

Abstract
Structural determination of membrane proteins by NMR, especially for helical membrane proteins remains a challenge. Here we report the NMR assignment and secondary structure of a 31 kDa helical membrane protein, the C-terminal domain of Stt3p. The C-terminal domain of Stt3p has been proposed to be the catalytic domain of the yeast Oligosaccharyl Transferase (OT), a multi-subunit membrane-associated enzyme complex catalyzing N-glycosylation, an essential and highly conserved protein modification. NMR assignment is the first critical step in the determination of high-resolution solution structure and for further structure-function studies.
Although predicted to constitute about 30% of the genome and 50% of current drug targets, membrane proteins remain one of the great challenges in structural biology.1 In the Protein Data Bank (PDB), less than 1% of the atomic structures represent that of membrane proteins, and this percentage is actually decreasing since more structures of soluble proteins are being deposited everyday.2 The primary reason for this disparity is the requirement of lipids (typically detergents) for the solublization of membrane proteins. For X-ray crystallography, detergents often interfere with crystallization,3 while for NMR, membrane proteins embedded in detergent micelles tumble slowly leading to rapid transverse relaxation rates, which broaden the resonance line widths and dramatically complicate the NMR spectra. As a result, membrane protein structure determination by NMR have only been possible for some small α-helical proteins,4 and relatively larger outer membrane bacterial porins having β-barrel folds that provide better amide proton chemical shift dispersion and ample long-range nuclear Overhauser effects (NOEs).5 The NMR assignment of a 13 kDa α-helical membrane protein that forms 40 kDa homotrimer has been reported.6 Recently, a 241-residue helical membrane protein backbone assignment was reported.7 Here, we show that it is possible to use NMR to assign the backbone resonances for a 274-residue (253 residues plus 21 His-tagged residues) monomeric α-helical membrane protein, the C-terminal domain of Stt3p, in detergent micelles.
Oligosaccharyl transferase (OT) is a remarkably complex enzyme that catalyzes N-glycosylation, the most ubiquitous protein modification in eukaryotic cells. In the case of the yeast S. cerevisiae, OT is composed of nine nonidentical membrane protein subunits, of which, five subunits including Stt3p are essential for the viability of the cell.8 Stt3p, the only conserved subunit in the three domains of life,9 has been shown to be the catalytic subunit of the OT complex by several research groups.10 To date, only the NMR structure of a 36-residue Ost4p,11 a non-essential subunit of OT, and the crystal structure of the C-terminal domain of Stt3p from an archaeal source,12 that has very limited sequence similarities to the eukaryotic Stt3p, have been reported. We have recently over-expressed the His-tagged C-terminal domain of Stt3p (residue 466-718) in E. coli and produced very pure homogeneous protein at very high level and have demonstrated that SDS is the optimal detergent for structure determination by high-resolution solution NMR.13 Here we report the NMR assignment and secondary structure of this 31.5 kDa membrane protein.
All NMR experiments were carried out at 55 °C using uniformly 2H, 13C, 15N-labeled C-terminal domain of Stt3p. Except the HN(CO)CACB which was acquired as constant time (CT) type experiment, all experiments, including HNCACB, HNCA, HN(CO)CA, HNCO, HN(CA)CO, were collected as TROSY-based. TROSY-HNCACB experiment was recorded with the 13Cα-13Cβ transfer times optimized for maximum sensitivity of 13Cβ peaks using delays which were less than 1/(2JCαCβ). This led to the appearance of typically weak 13Cα correlations in these spectra, but strong cross-peaks involving the 13Cβ as expected. Protein stability at elevated temperatures was verified by Circular Dichroism (CD) and HSQC NMR experiments. Protein sample was found to be stable at 55 °C for at least one month. It is noteworthy that 100% perdeuteration was essential for assigning many resonances. It was also found that the TROSY-based experiments offer significant improvements in both resolution and sensitivity in these 1HN-15N correlation based experiments.
