

Abstract
A concise synthesis of the ERK inhibitor FR180204 has been developed. The synthesis consists of six operationally simple steps and can be utilized to make multi-gram quantities of FR180204.
FR180204 is a potent and selective ATP-competitive inhibitor of extracellular signal-regulated kinases (ERK) 1and 2.1 Discovered by Ohori and co-workers, it has IC50 values of 0.51 and 0.33 μM in enzymatic assays against ERK1 and ERK2, respectively.2 It was also shown to inhibit TGFβ-induced AP-1 activation with an IC50 of 3.1 μM in a cellular assay. A crystal structure of FR180204 in human wild-type ERK2 revealed that it is bound to the kinase hinge region via the 3-NH2 and the 2-N of the pyrazolopyridazine.3 Besides in vitro activity, Ohori has shown that a greater than60% response can be achieved in a rheumatoid arthritis mouse model with an ip administration of 100 mg/kg bid dose of FR180204.4 Recently, Regan et al. have demonstrated that FR180204 can reduce iron-mediated neuronal damage caused by hemoglobin breakdown by a likely reduction in phosphorylation of hemeoxygenase by ERK.5
It is well-known that irregularities in the Ras-Raf-MEK-ERK signal transduction pathway play a crucial part in many cancers.6 The MAP kinase ERK is the last player in this pathway controlling the activation of transcription factors in the nucleus which control cellular processes such as proliferation, survival, and growth. The inhibition of ERK also holds promise as a therapy for inflammatory diseases.7 For instance, the signaling through the TGFβ pathway, an important cascade controlling inflammation, may lead to ERK activation.8 Thus, the modulation of this nodal downstream protein kinase might prove to be extremely beneficial in controlling disease states arising due to malfunctioning of various upstream proteins. With this background, it is surprising that a synthetic route to FR180204 is not available in the literature. To that end, we set out to devise a simple scalable synthetic route that would allow access to gram quantities. This will facilitate its use as an important tool compound for the study of ERK inhibition in various disease models.
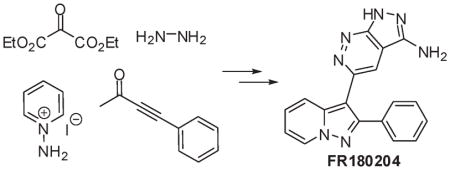
The FR180204 (1) chemical structure consists of two drug-like motifs, a pyrazolo[1,5-a]pyridine and a pyrazolo[3,4-c]-pyridazine. The disconnection that we envisioned passes though the synthesis of oxopyridazine carboxamide 2 and pyrazolopyridine ketone 3 as key intermediates. The pyrazolopyridine ring system can be accessed via a 3 + 2 cycloaddition of aminopyridium salts and alkynes. We decided to construct this ring system first, as alternate routes involved carrying a reactive alkyne throughout the synthesis of the pyrazolopyridazine ring or its late generation for the proposed cycloaddition. To that end, commercially available reagents 1-aminopyridinium iodide and 4-phenylbut-3-yn-2-one were condensed under basic conditions9 to provide the known ethanone 3 (Scheme 1).10 The latter contained the methyl ketone which would form a template for the stepwise construction of the pyrazolopyridazine ring system. An initial attempt of producing the oxopyridazine carboxamide intermediate involved the introduction of the remaining carbon atoms via the displacement of a α-haloketone with diethylmalonate.11 We found that treatment of 3 with bromine led to poor yields of α-bromoketone 4 with significant α,α-dibromination. Portionwise addition of 1 equiv of tetrabutylammonium tribromide did not circumvent this issue. However, this problem could be resolved via formation of the intermediate trimethylsilyl enol ether and subsequent treatment with NBS. Treatment of 4 with 3 equiv of diethylmalonate in refluxing acetone under basic conditions led to good conversion to 5. Reaction of 5 with 1 equiv of hydrazine led to practically no conversion to the desired ester 7 and recovery of starting material. Heating with multiple equivalents of hydrazine allowed for the ring closure but with uncontrollable simultaneous formation of the corresponding hydrazide 6. Heating with lesser equivalents of hydrazine in the microwave or the use of higher boiling solvents such as isoamyl alcohol did not resolve this problem.
Scheme 1.

