Google Scholar]
- 71.Olkhova E, Helms V, Michel H. Titration behavior of residues at the entrance of the D-pathway of cytochrome c oxidase from Paracoccus denitrificans investigated by continuum electrostatic calculations. Biophys J. 2005;89:2324–31. doi: 10.1529/biophysj.105.062091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Popovic DM, Stuchebryukhov AA. Proton pumping mechanism and catalytic cycle of cytochrome c oxidase: Coulomb pump model with kinetic gating. FEBS Lett. 2004;566:126–130. doi: 10.1016/j.febslet.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 73.Xu J, Voth GA. Computer simulation of explicit proton translocation in cytochrome c oxidase: The D-pathway. Proc Natl Acad Sci USA. 2005;102:6795–6800. doi: 10.1073/pnas.0408117102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Belevich I, Tuukkanen A, Wikström M, Verkhovsky MI. Proton-coupled electron equilibrium in soluble and membrane-bound cytochrome c oxidase from Paracoccus denitrificans. Biochemistry. 2006;45:4000–4006. doi: 10.1021/bi052458p. [DOI] [PubMed] [Google Scholar]
- 75.Qin L, Hiser C, Mulichak A, Garavito RM, Ferguson-Miller S. Identification of conserved lipid/detergent-binding sites in a high-resolution structure of the membrane protein cytochrome c oxidase. Proc Natl Acad Sci U S A. 2006;103:16117–16122. doi: 10.1073/pnas.0606149103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Medvedev DM, Medvedev ES, Kotelnikov AI, Stuchebrukhov AA. Analysis of the kinetics of the membrane potential generated by cytochrome c oxidase upon single electron injection. Biochim Biophys Acta. 2005;1710:47–56. doi: 10.1016/j.bbabio.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 77.Lepp H, Svahn E, Faxen K, Brzezinski P. Charge transfer in the K proton pathway linked to electron transfer to the catalytic site in cytochrome c oxidase. Biochemistry. 2008;47:4929–4935. doi: 10.1021/bi7024707. [DOI] [PubMed] [Google Scholar]
- 78.Sharpe MA, Ferguson-Miller S. A chemically explicit model for the mechanism of proton pumping in heme-copper oxidases. J Bioenerg Biomembr. 2008;40:541–549. doi: 10.1007/s10863-008-9182-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Verkhovsky MI, Tuukkanen A, Backgren C, Puustinen A, Wikström M. Charge translocation coupled to electron injection into oxidized cytochrome c oxidase from Paracoccus denitrificans. Biochemistry. 2001;40:7077–7083. doi: 10.1021/bi010030u. [DOI] [PubMed] [Google Scholar]
- 80.Gorbikova EA, Belevich I, Wikstrom M, Verkhovsky MI. The proton donor for O-O bond scission by cytochrome c oxidase. Proc Natl Acad Sci USA. 2008;105:10733–10737. doi: 10.1073/pnas.0802512105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Namslauer A, Lepp H, Branden M, Jasaitis A, Verkhovsky MI, Brzezinski P. Plasticity of proton pathway structure and water coordination in cytochrome c oxidase. J Biol Chem. 2007;282:15148–58. doi: 10.1074/jbc.M700348200. [DOI] [PubMed] [Google Scholar]
- 82.Siegbahn PE, Blomberg MR. Proton pumping mechanism in cytochrome c oxidase. J Phys Chem. 2008;112:12772–12780. doi: 10.1021/jp801635c. [DOI] [PubMed] [Google Scholar]
- 83.Qin L, Liu J, Mills DA, Proshlyakov DA, Hiser C, Ferguson-Miller S. Redox dependent conformational changes in cytochrome c oxidase suggest a gating mechanism for proton uptake. Biochemistry. 2009;48:5121–5130. doi: 10.1021/bi9001387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wiertz FGM, Richter OMH, Ludwig B, de Vries S. Kinetic resolution of a tryptophan-radical intermediate in the reaction cycle of Paracoccus denitrificans cytochrome c oxidase. J Biol Chem. 2007;282:31580–31591. doi: 10.1074/jbc.M705520200. [DOI] [PubMed] [Google Scholar]
- 85.Kaila VRI, Johansson MP, Sundholm D, Laakkonen L, Wikström M. The chemistry of the CuB site in cytochrome c oxidase and the importance of its unique His–Tyr bond. Biochim Biophys Acta. 2009;1787:221–233. doi: 10.1016/j.bbabio.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 86.Wikström M, Verkhovsky MI, Hummer G. Water-gated mechanism of proton translocation by cytochrome c oxidase. Biochim Biophys Acta. 2003;45238:1–5. doi: 10.1016/s0005-2728(03)00041-0. [DOI] [PubMed] [Google Scholar]
- 87.Sharpe MA, Krzyaniak MD, Xu S, McCracken J, Ferguson-Miller S. EPR evidence of cyanide binding to the Mn(Mg) center of cytochrome c oxidase: Support for Cua-Mg involvement in proton pumping. Biochemistry. 2009;48:328–335. doi: 10.1021/bi801391r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sugitani R, Stuchebrukhov AA. Molecular dynamics simulation of water in cytochrome c oxidase reveals two water exit pathways and the mechanism of transport. Biochim Biophys Acta. 2009;1787:1140–1150. doi: 10.1016/j.bbabio.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Michel H. Cytochrome c oxidase: Catalytic cycle and mechanism of proton pumping - a discussion. Biochemistry. 1999;38:15129–15140. doi: 10.1021/bi9910934. [DOI] [PubMed] [Google Scholar]
- 90.Proshlyakov DA, Pressler MA, Babcock GT. Dioxygen activation and bond cleavage by mixed-valence cytochrome c oxidase. Proc Natl Acad Sci USA. 1998;95:8020–8025. doi: 10.1073/pnas.95.14.8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Proshlyakov DA, Pressler MA, DeMaso C, Leykam JF, DeWitt DL, Babcock GL. Oxygen activation and reduction in respiration: Involvement of redox-active tyrosine 244. Science. 2000;290:1588–1591. doi: 10.1126/science.290.5496.1588. [DOI] [PubMed] [Google Scholar]
- 92.Fabian M, Wong WW, Gennis RB, Palmer G. Mass spectrometric determination of dioxygen bond splitting in the “peroxy” intermediate of cytochrome c oxidase. Proc Natl Acad Sci USA. 1999;96:13114–13117. doi: 10.1073/pnas.96.23.13114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gopta OA, Feniouk BA, Junge W, Mulkidjanian AY. The cytochrome bc1 complex of Rhodobacter capsulatus: Ubiquinol oxidation in a dimeric Q-cycle? FEBS Lett. 1998;431:291–296. doi: 10.1016/s0014-5793(98)00768-6. [DOI] [PubMed] [Google Scholar]
- 94.Mamedov MD, Tyunyatkina AA, Siletsky SA, Semenov AY. Voltage changes involving photosystem II quinone-iron complex turnover. Eur Biophys J. 2006;35:647–654. doi: 10.1007/s00249-006-0069-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.