The high content of α-helical secondary structure in this protein (see below), combined with the relatively large number of cross peaks (263 non-proline residues), resulted in severe overlap in the central part of the HSQC spectra (Figure 1), making NMR assignment extremely challenging. The sequential NMR spin system connectivities were established using [15N-1H]-TROSY-HNCACB and [15N-1H]-TROSY-HNCA (Figure 2S), which provided both intra- and inter-residue (sequential) cross-peaks of Cβ and Cα respectively. Ambiguities were resolved by [15N-1H]-TROSY-HN(CO)CA and CT-HN(CO)CACB, which provides only sequential cross-peaks. All the assignments were also confirmed by another complementary pair experiments: [15N-1H]-TROSY-HNCO and [15N-1H]-TROSY-HN(CA)CO. It is noteworthy that, the use of CT-HN(CO)CACB was found to be extremely useful not only for the improved resolution in the carbon dimension, but more importantly, it provided the phase information, which was used in the assignment process. In the CT-HN(CO)CACB experiment, the Cβ resonance of residues with an odd number of aliphatic carbons attached has opposite sign to that of the Cβ resonance of residues with an even number of attached aliphatic carbons.14 Based on the fact that the number of amino acids containing odd sets of aliphatic carbons attached to the Cβ is approximately same as that of those amino acids with even sets of aliphatic carbons attached to the Cβ, the sign of the cross peaks of Cβ significantly facilitates in resolving ambiguities during assignments. Approximately 93% (255 out of 274 residues) backbone resonance assignments were completed for the C-terminal domain of Stt3p.
Figure 1.
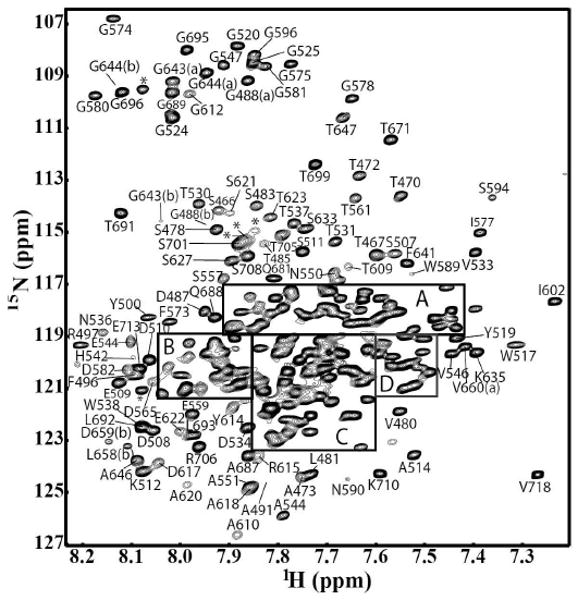
15N-1HN TROSY-HSQC spectrum of U-{15N, 13C, 2H}-labeled C-terminal domain of Stt3p recorded at 55°C. Four regions of the spectrum are enlarged (see Figure 1S) and peaks are labeled with residue numbers. Due to isoasparaginyl and proline cis/trans isomerizational linkage, there are two sets of assignment for a few residues, which are labeled with (a) and (b) respectively. The His-tagged residues are labeled with the symbol of *.
Interestingly, during the course of the assignment, an isoaspartyl linkage in the protein sequence, –IsoAsp642-Gly643, which is an isomerized form of deamidated Asn642-Gly643 connection (β-linked peptide), was unambiguously identified based on the fact that in the CT-HN(CO)CACB spectra, the cross peaks involving Cα and Cβ are of opposite signs (Figure 3S). Asparaginyl deamidation in proteins has been studied extensively, showing that this nonenzymatic post-translational modification may play an important role in protein stability and have significant impact on protein structure and/or function.15 Another interesting finding is the presence of proline cis/trans isomerizational linkage in this protein. In fact, there were two sets of cross peaks that were assigned for residues between Leu658 and Pro661 (Figure 4S); suggesting the peptide bond Val660-Pro661 adopts both cis and trans conformations. The question as to whether this proline cis/trans isomerization plays an important role, can only be addressed by further studies.