Ester 7 would have allowed for the introduction of unsaturation in the dihydropyridazinone 2 via a bromination and elimination sequence. Amidation followed by treatment with phosphorus oxychloride and cyclization with hydrazine would have then provided FR180204 (Scheme 2).
Scheme 2.

As an alternative route, we turned our attention to a related method for the construction of pyrazolopyridazines, which utilizes 2-oxodiethyl malonate as the source of the carbon atoms of the ring system (Scheme 3).12 Thus, methyl ketone 3 was heated with diethyl 2-oxomalonate to execute an aldol-like process to provide 8. The crude tertiary alcohol was then dissolved in ethanol and heated with excess hydrazine. This led to the precipitation of pure 9, where the pyridazin-3(2H)-one had formed via elimination of water and ring closure. Concomitant formation of the hydrazide had also taken place during the course of the reaction. Refluxing in concentrated hydrochloric acid led to the initial dissolution of 9 and the subsequent precipitation of acid 10. The latter was simply filtered off and washed with water to provide pure 10. A high yielding amidation of 10 was possible with isobutylchloroformate and aqueous ammonia. The pure amide 11 was simply filtered off from the reaction mixture. 11 had to be refluxed with neat phosphorus oxychloride to simultaneously convert the pyridazinone and the carboxamide to the chloropyridazine and nitrile, respectively. At this stage, we felt that purification of nonpolar 12 by flash silica gel chromatography would be useful in providing material with high purity for the last step. Purified 12 was refluxed with hydrazine in ethanol for2 h and then left standing for 16 h to cause the precipitation of FR180204 1. The solid was filtered to provide material which was of at least 95% purity as determined by 1H NMR spectroscopy and reverse-phase HPLC.
Scheme 3.

We evaluated the kinase selectivity of 1 in a panel of 442 kinases at 10 μM and found >99% inhibition of kinase activity for 12 out of 386 nonmutant kinases (>90% for 29/386).13 We also measured the dissociation constants against a few kinases of interest.13 Table 1 discloses some activities in the range of previously reported values. These studies confirm the activity and selectivity reported by Ohori.1
Table 1.
Kinase Activity Data
| enzyme | Kd (nM) |
|---|---|
| ERK1 | 160 |
| ERK2 | 420 |
| MEK1 | 8700 |
| EGFR | 1200 |
| P38-alpha | 1400 |
In summary, we have developed a scalable route toFR180204. This was used to prepare multi-gram quantities of FR180204 from known ketone 3 in six steps and 30%overall yield. Silica gel chromatography was done in only one step. This route will give access to gram quantities of FR180204, which will enable its use as a tool compound for studying the therapeutic effects of ERK inhibition in in vitro and in vivo disease models.
Experimental Section
General Methods
Chromatography on silica gel was performed using forced flow (liquid) of the indicated solvent system on Biotage KP-Sil prepacked cartridges and using the Biotage SP-1 automated chromatography system. 1H and 13C NMR spectra were recorded on a Varian Inova 400 MHz spectrometer. Chemical shifts are reported in δ with the solvent resonance as the internal standard (CDCl3 δ 7.26, 77.0, DMSO-d6 δ 2.50, 39.5 for 1H and 13C, respectively). Analytical analysis and retention times reported here were performed on an Agilent LC/MS (Agilent Technologies, Santa Clara, CA) with a 3 min gradient of 4–100% MeCN (containing 0.025% TFA) in water (containing 0.05% TFA) and was used with a 4.5 min run time at a flow rate of 1 mL/min. A Phenomenex Gemini phenyl column (3 μm, 3 × 100 mm) was used at a temperature of 50°C. Infrared spectra were recorded on a Perkin-Elmer ET-IR Spectrum 10 instument. Molecular weight was confirmed using a TOF mass spectrometer (Agilent Technologies, Santa Clara, CA). A 3 min gradient from 4 to 100% acetonitrile (0.1% formic acid) in water (0.1% formic acid) was used with a 4 min run time at a flow rate of 1 mL/min. A Zorbax SB-C18 column (3.5 μm, 2.1 × 30 mm) was used at a temperature of 50 °C. Confirmation of molecular formula was confirmed using electrospray ionization in the positive mode with the Agilent Masshunter software(version B.02).
