

Abstract
Background & Aims
Hepatocellular carcinoma (HCC) is an aggressive cancer with a poor prognosis mainly due to metastasis. MicroRNAs are endogenous small noncoding RNAs that regulate cellular gene expression and are functionally linked to tumourigenesis. Using microarray analysis, we recently identified 20 miRNAs associated with HCC metastasis. Here, we carried out further analyses on one of these microRNAs, let-7g, to determine whether it is functionally linked to HCC metastasis.
Methods
Quantitative real-time polymerase chain reaction was used to determine the level of mature let-7g transcript in HCC clinical specimens and its correlation with patient survival. Ectopic expression of let-7g was carried out in HCC cell lines to assess its influence on cell growth, migration and invasion.
Results
We confirmed that the level of let-7g was significantly lower in metastatic HCCs compared to metastasis-free HCCs. Moreover, low let-7g expression in a tumour was predictive of poor survival in HCC patients. Functional studies indicated that ectopic expression of let-7g significantly inhibits HCC cell migration and cell growth. In-silico analysis revealed members of soluble collagens as potential targets of let-7g. Consistently, the levels of type I collagen α2 (COL1A2) and let-7g were inversely correlated in HCC clinical specimens. COL1A2 was experimentally validated as a direct target of let-7g. Moreover, addition of COL1A2 counteracted the inhibitory effect of let-7g on cell migration.
Conclusions
These results suggest that let-7g may suppress HCC metastasis partially through targeting COL1A2.
Keywords: hepatocellular carcinoma, microRNA, let-7g, metastasis, cell mobility, prognosis
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common cancer and the third leading cause of cancer death worldwide. Its incidence continues to increase in most industrialised countries, including the United States [1;2]. Metastasis has been the major problem for the dismal outcome of HCC patients [3]. Recently, HCC metastasis-associated mRNA and microRNA signatures have been identified in our lab [4;5].
MicroRNAs (miRNAs) are a class of endogenous, small (21–23 nucleotides in length), noncoding but functional RNAs. Mature miRNAs can be generated from sequential processing of primary miRNA transcripts by Drosha and Dicer, and they then serve as posttranscriptional regulators of gene expression through complementary base pairing to messenger RNAs [6]. Let-7 family members were originally observed in C. elegans [7;8], and nine members of the let-7 family have been found in humans [9]. Recently, this family has been shown to function as tumour suppressors. For example, let-7 is down-regulated in lung cancer, melanoma and head and neck squamous carcinoma, while over-expression of let-7 can inhibit cancer cell growth [10–17]. Oncogenes such as RAS, MYC, and HMGA2 are direct targets of let-7 [10;18–20]. Moreover, LIN28B, a suppressor of let-7 expression, is highly expressed in HCC with poor clinical outcomes [21–23].
Recently, we identified a 20-miRNA HCC metastasis signature that contains let-7g [5]. This signature can significantly distinguish primary HCC tissues with venous metastasis from metastasis-free solitary tumours as well as predict survival. The current study was designed to validate our previous findings and to determine the role of let-7 in HCC progression.
Materials and Methods
Clinical samples and cell lines
MicroRNA expression was quantified in 55 HCC tissues and paired non-tumour tissues obtained with informed consent from patients who underwent radical resection between 2002 and 2003 at the Liver Cancer Institute and Zhongshan Hospital (Fudan University, Shanghai, China). Follow-up data were available for all cases, and metastasis status was known for 22 cases. HuH-1 and huH7 HCC cell lines were used for let-7g functional studies. The retroviral packaging cell line AmphoPack-293 was used to produce retroviruses expressing let-7g. These cells were cultured in 10% foetal bovine serum (FBS) supplemented Dulbecco’s modified essential medium (DMEM) (Invitrogen), containing L-glutamine (2mM) (Invitrogen) and penicillin sodium (100 U/ml)/streptomycin sulphate (100 µg/ml) (Invitrogen) in a humidified incubator at 37°C with 5% CO2. Soluble type I collagen (Southern Biotech) (0.2 µg/ml) was used in this study.
