

Summary

Maturation of red fluorescent proteins has been shown to proceed through a blue intermediate. We determined the 2.10 Å crystal structure of the red fluorescent protein TagRFP and the 1.95 Å crystal structure of its derivative, the blue fluorescent protein mTagBFP. The crystallographic analysis is consistent with a model in which TagRFP has the trans coplanar anionic chromophore with the conjugated π-electron system, similar to that of DsRed-like chromophores. Refined conformation of mTagBFP suggests the presence of an N-acylimine functionality in its chromophore and single Cα-Cβ bond in the Tyr64 side chain. Mass spectrum of mTagBFP chromophore-bearing peptide indicates a loss of 20 Da upon maturation, while tandem mass spectrometry reveals that the Cα-N bond in Leu63 is oxidized. These data, together with mutagenesis of mTagBFP, indicate that mTagBFP has a new type of the chromophore, N-[(5-hydroxy-1H-imidazole-2-yl)methylidene]acetamide.
Introduction
Crystal structures for a number of green fluorescent protein (GFP)-like proteins, including fluorescent proteins (FPs) and absorbing but non-fluorescent chromoproteins (CPs), have been reported (Stepanenko et al, 2008). Chromophores in both FPs and CPs result from the spontaneous cyclization of X-Tyr-Gly tripeptides (where X represents any amino acid) inside the characteristic 11-stranded β-barrel. Chromophore formation is an autocatalytic process that only requires molecular oxygen. The green-emitting tyrosine-based chromophore consists of an anionic conjugate of imidazolone with p-hydroxybenzylidene (Ormö et al., 1996). Chromophore protonation results in an absorbance shift to ∼400 nm (Brejc et al., 1997). Blue and cyan color variants of GFP contain the substitutions of tyrosine in the chromophore with histidine and tryptophan (Heim and Tsien, 1996; Heim et al., 1994). In red tyrosine-based chromophores, such as in the one observed in DsRed (Yabrough et al., 2001), the peptide bond, which precedes the first amino acid in the chromophore tripeptide, is oxidized. The oxidation reaction yields an N-acylimine derivative that results in an extended π-system conjugation and a shift of the absorbance and emission spectra to the red (Yabrough et al., 2001).
The majority of FPs exhibits a cis chromophore configuration with the hydroxyphenyl ring coplanar to the imidazolone ring and pointing away from its carbonyl group (Ormö et al., 1996; Yabrough et al., 2001). Weakly fluorescent FPs and CPs, in turns, have the hydroxyphenyl ring in a trans configuration, i.e., pointing towards a carbonyl group of the imidazolone ring, and exhibit a noncoplanar conformation relative to the imidazolone ring (Prescott et al., 2003). The only known FP featuring a trans coplanar chromophore is eqFP611 (Petersen et al., 2003).
A detailed maturation mechanism for the chromophores of red FPs (RFPs) remains unknown (Piatkevich and Verkhusha, 2010). It has been suggested, however, that formation of the DsRed red chromophore proceeds via a green GFP-like immature intermediate (Gross et al., 2000). In contrast, our group proposed that the DsRed chromophore can be formed from a blue protonated chromophore intermediate with an absorbance maximum at ∼400 nm, but not from the anionic GFP-like chromophore, which is a dead-end product (Verkhusha et al., 2004). Absorbance at ∼400 nm has also been observed in immature species of many red-shifted CPs including asulCP, cgigCP, hcriCP and HcRed1 (Verkhusha et al., 2004; Wilmann et al., 2005). Previously, we have successfully applied a rational mutagenesis to five genetically unrelated RFPs, such as TagRFP (Merzlyak et al., 2007), mCherry (Shu et al., 2006), HcRed1 (Wilmann et al., 2005), M355NA (Bulina et al., 2003) and mKeima (Kogure et al., 2006), in order to develop the respective mTagBFP, mCherry-blue, HcRed1-blue, M355NA-blue and mKeima-blue blue FP (BFP) variants (Subach et al., 2008). That was achieved by introducing a limited number of the site-specific amino acid substitutions in these RFPs. Furthermore, several so called Fluorescent Timers, which change fluorescence color from blue to red over time, have been developed on the basis of mCherry (Subach et al., 2009), and their crystal structures have been determined (Pletnev et al., 2010).
Based on all these data we hypothesized that a formation pathway for a red chromophore via a blue intermediate should be conserved, and a direct comparison of chromophores and their environments in the closely related BFP and RFP should provide insight into the pathway of the red chromophore formation. Here, we report the crystal structures of TagRFP and its derivative mTagBFP, mass-spectral data of the mTagBFP chromophore-bearing peptide, and an extensive spectral and biochemical characterization of mTagBFP mutants. These data have revealed, for the first time, a chemical structure of the blue acylimine-containing chromophore and allowed us to suggest a chemical pathway for chromophore transformations from the non-oxidized colorless to the blue fluorescent state, and subsequently from the blue to the red fluorescent state.
Results
Overall structures of TagRFP and mTagBFP
The final crystal structure of TagRFP was refined at a 2.1 Å resolution (Table 1). The asymmetric unit contains four TagRFP protein chains arranged into two identical dimers (AB and CD) with total solvent accessible area of 18,080 Å2 per dimer (Figure S1A). The interface area within each dimer buries 1,475 Å2 per chain, significantly larger than the interface area of 409 Å2 per chain between the dimers. Most of the residues of each chain (Glu3 – Leu228) are well defined in the electron density map with the exception of six N-terminal residues and five C-terminal residues of each chain, which are not visible (here and below we follow the amino acid numbering used for eqFP611 (Petersen et al., 2003) and mKate (Pletnev et al., 2008); see Figure S1). The four molecules in the asymmetric unit have similar overall temperature factors of 23.9, 23.9, 24.2, and 23.7 Å2, and almost identical structures, with RMS deviations between all the Cα atoms of subunit A and three other subunits being 0.26, 0.29 and 0.14 Å, respectively.
Table 1.
Data collection and refinement statistics.
| Data collection | TagRFP | mTagBFP |
|---|---|---|
| Beamline | NSLS-X29A | NSLS-X29A |
| Wavelength (Å) | 1.08 | 1.08 |
| Resolution limits (Å) | 20–2.1 | 20–1.95 |
| Observed reflections | 200664 | 399633 |
| Unique reflections | 44512 | 65329 |
| Completeness (%) | 85.5 (42.2)a | 73.5 (25.1)a |
| Rmergeb | 0.090 (0.472)a | 0.070 (0.315)a |
| Refinement statistics | ||
| Protein nonhydrogen atoms | 6527 | 6869 |
| Water molecules | 664 | 402 |
| Rcrystc | 0.173 (0.387)a | 0.191 (0.348)a |
| Rfreec | 0.200 (0.397)a | 0.244 (0.419)a |
| Average B-factor (Å2) | 23.7 | 35.8 |
| RMSD from ideality | ||
| Bond length, Å | 0.010 | 0.023 |
| Bond angles (°) | 1.67 | 2.17 |
| Torsion angles (°) | 21.8 | 23.1 |
| Ramachandran plot | ||
| Core (%) | 93.6 | 94.2 |
| Allowed (%) | 5.2 | 4.7 |
| Generous (%) | 1.2 | 1.1 |
Values in parentheses indicate statistics for the high resolution bin.