The secondary structure of the C-terminal domain of Stt3p was determined on the basis of the Chemical Shift Index (CSI), which is the deviations of Cα and Cβ chemical shifts from mean random coil values that have been corrected for deuterium isotope effects.16 In Figure 2, the parameter (ΔCα − ΔCβ) is plotted versus the protein sequence. The NMR-based secondary structure result is consistent with the far-UV CD spectroscopy data, which also indicates that the C-terminal domain of Stt3p is highly helical.13
Figure 2.
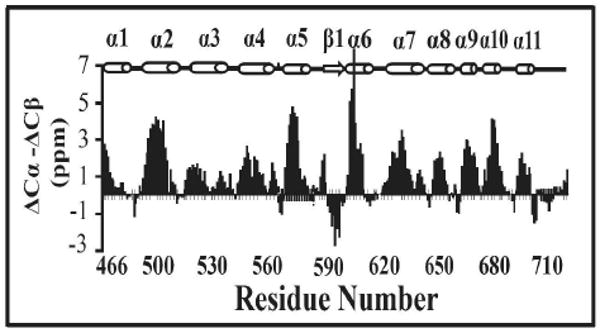
(A) ΔCα and ΔCβ are chemical shifts observed for the protein subtracted from the Cα and Cβ random-coil values respectively. The His-tagged residues were excluded for analysis.
In summary, we have presented nearly complete backbone (1HN, 15N, 13CO and 13Cα) and side-chain Cβ chemical shift assignments and the secondary structure for the His-tagged C-terminal domain of Stt3p, a 31.5 kDa, 274-residue helical membrane protein. The completion of the majority of the NMR resonance assignments demonstrates the feasibility of 3D structure determination for this 31.5 kDa monomeric helical membrane protein by solution NMR methods. Complete side-chain assignments and determination of the 3D structure of the C-terminal domain of Stt3p are progressing very well with the use of selectively protonated samples such as methyl-protonated {Ile(δ1 only), Leu(13CH3, 12CD3), Val(13CH3, 12CD3)} U-{15N, 13C, 2H} that were generated using biosynthetic precursors.17
The assigned backbone chemical shifts have been deposited in the BioMagResBank (BMRB accession number of 16701).
Supplementary Material
Acknowledgments
This research was financially supported by NIH grant DK082397 and NSF grant IBN-0628064 (to S. M). C. H. was financially supported by Alabama ACHE-GRSP Scholarship. The 900 MHz NMR data were collected at the NMR facility of the University of Georgia, funded by NIH grant GM66340.
Footnotes
Supporting Information Available: Figures 1S-4S, and experimental details for sample preparation and NMR data collection. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Korepanova A, Gao FP, Hua Y, Nakamoto RK, Cross TA. Protein Sci. 2005;14:148–158. doi: 10.1110/ps.041022305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao FP, Cross TA. Genome Biology. 2005;6:244. doi: 10.1186/gb-2005-6-13-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Sowadski JMJ. Bioenerg Biomembr. 1996;28:3–5. [PubMed] [Google Scholar]; (b) Ostermeier C, Michel H. Curr Opin Struct Biol. 1997;7:697–701. doi: 10.1016/s0959-440x(97)80080-2. [DOI] [PubMed] [Google Scholar]; (c) Roosild TP, Greenwald J, Vega M, Castronovo S, Riek R, Choe S. Science. 2005;307:1317–1321. doi: 10.1126/science.1106392. [DOI] [PubMed] [Google Scholar]
- 4.(a) Rastogi VK, Girvin ME. Nature. 1999;402:263–268. doi: 10.1038/46224. [DOI] [PubMed] [Google Scholar]; (b) MacKenzie KR, Prestegard JH, Engelman DM. Science. 1997;276:131–133. doi: 10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]