Constructs and luciferase assay
miRVec-control and miRVec-let7g plasmids were kindly provided by Prof. Reuven Agami. miRVec-let7g was used to produce a precursor form that is processed into mature let-7g in cultured cell lines. The wild-type and mutant let-7 binding sequence at the 3’-UTR of human COL1A2 and its complementary oligo were synthesised (IDT), denatured/renatured and then cloned into the Spe I/Hind III sites of a luciferase gene in the pMIR-REPORT luciferase vector (Ambion). The sense sequences were 5’-ctagtaacttccaaaggtttaaactacctcaaaaa-3’ (wild-type); 5’-ctagtaacttccaaaggtttaaactgttcgaaaaa-3’ (mutant). The reverse sequences were 5’-agcttttttgaggtagtttaaacctttggaagtta-3’ (wild-type); 5’-agcttttttcgaacagtttaaacctttggaagtta-3’ (mutant). The whole 3’UTR of COL1A2 with either the wild-type or a mutant let-7g binding site (the seed residues) was also cloned into Spe I/Hind III sites in the pMIR-REPORT vector. Luciferase assays were performed as previously described [24].
Retrovirus generation and infection
Retrovirus was generated as follows. Amphopack-293 cells were transfected with DNA by Lipofectamine 2000. Forty-eight hours later, the virus-containing medium was harvested. The retrovirus-containing medium was used to infect huH-1 or huH-7 cells, and blasticidine (2 µg/ml) was used to select positive cells. The surviving colonies from control and let-7g virus-infected cells, i.e., miRVec-control or miRVec-let7g, respectively, were trypsinised and pooled together in blasticidine-containing media to generate a mass culture. We used these cells within six passages to avoid cells adapting to let-7g.
RNA isolation and quantitative real time reverse transcriptase-polymerase chain reaction (qRT-PCR)
Total RNA isolation and qRT-PCR were performed as previously described [24]. RNU6B RNA was used as a control for let-7g detection, and actin was used as a control for COL1A2 detection.
Invasion and migration assay
For invasion assays, 4×104 cells in serum-free media were seeded into the upper chambers of a 24-well BioCoat Matrigel invasion chamber (Becton Dickinson Labware) with an 8-µm pore polycarbonate membrane coated with Matrigel. For migration assays, 4×104 cells were seeded into the upper chambers of 24-well BioCoat control inserts (BD Bioscience) with uncoated 8-µm pores in serum-free media. Medium with 5% FBS was added to the lower chambers as a chemoattractant. After 40 hours of incubation, cells remaining on the upper surface of the membrane were removed with a cotton swab, and cells that invaded through the membrane filter were fixed with 100% methanol, stained by haematoxylin and eosin, and photographed under a microscope (Olympus BX40 with a DP70 digital camera). The number of invading cells was manually counted per high-power field for each condition (eight fields on each membrane were randomly selected). The percentage of invasion was calculated using the following formula: # of cells invading through Matrigel inserts versus # of cells migrating through control inserts.
Wound healing assay
Cells were grown to confluence in 25 cm2 cell culture flasks. Artificial wound tracks were created by scraping confluent cell monolayers with a pipette tip. After removal of the detached cells by gently washing with PBS, the cells were fed with fresh complete medium and incubated over time to allow cells to migrate into the open area. The ability of the cells to migrate into the wound area was assessed at 1, 8, 24 and 32 hours after scratching by comparing the pixels of the wound tracks in micrographs of three randomly selected wounded areas.
Colony formation assay and cell proliferation assay
To assay colony formation, a total of 1,000 cells were plated onto a 100 mm-dish and cultured for 14 days in complete DMEM supplemented with blasticidine. The colonies were fixed with 100% methanol 2×10 min, stained with 0.2% crystal violet in 75% methanol for 30 min, and then visualised and counted. Cell proliferation assays were performed using a Calcein AM cell Viability Assay Kit (Biotium). Briefly, 3,000 cells/well were grown in 96-well plates and incubated for 1, 2, 3, 4, 5, 6 and 7 days. The fluorescence, representing relative cell number, was measured at each time point. The fluorescence readings at the first day were used as controls.
Protein extraction, collagen assay and western blotting
Cells were rinsed twice with PBS and lysed in RIPA buffer [25 mM Tris-HCl (pH 7.6), 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS] on ice for 5 minutes by vortexing. Insoluble components were removed by centrifugation, and protein concentration was measured. Cell extracts (100 µg) were used for collagen assays (Sircol Soluble Collagen Assay Kit, Biocolor) according to manufacturer’s instructions. Cellular proteins (40 µg) were adjusted to a total concentration of 4 µg/ul and digested for 1 hour at 37°C by adding pepsin to a final concentration 4 mg/ml in 10 mM HCL, followed by neutralising with NaOH. After boiling for 10 min in Loading Buffer (Bio-Rad), proteins were separated by 10% Tris-Glycine Gels (Invitrogen) and blotted onto nitrocellulose membrane (Invitrogen). Western blotting was performed using Rabbit anti-Human Collagen type I antibody (USbiological), and immunocomplexes were visualised by enhanced chemiluminescence (Amersham Pharmacia Biotech) according to the manufacturer's protocol. Total protein (40 µg) without pepsin digestion was used for actin detection (protein loading control).