Rmerge = ΣΣ j|Ij(hkl) – <I(hkl)>|/•ΣΣ j|<I(hkl)>|, where Ij is the intensity measurement for reflection j and <I> is the mean intensity over j reflections.
Rcryst/(Rfree) = Σ ‖Fo(hkl)| – |Fc(hkl)‖/•Σ |Fo(hkl)|, where Fo and Fc are observed and calculated structure factors, respectively. No σ-cutoff was applied. 5% of the reflections were excluded from refinement and used to calculate Rfree.
The crystal structure of mTagBFP was refined at a 1.95 Å resolution (Table 1). The asymmetric unit contains four mTagBFP protein chains (Figure S1C), which assemble into a tetramer identical to that observed in mKate (Pletnev et al., 2008). The tetramer demonstrates 222 point symmetry, its total solvent accessible area being 32,690 Å2 with the buried surface area of 2,240 Å2 per chain. The interface area between the dimers is 918 Å2 per chain, smaller than the interface of 1,450 Å2 per chain within a dimer. Within the mTagBFP tetramer, AB and CD dimers are very similar in overall structure to the TagRFP dimers. The overall temperature factors are 30.7, 33.6, 35.6, and 42.1 Å2 for the four molecules in the asymmetric unit. The RMS deviations between the subunit A and three other subunits are 0.24, 0.23, and 0.32 Å, respectively. The subsequent structural analysis will refer to the best defined A subunit of mTagBFP. Each chain of TagRFP and mTagBFP is folded into the characteristic 11-stranded β-barrel, described previously for GFP (Ormö et al., 1996), DsRed (Yabrough et al., 2001), eqFP611 (Petersen et al., 2003), HcRed (Wilmann et al., 2005), and mKate (Pletnev et al., 2008), where the central chromophores-containing α-helix is extended coaxially with the axes of the β-barrel (Figures S1B and S1D). Notably, gel filtration and semi-native gel electrophoresis experiments indicate that both TagRFP and mTagBFP remain monomeric in solution up to the concentration of 10 mg/ml (Merzlyak et al., 2007; Subach et al., 2008), indicating that oligomerization occurs only under very high protein concentration observed in the crystalline phase.
Chromophore structure in TagRFP
Electron density of the TagRFP Met63-Tyr64-Gly65 tripeptide suggests the formation of a 5-[(4-hydroxyphenyl)-methylene]-imidazolone chromophore. The chemical structure is the same as in mKate (Pletnev et al., 2008) and eqFP611 (Petersen et al., 2003). The electron density for Met63 can be interpreted as such that Cα is planar and characterized by sp2 hybridization, suggesting the formation of an N-acylimine double bond between Cα and N. As in other red and far-red FPs, this additional N-acylimine may extend the π-bonding system resulting in a red shift in excitation/emission spectra. Similar to other red FPs (Yabrough et al., 2001; Petersen et al., 2003), the Phe62-Met63 peptide bond in TagRFP has a cis conformation.
TagRFP has the almost coplanar trans chromophore with χ1 = -5±4° and χ2 = 5±4° (subunit A) (Figure 1A). The eqFP611 (Petersen et al., 2003) and mKate proteins at low pH (Pletnev et al., 2008) have the similar trans coplanar chromophore. The amino acid sequences of TagRFP and eqFP611 share 76% identity (Figure S1E). The immediate surroundings of the chromophores in these proteins are identical except for Arg67 in TagRFP, which is replaced by Lys67 in eqFP611. However, mKate carries three additional alterations in the immediate environment of the chromophore relative to TagRFP (Lys67, Ser143, Leu174, Arg197 in mKate vs. Arg67, Asn143, Phe174, His197 in TagRFP); and the mKate chromophore in the fluorescent state (high pH) has a cis configuration. The TagRFP chromophore is involved in multiple interactions within the β-barrel interior (Figure 2A).
Figure 1.

The structures of the chromophore-forming tripeptides superimposed onto their (2Fo-Fc) electron density maps. (A) TagRFP, subunit A. (B) mTagBFP, subunit A, two orthogonal views. Oxygen and nitrogen atoms are colored red and blue, respectively. The chromophore backbones for TagRFP and mTagBFP are shown in orange and cyan, respectively. See also Figure S1.
Figure 2.
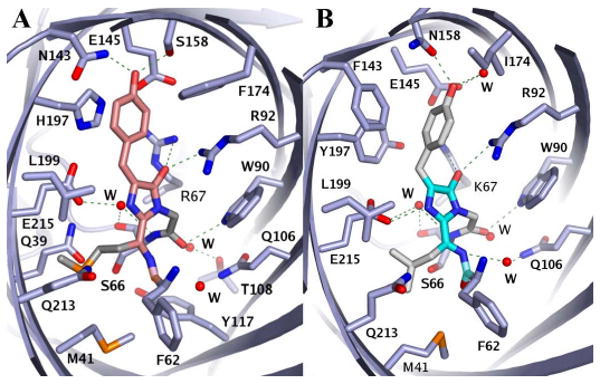
Chromophores and their environments in TagRFP (A) and mTagBFP (B). The chromophore backbones for TagRFP and mTagBFP are shown in orange and cyan, respectively. The hydrogen bonds are indicated with green dashed lines; the atoms are colored by atom type; and the water molecules are shown as red spheres. The Ala59, Thr60 and Ser61 residues are not included for figure clarity. See also description of contacts of the TagRFP and mTagBFP chromophores with the respective β-barrel interiors in the Supplemental Data.
Chromophore structure in mTagBFP
Electron density of mTagBFP can be interpreted as being consistent with the post-translational modification of the chromophore-forming tripeptide Leu63-Tyr64-Gly65 that results mainly in the formation of a 4-(4-hydroxybenzyl)-1H-imidazole-5-ol chromophore. Electron density at Leu63 Cα suggests sp2 hybridization, indicating that the blue fluorescent protein mTagBFP may have an N-acylimine double bond between Cα and N of Leu63, similar to that observed in red fluorescent proteins. Slightly diffuse electron density map might indicate presence of some sp3 hybridization at Leu63 Cα. This conformation could correspond to a minor fraction of immature mTagBFP molecules without acylimine. To further reveal chemical structure of the mTagBFP chromophore we subjected the chromophore-bearing peptide to mass spectrometry analysis (see below). It confirmed the presence of the N-acylimine C=N bond in the mTagBFP chromophore. Another chemical structure of the mTagBFP chromophore has not been detected. Therefore, a somewhat diffuse electron density at Leu63 Cα observed in the mTagBFP crystal is possibly the result of the limited resolution of mTagBFP structure. The Phe62-Leu63 peptide bond is close to the cis conformation that is characteristic of red FPs with the N-acylimine (Yabrough et al., 2001; Petersen et al., 2003).