Ketone 3
4-Phenylbut-3-yn-2-one (7.5 g, 52 mmol) and 1-aminopyridinium iodide (13.4 g, 60.4 mmol) were dissolved in DMSO (105 mL) and treated with K2CO3 (6.47 g, 46.8 mmol) and KOH (5.84 g, 104 mmol). The mixture was stirred for 3 h. Then the reaction was poured in water and extracted with EtOAc. The organic layer was dried (Mg2SO4), filtered, concentrated, and purified by flash silica gel chromatography (10–100% EtOAc/hexanes) to provide ketone 3 (11.1 g, 46.8 mmol, 90% yield): LC-MS rt (min) = 3.48; 1H NMR (400 MHz, CDCl3) δ ppm 2.15 (s, 3 H), 7.04 (td, J = 6.8, 1.6 Hz, 1 H), 7.47–7.54 (m, 4 H), 7.56–7.63 (m, 2 H), 8.46 (d, J = 9.0 Hz,1 H), 8.54 (d, J = 6.7 Hz, 1 H); C NMR (101 MHz, CDCl3) δ ppm 30.1, 111.6, 114.7, 120.2, 128.3, 128.4, 128.5, 129.1, 129.7, 133.4, 141.9, 156.8, 193.6; IR (neat, diamond/ZnSe) 3066, 3024, 1641, 1627, 1497, 1411, 1357, 1211 cm−1; HRMS (m/z) calcd forC15H13N2O (M + H)+ 237.1028, found 237.1021.
Hydrazide 9
Ketone 3 (4.30 g, 18.20 mmol) and diethyl 2-oxomalonate (6.34 g, 36.4 mmol) were taken in a microwave vial and heated at 200 °C for 2 h at a “high” setting. The vessel’s septum was carefully punctured with a needle to release pressure. The crude brown residue (tertiary alcohol) was diluted with EtOH, transferred to a RB flask, treated with excess hydrazine hydrate (10.0 g, 200 mmol), and refluxed overnight. The reaction was cooled, and the precipitate formed was filtered through a Büchner funnel, washed with ethanol, and air-dried to hydrazide 9 (6.30 g, 18.2 mmol, quantitative): LC-MS rt (min) = 2.92;1H NMR (400 MHz, DMSO-d6) δ ppm 4.80 (br s, 2 H), 7.09 (td, J = 6.8, 1.4 Hz, 1 H), 7.42–7.50 (m, 4 H), 7.57–7.65 (m, 2 H), 7.79 (s, 1 H), 7.91 (dt, J = 9.0, 1.3 Hz, 1 H), 8.83 (d, J = 7.0 Hz, 1H), 10.46 (br s, 1 H), 11.55 (br s, 1 H); 13C NMR (101 MHz, DMSO-d6) δ ppm 103.9, 113.9, 117.6, 126.2, 128.2, 128.8, 128.9, 129.0, 132.1, 133.6, 139.1, 141.6, 151.4, 159.3, 159.7; IR (neat, diamond/ZnSe) 3220, 2867, 1683, 1583, 1520, 1450, 1225 cm−1; HRMS (m/z) calcd for C18H15N6O2 (M + H)+ 347.1256, found 347.1253.
Acid 10
Hydrazide 9 (6.30 g, 18.2 mmol) was refluxed in concd HCl (100 mL). After initial dissolution, a precipitate was observed to form after 16 h of reflux. The reaction was cooled and filtered through a Büchner funnel. The residue was washed with water and diethyl ether and then air-dried to provide acid 10 (5.18 g, 15.6 mmol, 86% yield): LC-MS rt (min) = 3.31; 1H NMR (400 MHz, DMSO-d6) δ ppm 7.11 (td, J = 7.1, 1.6 Hz),7.44–7.52 (m, 4 H), 7.59–7.66 (m, 2 H), 7.70 (s, 1 H), 7.92 (d, J = 9.0 Hz, 1 H), 8.85 (d, J = 7.0 Hz, 1 H); 13CNMR (101 MHz, DMSO-d6) δ ppm 103.5, 114.1, 117.6, 126.5, 127.0, 128.9, 128.9, 129.1, 129.1, 132.1, 135.2, 139.2, 142.4, 151.6, 160.8, 163.5; IR (neat, diamond/ZnSe) 2739, 1741, 1640, 1591, 1422, 1401 cm−1; HRMS (m/z) calcd for C18H13N4O3 (M + H)+ 333.0988, found 333.0982.