Statistical analysis
Kaplan-Meier survival analysis was performed in GraphPad Prism software 5.0 with statistical P values generated by the Cox-Mantel log-rank test. P values were generated by Student’s t-test for qRT-PCR data, migration/invasion assays and colony formation assays and by two-way ANOVA for cell proliferation assays. All experiments were performed in triplicate.
Results
Let-7g is associated with HCC metastasis and is predictive of survival
Using a microarray-based approach, we recently found that a microRNA signature containing let-7g was associated with HCC metastasis [5]. To further validate the microarray data, we performed qRT-PCR to quantify mature let-7g expression in 11 HCC cases with clinically confirmed metastases and 11 cases without metastasis at the time of resection and with no recurrence after five years of follow-up. Consistent with the microarray data, the qRT-PCR analysis revealed that let-7g was significantly down-regulated in HCCs with metastasis compared to those without (Fig. 1A, B).
Figure 1. Let-7g was uniquely down-regulated in HCC cases with metastasis and was predictive of survival.
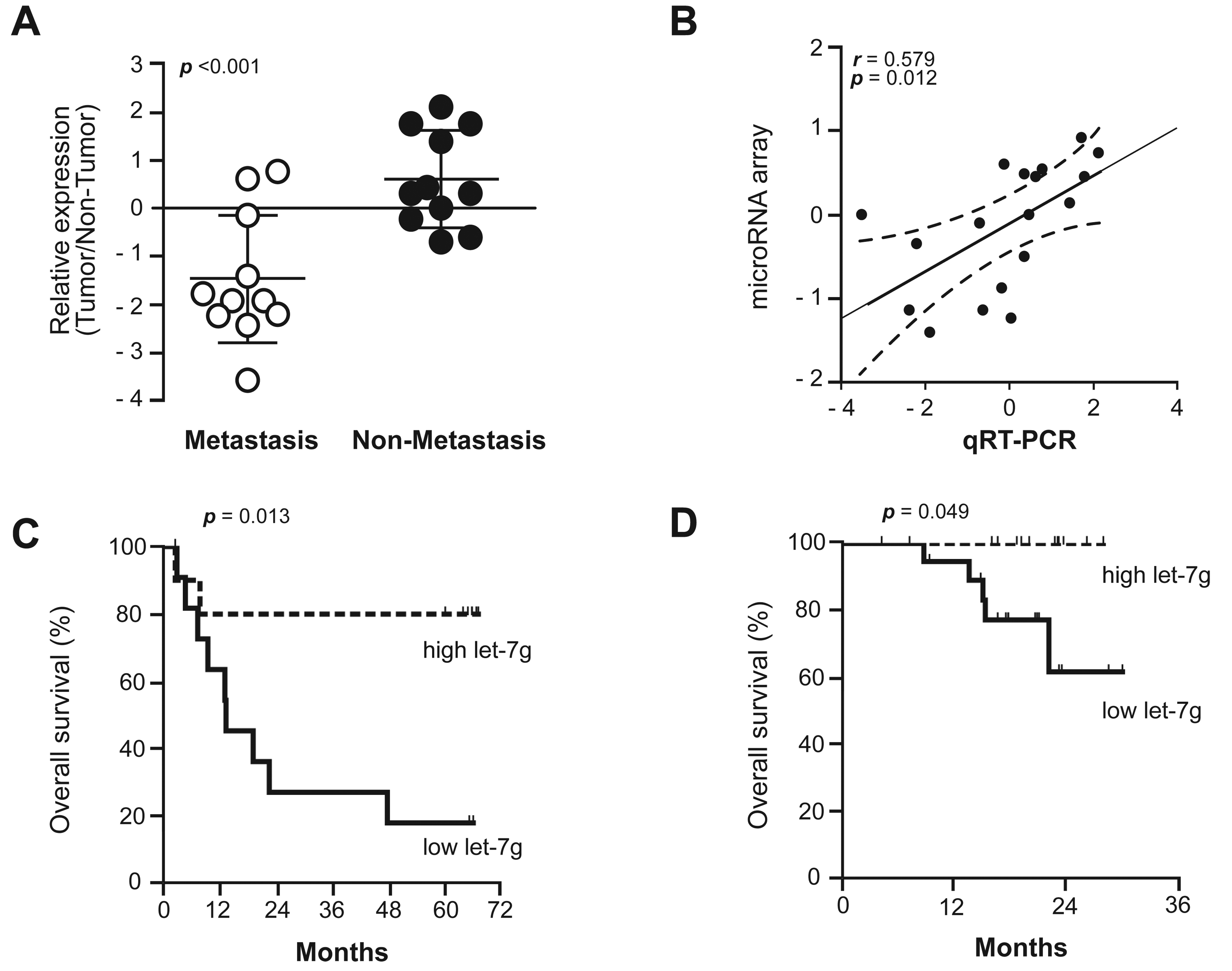
(A) The relative expression levels of let-7g from metastatic HCC cases (n = 11) and non-metastastic HCC cases (n = 11) were determined by qRT-PCR. Un-paired t-tests were used. (B) A linear regression and correlation among data from qRT-PCR versus microarray is shown with r (spearman) and p-values indicated (right panel). Expression status is shown as the tumour/non-tumour ratio in a log2 scale. (C) The association of tumour vs. non-tumour let-7g expression with overall survival of 22 HCC patients (high let-7g (tumour/non-tumour ≥ 1), n = 10; low let-7g (tumour/nontumour <1), n = 12). (D) The association of tumour vs. non-tumour let-7g expression with overall survival of 33 HCC patients (high let-7g, n = 12; low let-7g, n = 21).