The hydroxyphenyl group of Tyr64 has the noncoplanar conformation with the 1 = 136±4° and 2 = -66±4° (Figure 1B). A similar noncoplanar conformation has been observed in the dark state of photoswitchable FPs, in chromoproteins and in FPs with low quantum yields (Prescott et al., 2003; Quillin et al., 2005; Wilmann et al., 2006; Stiel et al., 2007; Andresen et al., 2005). The presence of this conformation in mTagBFP is remarkable, given the high quantum yield of this FP. Since none of the torsion angles is 0 or 180 degrees, Cβ of Tyr64 has sp3 hybridization. Based on these data, we suggest that in mTagBFP the hydroxyphenyl group of Tyr64 is not in conjugation with the imidazole ring, and that the Cα-Cβ bond of the Tyr64 is fully reduced. At the same time, the electron density of mTagBFP suggests that the Cα atom of Tyr64 has mainly planar geometry. In this conformation the imidazole ring is coplanar with the C=N bond of the acylimine in all four molecules of the asymmetric unit. This is consistent with the aromatic character of the imidazole ring and suggests sp2 hybridization of Tyr64 Cα. We cannot exclude, however, that there may be an alternative conformation of the chromophore, in which the imidazolone ring is present. Since the mTagBFP crystal structure mainly shows the planar geometry of the imidazole ring, the keto-enol equilibrium between imidazolone and imidazole-5-ol is shifted toward the imidazole-5-ol heterocycle. This shift possibly occurs because the delocalization of electron density in the aromatic imidazole-5-ol system is more energetically advantageous than that in the non-aromatic (non-conjugated) imidazolone ring. The mTagBFP chromophore is involved in multiple interactions within the β-barrel interior (Figure 2B). Contacts of the TagRFP and mTagBFP chromophore-tripeptides with the respective β-barrel interiors are described in the Supplemental Data.
Spectral properties of TagRFP and mTagBFP
Spectral variety of FPs is determined by different types of chromophores but also by chromophore interactions with the surrounding amino acids within the β-barrels. After denaturation in acid or alkali, absorbance spectrum reflects the type of the chromophore structure free from interactions with other residues. In acidic conditions (0.2 M HCl), TagRFP showed a single maximum at 387 nm. In alkaline conditions (0.7 M NaOH) it had a single maximum at 449 nm (Figure 3A). These spectral forms of the denatured TagRFP were interconvertible with a change in conditions from acidic to alkaline and vice versa. The maxima were similar to those observed for GFP in acid and alkali (Niwa et al., 1996). The DsRed-like chromophore in TagRFP converted into the GFP-like chromophore under acidic or alkaline conditions. Possibly, a hydration of the N-acylimine C=N bond in the TagRFP chromophore has occurred. Similar hydration of the acylimine bond has been observed for the chromophores in DsRed itself (Gross et al., 2000) and in gtCP (Martynov et al., 2003). In contrast, mTagBFP exhibited a single absorbance maximum at 335 nm in acid, and single maximum at 423 nm in alkali (Figure 3B). The mTagBFP forms, corresponding to these maxima, converted to each other with a change of the pH conditions as well. The maxima differed from those for GFP in acidic and alkaline conditions (Niwa et al., 1996). These data suggested that mTagBFP contains a non-GFP-like and non-DsRed-like chromophore.
Figure 3.

Absorbance spectra of TagRFP (A) and mTagBFP (B) in PBS buffer (solid lines), denatured in 0.7 M NaOH (dashed lines) and denatured in 0.2 M HCl (dotted lines).
Mutants of mTagBFP affecting chromophore formation
To study properties of the chromophore structure in mTagBFP, a site-directed mutagenesis of the amino acid residues of the chromophore-forming tripeptide and of its immediate environment was carried out next.
Position 64
To test whether a side chain of Tyr64 is necessary for the formation of the mTagBFP chromophore, we first substituted Tyr64 with several aliphatic residues such as Leu, Val, Ala and Gln. The substitution resulted in blue-emitting FP variants with the absorbance maxima at 398-400 nm and the emission maxima at 453-459 nm, which were very similar to those observed for mTagBFP (Table 2, Figure S4A-E). The spectral data suggested that Tyr64 in mTagBFP does not form a Cα-Cβ double bond and, therefore, the side chain of Tyr64 is not involved in the conjugation with the imidazole-5-ol residue.
Table 2.
Spectral properties of the mTagBFP mutants (see also Figure S2).
| mTagBFP mutant | Absorbance maximum in PBS, nm | Absorbance maximum in 0.7 M NaOH, nm | Absorbance maximum in 0.2 M HCl, nm | Fluorescence maximum, nm (excitation at 400 nm) | Extinction coefficient, M-1 cm-1 | Quantum yield | Brightness relative to mTagBFP, % * |
|---|---|---|---|---|---|---|---|
| mTagBFP | 400 | 423 | 335 | 455 | 52,000 | 0.63 | 100 |
| Y64L | 399 | ND ** | ND | 459 | 22,000 | 0.68 | 46 |
| Y64V | 400 | ND | ND | 457 | 13,000 | 0.68 | 27 |
| Y64A | 398 | ND | ND | 453 | 6,000 | 0.47 | 9 |
| Y64Q | 399 | ND | ND | 457 | 23,000 | 0.77 | 54 |
| F143S | 395, 515, 581 | 447 | 383 | 462 | 5,000 | 0.01 | 2 |
| 631 *** | 11,000 | 0.003 | 0.4 **** | ||||
| S66A | 400 | 419 | ND | 456 | 6,000 | 0.22 | 4 |
| Q106L | 380, 400 | 379, 428 | ND | 452 | 2,000 | 0.20 | 1 |
| E215A | 386 | ND | ND | not detectable | ∼0 | ∼0 | ∼0 |
Brightness of mTagBFP mutants was determined as a product of quantum yield and extinction coefficient and normalized to the brightness of mTagBFP, which was assumed 100%.
ND: not determined.
Fluorescence maximum at 631 nm was obtained with excitation at 581 nm.
Brightness relative to mKate.