- 5.(a) Fernandez C, Hilty C, Wider G, Guntert P, Wuthrich K. J Mol Biol. 2004;336:1211–1221. doi: 10.1016/j.jmb.2003.09.014. [DOI] [PubMed] [Google Scholar]; (b) Hwang PW, Choy W, Lo EI, Chen L, Forman-Kay JD, Raetz CR, Prive GG, Bishop RE, Kay LE. Proc Natl Acad Sci. 2002;99:13560–13565. doi: 10.1073/pnas.212344499. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Arora A, Abildgaard F, Bushweller JH, Tamm LK. Nature Struct Biol. 2001;8:334. doi: 10.1038/86214. [DOI] [PubMed] [Google Scholar]
- 6.Oxenoid K, Kim HJ, Jacob J, Snnichsen FD, Sanders CR. J Am Chem Soc. 2004;126:5048–5049. doi: 10.1021/ja049916m. [DOI] [PubMed] [Google Scholar]
- 7.Gautier A, Kirkpatrick JP, Nietlispach D. Angew Chem Int Ed. 2008;47:7297–7300. doi: 10.1002/anie.200802783. [DOI] [PubMed] [Google Scholar]
- 8.Yan A, Lennarz WJ. J Biol Chem. 2005;280:3121–3124. doi: 10.1074/jbc.R400036200. [DOI] [PubMed] [Google Scholar]
- 9.Zufferey R, Knauer R, Burda P, Stagljar I, te Heesen S, Lehle L, Aebi M. EMBO J. 1995;14:4949–4960. doi: 10.1002/j.1460-2075.1995.tb00178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Yan Q, Lennarz WJ. J Biol Chem. 2002;277:47692–47700. doi: 10.1074/jbc.M208136200. [DOI] [PubMed] [Google Scholar]; (b) Nilsson I, Kelleher DJ, Miao Y, Shao Y, Kreibich G, Gilmore R, von Heijne G, Johnson AE. J Cell Biol. 2003;161:715–725. doi: 10.1083/jcb.200301043. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Glover KJ, Weerapana E, Numao S, Imperiali B. Chem Biol. 2005;12:1311–1315. doi: 10.1016/j.chembiol.2005.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Feldman MF, Wacker M, Hernandez M, Hitchen PG, Marolda CL, Kowarik M, Morris HR, Dell A, Valvano MA, Aebi M. Proc Natl Acad Sci. 2005;102:3016–3021. doi: 10.1073/pnas.0500044102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Nasab FP, Schulz BL, Gamarro F, Parodi AJ, Aebi M. Mol Biol Cell. 2008;19:3758–3768. doi: 10.1091/mbc.E08-05-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zubkov S, Lennarz WJ, Mohanty S. Proc Natl Acad Sci. 2004;101:3821–3826. doi: 10.1073/pnas.0400512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Igura M, Maita N, Kamishikiryo J, Yamada M, Obita1 T, Maenaka K, Kohda D. EMBO J. 2008;27:234–243. doi: 10.1038/sj.emboj.7601940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang C, Mohanty S, Banerjee M. Biochemistry. 2010 doi: 10.1021/bi902181v. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shan X, Gardner KH, Muhandiram DR, Rao NS, Arrowsmith CH, Kay LE. J Am Chem Soc. 1996;118:6570–6579. [Google Scholar]
- 15.(a) Manning MC, Patel K, Borchard RT. Pharm Res. 1989;6:903–918. doi: 10.1023/a:1015929109894. [DOI] [PubMed] [Google Scholar]; (b) Aswad DW, Paranandi MV, Schuter BT. J Pharm Biomed Anal. 2000;21:129–1136. doi: 10.1016/s0731-7085(99)00230-7. [DOI] [PubMed] [Google Scholar]
- 16.(a) Venters RA, Farmer BT, Fierke CA, Spicer LD. J Mol Biol. 1996;264:1101–1116. doi: 10.1006/jmbi.1996.0699. [DOI] [PubMed] [Google Scholar]; (b) Wishart DS, Sykes BD. J Biomol NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 17.Tugarinov V, Kay LE. J Am Chem Soc. 2003;125:13868–13878. doi: 10.1021/ja030345s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.