As metastasis contributes significantly to HCC-related mortality, the observed deregulation of let-7g may be linked to HCC survival. Kaplan-Meier survival analysis of the 22 cases described above showed that HCC cases with low let-7g levels (tumour/non-tumour (T/N) <1) had poorer outcomes compared to those with high let-7g levels (T/N≥1) (p=0.013, log-rank test, Fig. 1C). To validate the data above, we quantified the level of mature let-7g in an independent cohort containing 33 HCC cases with unknown metastasis status at the time of resection. Consistent with the data from the 22 well-defined HCC cases, low let-7g was significantly associated with poor survival in these independent cases (p=0.049, log-rank test, Fig. 1D). Thus, let-7g expression status in tumours can predict HCC survival.
Inhibition of cell mobility by let-7g
We sought to determine whether let-7g affects HCC cell migration and invasion, the two key steps contributing to metastasis [25]. HuH-1 cells stably expressing let-7g (huH1-let7g) were generated using a retroviral construct encoding let-7g. huH1-miRCtrl cells were also generated to serve as a control using miRVec-control virus infection. Over-expression of let-7g was validated in huH1-let7g cells compared to both parental huH-1 cells and huH1-miRCtrl cells (Fig. 2A). A chamber-based cell migration assay revealed that the number of huH1-let7g cells migrating through the membrane was significantly lower than that of huH1-miRCtrl cells (28.42 ± 9.83 vs.188.8 ± 66.05, P<0.001), indicating that let-7g over-expression suppresses HCC cell migration. We next determined the ability of huH1-miRCtrl and huH1-let7g cells to invade through extracellular matrix (ECM) in a Boyden chamber invasion assay. We found that there was no significant difference in the number of invading cells between huH1-let7g and huH1-miRCtrl cells after normalising for the difference in cell migration (17.74 ± 2.65% vs. 19.78 ± 1.85%; P=0.336; Fig. 2C). Similar results were observed with huH-7 cells (data not shown). Thus, it appears that let-7g affects cell migration but not invasion.
Figure 2. Inhibition of cell migration in huH1 cells by let-7g in an in vitro cell migration assay.

(A) The expression levels of let-7g in huH-1, huH1-miRCtrl and huH1-let7g cells determined by qRT-PCR. (B) huH1-miRCtrl and huH1-let7g cells were subjected to a cell migration assay. Representative images of migrated cells are shown in the left panel and quantitative data are shown in the right panel. (C) A cell invasion assay was performed on huH1-miRCtrl and huH1-let7g cells. Representative images of migrated cells are shown in the left panel and quantitative data are shown in the right panel. Data from A-C are shown as the mean ± SD derived from three independent experiments, and Student’s t-test was used.
We also used an in vitro wound healing assay to determine the effect of let-7g over-expression on HCC cell migration. Figure 3A shows that the healing speed was slower and that the gaps were wider in huH1-let7g cells at each time point than in the huH1-miRCtrl cells. At 32 h, most of the gaps were completely closed in huH1-miRCtrl cells, whereas the gaps in the huH1-let7g cells remained open (Fig. 3B). These data are consistent with the results of the chamber-based cell migration assay, indicating that let-7g had an inhibitory effect on HCC cell migration. Similar results were observed in huH-7 cells expressing let-7g (data not shown). Thus, let-7g plays a role in inhibiting HCC cell mobility but not invasion.