Position 143
According to the mTagBFP crystal structure, the hydroxyphenyl group of Tyr64 is noncoplanar with the imidazole-5-ol. This occurs because the bulky side chain of Phe143 pushes the hydroxyphenyl group out of plane of the imidazole-5-ol ring. The Phe143Ser substitution resulted in the mutant with three absorbance maxima: at 395, 515 and 582 nm (Table 2, Figure S2F). Excitation of the mTagBFP/F143S mutant at 395 and 582 nm resulted in fluorescence with maxima at 462 and 631 nm, respectively (Table 2, Figure S2F). At the same time, the mTagBFP/F143S mutant showed a single absorbance maximum in acidic (at 383 nm) or alkaline (at 447 nm) conditions. Both absorbance maxima are characteristic of the GFP-like (Niwa et al., 1996) and DsRed-like (Gross et al., 2000) chromophores. The data suggest that the 395 nm, 515 nm, and 581 nm mTagBFP/F143S forms correspond to the protonated and anionic states of GFP-like chromophore, and DsRed-like chromophore, respectively. Because absorbance and fluorescence maxima of the mTagBFP/F143S are at 581 and 631 nm, respectively, its DsRed-like chromophore possibly has a cis configuration similar to that of mKate (Pletnev et al., 2008). Apparently Ser143 in the mTagBFP/F143S stabilizes the cis configuration of its chromophore. At the same time, Ser158 in TagRFP stabilizes the trans configuration of its chromophore. Thus, the Phe143 bulky side chain sterically inhibits oxidation of the Cα-Cβ bond in the Tyr64 side chain, by preventing the Tyr64 side chain to acquire conformation coplanar with the imidazole residue. The replacement of the phenyl group in the position 143 allowed oxidation and the formation of the red coplanar chromophore.
Positions 66, 106 and 215
To study whether an N-acylimine is formed in mTagBFP, we further mutated amino acid residues that are in close proximity to the N-acylimine in RFPs, such as the conserved among RFPs residues Ser66, Gln106 and Glu215. Earlier, the role of Oγ of the Ser69 (corresponding to Ser66 in mTagBFP) side chain in formation of the N-acylimine C=N bond in the DsRed chromophore has been proposed (Sniegowski et al., 2005). The Ser66Ala substitution resulted in the reduced fluorescence brightness at 456 nm with the absorbance peak at 400 nm (Table 2, Figure S2G). In other words, although the formation of the blue chromophore was deteriorated, it was still possible in the absence of the hydroxyl group of Ser66.
According to the crystal structure of mTagBFP, the Nε2 atom of Gln106 forms a hydrogen bond with a water molecule that, in turns, makes a hydrogen bond with the N atom of Leu63 (Figure 2B). The Gln106Leu mutation resulted in substantially reduced fluorescence brightness of mTagBFP at 452 nm with absorbance peak at 400 nm (Table 2, Figure S2H). mTagBFP/Q106L had an additional absorbance maximum at 380 nm. Similarly to the parental mTagBFP, in alkali, the 400 nm maximum converted into the red-shifted 428 nm. In the same conditions, however, the 380 nm peak converted into 379 nm. SDS-PAGE analysis showed that about 50% of the polypeptide chain of the mTagBFP/Q106L mutant were cleaved in the N-acylimine region. We suggest that the absorbance maximum at 380 nm corresponds to the hydrolyzed N-acylimine C=N bond similar to that observed for denatured DsRed at neutral or alkaline pH (Gross et al., 2000). These data indicate that Gln106Leu substitution reduced efficiency of the N-acylimine formation or its stability.
A catalytic role of Glu215 in the mechanism of the N-acylimine C=N bond formation in the DsRed chromophore has been suggested (Sniegowski et al., 2005). The Glu215Ala substitution in mTagBFP resulted in the complete absence of fluorescence with only a weak absorbance peak at 386 nm, suggesting very inefficient chromophore maturation. Nevertheless, we propose that the formation of the imidazole moiety of the chromophore still occurs. That was also the case for EGFP/E222A (Barondeau et al., 2007) and mKO/E212A (Kikuchi et al., 2008) mutants (equivalent to mTagBFP/E215A variant). Therefore, our data indicate the important role of the Glu215 side chain in the formation of the mTagBFP chromophore. Altogether, these data suggested an existence of an N-acylimine in the mTagBFP chromophore and indicated that the surrounding Ser66, Gln106 and Glu215 residues affect either the N-acylimine formation efficiency or its stability.
Mass-spectral analysis of the mTagBFP chromophore-bearing peptide
To confirm the chemical structure of the mTagBFP chromophore, we performed mass-spectral analysis. Denatured mTagBFP was digested by chymotrypsin. The mTagBFP peptide mixture was then desalted and subjected to the mass-spectral analysis. The mass spectrum of mTagBFP chymotrypsin-digested fragments has revealed two monoisotopic masses of 795.4 Da and 1201.6 Da that correspond to chromopeptides. These masses are exactly what would be expected from cyclization (loss of H2O) and oxidation (loss of H2) of the chromophore inside the fragments LYGSKTF (cleavage after Phe62 and Phe69) and ATSFLYGSKTF (cleavage after Leu58 and Phe69). Both fragments were separately subjected to further MS/MS fragmentation. The daughter ions for the fragment LYGSKTF are present in Figure 4. Analysis of the MS/MS fragments showed that the dehydrogenation site was located between nitrogen and α-carbon of Leu63. The most prominent peak y6(I) originated from the cleavage in the vicinity of the N-acylimine C=N bond inside Leu63. The same fragment was observed upon MS/MS fragmentation of the species ATSFLYGSKTF. Fragments with the same cleavage site near the C=N bond were observed in the case of the N-acylimine-containing chromopeptides derived from proteolysis of DsRed (Gross et al., 2000) and chromoprotein gtCP (Martynov et al., 2003). Therefore, mass-spectral data are consistent with the presence of the acylimine C=N bond and with the absence of the double bond between hydroxyphenyl ring of Tyr64 and imidazole-5-ol ring formed by the Leu63-Tyr64-Gly65 tripeptide.
Figure 4.

Mass-spectral analysis of the chromophore-bearing peptide of mTagBFP. Table representing the distinctive peaks in MS/MS spectra of the chromophore-bearing peptide ions and structure of the mTagBFP chymotrypsin-derived chromopeptide are shown. The most prominent MS/MS fragments are presented in brackets.