Amide 11
Acid 10 (5.13 g, 15.4 mmol) was taken in DME (200 mL), cooled to 0 °C, treated with 4-methylmorpholine (20.0 mL, 182 mmol), stirred for 5 min, and then treated with isobutylchloroformate (5.0 mL, 38 mmol). The mixture was stirred for 4 h and then treated with excess ammonium hydroxide (50 mL, 1.28 mol). The reaction was stirred for 16 h. The yellow precipitate formed was filtered through a Büchner funnel, washed with water and diethyl ether, and air-dried to provide amide 11 (4.37 g, 13.2 mmol, 85% yield): LC-MS rt (min) = 3.09; 1H NMR (400 MHz, DMSO-d6) δ ppm 7.09 (td, J = 6.9, 1.5 Hz, 1 H), 7.39–7.51 (m, 4 H), 7.57–7.65 (m, 2 H), 7.82 (s, 1 H), 7.89 (dt, J = 8.9, 1.3 Hz, 1 H), 7.95 (d, J = 4.1 Hz,1 H), 8.79–8.89 (m, 2 H), 13.86 (s, 1 H); 13C NMR (101 MHz, DMSO-d6) δ ppm 103.9, 113.9, 117.5, 126.1, 128.8, 128.8, 128.9, 129.0, 129.3, 132.1, 134.7, 139.1, 141.4, 151.3, 160.1, 162.3; IR (neat, diamond/ZnSe) 3312, 2836, 1692, 1580, 1519, 1449, 1419, 1362 cm−1; HRMS (m/z) calcd for C18H14N5O2 (M + H)+332.1147, found 332.1142.
Nitrile 12
Amide 11 (4.35 g, 13.1 mmol) was refluxed in phosphorus oxychloride (30.0 mL, 322 mmol) for 1 h. Dissolution of the yellow suspension to an orange colored solution was observed. The reaction was cooled and poured over ice. Precipitation of a yellow solid was observed. This was filtered. This was dissolved in DCM/MeOH mixture, adsorbed on silica gel, and purified by flash silica gel chromatography (0–5% EtOAc/DCM) to provide nitrile 12 (3.4 g, 10.3 mmol, 78% yield): LC-MS rt (min) = 3.77; 1H NMR (400 MHz, DMSO-d6) δ ppm 7.18 (td, J = 6.8, 1.6 Hz, 1 H), 7.44–7.53 (m, 3 H), 7.53–7.59(m, 1 H), 7.59–7.65 (m, 2 H), 8.02 (s, 1 H), 8.16 (d, J = 9.0 Hz,1 H), 8.91 (d, J = 7.0 Hz, 1 H); 13C NMR (101 MHz, CDCl3) δ ppm 103.5, 112.6, 113.8, 114.5, 119.7, 127.6, 128.9, 129.1, 129.4, 129.4, 129.9, 131.9, 140.4, 150.1, 154.0, 155.7; IR (neat, diamond/ZnSe) 3077, 3030, 2240, 1733, 1629, 1577, 1511, 1467, 1419, 1351, 1327, 1189 cm−1 HRMS (m/z) calcd for C18H11ClN5 (M + H)+ 332.0703, found 332.0700.
FR108204 1
Nitrile 12 (4.80 g, 14.5 mmol) was taken in ethanol (150 mL) and treated with excess hydrazine hydrate (7.00 mL, 143 mmol) and then heated to reflux. The suspension dissolved to an orange solution. After refluxing for 3 h, the reaction was cooled and left standing for 2.5 days. A yellow solid had precipitated out. This was filtered via a Büchner funnel. The solid was washed with EtOH and water and dried to provide FR180204 1 (4.00 g, 12.2 mmol, 84% yield): LC-MS rt (min) = 2.98; 1H NMR (400 MHz, DMSO-d6) δ ppm 5.90 (br s, 2 H), 7.04 (td, J = 6.8, 1.4 Hz, 1 H), 7.22–7.46(m, 4 H), 7.47–7.64 (m, 2 H), 7.76 (dt, J = 8.8, 1.2 Hz, 1 H), 7.96 (s, 1 H), 8.82 (dt, J = 6.9, 1.1 Hz, 1 H), 12.67 (s, 1 H); 13C NMR (101 MHz, DMSO-d6) δ ppm 107.3, 108.6, 113.3, 117.4, 119.5, 125.1, 128.3, 128.5, 128.5 (two signals overlapped at 128.5), 128.8, 132.7, 139.8, 144.8, 148.2, 150.6, 154.0; IR (neat, diamond/ZnSe) 3302, 3161, 1630, 1529, 1458, 1422, 1352,1108 cm−1; HRMS (m/z) calcd for C18H14N7 (M + H)+ 328.1311, found 328.1309.