Figure 3. Inhibition of cell migration in huH1 cells by let-7g in an in vitro wound healing assay.

Scratches were generated in a confluent monolayer of huH1-miRCtrl or huH1-let7g cells and the degree of “wound remaining” was measured after 1 hr, 8 hr, 24 hr and 32 hr. (A) Representative images of the wound gaps in controls and huH1-let7g cells at each time point. (B) Percent wound remaining is shown as the mean ± SD of three experiments.
Because let-7g was shown to inhibit lung adenocarcinoma cell growth [11,13], we sought to test whether let-7g could inhibit HCC cell growth. Using a colony formation assay, we found that the ability of huH1-let7g cells to form colonies was significantly lower than that of huH1-miRCtrl cells (Fig. 4A). The cell proliferation assay also showed growth inhibition in huH1-let7g cells compared to huH1-miRCtrl cells (Fig. 4B). Similar results were observed in huH-7 cells (data not shown). These data indicate that let-7g over-expression reduces the growth and clonogenic capacity of HCC cells.
Figure 4. Inhibition of HCC cell growth by let-7g.

(A) A colony formation assay in huH1-miRCtrl and huH1-let7g cells was performed, and representative dishes of huH1-miRCtrl and huH1-let7g cells are shown in the left panel and quantitative data are in the right panel. (B) A cell proliferation assay in huH1-miRCtrl and huH1-let7g cells. The percentage of cells forming colonies (A) and the relative cell number (B) are shown as the mean ± SD of three experiments.
COL1A2 as a direct target of let-7g-mediated silencing
As RAS, MYC and HMGA2 have been shown to be direct target genes for members of the let-7 family, we first determined whether the levels of these proteins were altered in huH-1 cells expressing let-7g. Interestingly, none of the reported let-7 target genes showed altered expression in huH1-let7g cells (data not shown). To identify new let-7g targets, we performed in-silico screening using TargetScan [26] with a recently described strategy [24]. We found that, among the predicted let-7g target genes, the 3’UTR of 11 collagen genes contained binding sites for let-7g with reasonable scores (Supplementary Table 1). Some of these collagens have been implicated in cancer metastasis and proliferation [27–29]. To experimentally validate whether collagens are possible targets of let-7g, we performed a collagen assay to detect the total amount of soluble collagens (i.e., types I to V) in huH1-let7g vs. huH1-miRCtrl cells. Figure 5A shows that over-expression of let-7g significantly decreased the production of soluble collagens. We selected collagen type I a2 (COL1A2) for further validation because it ranked at the top of the candidate list (Supplementary Table 1). Sequence analyses revealed that the 3’-UTR of COL1A2 mRNA contains a putative site partially complementary to let-7g (Fig. 5B). Consistently, COL1A2 expression was significantly down-regulated in huH1-let7g cells at both the mRNA and protein levels (Fig. 5C). To further determine whether COL1A2 was a bona fide target of let-7g-mediated siRNA silencing, the entire 3’-UTR of COL1A2 mRNA and the let-7g binding region within 3’UTR of COL1A2 mRNA were cloned into a luciferase reporter. We found that forced expression of let-7g led to a significant decrease in luciferase activity when the reporter contained a wild-type sequence (WT), but not when it contained a mutant sequence (MT) within the let-7g binding site (five nucleotides within the complementary seed sequence) (Fig. 5D). Taken together, these results indicate that let-7g directly targets COL1A2, and that a single site within COL1A2 mRNA is sufficient for let-7g-mediated gene silencing.
Figure 5. Direct targeting of COL1A2 by let-7g.

(A) Collagen assays were performed in huH1-miRCtrl and huH1-let7g cells. Relative expression of collagens type I to V is shown as the mean ± SD. (B) Predicted duplex formation between the 3’-UTR sequences of human COL1A2 and let-7g. The underlined bases of let-7g highlight the seed sequences of this miRNA. The underlined bases in the COL1A2 3’-UTR were targeted for mutation analysis when inserted into a luciferase reporter. (C) The expression of COL1A2 at the mRNA (right) and protein (left) levels in huH1-miRCtrl and huH1-let7g cells. (D) Luciferase activity of various reporter plasmids in huH1-miRCtrl and huH1-let7g cells. The luciferase activity is shown as mean ± SD. (E) A wound healing assay was performed in huH1-miRCtrl and huH1-let-7g cells with and without the presence of 0.2 µg/ml of type I collagen. The wound healing assay was done in 20 hours. Statistical significance was calculated using Student’s t-test. * refers to p <0.05.