Discussion
An understanding of how specific chemical environments define the distinctive spectral properties of red and blue chromophores might reveal a pathway for their formation. Our data show that TagRFP has the trans coplanar anionic chromophore (Figure 1A). The side chains of Asn143 and Ser158 make hydrogen bonds with the hydroxyl of the hydroxyphenyl moiety (Figure 2A). These two hydrogen bonds, together with the π-π interaction between the hydroxyphenyl group and His197, are probably the most important determinants for stabilizing the hydroxyphenyl moiety in a trans coplanar anionic configuration. It has been shown in eqFP611 (Nienhaus et al., 2008) and mKate (Pletnev et al., 2008) that trans or cis anionic configurations of the chromophore are determined by residues 143 and 158. When Ser was introduced at the position 143, a cis configuration was stabilized; and when Ser was introduced at the position 158, a trans configuration was stabilized.
The comparison of TagRFP and mTagBFP structures, as well as the analysis of mTagBFP/F143S mutant, revealed the main driving force favoring the formation of the blue chromophore in mTagBFP. Phe143 pushes the hydroxyphenyl group of Tyr64 out of the plane of the imidazole-5-ol ring and prevents the formation of the Cα-Cβ double bond in the Tyr64 side chain. We suggest a chemical structure of the mTagBFP chromophore (B-form) and mechanism of its formation in Figure 5. This structure is based on several lines of evidence. First of all, the electron density in the mTagBFP crystal is consistent with the mainly planar geometry of Leu63 Cα and tetrahedral geometry of Tyr64 Cβ. This implies that the N-acylimine is possibly present in the mTagBFP chromophore, however, there is no oxidation of the Cα-Cβ bond in the Tyr64 side chain. Moreover, electron density indicates mainly planar geometry of the Tyr64 Cα and coplanar imidazole ring with the C=N acylimine bond. This observation suggests that mTagBFP contains the imidazole-5-ol but not the imidazolone in its chromophore. Secondly, the mTagBFP denaturation in alkali or acid resulted in the absorbance spectra that are not characteristic of the GFP-like or DsRed-like chromophores (Figure 3B), which contain the Cα-Cβ double bond in the Tyr64 side chain. Thirdly, mutagenesis of Ser66, Gln106 and Glu215 surrounding the N-acylimine influenced either formation or stability of the N-acylimine C=N bond. Moreover, mTagBFP and its mutants with the aliphatic residues at position 64 had almost identical absorbance and emission spectra. This confirms that there is no Cα-Cβ double bond in the Tyr64 side chain. Lastly, the mass spectrum analysis provides the mass of the chromophore-bearing peptide to be 20 Da less than that calculated from the amino acid content. Tandem mass spectrometry indicates that the dehydrogenation site in the mTagBFP chromophore is located between N and Cα of Leu63, which is similar to the site of the N-acylimine C=N bond in the DsRed-like chromophore. Altogether, these data are in agreement with N-[(5-hydroxy-1H-imidazole-2-yl)methylidene]acetamide, which we propose to be the mTagBFP chromophore (B-form in Figure 5).
Figure 5.

Proposed pathways for the formation of the mTagBFP and TagRFP chromophores. The C-form is the chromophore precursor; the B-form is the blue-emitting chromophore form; and the R-form is the red-emitting chromophore form.
It has been suggested that the first step in maturation of a DsRed chromophore is the formation of the GFP-like chromophore containing the Cα-Cβ double bond in the Tyr64 side chain (Gross et al., 2000; Yabrough et al., 2001). The second step involves an oxidation of the bond between the Cα and N of the first amino acid in the chromophore tripeptide to form the N-acylimine. The crystallographic analysis, mass spectrometry data and properties of the site-specific mutants allowed for the proposal of a hypothetical chemical pathway for the formation of the TagRFP chromophore (R-form) (Figure 5). The key feature of the pathway is that the formation of the N-acylimine C=N bond precedes the oxidation of the Cα-Cβ bond in the Tyr64 side chain. It is in agreement with the observation that the DsRed-like red chromophores are formed after the blue-emitting precursors (Verkhusha et al., 2004) and with the chemical structure of the mTagBFP blue chromophore discussed above.
There are two possible pathways for the B-form formation (Figure 5). In one pathway, the first step consists of cyclization of the chromophore tripeptide, followed by dehydration, resulting in the formation of a colorless non-hydroxylated C-form. The next oxidation step results in the formation of an N-acylimine containing the blue-emitting chromophore (B-form). In an alternative pathway for the B-form formation, the oxidation step precedes dehydration of the tripeptide. Oxidation results in the formation of the hydroxylated C-form. Dehydration of the hydroxylated C-form can then occur with the formation of the B-form. It has been shown for GFP maturation that the oxidation reaction, accompanied by the hydrogen peroxide release, takes place before the chromophore dehydration (Zhang et al., 2006) that is similar to the second proposed pathway. Further oxidation of the Cα-Cβ bond in the Tyr64 side chain of the B-form results in the formation of the red chromophore (R-form).
We suggest that the formation pathway for the TagRFP's red chromophore via the mTagBFP-like intermediate proposed in this work may also occur in other RFPs (Piatkevich and Verkhusha, 2010). This suggestion is based on several observations. First, the blue intermediates have been observed in the courses of maturation of many RFPs and red-shifted CPs (Verkhusha et al., 2004; Wilmann et al., 2005). Second, several Fluorescent Timer proteins, which change color from blue to red over time, were developed from mCherry RFP (Subach et al., 2009; Pletnev et al., 2010). Lastly, several BFPs have been obtained by introducing the similar mutations into RFPs of different genetic background (Subach et al., 2008).
A model for the formation of the red chromophore via a GFP-like intermediate has been suggested on the basis of the observation of green and red emitting species in DsRed (Gross et al., 2000). Later, however, it was shown that the GFP-like chromophore is not an intermediate in the maturation pathway of the DsRed-like red chromophore but the dead-end product (Verkhusha et al., 2004). A possible reason why the red chromophore can not formed from the green intermediate is that the GFP-like chromophore is anionic (Niwa et al., 1996). A carbanion state has been proposed for the formation pathway of the DsRed-like chromophore (Yabrough et al., 2001). However, formation of the carbanion from the anionic GFP-like chromophore is energetically unfavorable. In contrast, the proposed mTagBFP-like chromophore does not contain a negative charge and allows formation of its carbanion derivative; consequently, the DsRed-like red fluorescent chromophore can be formed from it.
Figure 5 can be expanded further to explain formation of different fluorescent colors observed in FPs. After cyclization of the chromophore-bearing tripeptide, there are two chemical pathways. The first one is the formation of the N-acylimine, and the second one is oxidation of the Cα-Cβ bond in the Tyr64 side chain. These two reactions are competitive processes. In GFPs, reaction of the Tyr64 Cα-Cβ bond dehydrogenation occurs first and results in the anionic GFP-like chromophore (Niwa et al., 1996). The second oxidation, which leads to the N-acylimine formation, becomes energetically unfavorable. It can occur, nevertheless, with a GFP-like chromophore in an excited state in the presence of a strong oxidant (Bogdanov et al., 2009). In contrast, in RFPs, the N-acylimine formation occurs first, followed by oxidation of the Cα-Cβ bond in Tyr64. In BFPs, the chemical transformations are stopped right after the N-acylimine formation.