Supplementary Material
Figure 1.

Retrosynthesis.
Acknowledgments
S.P. would like to thank Dr. Craig Thomas and Dr. Jingbo Xiao for useful suggestions.
Footnotes
Supporting Information Available: 1H and 13C NMR data for 3, 9, 10, 11, 12, and 1. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ohori M, Kinoshita T, Okubo M, Sato K, Yamazaki A, Arakawa A, Nishimura S, Inamura N, Nakajima H, Neya M, Miyake H, Fujii T. Biochem Biophys Res Commun. 2005;336:357–363. doi: 10.1016/j.bbrc.2005.08.082. [DOI] [PubMed] [Google Scholar]
- 2.For another enzyme-potent small-molecule inhibitor of ERK, see: Aronov AM, Baker C, Bemis GW, Cao J, Chen G, Ford PJ, Germann UA, Green J, Hale MR, Jacobs M, Janetka JW, Maltais F, Martinez-Botella G, Namchuk MN, Straub J, Tang Q, Xie X. J Med Chem. 2007;50:1280–1287. doi: 10.1021/jm061381f.
- 3.Kinoshita T, Warizaya M, Ohori M, Sato K, Neya M, Fujii T. Bioorg Med Chem Lett. 2006;16:55–58. doi: 10.1016/j.bmcl.2005.09.055. [DOI] [PubMed] [Google Scholar]
- 4.Ohori M, Takeuchi M, Maruki R, Nakajima H, Miyake H. Naunyn-Schmiedeberg’s Arch Pharmacol. 2007;374:311–316. doi: 10.1007/s00210-006-0117-7. [DOI] [PubMed] [Google Scholar]
- 5.Chen-Roetling J, Li Z, Chen M, Awe OO, Regan RF. Neuropharmacology. 2009;56:922–928. doi: 10.1016/j.neuropharm.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hilger RA, Scheulen ME, Strumberg D. Onkologie. 2002;25:511–518. doi: 10.1159/000068621. [DOI] [PubMed] [Google Scholar]
- 7.Hitti E, Kotlyarov A. Anti-Inflamm Anti-Allergy Agents Med Chem. 2007;6:85–97. [Google Scholar]
- 8.Mulder KM. Cytokine Growth Factor Rev. 2000;11:23–35. doi: 10.1016/s1359-6101(99)00026-x. [DOI] [PubMed] [Google Scholar]
- 9.Uehling D, et al. 2006/068826 WO.
- 10.Ha H-H, Kim JS, Kim BM. Bioorg Med Chem Lett. 2000;18:653–656. doi: 10.1016/j.bmcl.2007.11.064. [DOI] [PubMed] [Google Scholar]
- 11.(a) Brana MF, Cacho M, Garcia ML, Mayoral EP, Lopez B, Pascual-Teresa B, Ramos A, Acero N, Llinares F, Munoz-Mingarro D, Lozach O, Meijer L. J Med Chem. 2005;48:6843–6854. doi: 10.1021/jm058013g. [DOI] [PubMed] [Google Scholar]; (b) Wermuth CG, Schlewer G, Bourguignon JJ, Maghioros G, Bouchet MJ, Moire C, Kan JP, Worms P, Biziere K. J Med Chem. 1989;32:528–537. doi: 10.1021/jm00123a004. [DOI] [PubMed] [Google Scholar]; (c) Pinna GA, Curzu MM, Gavini E, Mule A, Pirisino G, Satta M, Peana G. Il Farmaco. 1993;48:1239–1248. [PubMed] [Google Scholar]
- 12.(a) Sircar I, Duell BL, Bobowski G, Bristol JA, Evans DB. J Med Chem. 1985;28:1405–1413. doi: 10.1021/jm00148a006. [DOI] [PubMed] [Google Scholar]; (b) Singh B, Lesher GY. Heterocycles. 1990;31:2163–2172. [Google Scholar]
- 13.These data were generated by Ambit Biosciences.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.