To determine whether COL1A2 functions downstream of let-7g, we performed a rescue experiment by culturing HCC cells in the presence of purified type I collagen. We found that, while huH1-let-7g cells showed a significant decrease in cell migration when compared to huH1-miRCtrl cells, addition of type I collagen completely eliminated this effect (Fig. 5E). Consistently, we found that the COL1A2 expression was significantly up-regulated in metastatic HCC cases compared to non-metastatic cases (Fig. 6A), and it was inversely correlated with the level of let-7g (p<0.05) (Fig. 6B).
Figure 6. COL1A2 expression was negatively correlated with let-7g level in HCC cases.

(A) The relative expression levels of COL1A2 from metastatic HCC cases (n = 11) and non-metastatic HCC cases (n = 11) from microarray data. (B) A linear regression and correlation among let-7g versus COL1A2 data is shown with r (spearman) and p-value indicated (right panel). Expression status is shown as the tumour/non-tumour ratio in a log2 scale.
Discussion
Intra-hepatic metastasis is the major characteristic of HCC malignance. The identification of molecular events contributing to HCC metastasis offers hope of finding a cure for metastatic HCC. Recently, we have identified a 20-miRNA signature that is associated with HCC intra-hepatic metastasis [5]. We hypothesised that some of these microRNAs may functionally contribute to HCC progression. In the current study, we demonstrated that let-7g, one of 20 metastasis-associated miRNAs, is functionally involved in suppressing HCC cell migration. This finding was supported by both clinical data and cell culture studies. In clinical samples, let-7g was uniquely down-regulated in HCC with metastasis but not in HCC without metastasis, and low let-7g in tumours was associated with poor survival of HCC patients. In cell culture experiments, over-expression of let-7g led to decreased HCC cell motility, possibly through targeting the extracellular matrix by degrading soluble collagens such as COL1A2. Our results suggest that leg-7g may act as an HCC metastasis suppressor, in part by suppressing cell migration and metastatic colonisation by targeting collagens that contribute to metastatic progression.
Our results are consistent with several recent studies indicating that let-7 family members may act as tumour suppressors and suppress metastasis [10–12;18;22;30]. One recent paper reports that HCCs with a high level of LIN28B have poor clinical prognoses and that let-7 expression is low in these cases [22]. Several known oncogenes, i.e., Ras [10], Myc [18] and HMGA2 [19,20], have been reported as targets of the let-7 microRNA family. These studies also showed that let-7 can suppress cell proliferation and induce apoptosis. Interestingly, we could not detect any change in Ras, Myc or HMGA2 expression levels in HCC cells over-expressing let-7g. The reason that no change was detected in HCC cells is unclear. Cell type specificity could contribute to this discrepancy. Another possible mechanism is that the presence of single nucleotide polymorphisms (SNPs) may alter microRNA binding to the target genes, resulting in a loss or gain of its regulatory function. Such SNPs have recently been identified in the 3’ UTRs of KRAS and HMGA2, which interact with let-7 [31–33]. Because each microRNA is expected to target many cellular genes, it is currently unclear how an individual microRNA can carry out its activity through discriminating various targets. This is a challenging task requiring future exploration.
Collagens have been reported to promote migration and proliferation. Encouragingly, we found that let-7g over-expression significantly reduced the levels of soluble collagens, including types I to V. We subsequently validated COL1A2 as a direct target gene of let-7g. Importantly, we have established a functional link between let-7g and COL1A2 in HCC cell motility because exogenous collagen I can counteract the suppression of cell migration by let-7g. Consistently, COL1A2 expression was negatively correlated with let-7g levels in HCC clinical specimens. Both cell motility and cell invasion (the ability of a cell to migrate through an ECM barrier) contribute to HCC metastasis. Interestingly, let-7g appears to only interfere with cell motility in the HCC cell lines tested.
Let-7 is the second miRNA discovered that acts as a key regulator in developmental processes. Recently, this family was found to be involved in regulating the self-renewal and differentiation of neural stem cells and breast cancer cells by targeting HMGA2 and H-Ras [12,34]. Let-7b, but not other let-7 homologues, was found to regulate self-renewal of neural stem cells [34], while let-7a mainly contributed to breast cancer cells [12]. We recently identified a subgroup of HCC, EpCAM+AFP+ HCC, also known as HpSC-HCC, with stem cell features (i.e., the abilities to self-renew, differentiate, and initiate aggressive tumours in NOD/SCID mice) and poor clinical outcome [24,35,36]. We found that let-7g was significantly down-regulated in HpSC-HCC-related clinical specimens as well as in isolated HpSC-HCC cells [24] (Supplementary Fig. 1). Interestingly, the level of let-7g appeared to be unaffected following hepatic CSC self-renewal and differentiation (data not shown). These results suggest that down-regulation of let-7g in HpSC-HCC mainly contributes to certain aspects of HCC metastasis. It is possible that different let-7 homologues may have similar but not identical functions due to the presence of their own preferred targets.