Significance
In this work the crystal structures of the red fluorescent protein TagRFP and its blue variant mTagBFP have been determined. Based on the crystal structure, mass spectra, and analysis of the site-specific mutants, we have shown that mTagBFP has a novel type of the chromophore, which contains the N-acylimine but does not have the Cα-Cβ double bond in the Tyr64 side chain. We have proposed the chemical pathway for the formation of the DsRed-like chromophore. This pathway may provide a basis for the rational design of FPs with novel spectral and photophysical properties, and for the development of molecular FP-based biosensors.
Experimental Procedures
Cloning, expression, and protein purification
For bacterial expression of TagRFP, the pBAD/HisB vector (Invitrogen) was modified by inserting the DNA sequence of the Tobacco Etch Virus (TEV) protease site (-ENLYPQG- amino acids) (Carrington and Dougherty, 1988) between the BglII site and the sequence encoding His6-tag. The polymerase chain reaction (PCR) amplified BglII/EcoRI fragment encoding TagRFP was then cloned into the modified pBAD/HisB. The recombinant TagRFP protein with the N-terminal His6-tag was expressed in the LMG194 bacterial strain (Invitrogen) by overnight culture in RM minimal medium at 37°C in the presence of 0.005% arabinose. The culture was then centrifuged at 5,000 rpm at 4°C for 15 min, the cell pellet resuspended in 50 mM NaH2PO4, 300 mM NaCl, pH 8.0 buffer and lysed by sonication on ice. The recombinant protein was purified with Ni-NTA resin (Qiagen) followed by dialysis for 3 h against the TEV protease cleavage buffer consisting of 50 mM Tris-HCl, 1 mM EDTA, 1 mM DTT, 150 mM NaCl at pH 7.5. The His6-tag was cleaved with TEV protease at a TagRFP:TEV protease ratio of 10:1 at room temperature for 20 h. The released TagRFP was dialysed against 10 mM NaH2PO4, pH 6.0 followed by purification with cation-exchange chromatography using SP-sepharose (GE Healthcare).
For bacterial expression of mTagBFP, the pBAD/HisB vector (Invitrogen) was modified by shortening the N-terminal His6-tag to the MGSHHHHHHGRS- amino acids. The PCR-amplified BglII/EcoRI fragment encoding mTagBFP was cloned into the modified pBAD/HisB vector and expressed in LMG194 host (Invitrogen). The bacterial culture in RM minimal medium supplemented with 0.005% arabinose was grown overnight at 37°C. The culture was centrifuged at 5,000 rpm at 4°C for 15 min. The cell pellet was resuspended in 50 mM NaH2PO4, 300 mM NaCl, pH 8.0 buffer and lysed by sonication on ice. The recombinant protein was purified using Ni-NTA resin (Qiagen) followed by dialysis for 3 h against 10 mM NaH2PO4, pH 6.0. The mTagBFP protein was then purified by cation-exchange chromatography using the SP-sepharose (GE Healthcare).
Protein crystallization
Before crystallization, TagRFP protein without His6-tag and mTagBFP protein containing a short N-terminal His6-tag were dialyzed against 10 mM Tris-HCl, 100 mM NaCl, pH 8.0 and concentrated to 15 mg/ml using the 3,000 MWCO Amicon (Millipore) centrifugal concentrator. Diffraction quality crystals were grown using the sitting drop vapor diffusion method by mixing 1 μl of protein and 1 μl of reservoir solution and equilibrating the samples against reservoir solution. For TagRFP, the reservoir solution contained 30% PEG 4000, 0.1 M Tris-HCl, pH 8.5, 0.2 M MgCl2, 0.1 M TCEP. Diffraction from the TagRFP crystals was consistent with the space group I41 with unit cell dimensions a= b= 130.99 Å, c= 105.97 Å, α= β= γ= 90° and four TagRFP chains in the asymmetric unit. In the case of mTagBFP, the reservoir solution contained 2.0 M ammonium sulfate. Diffraction was consistent with the space group P212121, with unit cell dimensions a= 82.95 Å, b= 105.94 Å, c= 137.16 Å, α= β= γ= 90° and four mTagBFP chains in the asymmetric unit.
X-ray diffraction data collection and crystallographic refinement
Crystals of TagRFP with dimensions 0.05 × 0.05 × 0.4 mm3 were mounted in cryo-loops directly from the crystallization droplet and flash-cooled in liquid nitrogen. Prior to freezing, 1 μl of 2 M lithium sulfate was added (as cryo-protectant) to the droplets containing mTagBFP crystals, then crystals were flash-cooled in liquid nitrogen. Diffraction data were collected on a Quantum 315 CCD detector (Area Detector Systems Corporation, Poway) with 1.08 Å wavelength radiation on the X29A beamline (National Synchrotron Light Source, Brookhaven. Intensities were integrated using the program HKL2000 and reduced to amplitudes using the program TRUNCATE (see Table 1 for statistics) (Otwinowski and Minor, 1997; Storoni et al., 2004). The structures were determined using the molecular replacement method with PHASER (Collaborative Computational Project, 1994). Model building and refinement were performed with the programs REFMAC and COOT (Storoni et al., 2004; Emsley and Cowtan, 2004). The quality of the final structure was verified with composite omit maps, and the stereochemistry was checked with the programs WHATCHECK (Hooft et al., 1996) and PROCHEK (Laskowski et al., 1993). The LSQKAB and SSM algorithms were used for structural superimpositions (Storoni et al., 2004; Krissinel and Henrick, 2004). Quaternary structures were analyzed using the PISA server (Krissinel and Henrick, 2007). No rotational order-disorder structure was observed (Pletnev et al., 2009). The atomic coordinates and structural factors have been deposited in the Protein Data Bank (entry 3M22 for TagRFP and entry 3M24 for mTagBFP).
Mutagenesis of mTagBFP and TagRFP
mTagBFP and TagRFP mutants were produced by site-directed mutagenesis using the mTagBFP and TagRFP genes in the pBAD/HisB vector as the templates. The mutant proteins were expressed in LMG host (Invitrogen) and purified as described above for TagRFP and mTagBFP.
Spectroscopy
Absorbance spectra were recorded on a U-3010 spectrophotometer (Hitachi). The excitation and emission spectra were measured using a FluoroMax-3 spectrofluorometer (Jobin Yvon). For measurements the protein samples in phosphate buffered saline (PBS) were used. Absorbance and fluorescence of acid or alkali denatured FPs were measured right after the mixing of the protein sample with HCl (to the final concentration of 0.2 M) or NaOH (to the final concentration of 0.7 M) in the optical cuvette. Brightness of proteins was calculated as a product of quantum yield and extinction coefficient. Quantum yield was measured using mTagBFP (quantum yield is 0.63 (Subach et al., 2008)) or TagRFP (quantum yield is 0.48 (Merzlyak et al., 2007)) as the reference standards, respectively. Protein concentrations used in the calculation of extinction coefficients were determined by the BCA assay (Pierce).
Enzymatic Proteolysis
Aliquot of 20 μg of His6-tag mTagBFP was reduced with 5 mM DTT and alkylated with 10 mM iodoacetamide and then subjected to an in-solution digestion with chymotrypsin as described (Gross et al., 2000). The sample was evaporated to near dryness in a Speedvac and resuspended in 3 μl of 6 M guanidinium chloride and heated to 80°C for 30 s. The sample was cooled to room temperature and 1.0 μl of 0.33 M hydrochloric acid was added to neutralize the digestion buffer. 30 μl of chymotrypsin buffer containing 0.1 M Tris-HCl, pH 7.8 and 10 mM CaCl2 was added, and then chymotrypsin was added at an enzyme/protein ratio of 1:60. The digests were incubated at room temperature for 22 h and quenched with 0.1% TFA. The peptides were desalted and isolated using a C18 ZipTip (Millipore).
Peptide Analysis
The chymotryptic peptides were mixed (1:1) with α-cyano-4-hydroxycinnamic acid solution (50% acetonitrile and water containing 0.1% tri-fluoroacetic acid). An aliquot of 1 μl of the mixture was put on a MALDI target and air-dried. Mass spectra were acquired on a 4800 MALDI TOF/TOF mass spectrometer (Applied Biosystems). The instrument was equipped with a Nd:YAG laser (PowerChip, JDS Uniphase) operating at 200 Hz and controlled by Applied Biosystems 4000 Series Explorer version 3.6 software. Each spectrum was accumulated with 500 shots in positive ion mode. MS/MS were acquired in PSD mode with mass isolation window +/-3 Da.
Supplementary Material
Acknowledgments
This work was supported by the grant GM073913 from the National Institutes of Health to V.V.V. and by the Albert Einstein Cancer Center. We thank Dr. H.Xiao for measuring mass spectra and Dr. U.Ramagopal for assistance with structure determination. We also thank the staff of the X29 beamline at the National Synchrotron Light Source, Brookhaven National Laboratory.
Footnotes
SUPPLEMENTAL DATA: Supplemental Data include description of contacts of the TagRFP and mTagBFP chromophores with the respective β-barrel interiors and two Figures and can be found with this article online.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andresen M, Wahl MC, Stiel AC, Gräter F, Schäfer LV, Trowitzsch S, Weber G, Eggeling C, Grubmüller H, Hell SW, Jakobs S. Structure and mechanism of the reversible photoswitch of a fluorescent protein. Proc Natl Acad Sci USA. 2005;102:13070–13074. doi: 10.1073/pnas.0502772102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barondeau DP, Kassmann CJ, Tainer JA, Getzoff ED. The case of the missing ring: radical cleavage of a carbon-carbon bond and implications for GFP chromophore biosynthesis. J Am Chem Soc. 2007;129:3118–3126. doi: 10.1021/ja063983u. [DOI] [PubMed] [Google Scholar]
- Bogdanov AM, Mishin AS, Yampolsky IV, Belousov VV, Chudakov DM, Subach FV, Verkhusha VV, Lukyanov S, Lukyanov KA. Green fluorescent proteins are light-induced electron donors. Nat Chem Biol. 2009;5:459–461. doi: 10.1038/nchembio.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brejc K, Sixma TK, Kitts PA, Kain SR, Tsien RY, Ormö M, Remington SJ. Structural basis for dual excitation and photoisomerization of the Aequorea victoria green fluorescent protein. Proc Natl Acad Sci U S A. 1997;94:2306–2311. doi: 10.1073/pnas.94.6.2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulina ME, Verkhusha VV, Staroverov DB, Chudakov DM, Lukyanov KA. Hetero-oligomeric tagging diminishes non-specific aggregation of target proteins fused with Anthozoa fluorescent proteins. Biochem J. 2003;371:109–114. doi: 10.1042/BJ20021796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrington JC, Dougherty WG. A viral cleavage site cassette: identification of amino acid sequences required for tobacco etch virus polyprotein processing. Proc Natl Acad Sci USA. 1988;85:3391–3395. doi: 10.1073/pnas.85.10.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Gross LA, Baird GS, Hoffman RC, Baldridge KK, Tsien RY. The structure of the chromophore within DsRed, a red fluorescent protein from coral. Proc Natl Acad Sci USA. 2000;97:11990–11995. doi: 10.1073/pnas.97.22.11990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim R, Prasher DC, Tsien RY. Wavelength mutations and posttranslational autooxidation of green fluorescent protein. Proc Natl Acad Sci USA. 1994;91:12501–12504. doi: 10.1073/pnas.91.26.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim R, Tsien RY. Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence energy transfer. Curr Biol. 1996;6:178–182. doi: 10.1016/s0960-9822(02)00450-5. [DOI] [PubMed] [Google Scholar]
- Hooft RW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- Kikuchi A, Fukumura E, Karasawa S, Mizuno H, Miyawaki A, Shiro Y. Structural characterization of a thiazoline-containing chromophore in an orange fluorescent protein, monomeric Kusabira Orange. Biochemistry. 2008;47:11573–11580. doi: 10.1021/bi800727v. [DOI] [PubMed] [Google Scholar]
- Kogure T, Karasawa S, Araki T, Saito K, Kinjo M, Miyawaki A. A fluorescent variant of a protein from the stony coral Montipora facilitates dual-color single-laser fluorescence cross-correlation spectroscopy. Nat Biotechnol. 2006;24:577–581. doi: 10.1038/nbt1207. [DOI] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline protein state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Laskowski R, MacArthur M, Moss D, Thornton J. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- Martynov VI, Maksimov BI, Martynova NY, Pakhomov AA, Gurskaya NG, Lukyanov SA. A purple-blue chromoprotein from Goniopora tenuidens belongs to the DsRed subfamily of GFP-like proteins. J Biol Chem. 2003;278:46288–46292. doi: 10.1074/jbc.M306810200. [DOI] [PubMed] [Google Scholar]