In sum, our results indicate that the ability of let-7g to suppress HCC metastasis may be partially due to its ability to inhibit cell motility and inhibit colony formation through targeting COL1A2, and that down-regulation of let-7g could be used as a tool to predict HCC poor survival. The mechanism of let-7g down-regulation in metastatic HCC is unknown. One plausible mechanism is through Lin-28 because it is over-expressed in HCCs with poor outcome [21,22] and can mediate terminal uridylation leading to let-7 down-regulation [23]. Future studies are required to further evaluate the ability of let-7g to suppress metastasis, its functional link to Lin-28 in regulating HCC metastasis in mouse models, and to determine whether one can use let-7g-based therapy for metastatic HCC, a strategy that was recently described [37].
Supplementary Material
Acknowledgements
The authors who have taken part in this study declared that they do not have anything to declare regarding funding from industry or conflict of interest with respect to this manuscript. We thank the NIH Fellows Editorial Board for editing the manuscript. This work was supported by the Intramural Research Program of the Centre for Cancer Research, the National Cancer Institute (Z01 BC 010313 and Z01 BC 010876). LZ was also supported by China National Natural Scientific Fund (30801383).
Abbreviations
- HCC
hepatocellular carcinoma
- miRNAs
microRNAs
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial disclosure: the authors declare that they do not have anything to disclose regarding funding from industries or conflict of interest with respect to this manuscript.
References
- 1.Reference List
- 2.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 3.El-Serag HB, Mason AC. Rising incidence of hepatocellular carcinoma in the United States. N Engl J Med. 1999;340:745–750. doi: 10.1056/NEJM199903113401001. [DOI] [PubMed] [Google Scholar]
- 4.Budhu A, Wang XW. Human hepatocellular carcinoma : new insights from gene expression profiling. In: Jeffreis LP, editor. New Developments in Cancer Research. Nova Science Publishers Inc; 2006. pp. 1–32. [Google Scholar]
- 5.Ye QH, Qin LX, Forgues M, He P, Kim JW, Peng AC, et al. Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat Med. 2003;9:416–423. doi: 10.1038/nm843. [DOI] [PubMed] [Google Scholar]
- 6.Budhu A, Jia HL, Forgues M, Liu CG, Goldstein D, Lam A, et al. Identification of metastasis-related microRNAs in hepatocellular carcinoma. Hepatology. 2008;47:897–907. doi: 10.1002/hep.22160. [DOI] [PubMed] [Google Scholar]
- 7.Ji J, Wang XW. New kids on the block: Diagnostic and prognostic microRNAs in hepatocellular carcinoma. Cancer Biol Ther. 2009;8 doi: 10.4161/cbt.8.18.8898. [DOI] [PubMed] [Google Scholar]
- 8.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 9.Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- 10.Roush S, Slack FJ. The let-7 family of microRNAs. Trends Cell Biol. 2008;18:505–516. doi: 10.1016/j.tcb.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 11.Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 12.11. Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004;64:3753–3756. doi: 10.1158/0008-5472.CAN-04-0637.
- 13.Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–1123. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
- 14.Esquela-Kerscher A, Trang P, Wiggins JF, Patrawala L, Cheng A, Ford L, et al. The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle. 2008;7:759–764. doi: 10.4161/cc.7.6.5834. [DOI] [PubMed] [Google Scholar]
- 15.Chang SS, Jiang WW, Smith I, Poeta LM, Begum S, Glazer C, et al. MicroRNA alterations in head and neck squamous cell carcinoma. Int J Cancer. 2008;123:2791–2797. doi: 10.1002/ijc.23831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Torrisani J, Bournet B, Chalret du RM, Bouisson M, Souque A, Escourrou J, et al. Let-7 microRNA transfer in pancreatic cancer-derived cells inhibits in vitro cell proliferation but fails to alter tumor progression. Hum Gene Ther. 2009 doi: 10.1089/hum.2008.134. [DOI] [PubMed] [Google Scholar]
- 17.Peng Y, Laser J, Shi G, Mittal K, Melamed J, Lee P, et al. Antiproliferative effects by Let-7 repression of high-mobility group A2 in uterine leiomyoma. Mol Cancer Res. 2008;6:663–673. doi: 10.1158/1541-7786.MCR-07-0370. [DOI] [PubMed] [Google Scholar]
- 18.Schultz J, Lorenz P, Gross G, Ibrahim S, Kunz M. MicroRNA let-7b targets important cell cycle molecules in malignant melanoma cells and interferes with anchorage-independent growth. Cell Res. 2008;18:549–557. doi: 10.1038/cr.2008.45. [DOI] [PubMed] [Google Scholar]
- 19.Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet. 2007;39:673–677. doi: 10.1038/ng2003. [DOI] [PubMed] [Google Scholar]
- 20.Lee YS, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. :2007. doi: 10.1101/gad.1540407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–1579. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo Y, Chen Y, Ito H, Watanabe A, Ge X, Kodama T, et al. Identification and characterization of lin-28 homolog B (LIN28B) in human hepatocellular carcinoma. Gene. 2006;384:51–61. doi: 10.1016/j.gene.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 23.Viswanathan SR, Powers JT, Einhorn W, Hoshida Y, Ng TL, Toffanin S, et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet. 2009;41:843–848. doi: 10.1038/ng.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heo I, Joo C, Cho J, Ha M, Han J, Kim VN. Lin28 mediates the terminal uridylation of let-7 precursor MicroRNA. Mol Cell. 2008;32:276–284. doi: 10.1016/j.molcel.2008.09.014. [DOI] [PubMed] [Google Scholar]
- 25.Ji J, Yamashita T, Budhu A, Forgues M, Jia HL, Li C, et al. Identification of microRNA-181 by genome-wide screening as a critical player in EpCAM-positive hepatic cancer stem cells (p NA) Hepatology. 2009 doi: 10.1002/hep.22989. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 27.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 28.Shintani Y, Hollingsworth MA, Wheelock MJ, Johnson KR. Collagen I promotes metastasis in pancreatic cancer by activating c-Jun NH(2)-terminal kinase 1 and up-regulating N-cadherin expression. Cancer Res. 2006;66:11745–11753. doi: 10.1158/0008-5472.CAN-06-2322. [DOI] [PubMed] [Google Scholar]
- 29.Tanjore H, Kalluri R. The role of type IV collagen and basement membranes in cancer progression and metastasis. Am J Pathol. 2006;168:715–717. doi: 10.2353/ajpath.2006.051321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ogawa M, Ikeuchi K, Watanabe M, Etoh K, Kobayashi T, Takao Y, et al. Expression of matrix metalloproteinase 7, laminin and type IV collagen-associated liver metastasis in human colorectal cancer: immunohistochemical approach. Hepatogastroenterology. 2005;52:875–880. [PubMed] [Google Scholar]
- 31.Kumar MS, Erkeland SJ, Pester RE, Chen CY, Ebert MS, Sharp PA, et al. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc Natl Acad Sci U S A. 2008;105:3903–3908. doi: 10.1073/pnas.0712321105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chin LJ, Ratner E, Leng S, Zhai R, Nallur S, Babar I, et al. A SNP in a let-7 microRNA complementary site in the KRAS 3' untranslated region increases non-small cell lung cancer risk. Cancer Res. 2008;68:8535–8540. doi: 10.1158/0008-5472.CAN-08-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Christensen BC, Moyer BJ, Avissar M, Ouellet LG, Plaza S, McClean MD, et al. A let-7 microRNA binding site polymorphism in the KRAS 3' UTR is associated with reduced survival in oral cancers. Carcinogenesis. 2009 doi: 10.1093/carcin/bgp099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lettre G, Jackson AU, Gieger C, Schumacher FR, Berndt SI, Sanna S, et al. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat Genet. 2008;40:584–591. doi: 10.1038/ng.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nishino J, Kim I, Chada K, Morrison SJ. Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf Expression. Cell. 2008;135:227–239. doi: 10.1016/j.cell.2008.09.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamashita T, Forgues M, Wang W, Kim JW, Ye Q, Jia H, et al. EpCAM and alpha-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res. 2008;68:1451–1461. doi: 10.1158/0008-5472.CAN-07-6013. [DOI] [PubMed] [Google Scholar]
- 37.Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang HY, et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136:1012–1024. doi: 10.1053/j.gastro.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kota J, Chivukula RR, O'donnell KA, Wentzel EA, Montgomery CL, Hwang HW, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005–1017. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.