- Merzlyak EM, Goedhart J, Shcherbo D, Bulina ME, Shcheglov AS, Fradkov AF, Gaintzeva A, Lukyanov KA, Lukyanov S, Gadella TW, Chudakov DM. Bright monomeric red fluorescent protein with an extended fluorescence lifetime. Nat Methods. 2007;4:555–557. doi: 10.1038/nmeth1062. [DOI] [PubMed] [Google Scholar]
- Nienhaus K, Nar H, Heilker R, Wiedenmann J, Nienhaus GU. Trans-cis isomerization is responsible for the red-shifted fluorescence in variants of the red fluorescent protein eqFP611. J Am Chem Soc. 2008;130:12578–12579. doi: 10.1021/ja8046443. [DOI] [PubMed] [Google Scholar]
- Niwa H, Inouye S, Hirano T, Matsuno T, Kojima S, Kubota M, Ohashi M, Tsuji FI. Chemical nature of the light emitter of the Aequorea green fluorescent protein. Proc Natl Acad Sci USA. 1996;93:13617–13622. doi: 10.1073/pnas.93.24.13617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormö M, Cubitt AB, Kallio K, Gross LA, Tsien RY, Remington SJ. Crystal structure of the Aequorea victoria green fluorescent protein. Science. 1996;273:1392–1395. doi: 10.1126/science.273.5280.1392. [DOI] [PubMed] [Google Scholar]
- Otwinowski W, Minor F. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Petersen J, Wilmann PG, Beddoe T, Oakley AJ, Devenish RJ, Prescott M, Rossjohn J. The 2.0 Å crystal structure of eqFP611, a far red fluorescent protein from the sea anemone Entacmaea quadricolor. J Biol Chem. 2003;278:44626–44631. doi: 10.1074/jbc.M307896200. [DOI] [PubMed] [Google Scholar]
- Piatkevich KD, Verkhusha VV. Advances in engineering of fluorescent proteins and photoactivatable proteins with red emission. Curr Opin Chem Biol. 2010;14:23–29. doi: 10.1016/j.cbpa.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletnev S, Shcherbo D, Chudakov D, Pletneva N, Merzlyak E, Wlodawer A, Dauter Z, Pletnev V. A crystallographic study of bright far-red fluorescent protein mKate reveals pH-induced cis-trans isomerization of the chromophore. J Biol Chem. 2008;283:28980–28987. doi: 10.1074/jbc.M800599200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletnev S, Morozova KS, Verkhusha VV, Dauter Z. Rotational order-disorder structure of fluorescent protein FP480. Acta Crystallogr D Biol Crystallogr. 2009;65:906–912. doi: 10.1107/S0907444909020927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletnev S, Subach FV, Dauter Z, Wlodawer A, Verkhusha VV. Understanding blue-to-red conversion in monomeric fluorescent timers and hydrolytic degradation of their chromophores. J Am Chem Soc. 2010;132:2243–2253. doi: 10.1021/ja908418r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott M, Ling M, Beddoe T, Oakley AJ, Dove S, Hoegh-Guldberg O, Devenish RJ, Rossjohn J. The 2.2 Å crystal structure of a pocilloporin pigment reveals a nonplanar chromophore conformation. Structure. 2003;11:275–284. doi: 10.1016/s0969-2126(03)00028-5. [DOI] [PubMed] [Google Scholar]
- Quillin ML, Anstrom DM, Shu X, O'Leary S, Kallio K, Chudakov DM, Remington SJ. Kindling fluorescent protein from Anemonia sulcata: dark-state structure at 1.38 Å resolution. Biochemistry. 2005;44:5774–5787. doi: 10.1021/bi047644u. [DOI] [PubMed] [Google Scholar]
- Shu X, Shaner NC, Yarbrough CA, Tsien RY, Remington SJ. Novel chromophores and buried charges control color in mFruits. Biochemistry. 2006;45:9639–9647. doi: 10.1021/bi060773l. [DOI] [PubMed] [Google Scholar]
- Sniegowski JA, Lappe JW, Patel HN, Huffman HA, Wachter RM. Base catalysis of chromophore formation in Arg96 and Glu222 variants of green fluorescent protein. J Biol Chem. 2005;280:26248–26255. doi: 10.1074/jbc.M412327200. [DOI] [PubMed] [Google Scholar]
- Stepanenko OV, Verkhusha VV, Kuznetsova IM, Uversky VN, Turoverov KK. Fluorescent proteins as biomarkers and biosensors: throwing color lights on molecular and cellular processes. Curr Protein Pept Sci. 2008;9:338–369. doi: 10.2174/138920308785132668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiel AC, Trowitzsch S, Weber G, Andresen M, Eggeling C, Hell SW, Jakobs S, Wahl MC. 1.8 Å bright-state structure of the reversibly switchable fluorescent protein Dronpa guides the generation of fast switching variants. Biochem J. 2007;402:35–42. doi: 10.1042/BJ20061401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- Subach FV, Subach OM, Gundorov IS, Morozova KS, Piatkevich KD, Cuervo AM, Verkhusha VV. Monomeric fluorescent timers that change color from blue to red report on cellular trafficking. Nat Chem Biol. 2009;5:118–126. doi: 10.1038/nchembio.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subach OM, Gundorov IS, Yoshimura M, Subach FV, Zhang J, Grünwald D, Souslova EA, Chudakov DM, Verkhusha VV. Conversion of red fluorescent protein into a bright blue probe. Chem Biol. 2008;15:1116–1124. doi: 10.1016/j.chembiol.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhusha VV, Chudakov DM, Gurskaya NG, Lukyanov S, Lukyanov KA. Common pathway for the red chromophore formation in fluorescent proteins and chromoproteins. Chem Biol. 2004;11:845–854. doi: 10.1016/j.chembiol.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Wilmann PG, Petersen J, Pettikiriarachchi A, Buckle AM, Smith SC, Olsen S, Perugini MA, Devenish RJ, Prescott M, Rossjohn J. The 2.1 Å crystal structure of the far-red fluorescent protein HcRed: inherent conformational flexibility of the chromophore. J Mol Biol. 2005;349:223–237. doi: 10.1016/j.jmb.2005.03.020. [DOI] [PubMed] [Google Scholar]
- Wilmann PG, Turcic K, Battad JM, Wilce MC, Devenish RJ, Prescott M, Rossjohn J. The 1.7 Å crystal structure of Dronpa: a photoswitchable green fluorescent protein. J Mol Biol. 2006;364:213–224. doi: 10.1016/j.jmb.2006.08.089. [DOI] [PubMed] [Google Scholar]
- Yarbrough D, Wachter RM, Kallio K, Matz MV, Remington SJ. Refined crystal structure of DsRed, a red fluorescent protein from coral, at 2.0 Å resolution. Proc Natl Acad Sci USA. 2001;98:462–467. doi: 10.1073/pnas.98.2.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Patel HN, Lappe JW, Wachter RM. Reaction progress of chromophore biogenesis in green fluorescent protein. J Am Chem Soc. 2006;128:4766–4772. doi: 10.1021/ja0580439. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.